

Abstract
As the differential diagnosis of dementias based on established clinical criteria is often difficult, biomarkers for applicable diagnostic testing are currently under intensive investigation. Amyloid plaques deposited in the brain of patients suffering from Alzheimer's disease, dementia with Lewy bodies (DLB) and Parkinson's disease dementia (PDD) mainly consist of carboxy-terminally elongated forms of amyloid-beta (Aβ) peptides, such as Aβ1–42. Absolute Aβ1–42 levels in CSF have shown diagnostic value for the diagnosis of Alzheimer's disease, but the discrimination among Alzheimer's disease, DLB and PDD was poor. A recently established quantitative urea-based Aβ-sodium-dodecylsulphate–polyacrylamide-gel-electrophoresis with Western immunoblot (Aβ-SDS–PAGE/immunoblot) revealed a highly conserved Aβ peptide pattern of the carboxy-terminally truncated Aβ peptides 1–37, 1–38, 1–39 in addition to 1–40 and 1–42 in human CSF. We used the Aβ-SDS–PAGE/immunoblot to investigate the CSF of 23 patients with Alzheimer's disease, 21 with DLB, 21 with PDD and 23 non-demented disease controls (NDC) for disease-specific alterations of the Aβ peptide patterns in its absolute and relative quantities. The diagnostic groups were matched for age and severity of dementia. The present study is the first attempt to evaluate the meaning of Aβ peptide patterns in CSF for differential diagnosis of the three neurodegenerative diseases—Alzheimer's disease, DLB and PDD. The Aβ peptide patterns displayed disease-specific variations and the ratio of the differentially altered Aβ1–42 to the Aβ1–37 levels subsequently discriminated all diagnostic groups from each other at a highly significant level, except DLB from PDD. Additionally, a novel peptide with Aβ-like immunoreactivity was observed constantly in the CSF of all 88 investigated patients. The pronounced percentage increase of this peptide in DLB allowed a highly significant discrimination from PDD. Using a cut-off point of 0.954%, this marker yielded a diagnostic sensitivity and specificity of 81 and 71%, respectively. From several lines of indication, we consider this peptide to represent an oxidized α-helical form of Aβ1–40 (Aβ1–40*). The increased abundance of Aβ1–40* probably reflects a disease-specific alteration of the Aβ1–40 metabolism in DLB. We conclude that Aβ peptide patterns reflect disease-specific pathophysiological pathways of different dementia syndromes as distinct neurochemical phenotypes. Although Aβ peptide patterns failed to fulfil the requirements for a sole biomarker, their combined evaluation with other biomarkers is promising in neurochemical dementia diagnosis. It is noteworthy that DLB and PDD exhibit distinct clinical temporal courses, despite their similar neuropathological appearance. Their distinct molecular phenotypes support the view of different pathophysiological pathways for each of these neurodegenerative diseases.
Keywords: Alzheimer's dementia, Lewy-body dementia, Parkinson's disease dementia, cerebrospinal fluid, amyloid-β peptides
Keywords: Aβ peptides = amyloid-beta peptides; Aβ-SDS–PAGE/immunoblot = amyloid-beta-sodium-dodecyl-sulphate–polyacrylamide-gel-electrophoresis with Western immunoblot; APP = beta-amyloid precursor protein; bicine = N,N′-bis-[2-hydroxyethyl]glycine; C% = percentage of N,N′methylenebisacrylamide (bis) of the total of bis plus acrylamide; DLB = dementia with Lewy bodies; IP = immunoprecipitation; IPG = immobilized pH gradients; MALDI–TOF = matrix-assisted laser desorption ionization mass analysis–time-of-flight modus; MMSE = Mini-Mental-Status Examination; NINCDS–ADRDA = National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association; ND = non-demented; PDD: Parkinson's disease dementia; SKT = Syndrom-Kurz-Test; SPE = solid-phase extraction; T% = percentage of acrylamide of the total of bis plus acrylamide
Introduction
Amyloid-beta (Aβ) peptides form the major component of amyloid plaques deposited in the brain of patients suffering from neurodegenerative diseases such as Alzheimer's disease (Glenner and Wong, 1984), dementia with Lewy bodies (DLB) (Jendroska et al., 1997) and Parkinson's disease dementia (PDD) (Jendroska et al., 1996).
Aβ peptides derive from a transmembrane amyloid precursor-protein (APP), when cleaved by two enzymes, β- and γ-secretase (Haas and Selkoe, 1993). Distinct γ-secretase activities are hypothesized to be responsible for generation of either carboxy-terminally truncated (Ct-truncated) or elongated (Ct-elongated) Aβ peptides as referenced to Aβ1-40 (Citron et al., 1996). Cleavage of the APP by the δ-secretase results in amino-terminally elongated (At-elongated) Aβ peptide species (Simons et al., 1996).
The differential diagnosis of dementias based on established clinical criteria is often difficult during lifetime and the selective reduction of Aβ1–42 in the CSF of Alzheimer's disease patients has widely been investigated as a diagnostic biomarker to support the diagnostic accuracy during lifetime. An expert review recently considered it adequate for applicable Alzheimer's disease diagnostic testing in addition to clinical criteria (Andreasen et al., 2003). Decreased Aβ1–42 levels have also been reported for DLB (Andreasen et al., 2001; Mollenhauer et al., 2005) and PDD (Mollenhauer et al., 2005) patients. Thus, the specificity of this finding and consequently its differential diagnostic value in distinguishing between different subtypes of dementias was low (Andreasen et al., 2001, 2003; Mollenhauer et al., 2005).
A quantitative urea-based Aβ-sodium-dodecylsulphate-polyacrylamide-gel-electrophoresis with Western immunoblot (Aβ-SDS–PAGE/immunoblot) recently revealed the regular abundance of the Ct-truncated Aβ peptides 1–37, 1–38, 1–39 in addition to 1–40 and 1–42 in CSF. This Aβ peptide pattern displayed disease-specific variations in its absolute and relative quantities in the CSF of patients with Alzheimer's disease, Creutzfeldt–Jakob disease (CJD), chronic inflammatory diseases and other neuropsychiatric diseases (Wiltfang et al., 2002, 2003).
To evaluate the meaning of this finding for the differential diagnosis of dementias, we investigated 88 age-matched patients suffering from Alzheimer's disease, DLB, PDD and various neuropsychiatric diseases for disease-specific Aβ peptide patterns.
We were able to demonstrate disease-specific variations of the Aβ peptide patterns in CSF for each of the diagnostic groups, which allow a highly significant discrimination and may reflect pathophysiological pathways of dementia subtypes.
Patients and methods
Aβ peptide patterns were analysed by Aβ-SDS–PAGE/immunoblot (Wiltfang et al., 2002) in the CSF of patients with Alzheimer's disease, DLB, PDD and non-demented disease controls (NDC). A total of 88 patients were divided into four diagnostic groups according to their clinical diagnosis and tested for significant differences in absolute and relative Aβ peptide values. The patients were selected between 1999 and 2004 onward and from the dementia outpatient clinic. The mean age did not significantly differ between the diagnostic groups. Mini-Mental-Status-Examination (MMSE) (Folstein et al., 1975) was performed on patients suffering from cognitive impairments at the time of lumbar puncture. The MMSE score did not significantly differ between the diagnostic groups of dementias (Alzheimer's disease, DLB, PDD) and was significantly different for the NDC group (P = 2.0 × 10−6).
The study was conducted under the guidelines of the Declaration of Helsinki (World Medical Organisation, 1996) and approved by the ethics committee of the University of Goettingen. Investigations were carried out with the informed consent of all patients or, for patients with severe dementia, their next of kin.
