

Abstract
Current computational and mathematical tools are demonstrating the high value of using systems modeling approaches (e.g. Quantitative Systems Pharmacology) to understand the effect of a given compound on the biological and physiological mechanisms related to a specific disease. This review provides a short survey of the evolution of the mathematical approaches used to understand the effect of particular cholesterol-lowering drugs, from pharmaco-kinetic (PK) / pharmaco-dynamic (PD) models, through physiologically based pharmacokinetic models (PBPK) to QSP. These mathematical models introduce more mechanistic information related to the effect of these drugs on atherosclerosis progression and demonstrate how QSP could open new ways for stratified medicine in this field.
Keywords: Quantitative Systems Pharmacology (QSP), cholesterol-lowering drugs, statins, atherosclerosis, mathematical modeling, simulation, personalized medicine
1. INTRODUCTION
It is no secret that the pharmaceutical industry is facing enormous challenges in terms of their efficacy to convert fundamental R&D into drugs. The empirical approach of hypothesis generation and testing has proven successful over the last half-century; resulting in tremendous increases in productivity and rates of approval for new drug applications at the Food and Drug Administration [1]. However, the truth is that given the poor conversion ratio between ‘potential’ targets into products, the difficulty in finding new targets for therapies and the increasing cost required to develop new drugs, the development and standardization of new tools and novel methods that take into account a larger, fuller, more challenging contextual setup for drug design, i.e., a ‘systems’ approach, where a surrogate closer to the biological organism can be interrogated, its complexity better captured and analysed, have become pressing. This is an area where ‘in-silico’ tools have a significant role to play, as modelling and simulation allows numerical testing of diverse potential scenarios, avoiding the immense costs of testing in-vitro or in-vivo scenarios that are by their own nature ‘risky’ and highly likely to fail. The need to produce these novel, risk-absorbing methodologies is evidenced by the emergence of Quantitative Systems Pharmacology (QSP), an approach that integrates knowledge coming from multiple disciplines including drug pharmacology, systems biology, physiology, mathematics and biochemistry.
QSP can even be used to better understand how certain, approved and successful drugs affect some patient groups in some ways and not others, which is in fact a case of patient stratification after treatment. It makes economic and medical sense to optimize the number of ‘interventions’ (drug administrations) to those patients that effectively need them, for whom these drugs will make a difference, still minimizing secondary or polypharmacology effects. Statins are a case in point. Hailed as ‘wonder drugs’, they have had much publicity and undeniable success but have also created no small amount of controversy [2, 3]. While statins are considered ‘safe’ in general, each individual statin has its own safety profile. For example, Baycol (cerivastatin) was removed from the market in 2003 due to an unfavorable risk-benefit profile [4].
Statins are the most widely-prescribed drugs in the world [3]. And yet, the fact that a statin is ‘safe’ (no toxicity) and ‘working’, understood as the lowering of cholesterol levels in blood, is a ‘check’ still quite far removed from the clinical endpoint, which is in the end, the one that matters. Is this patient going to die of a myocardial infarction due to plaque blockage/break-up even under this statin? Is this patient at risk because of all the medication he/she is taking together with his/her statin? Can we make it safer for patient X to take statins if we optimize and personalize dosage? In terms of efficacy, these are the questions that matter. An interesting medical fact is that highly obtrusive plaque (95% stenosed or so) can be perfectly asymptomatic and some plaques well before reaching a significant clinically meaningful threshold can become unstable and break-up. It is very difficult to assess via imaging alone which plaques are unstable; atherosclerotic plaques are a challenging imaging target and as of today many of the currently available imaging tools for clinical use still provide minimal information about the biological characteristics of plaques, because they are limited with respect to spatial and temporal resolution. Moreover, many of these imaging tools are invasive [5]. Preventive screening is not really a cost-effective or practical option. So, the question remains: is statins treatment for each patient effective? If so, by which measure? Can statins be effective if the dose is increased or combined with other drugs? The ‘effectiveness’ of the treatment is linked to each individual’s plaque development and the characteristics of each patient. This is what stratified medicine is about.
In this paper, we will review the (successful) story of statins and how computational models using different modelling techniques have evolved and been used to understand specific aspects of the drug, to treat patients better, to assist in costly drug-design decisions, to understand intervariability and finally why a QSP approach to understand their effect on atherosclerosis as the next step, is still important in the context of personalized treatment and stratified medicine.