Patients with Alzheimer's disease
All 23 patients (8 men and 15 women) of this group fulfilled the Diagnostic and Statistical Manuals (DSM) IV criteria for Alzheimer's disease and the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association (NINCDS–ADRDA) criteria for clinical diagnosis of probable Alzheimer's disease (McKhann et al., 1984). Age of this group was 69.5 ± 11.5 years (mean ± standard deviation). MMSE was available for 22 patients. One patient underwent the Syndrom-Kurz-Test (SKT) instead of the MMSE. The SKT score was 20, indicating a moderate form of dementia. The mean MMSE score was 18.4 ± 5.1 (mean ± standard deviation) in this group.
Patients with dementia with Lewy bodies
All 21 patients (15 men and 6 women) of this group fulfilled the DSM IV criteria for dementia and the McKeith criteria for clinical diagnosis of probable DLB (McKeith et al., 1996). Age of this group was 71.5 ± 6.6 years (mean ± standard deviation). MMSE was available for 20 patients. One patient rejected the cognitive testing. He displayed moderate cognitive impairments at the time of lumbar puncture, and neuropathological postmortem analysis confirmed this patient to be a case of DLB. The mean MMSE score was 18.1 ± 5.3 (mean ± standard deviation) in this group.
Parkinson's disease dementia patients
All 21 patients (16 men and 5 women) of this group fulfilled the DSM IV criteria for dementia and the UK Parkinson's Disease Society Brain Bank clinical diagnostic criteria for idiopathic Parkinson's disease (Gibb and Lees, 1988). All patients in this group presented parkinsonism at least one year before onset of dementia according to the criteria of McKeith et al. (1996). Age of this group was 72.4 ± 6.8 years (mean ± standard deviation). The MMSE score was 17.7 ± 7.3 (mean ± standard deviation) in this group.
Non-demented disease controls
This group consisted of 23 non-demented patients (4 men and 19 women), who underwent lumbar puncture for other differential diagnostic reasons; none of these patients displayed clinical features of neurodegenerative disease. Age of this group was 68.5 ± 9.0 years (mean ± standard deviation). A subgroup of 11 patients suffering from depression with cognitive complaints was assessed by MMSE. The score was 27.7 ± 3.1 (mean ± standard deviation). One patient suffering from major depression rejected cognitive testing. The cognitive impairments of all depressive patients improved after antidepressant medication. The group further included patients suffering from temporal lobe epilepsy (n = 1), normal pressure hydrocephalus (n = 2), cerebral transient ischaemic attacks (n = 1), brain metastasis (n = 1), peripheral herpes zoster infection (n = 1), breast cancer (n = 1), peripheral facial nerve palsy (n = 1), systemic vasculitis (n = 1), spinal cord compression (n = 1) and intervertebral disk herniation (n = 1).
Pre-analytical treatment of CSF for Aβ-SDS–PAGE immunoblot
CSF was drawn from patients by lumbar puncture, sampled in polypropylene vials, and centrifuged (1000g, 10 min, 4°C), and aliquots of 200 μl were stored at −80°C within 24 h for subsequent one- and two-dimensional Aβ-SDS–PAGE/immunoblot analysis.
Pre-analytical concentration of CSF by immunoprecipitation (IP) was performed as recorded previously (Wiltfang et al., 2002). The amino-terminal-selective mouse monoclonal antibodies 6E10 (Senetec Drug Delivery Technologies Inc, USA) and 1E8 were used in comparison with the carboxy-terminal-selective 13E9 and 6D5 directed against the carboxy-terminus of Aβ1–40 and Aβ1–42, respectively. The latter three antibodies were provided by Schering AG, Berlin, Germany.
One-dimensional Aβ-SDS–PAGE/immunoblot
For separation of Aβ peptides and subsequent detection, 10 μl of unconcentrated CSF was boiled in a sample buffer for SDS–PAGE, and Aβ-SDS–PAGE/immunoblot was conducted as published elsewhere (Wiltfang et al., 1997, 2002).
Samples were run as triplicates and each gel carried a four-step dilution series of the synthetic Aβ peptides Aβ1–37, Aβ1–38, Aβ1–39, Aβ1–40 and Aβ1–42. Synthetic peptides Aβ1–38, Aβ1–40 and Aβ1–42 were obtained from Bachem (Bubendorf, Switzerland); Aβ1–37 and Aβ1–39 were synthesized automatically according to Janek et al. (Janek et al., 2001). Standard preparations of synthetic Aβ peptide mixture were created as described previously (Bibl et al., 2004) and bands were quantified from individual blots of each patient relative to this dilution series using a charge coupled device camera (CCD-camera) and the Quantity-ONE software (BIORAD).
The inter- and intra-assay coefficients of variation and sensitivity of detection of the Aβ-SDS–PAGE/immunoblot have been published elsewhere (Wiltfang et al., 2002; Bibl et al., 2004).
Two-dimensional Aβ-SDS–PAGE/immunoblot-Aβ-IPG-2D-PAGE
For isoelectric focussing (IEF) on immobilized pH gradients (IPG) followed by Aβ-SDS–PAGE/immunoblot (Aβ-IPG-2D-PAGE), 25 μl of immunoprecipitated CSF were equilibrated in IPG sample buffer, and Aβ-IPG-2D-PAGE was performed as published previously (Wiltfang et al., 2002).
Two-dimensional non-urea/urea-SDS–PAGE
Bicine/bistris/tris/sulphate SDS–PAGE (without urea) on 12% T (acrylamide)/5% C (N,N′methylenebisacrylamide) gels (Wiltfang et al., 1991) was used for the first dimension to achieve a separation that solely depends on the effective molecular radii of the peptides. In this separation gel, monomeric Aβ peptides migrate in one single band close to the moving boundary, whereas oligomeric forms can be separated as a result of their higher molecular radii. Five microlitres of immunoprecipitated CSF were applied per lane. After electrophoresis at a constant current of 12 mA/gel, the whole lane was cut out and placed horizontally on a 0.75 mm thick separation gel of the same composition but containing 8 M urea. The gel was run at 18 mA/gel constant current for separating the different Aβ peptide species on the basis of urea-induced peptide-specific shifts in binding of SDS.
Solid-phase extraction of Aβ 1–40
HLB Extraction Cartridges (Waters, Nr. 186000115) were activated with 50% v/v methanol and subsequently washed with 8 ml 5% methanol prior to sample loading. Ten microlitres of Aβ peptide (0.1 mg/ml) in SDS–PAGE sample buffer were mixed with 490 μl of phosphate-buffered saline (PBS) and loaded into one cartridge. The Aβ peptide was eluted with 83% acetonitrile/0.08% trifluoroacetic acid (TFA). Four fractions of 1.5 ml each were collected in polypropylene vials. From each fraction, 50 μl were vacuum-dried and re-dissolved in binding buffer for further IP according to the protocol of the manufacturer (Bruker Immunocapturing Kit #233794) and subsequent matrix-assisted laser desorption ionization mass analysis–time-of-flight modus (MALDI–TOF) analysis. Samples for Aβ-SDS–PAGE were dissolved in SDS–PAGE buffer according to Wiltfang et al. (1997, 2002).
Localization and excision of bands corresponding to Aβ 1–40 and Aβ 1–40* from unstained SDS–PAGE
To localize Aβ 1–40 and Aβ 1–40* in unstained gels, contact blots were produced immediately after electrophoresis by placing pre-wetted PVDF blotting membranes on each gel for 5 min at room temperature. After a brief rinse in double distilled water (H2Odd), peptides and proteins were visualized on the PVDF membrane by colloidal silver-staining [2% (w/v) trisodium citrate dihydrate, 0.8% ferrum(II) sulphate heptahydrate, 0.2% nitrated silver] according to van Oostveen et al. (1997). The stained membranes served as templates for cutting Aβ 1–40 and Aβ 1–40* out of the gel.