1.1. Atherosclerosis
Atherosclerosis, the main cardiovascular disease (affecting large arteries), is the primary cause of heart disease and stroke accounting for about 50% of all deaths in developed countries [6]. Over the past three decades much progress has been made to understand the molecular dynamics of cholesterol mechanisms and the development of atherosclerotic plaque. What was once viewed as an inevitable consequence of ageing is now understood to be a chronic inflammatory condition that can be converted into an acute clinical event by plaque rupture and thrombosis [7]. Atherosclerosis is a long-term, progressive, multifactorial disease, characterized by the accumulation of lipids and fibrous elements in the large arteries. Atherosclerotic disease progresses in stages, with multiple changes in the arterial wall. Early lesions are mainly subendothelial accumulations of foam cells, which are the result of macrophages uptake of low-density-lipoprotein cholesterol (LDL-C); in time, they will become more fibrotic in nature. The initiation occurs when LDL-C molecules penetrate the endothelial barrier triggering an inflammation/immune response within the arterial wall.
The disease is systemic and spans decades, with the first visible atherosclerotic lesions (also called the ‘fatty streak’) appearing in different anatomical sites, first in the aorta, then the coronaries, finally the cerebral arteries, in around 40 years.
The disease has molecular, cellular, metabolic, genetic and environmental factors associated to increased risk. To compound to its complexity, blood flow dynamics also play a crucial role in the development of plaque, with preferred sites of lesion formation within the arteries depending on mechanical stimuli on the arterial wall as these affect endothelial behaviour and its capacity to prevent LDL-penetration [6]. Fig. (1) presents a schematic explanation of this process.
Fig. (1).
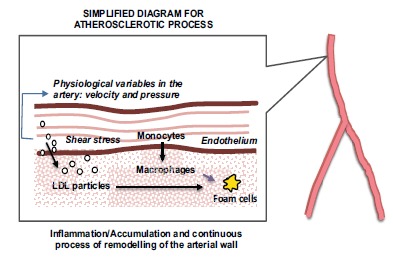
Atherosclerosis: A Simplified Diagram.
It is quite straightforward to see the clinical and industrial interest to develop or optimise cholesterol-lowering drugs since the clinical manifestations of the disease are life defining at best, with huge cost implications national health systems. In the next section we will focus our review on two types of cholesterol lowering drugs: statins and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors.
1.2. Cholesterol-Lowering Drugs, Some Definitions
1.2.1. Statins
Statins are inhibitors of HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase) which is the rate-controlling enzyme of the main metabolic, nicotinamide adenine dinucleotide (NADH) dependent, pathway for the production of cholesterol (mevalonate pathway). Inhibition of the reductase causes upregulation of LDL receptors (LDLR) in the liver, which increases the catabolism of plasma LDL and lowers the plasma concentration of cholesterol thus reducing the risk of cardiovascular diseases (i.e. atherosclerosis) [8, 9].
1.2.2. PCSK9 Inhibitors
PCSK9 is an enzyme playing an important role in lipoprotein metabolism. Some evidence has suggested that decreasing the levels of PCSK9 reduces LDL-C levels, decreasing the risk of developing cardiovascular diseases. The PCSK9 mechanism of action is believed to be related to its binding to LDLR on the surface of the cell shifting the equilibrium to lysosomal-dependent degradation rather than recycling LDLR from the endosome to the cell membrane, which suggests PCSK9 function can be quite significant in targeting LDLR degradation [10].
1.3. In Brief: Mathematical Modelling in Pharmacology
1.3.1. PKPD Modelling
Pharmacokinetic (PK) modelling is based in the application of a classical mass balance to quantify (and understand) the absorption, distribution and elimination of a drug in the body after a given dose. The simplest (and probably most used) approach in PK modelling is considering the volume of the studied species (e.g. mouse, rat, dog, human, etc.) as a well-stirred constant volume, normally associated to the blood or plasma volume where the drug is going to be distributed after a specific dose (some graphical examples are shown in Figs. 2, 3 and 4). Under these assumptions, a PK model can be written based on Ordinary Differential Equations (ODEs) describing the mass balance of the drug:
Fig. (2).