Preparation of gel pieces for MALDI–TOF analysis
Pre-analytical concentration of CSF by IP with the amino-terminal-selective mouse monoclonal antibody 1E8 non-covalently coupled to M280-Dynal-Beads was performed as described previously (Wiltfang et al., 2002). In brief, excised gel pieces were incubated overnight at 4°C in 40 μl of 5-fold concentrated RIPA detergent buffer (RIPA5×: 2.5% Nonidet P-40, 1.25% sodium desoxycholate, 0.25% SDS, 750 mM NaCl, 250 mM HEPES, one tablet of Protease Inhibitor Cocktail Complete Mini per 2 ml of RIPA5×, pH adjusted to 7.4 with NaOH) and 160 μl H2Odd in addition to 20 μl of 1E8-M280-Dynal-Beads. The magnetic beads were then captured on a magnetic stand and washed twice with PBS/0.1% bovine serum albumin (BSA) and once with 10 mM Tris-HCL (pH 7.4). Finally, the bound proteins were eluted in 10 μl of elution buffer (Bruker Immunocapturing Kit #233794) under constant agitation at 37°C for 1 h and for an additional 2 min in an ultrasonic bath.
MALDI–TOF mass spectrometry
A thin layer of saturated α-cyano-hydroxycinnamic acid in acetone containing 3% H2O and 10 mM NH4H2PO4 was prepared on a ground steel target. An aliquot of 0.5 μl sample was directly spotted onto the matrix and dried. Subsequently, the preparation was washed twice by adding 5 μl 10 mM NH4H2PO4/0.1%TFA and removing it after 10 s, dried and analyzed using an autoflex II TOF/TOF mass spectrometer (Bruker Daltonics, Germany) in the positive reflectron mode. Three hundred individual shots were aggregated for each spectrum. Spectra were calibrated using ClinProt Standard (Bruker Daltonics, Germany).
Statistical analysis
Aβ peptide levels were scanned and calculated as absolute values (ng/ml) as well as percentages of the total Aβ peptide concentration of all Aβ peptide species that were detected. The data on peptide levels were obtained from individual blots of each patient. For comparison of the patient groups, mean concentrations and standard deviation (SD) were calculated.
The Mann–Whitney U-test was applied to evaluate the significance of differences between the groups.
The two-sided level of significance was defined as P < 0.05. A P-value < 0.01 was considered as highly significant.
Receiver operating characteristic (ROC) curve analysis was used to determine cut-off points. The cut-off level for dichotomizing values was selected as the situation optimizing sensitivities, specificities and the Youden index.
Computations were performed using the statistical software package SPSS, version 10.0.
Results
An additional peptide with Aβ-like immunoreactivity: its specification and quantification
A highly conserved pattern of three Ct-truncated Aβ peptides in addition to Aβ1–40, 1–42 could be shown by the Aβ-SDS–PAGE/immunoblot in the CSF of all investigated patients. Additionally, a previously undescribed peptide with Aβ-like immunoreactivity was found to regularly migrate cathodically of Aβ1–37 in all CSF samples analysed (see Supplementary Fig. 1). All peptides migrate as a single band of approximately 4 kDa in a conventional SDS–PAGE, where urea is absent in otherwise unchanged separation gels (Fig. 1). In contrast, synthetic α-synuclein was found to migrate at a molecular mass of approximately 16 kDa in a non-urea SDS–PAGE (data not shown).
Fig. 1.
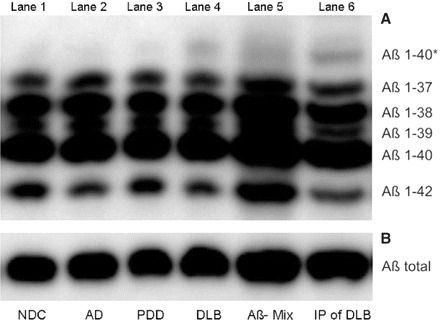
Urea-based Aβ-SDS–PAGE/immunoblot (A) and conventional SDS–PAGE (B) of CSF (lane 1–4,6) and synthetic Aβ-peptides 1–37, 1–38, 1–39, 1–40, 1–42 (lane 5). Ten microlitres of an unconcentrated CSF pool of seven NDC (lane 1), Alzheimer's disease (lane 2), PDD (lane 3) and DLB (lane 4) patients were applied. 1E8 immunoprecipitated CSF pool of seven DLB patients was applied to lane 6. Quantifications have been obtained from individual blots of each patient.
The novel peptide displayed no electrophoretic co-migration with the synthetic Aβ peptides Aβ1–43, Aβ1–36, Aβ1–35, Aβ1–34, Aβ1–33 and Aβ1–28 in the Aβ-SDS–PAGE/immunoblot (data not shown).
The peptide was detected by the anti-Aβ peptide-specific monoclonal antibody 1E8 during immunoblot and was not visualized on account of unspecific reactions of the secondary antibody or biotin-mediated affinity to the streptavidin complex (data not shown). A cross-reactivity of the 1E8 to synthetic α-synuclein could be ruled out also in the immunoblot (see Supplementary Fig. 2). During IP, the novel peptide was enriched by the amino-terminal-specific antibodies 1E8 (Fig. 1) and 6E10 against Aβ peptides and by the 13E9, directed against the carboxy-terminus of Aβ1–40, respectively. The carboxy-terminal-specific antibody against Aβ1–42 6D5 did not recognize the peptide (data not shown).
A two-dimensional combination of a conventional SDS–PAGE followed by urea-based Aβ-SDS–PAGE/immunoblot (non-urea/urea-SDS–PAGE) revealed that the novel peptide migrated in one band with the other five Aβ peptides in the conventional SDS–PAGE and was subsequently separated cathodically of Aβ1–37 in the urea-based Aβ-SDS–PAGE/immunoblot (Fig. 2A and B).
Fig. 2.
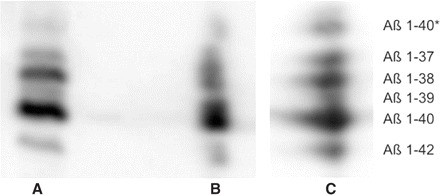
One- and two-dimensional Aβ-SDS–PAGE/immunoblot of 1E8 immunoprecipitated CSF pool of seven DLB patients: one-dimensional separation in the urea-based Aβ-SDS–PAGE/immunoblot (A) in comparison with urea-based Aβ-SDS–PAGE/immunoblot after conventional SDS–PAGE [non-urea/urea SDS–PAGE (B)] and electrofocussing by IPG [Aβ-IPG-2D-PAGE (C)], respectively.
The Aβ-IPG-2D-PAGE revealed an identical isoelectric point for the Aβ peptide quintet and the novel peptide (Fig. 2C).
A band co-migrating with Aβ1–40* was detected after the application of synthetic Aβ1–40 to solid-phase extraction (SPE) and subsequent acetonitrile/TFA elution in the same fractions as Aβ1–40 (synthetic Aβ1–40*) as shown by Aβ-SDS–PAGE/immunoblot analysis (see Supplementary Fig. 3). Moreover, synthetic Aβ1–40* was present after picking of Aβ1–40 from a coomassie stained gel, but was not found when Aβ1–40 was picked from unstained gels and analysed subsequently by one-dimensional Aβ-SDS–PAGE/immunoblot (data not shown). The synthetic Aβ1–40* was purified through separation in Aβ-SDS–PAGE/immunoblot and subsequent picking (purified synthetic Aβ1–40*). This purified synthetic Aβ1–40*, an SPE fraction of synthetic Aβ1–40, synthetic standard preparations of Aβ peptides (mixture of five Aβ peptides) and an immunoprecipitated CSF pool of five representative DLB patients were comparatively applied to the urea-based Aβ-SDS–PAGE/immunoblot, where the band corresponding to Aβ1–40* co-migrated among all four samples (see Supplementary Fig. 3). In the absence of urea and in otherwise unchanged separation gels, one single band in the molecular weight range of monomeric Aβ (∼4 kDa) was detected in all samples. The SPE fraction, synthetic standard mixture of five Aβ peptides and an immunoprecipitated CSF pool of five representative DLB patients were applied to IEF by IPG. After IEF, the region corresponding to the isoelectric point of N-terminally unchanged Aβ peptides (e.g. 5.37) was cut out of each strip and analysed in the Aβ-SDS–PAGE/immunoblot on parallel lanes. This approach revealed one- and two-dimensional co-migration of the peptide generated from Aβ1–40 during SPE, with Aβ1–40* occurring in CSF and the respective band detected in standard preparations (see Supplementary Fig. 3). Moreover, the synthetic Aβ1–40* was captured by the carboxy-terminally-specific antibody against Aβ1–40 (13E9) during IP and detected by the amino-terminally-specific antibody against Aβ1–40 (1E8) in the immunoblot (see Supplementary Fig. 3). Thus, the synthetic Aβ1–40* exhibited similar electrophoretic and immunological features to Aβ1–40* that occurs in vivo. The direct mass analysis of the SPE fractions using MALDI–TOF analysis revealed two mass peaks of 4329.9 and 4345.2 Da, respectively. The expected mass of Aβ1–40 and oxidized Aβ1–40 (Aβ1–40ox) would be 4329.6 and 4345.6 Da, respectively. MALDI–TOF analysis of each respective band, picked from the Aβ-SDS–PAGE and enriched by IP revealed that the Aβ1–40* band comprises exclusively a mass peak corresponding to the expected mass of Aβ1–40ox (see Supplementary Fig. 4). Otherwise, the Aβ1–40 band exhibits predominantly a mass peak corresponding to the expected mass of unoxidized Aβ1–40 (see Supplementary Fig. 4). The expected mass for Aβ1–40ox could be inconsistently detected herein and, if present, its intensity was minor as compared with the one expected for Aβ1–40.
We conclude from the data that the novel peptide is a monomeric Aβ peptide with electrophoretic and immunological features of Aβ1–40, but it migrates at a different position in the Aβ-SDS–PAGE/immunoblot. A similar band originates from Aβ1-40 in vitro and exhibits a mass corresponding to Aβ1–40ox. We consequently named the novel peptide Aβ1–40*
Another unknown peptide migrated cathodically of Aβ1–40* and exhibited similar electrophoretic properties to Aβ1–40*. We consequently named this peptide Aβ1–40**. Aβ1–40** was not consistently detectable in all investigated samples and, if present, its concentration was close to the level of detection. Therefore, Aβ1–40** was not systematically quantified in CSF.
The absolute values of Aβ1–40* given below were measured arbitrarily relative to the Aβ1–37 standard peptide. The abundance of Aβ1–40* in absolute and relative forms was the lowest of all Aβ peptides quantified.
Aβ-SDS–PAGE/immunoblot: Aβ peptide patterns and their use for neurochemically supported differential diagnosis of dementias
Each dementia group, DLB, Alzheimer's disease and PDD, displayed decreased absolute levels of Aβ1–42 relative to the NDC group, whereas Aβ1–37 levels increased slightly in Alzheimer's disease as compared with the other dementia groups (Fig. 3). The decrease of Aβ1–42 was most pronounced for the Alzheimer's disease group and not statistically significant for the PDD group (P > 0.05).
Fig. 3.
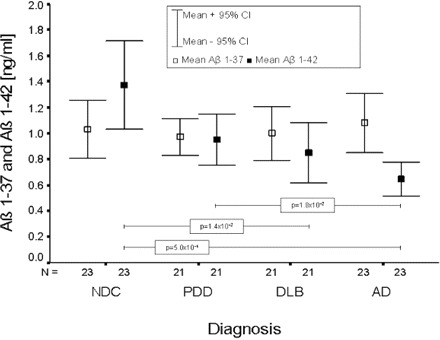
Mean and 95% confidence interval (CI) of absolute Aβ1–37 and Aβ1–42 levels for each diagnostic group. Only significant differences are indicated within the figure.
By introducing the ratio of Aβ1–42 to Aβ1–37, the NDC group could be differentiated at a highly significant level from Alzheimer's disease (P = 5.2 × 10−7), DLB (P = 2.0 × 10−4) and PDD (P = 1.2 × 10−3). The Alzheimer's disease group was highly significantly differentiated from the DLB group (P = 3.2 × 10−3) and the PDD group (P = 1.1 × 10−4), respectively (Fig. 4). Using a cut-off value of 0.848, Alzheimer's disease could be discriminated from NDC with a sensitivity and specificity of 87%.
Fig. 4.
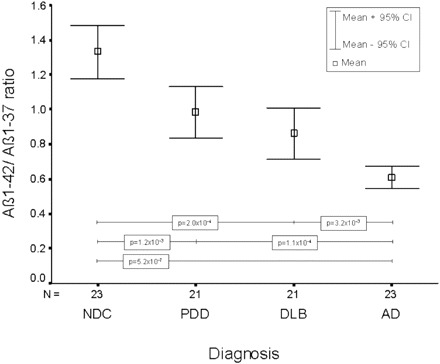
Mean and 95% confidence interval (CI) of the Aβ1–42 : Aβ1–37 ratio for each diagnostic group. Only significant differences are indicated within the figure.
The DLB group could be differentiated from the PDD group by a percentage increase of Aβ1–40* relative to the sum of all Aβ peptides (Aβ1–40*%) in the DLB group at a highly significant level (P = 6.0 × 10−4) (Fig. 5). Aβ1–40* was also elevated in the Alzheimer's disease group, but failed the level of significance.
Fig. 5.
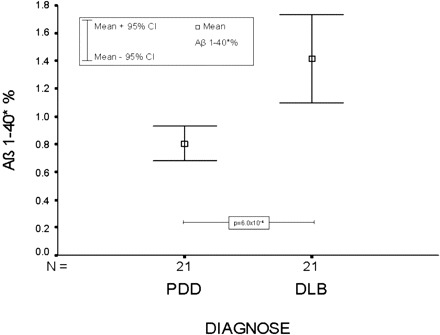
Mean and 95% confidence interval (CI) of the relative abundance of Aβ1–40* (Aβ 1–40*%) for PDD and DLB. The difference between PDD and DLB was highly significant (P = 6.0 × 10−4).
The absolute and relative abundances of Aβ peptides of each diagnostic group are summarized in Table 1. The cut-off points, sensitivities and specificities of the best discriminating Aβ peptide ratio (i.e. Aβ1–42/37 or Aβ1–40*%, respectively) for each differential diagnostic testing are summarized in Table 2.
Table 1.
Absolute and relative abundances of Aβ peptide patterns in the CSF of the diagnostic groups (mean ± standard deviation).
| Diagnosis | NDC (n = 23) |
AD (n = 23) |
DLB (n = 21) |
PDD (n = 21) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Mean |
±SD |
mean |
±SD |
mean |
±SD |
mean |
±SD |
||||
| Age | 68.5 | 9.0 | 69.5 | 11.5 | 71.5 | 6.6 | 72.4 | 6.8 | ||||
| MMSE | 27.7 | 3.1 | 18.4 | 5.1 | 18.1 | 5.3 | 17.7 | 7.3 | ||||
| Aβ1–40* | 0.08 | 0.03 | 0.10 | 0.06 | 0.16 | 0.15 | 0.08 | 0.04 | ||||
| Aβ1–37 | 1.03 | 0.53 | 1.08 | 0.53 | 1.00 | 0.45 | 0.97 | 0.32 | ||||
| Aβ1–38 | 1.66 | 0.75 | 1.81 | 0.83 | 1.58 | 0.74 | 1.45 | 0.51 | ||||
| Aβ1–39 | 0.83 | 0.40 | 0.87 | 0.40 | 0.82 | 0.40 | 0.85 | 0.31 | ||||
| Aβ1–40 | 6.38 | 2.53 | 6.27 | 2.50 | 5.93 | 2.22 | 5.97 | 1.59 | ||||
| Aβ1–42 | 1.37 | 0.80 | 0.65 | 0.30 | 0.85 | 0.51 | 0.95 | 0.44 | ||||
| total A↠| 11.34 | 4.83 | 10.78 | 4.30 | 10.34 | 4.25 | 10.27 | 2.92 | ||||
| Aβ1–40*‡ | 0.73 | 0.38 | 1.04 | 0.62 | 1.42 | 0.70 | 0.80 | 0.27 | ||||
| Aβ1–37‡ | 8.85 | 1.00 | 9.81 | 1.61 | 9.52 | 1.21 | 9.37 | 1.11 | ||||
| Aβ1–38‡ | 14.63 | 2.51 | 16.68 | 3.07 | 14.93 | 2.30 | 13.82 | 2.26 | ||||
| Aβ1–39‡ | 7.30 | 1.52 | 8.05 | 1.49 | 7.85 | 0.92 | 8.15 | 1.23 | ||||
| Aβ1–40‡ | 56.80 | 4.45 | 58.46 | 5.11 | 58.24 | 4.43 | 58.73 | 5.13 | ||||
| Aβ1–42‡ | 11.68 | 3.13 | 5.97 | 1.70 | 8.05 | 2.75 | 9.13 | 2.90 | ||||
| Aβ1–42/37§ | 1.33 | 0.36 | 0.61 | 0.15 | 0.86 | 0.33 | 0.98 | 0.32 | ||||
Total Aβ peptide concentration; ‡percentage abundance of Aβ peptides relative to the total Aβ peptide concentration; §ratio of absolute Aβ 1–42 to Aβ 1–37 levels. AD = Alzheimer's disease
Table 2.
Cut-off points, sensitivities and specificities of the best discriminating Aβ-peptide ratio (i.e. Aβ1–42/37 or Aβ1–40*%, respectively) for each differential diagnostic testing
| Differential diagnosis |
Parameter |
Cut off |
Sensitivity (%) |
Specificity (%) |
|---|---|---|---|---|
| AD versus NDC | Aβ1–42/37 | 0.998 | 100 | 83 |
| AD versus DLB | Aβ1–42/37 | 0.659 | 74 | 71 |
| AD versus PDD | Aβ1–42/37 | 0.772 | 83 | 76 |
| DLB versus NDC | Aβ1–42/37 | 1.264 | 86 | 74 |
| DLB versus PDD | Aβ1–40*% | 0.954 | 81 | 71 |
| PDD versus NDC | Aβ1–42/37 | 1.232 | 76 | 74 |
Discussion
Differentially expressed Aβ peptide patterns in Alzheimer's disease, DLB and PDD—pathophysiological implications
A previously undescribed peptide with Aβ-like immunoreactivity (Aβ1–40*) aside a highly conserved pattern of the Aβ peptides 1–37, 1–38, 1–39, 1–40 and 1–42 was constantly observed in all 88 CSF samples investigated. In summary, Aβ1–40* exhibits electrophoretic and immunological features of Aβ1–40, but migrates at a different position in the Aβ-SDS–PAGE/immunoblot. A similar band originates from synthetic Aβ1–40 during SPE under hydrophobic conditions (synthetic Aβ1–40*) and exhibits a mass corresponding to the expected mass of Aβ1–40ox.
The percentage abundance of Aβ1–40* relative to the sum of all investigated Aβ peptides (Aβ1–40*%) was prominently increased in DLB and to a lesser degree also in Alzheimer's disease as compared with PDD and NDC, respectively.
Significantly decreased CSF Aβ1–42 levels relative to NDC were most prominent in Alzheimer's disease and could also be shown for DLB, but not for PDD. In contrast, Aβ1–37, Aβ1–38 and Aβ1–40 were slightly elevated in Alzheimer's disease as compared with the other dementia groups. The introduction of ratios of Aβ1–42 to Aβ1–37, 1–38, 1–39 and 1–40, respectively, improved the diagnostic test accuracy for each differential diagnostic question relative to the Aβ1–42 levels alone. First, this may be due to disease-specific interactions of each ongoing neurodegenerative dementia process with APP metabolism, which cannot be adequately represented by the sole measurement of absolute Aβ1–42 levels (Wiltfang et al., 2001). Second, the percentage abundance of each Aβ peptide species displayed a lower inter-individual variance of values than its absolute levels (Wiltfang et al., 2003). This corresponds to the observation that the abundances of single Aβ peptide species are closely correlated to each other and regulated in narrow limits, whereas the total amount of Aβ peptides varies inter-individually (Wiltfang et al., 2002, 2003). The ratio of the differentially altered Aβ1–42 to the Aβ1–37 levels enabled the best test accuracies and a highly significant differentiation of all diagnostic groups from each other, with the exception of DLB versus PDD. DLB and PDD could then be discriminated at a highly significant level by the specifically increased Aβ1–40*% in DLB.
We can only speculate on the pathophysiological implications of these disease-specific Aβ peptide patterns
Decreased levels of CSF Aβ1–42 have been reported for patients with Alzheimer's disease (Motter et al., 1995; Andreasen et al., 2001, 2003; Wiltfang et al., 2002, 2003; Mollenhauer et al., 2005) and DLB (Andreasen et al., 2001; Mollenhauer et al., 2005). The reduction of Aβ1–42 levels in Alzheimer's disease has been explained by an increased clearance of the peptide from CSF into senile amyloid plaques for a long time (Motter et al., 1995). Other studies indicate the existence of alternative mechanisms, including the formation of SDS-stable oligomers (Podlisny et al., 1995) and chaperone complexes of Aβ peptides with specific carrier proteins (Wiltfang et al., 2002, 2003; Bibl et al., 2004). However, misfolding and subsequent deposition of proteins is considered to be a major pathological event in both neurodegenerative diseases and amyloid pathology has also been reported for DLB (Merdes et al., 2003).
Alpha-synuclein, the major component of Lewy bodies, has been reported to facilitate the aggregation of Aβ peptides, and the interactions between the two peptides essentially involve their respective hydrophobic domains (Yoshimoto et al., 1995). Moreover, the deposition of amyloid plaques in DLB has been most recently shown to be related to the amount of cortical Lewy bodies (Pletnikova et al., 2005). Interactions of hydrophobic templates or domains with Aβ can induce a conformational shift of the peptide into an α-helix in vitro (Giacomelli and Norde, 2005). Similar interactions of Aβ, for example, with α-synuclein (Yoshimoto et al., 1995) in the case of DLB, may lead to α-helical Aβ peptide species in vivo. The transient formation of an α-helix has been reported to play a major role in the assembly of toxic oligomers (Klimov and Thirumalai, 2003) and β-sheet formation, suggesting it to be an on-pathway to aggregation of Aβ (Kirkitadze et al., 2001).
Additionally, the α-helical structure alters the electronic environment around the sulphur of methionine residue 35 (met-35), making it prone to oxidation (Butterfield, 2003) by a broad variety of oxidizing agents that are abundant in biological systems. The pathogenic role of oxidative stress (e.g. membrane damage and disruption of cellular calcium homeostasis) is well documented for Alzheimer's disease (Butterfield et al., 2003) and DLB (Giasson et al., 2000). On the one hand, oxidation of met-35 to methionine sulphoxide (two-electron oxidation) decreases the cellular toxicity and pro-oxidative potential of Aβ (Varadarajan et al., 2001; Butterfield, 2003) and also prevents its fibril aggregation (Watson et al., 1998; Hou et al., 2002; Palmblad et al., 2002). On the contrary, metal ions, such as copper, may react with met-35 to form a sulphuramyl free radical on the sulphur atom (one-electron oxidation), which causes enhanced oxidative stress via DNA/RNA and protein oxidation, lipid peroxidation and formation of reactive oxygen species, respectively (Butterfield, 2003). Moreover, the metal-dependent aggregation of Aβ is not affected by the formation of methionine sulphoxide (Barnham et al., 2003) and met-35-oxidized Aβ comprises a major component of total brain Aβ in senile amyloid plaques (Atwood et al., 2002; Dong et al., 2003). Whilst met-35-oxidized Aβ is more hydrophilic, its enhanced release from the neuronal membrane into the synaptic cleft may mediate frequent contact with metal ions, such as zinc and copper, released during neural transmission (Barnham et al., 2003). This may contribute to metal-dependent aggregation and trigger Aβ precipitation as a kind of seed at a quite sensitive site of the neuron.
The initial overexpression of α-helical Aβ may thus promote both the fibril formation and the metal-dependent aggregation of the peptide. Taken together, the increased abundance of Aβ1–40* points to a disease-specific mechanism of amyloid deposition in DLB triggered by conformational transition of Aβ upon hydrophobic interactions, probably mediated by α-synuclein, and enhanced posttranslational peptide oxidation.
Although neuropathological similarities between DLB and PDD have been reported (Iseki, 2004), one major difference remains that Lewy bodies in PDD are predominantly localized in the brainstem (Jendroska et al., 1996). The less frequent occurrence of cortical Lewy bodies may contribute to the lower extent of cortical amyloid deposition in PDD as compared with DLB (Mastaglia et al., 2003; Pletnikova et al., 2005), which might be reflected by a distinct neurochemical phenotype among both neurodegenerative diseases in CSF. These findings correspond to the observation that the majority of DLB patients show similar clinical features to PDD at an earlier stage of disease with a pronounced dementia syndrome.
Despite some overlap, especially with regard to Alzheimer's disease and DLB, the reported disease-specific neurochemical phenotypes in CSF indicate the existence of distinct pathophysiological mechanisms in the Aβ peptide metabolism for Alzheimer's disease, DLB and PDD. We propose the investigation of Aβ peptide patterns in CSF and brain homogenates of neuropathologically defined patient groups to further elucidate this aspect.
The specification of a novel peptide with Aβ-like immunoreactivity in CSF
The amino acid sequence of the novel peptide and its secondary structure is currently under evaluation, but remains unclear. Nonetheless, four different lines of indication point to an oxidized and α-helical form of monomeric Aβ1–40 as a probable candidate for Aβ1–40*:
First, the two-dimensional combination of a non-urea SDS–PAGE followed by a urea-based Aβ-SDS–PAGE/immunoblot (non-urea/urea-SDS–PAGE) demonstrated Aβ1–40* to migrate in one single band with other monomeric Aβ peptide species at a molecular mass of approximately 4 kDa in the absence of urea and its subsequent electrophoretic separation cathodically of Aβ1–37 in the Aβ-SDS–PAGE/immunoblot. The same held true for the synthetic Aβ1–40* generated from synthetic Aβ1–40 via SPE. Like other monomeric Aβ peptide species, the peptide can obviously be separated only as a result of urea-induced peptide-specific shifts in binding of SDS during the Aβ-SDS–PAGE (Kawooya et al., 2003). Oligomerized Aβ peptides can be separated from the monomeric band as a result of their significantly higher mass and correspondingly larger effective molecular radii during a non-urea SDS–PAGE. In accordance with the current literature, synthetic full-length α-synuclein was found to migrate at a molecular mass of approximately 16 kDa during a non-urea SDS–PAGE. In brain homogenates from DLB patients, two additional peptides have been reported to migrate in the 12 and 6 kDa range, respectively, in 10% Tris/Tricine gels (Culvenor et al., 1999). These peptides could only be stained with an antibody directed against the NAC region (i.e. non-Aβ-component of Alzheimer's disease amyloid) of α-synuclein (Culvenor et al., 1999), suggesting that the NAC peptide exhibits a molecular mass of approximately 6 kDa at minimum. These data indicate a monomeric form of Aβ peptide to be visualized cathodically of Aβ1–37 and make SDS-stable oligomers of Aβ peptides or α-synuclein and NAC, respectively, unlikely to provoke this band.
Second, the peptide exhibits properties of an amino-terminally unmodified Aβ peptide. The Aβ-IPG-2D-PAGE revealed an identical isoelectric point for Aβ1–40* and the other five Aβ peptides. During an IEF, the amino-terminally truncated Aβ peptides (Aβ2-X, Aβ3-X) show a shift in their isoelectric point of one pH unit (5.37 to 6.37) (Wiltfang et al., 2002), whereas amino-terminally elongated Aβ peptides (Aβ-12-X), which are generated after a cleavage of APP by δ-secretase, would shift to a more acidic isoelectric point owing to an additional negatively charged amino acid. In contrast, Ct-truncation down to Aβ1–28 or Ct-elongation up to Aβ1–49 will not influence the isoelectric point. Additionally, a co-migration of Aβ1–40* with various carboxy-terminally modified synthetic Aβ peptides that have been reported to occur in vivo was excluded in the urea-based Aβ-SDS–PAGE/immunoblot.
Third, the amino- and carboxy-terminus of Aβ1–40* immunologically react like Aβ1–40. Aβ1–40* immunoprecipitates with amino-terminally-specific antibodies against Aβ peptides (1E8 and 6E10) and with a carboxy-terminally-specific antibody against Aβ1–40 (13E9), but not with one against Aβ1–42 (6D5). Otherwise, we have ruled out cross-reactions of the detection antibody 1E8 with synthetic α-synuclein during immunoblot procedures. As the NAC peptide is considered to be a cleavage product of α-synuclein, a cross-reaction with NAC is also unlikely. Neither is there any evidence from the literature that the mAb 1E8 cross-reacts with either α-synuclein or the NAC peptide (Culvenor et al., 1999).
Fourth, a band with electrophoretic and immunological features similar to Aβ1–40* that occurs in vivo was found after SPE of synthetic Aβ1–40 (synthetic Aβ1–40*) under hydrophobic conditions. MALDI–TOF analysis of the respective fractions revealed two mass peaks corresponding to the expected molecular mass of Aβ1-40 and oxidized Aβ1–40 (Aβ1–40ox), respectively. MALDI–TOF analysis of each respective band, picked from the Aβ-SDS–PAGE and enriched by IP, revealed that the synthetic Aβ1–40* band comprises exclusively a mass peak corresponding to the expected mass of Aβ1–40ox. The synthetic Aβ1–40 band exhibits predominantly a mass peak corresponding to the expected mass of unoxidized Aβ1–40, although minor amounts of Aβ1–40ox could inconsistently also be detected herein. Accordingly, the oxidation of Aβ1–40 probably contributes to its altered migration behaviour in the Aβ-SDS–PAGE. Additionally, the above findings strongly suggest that hydrophobic interactions and probably the subsequent formation of a stable α-helix of Aβ1–40 (Giacomelli et al., 2003, 2005) are thoroughly involved in the generation of Aβ1–40*. Two α-helices covering the positions 16–24 and 28–36, respectively, have been detected within Aβ1–40 (Coles M et al., 1998), the first of which is reportedly discordant and, without sufficient stabilization, is particularly prone to forming β-stranded structures (Kallberg et al., 2001; Päiviö et al., 2003). The second α-helix around position 28–36 becomes destabilized and is abolished completely in the case of met-35 oxidation, whereas the first α-helix remains unaffected (Watson et al., 1998). The oxidation and secondary structural transition of Aβ1–40 may change the peptide-specific binding of SDS and thus explain its altered migration behaviour during urea-based electrophoresis (Kawooya et al., 2003).
We currently apply circular dichroism spectroscopy to validate our hypothesis.
Aβ peptide patterns and their use for neurochemically supported differential diagnosis of dementias
The data clearly demonstrate that CSF Aβ peptide patterns vary in a disease-specific manner between Alzheimer's disease, DLB, PDD and neuropsychiatric diseases. However, there is insufficient evidence to suggest Aβ peptide patterns as a sole biomarker for differential diagnosis among the three investigated dementias. According to the criteria recommendations of an international consensus group (Wiltfang et al., 2005), Aβ peptide patterns come closest, but fail to fulfil the requirements (e.g. both sensitivity and specificity beyond 85%). Nevertheless, using a cut-off value of 0.848 for the ratio of Aβ1–42/Aβ1–37, a reasonable accuracy in discriminating Alzheimer's disease from NDC (i.e. 87% sensitivity and specificity each) could be obtained. The previously reported sensitivities for Alzheimer's disease detection and specificities for DLB exclusion, respectively, did not exceed 75% in a combined assay of tau and Aβ 1–42 enzyme-linked immunosorbent assay (ELISA) (Andreasen et al., 2001). Thus, the actual differential diagnostic value of Aβ peptide patterns can be considered to be as relevant as the established ELISAs for tau and Aβ1–42. Moreover, since multiparametric approaches are gaining increasing importance in the early and differential diagnosis of dementias (Lewczuk et al., 2004; Wiltfang et al., 2005), the evaluation of Aβ peptide patterns may aid neurochemical dementia diagnosis in combination with other biomarkers (e.g. tau and phospho-tau) (Wiltfang et al., 2005).
Although the Aβ-SDS–PAGE/immunoblot is a highly sensitive method (Wiltfang et al., 2002), the very low CSF concentration of Aβ1–40* comes close to the level of detection in some cases. This can be considered as a major drawback of the test, which most probably contributes to an increased variance of values and consequently to a loss of accuracy. In respect of this concern, the pre-concentration of Aβ peptides from CSF using the highly valid and reproducible N-terminally-specific IP (Wiltfang et al., 2002) prior to the Aβ-SDS–PAGE/immunoblot promises improved test accuracy. Additionally, it must be taken into consideration that the investigated PDD patients all presented with two or three core features demanded for the diagnosis of probable DLB (McKeith et al., 1996). The differentiation between probable DLB and PDD that lacks further core features of DLB (e.g. fluctuations or hallucinations) might have revealed higher test accuracy.
Hence, the test will have to be re-evaluated using immunoprecipitated CSF samples of neuropathologically defined Alzheimer's disease, DLB and PDD patients to determine whether Aβ1–40* will indeed be applicable as a novel neurochemical dementia marker.
Supplementary material
Supplementary data are available at Brain online.
Supplementary Material
Acknowledgments
M.B., P.L., H.E., J.K., M.O. and J.W. are supported by the BMBF (German Federal Ministry of Education and Science) funded grant, Competence Net Dementia (01GI0102); J.W. and P.L. are supported by University of Erlangen-Nuremberg ELAN-Program Funds; M.B. is supported by the Research program, Faculty of Medicine, Georg-August-Universität Göttingen; and M.O. and J.W. are supported by the CMPB/DFG research center. P.L. and J.W. are supported by the BMBF-funded grant NGFN2 (Project No. PPO-S10T10).
The authors would like to thank Sabine Paul, Birgit Otte, Heike Zech and Nikolaus Kunz for excellent technical assistance.
Funding to pay the Open Access publication charges for this article was provided by German Competence Net Dementias (CND, http://www.kompetenznetz-demenzen.de/) and Research program. Faculty of Medicine, Georg-August-Universität, Göttingen.
Footnotes
These authors contributed equally to this work.
References
- Andreasen N, Minthon L, Davidsson P, Vanmechelen E, Vanderstichele H, Winblad B, et al Evaluation of CSF-tau and CSF-Aβ42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol 2001; 58: 373–9. [DOI] [PubMed] [Google Scholar]
- Andreasen N, Sjogren M, Blennow K. CSF markers for Alzheimer's disease: total tau, phospho-tau and Abeta42. World J Biol Psychiatry 2003; 4: 147–55. [DOI] [PubMed] [Google Scholar]
- Atwood CS, Martins RN, Smith MA, Perry G. Senile plaque composition and posttranslational modification of amyloid-β peptide and associated proteins. Peptides 2002; 23: 1343–50 [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Ciccotosto GD, Tickler AK, Ali FE, Smith DG, Williamson MA, et al Neurotoxic, redox-competent Alzheimer's β-Amyloid is released from lipid membrane by methionine oxidation. J Biol Chem 2003; 278: 42959–65. [DOI] [PubMed] [Google Scholar]
- Bibl M, Esselmann H, Otto M, Lewczuk P, Cepek L, Rüther E, et al Cerebrospinal fluid amyloid beta peptide patterns in Alzheimer's disease patients and nondemented controls depend on sample pre-treatment: Indication of carrier-mediated epitope masking of amyloid beta peptides. Electrophoresis 2004; 25: 2912–8. [DOI] [PubMed] [Google Scholar]
- Butterfield DA. Amyloid β-peptide (1-42)-associated free radical-induced oxidative stress and neurodegeneration in Alzheimer's disease brain: mechanisms and consequences. Curr Med Chem 2003; 10: 2651–9. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D, Castegna A. Proteomics in Alzheimer's diesease: insights into potential mechanisms of neurodegradation. J Neurochem 2003; 86: 1313–27. [DOI] [PubMed] [Google Scholar]
- Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid beta protein are generated from the beta-amyloid precursor protein by different protease activities. Proc Natl Acad Sci USA 1996; 93: 13170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles M, Bricknell W, Watson A, Fairlie D, Craig D. Solution structure of amyloid β-peptide (1-40) in water-micelle environment: is the membrane spanning domain where we think it is? Biochemistry 1998; 37: 12700–6. [DOI] [PubMed] [Google Scholar]
- Culvenor JG, McLean CA, Cutt S, Campbell BCV, Maher F, Jäkälä P, et al Non-Ab component of Alzheimer's disease amyloid (NAC) revisited. Am J Pathol 1999; 155: 1173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, et al. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 2003; 42: 2768–73. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’ A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–98. [DOI] [PubMed] [Google Scholar]
- Giacomelli CE, Norde W. Structural changes in proteins resulting from homomolecular exchange at solid surfaces. In: Encyclopedia of surface and colloid science, New York: Marcel Dekker; 2003. p. 1.
- Giacomelli CE, Norde W. Conformational changes in the amyloid β peptide (1-40) adsorbed on solid surfaces. Macromol Biosci 2005; 5: 401–7. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000; 290: 985–9. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 1988; 51: 745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120: 885–90. [DOI] [PubMed] [Google Scholar]
- Haas C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993; 75: 1039–42. [DOI] [PubMed] [Google Scholar]
- Hou L, Kang I, Marchant RE, Zagorski MG. Methionine 35 oxidation reduces fibril assembly of the amyloid Aβ-(1-42) peptide of Alzheimer's disease. J Biol Chem 2002; 277: 40173–6. [DOI] [PubMed] [Google Scholar]
- Iseki E. Dementia with Lewy bodies: reclassification of pathological subtypes and boundary with Parkinson's disease or Alzheimer's disease. Neuropathology 2004; 24: 72–8. [DOI] [PubMed] [Google Scholar]
- Janek K, Rothemund S, Gast K, Beyermann M, Zipper J, Fabian H, et al Study of the conformational transition of A beta(1-42) using D-amino acid replacement analogues. Biochemistry 2001; 40: 5457–63. [DOI] [PubMed] [Google Scholar]
- Jendroska K, Lees A, Poewe, W, Daniel S. Amyloid β-peptide and the dementia of Parkinson's disease. Mov Disord 1996; 11: 647–53. [DOI] [PubMed] [Google Scholar]
- Jendroska K, Kashiwagi M, Sassoon J, Daniel SE. Amyloid beta-peptide and its relationship with dementia in Lewy body disease. J Neural Transm Suppl 1997; 51: 137–44. [DOI] [PubMed] [Google Scholar]
- Kallberg Y, Gustafsson M, Perrson B, Thyberg J, Johansson J. Prediction of amyloid forming proteins. J Biol Chem 2001; 276: 12945–50. [DOI] [PubMed] [Google Scholar]
- Kawooya JK, Emmons TL, Gonzales-DeWhitt PA, Camp MC, D'Andrea SC. Electrophoretic mobility of Alzheimer's amyloid-β peptides in urea-sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Anal Biochem 2003; 323: 103–13. [DOI] [PubMed] [Google Scholar]
- Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol 2001; 312: 1103–19. [DOI] [PubMed] [Google Scholar]
- Klimov DK, Thirumalai D. Dissecting the assembly of Aβ16-22 amyloid peptides into antiparallel β sheets. Structure 2003; 11: 295–307. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, Esselmann H, Otto M, Maler JM, Henkel AW, Henkel MK, et al Neurochemical diagnosis of Alzheimer's dementia by CSF Abeta 42, Abeta42/Abeta40 ratio and total tau. Neurobiol Aging 2004; 25: 273–81. [DOI] [PubMed] [Google Scholar]
- Mastaglia FL, Johnsen RD, Byrnes ML M, Kakulas BA. Prevalence of amyloid-beta deposition in the cerebral cortex of Parkinson's disease. Mov Disord 2003; 18: 81–86. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996; 47: 1113–24. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984; 34: 939–44. [DOI] [PubMed] [Google Scholar]
- Merdes AR, Hansen MD, Jeste DV, Galasko D, Hofstetter CR, et al Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy-bodies. Neurology 2003; 60: 1586–90. [DOI] [PubMed] [Google Scholar]
- Mollenhauer B, Cepek L, Bibl M, Wiltfang J, Schulz-Schaeffer W, Ciesielczyk B, et al Tau protein, beta-amyloid (1-42) and S100B protein in cerebrospinal fluid of patients with dementia with Lewy bodies. Dement Geriatr Cogn Disord 2005; 19: 164–70. [DOI] [PubMed] [Google Scholar]
- Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, et al Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol 1995; 38: 643–8. [DOI] [PubMed] [Google Scholar]
- Palmblad M, Westlind-Danielsson A, Bergquist J. Oxidation of methionine 35 attenuates formation of amyloid β-peptide 1-40 oligomers. J Biol Chem 2002; 277: 19506–10. [DOI] [PubMed] [Google Scholar]
- Päiviö A, Nordling E, Kallberg Y, Thyberg J, Johansson J. Stabilization of discordant helices in amyloid fibril-forming proteins. Protein sci 2004; 13: 1251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, et al Aβ deposition is associated with enhanced cortical α-synuclein lesions in Lewy body diseases. Neurobiol Aging 2005; 26: 1183–92. [DOI] [PubMed] [Google Scholar]
- Podlisny M, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, et al Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem 1995; 270: 9564–70. [DOI] [PubMed] [Google Scholar]
- Simons M, De Strooper B, Multhaup G, Tienari PJ, Dotti CG, Beyreuther K. Amyloidogenic processing of the human amyloid precursor protein in primary cultures of rat hippocampal neurons. J Neurosci 1996; 16: 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oostveen I, Ducret A, Aerbersold R. Colloidal silver staining of electroblotted proteins for high sensitivity peptide mapping by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Biochem 1997; 247: 310–8. [DOI] [PubMed] [Google Scholar]
- Varadarajan S, Kanski J, Askenova M, Lauderback CM, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's Aβ(1-42) and Aβ(25-35). J Am Chem Soc 2001; 123: 5625–31. [DOI] [PubMed] [Google Scholar]
- Watson AA, Fairlie DP, Craik DJ. Solution structure of methionine-oxidized amyloid β-peptide (1-40). Does oxidation affect conformational switching? Biochemistry 1998; 37: 12700–6. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Arold N, Neuhoff V. A new multiphasic buffer system for sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100,000–1000, and their detection with picomolar sensitivity. Electrophoresis 1991; 12: 352–66. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Smirnov A, Schnierstein B, Kelemen G, Matthies U, Klafki H-W, et al Improved electrophoretic separation and immunoblotting of beta-amyloid (Aβ) peptides 1–40, 1–42 and 1–43. Electrophoresis 1997; 18: 527–32. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Esselmann H, Cupers P, Neumann M, Kretzschmar H, Beyermann M, et al Elevation of beta-amyloid peptide 2-42 in sporadic and familial Alzheimer's disease and its generation in PS1 knockout cells. J Biol Chem 2001; 276: 42645–57. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Esselmann H, Bibl M, Smirnov A, Otto M, Paul S, et al Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer's disease and patients with chronic neuroinflammation. J Neurochem 2002; 81: 481–96. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Esselmann H, Smirnov A, Bibl M, Cepek L, Steinacker P, et al Beta-amyloid peptides in cerebrospinal fluid of patients with Creutzfeldt–Jakob disease. Ann Neurol 2003; 54: 263–7. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Lewczuk P, Maler JM, Svitek J, Miertschischk J, Smirnov A, et al Neurochemical dementia diagnostics: current status and perspectives. World J Biol Psychiatry 2005; 6 (Suppl 1): 133. [Google Scholar]
- Wiltfang J, Lewczuk P, Riederer P, Grunblatt E, Hock C, Scheltens P, et al Consensus paper of the WFSBP Task Force on Biological Markers of Dementia: the role of CSF and blood analysis in the early and differential diagnosis of dementia. World J Biol Psychiatry 2005; 6: 69–84. [DOI] [PubMed] [Google Scholar]
- World Medical Organisation. Declaration of Helsinki. Br Med J 1996; 313: 1448–9. [Google Scholar]
- Yoshimoto M, Iwai A, Kang D, Otero DA, Xia Y, Saitoh T. NACP, the precursor of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc Natl Acad Sci USA 1995; 92: 9141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.