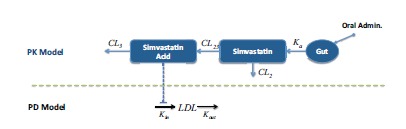
Fig. (3).
PKPD Model for a PCSK9 inhibitor [16].
Accumulation of Drug in the Body =
![]()
dDrug/dt = Dose – Kel Drug (2)
Where Dose will depend on how the dose is administered (e.g. intravenous, oral, etc), which can depend on the volume of distribution of the drug and Kel represents the kinetic rate constant related to the elimination (or clearance) of the drug. PK models could include additional compartments in order to consider specific physiological mechanisms or additional non-linearities that one compartment model cannot capture from the original data.
Pharmacodynamics models are related to mathematical models used to estimate the effect of a drug on a given species, which can include secondary (or adverse) effects and/or specific biomarkers. Classical examples of PD models are based on sigmoidal functions, assuming the drug has a threshold (or saturation limit) when having an effect in a specific biomarker for example. Thus, making use of the information about the temporal variations of drug using a given PK model a PD model can be written using the classical hill equation (or Emax model):
![]()
Where Effect represents the magnitude of the effect produced by a given amount of Drug; Emax is the maximal effect produced by the drug; EC50 is the concentration of drug producing half of the maximal effect and n is the hill coefficient.
1.3.2. PBPK Modelling
As mentioned previously, simple PK models (e.g. 1-2 compartments) can be used to replicate dynamic data of a drug in the body, however if additional physiological mechanisms are needed in the model in order to understand specific links of the drug with the physiology, additional compartments need to be added to this initial PK approach. This is probably the main motivation of Physiologically-Based Pharmacokinetic (PBPK) models.
A PBPK model is basically a multi-compartmental PK model where each of the compartments represents real tissue and physiological volume (e.g. organs). An example of this kind of models is shown in Fig. (5).
Fig. (5).
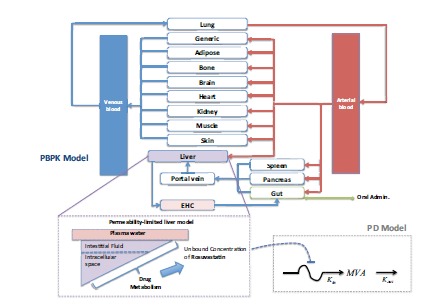
Simplified Diagram of a PBPK model: Understanding the Pleiotropic effect of Statins [21].
The initial (and simplest) PBPK models consider these physiological volumes to be homogenous. However, more advanced models describe specific organs and tissues (e.g. gut, liver) with more details in order to provide a more realistic representation of these organs.
A general mathematical representation for PBPK models can be written based on the mass balance of the drug for a given compartment, as shown in equation (4):
![]()
Where i refers to a given compartment (organ) in the model; Input drug and Output drug represent the drug entering or being eliminated in this compartment and Transformation is any process related to the transformation (e.g. metabolism) of the drug in this compartment.
1.3.3. Quantitative Systems Pharmacology
PBPK approaches clearly demonstrate the utility of incorporating additional physiological mechanisms for the quantification of drug pharmacokinetics. Applying the same approach to understand the effect of the drug in a specific biomarker or disease progression led us to the development of new emergent mathematical models combining the classical concepts of PKPD and Systems Biology or Quantitative Systems Pharmacology (QSP).
Under this perspective, QSP models are also based on the development of mathematical models based on ODEs representing biological/physiological mechanisms of a given disease in conjunction with biochemical interactions of specific biomarkers and a quantitative model the drug under study. The differential equation of the drug could take the form of any PK model (e.g. equations 2 or 4). The rest of the QSP model will describe the dynamics of the different biomarkers associated to the disease, which could be represented with equation (5):
![]()
Where i refers to a given compartment in the QSP model (e.g. cell, organ, etc.); Input BioMj and Output BioMj represent the biomarker entering or being eliminated in this compartment and “Transformation” is any process related to the transformation (e.g. metabolism) of the biomarker in this compartment.
2. CATCHING-UP AFTER MORE THAN THREE DECADES OF RESEARCH: FROM PKPD TO QSP MODELS
It might seem odd now, but as recently as the 1980s, there was debate in the medical community about whether cholesterol was really a cause of coronary arterial disease (CAD). In fact, results of the Coronary Primary Prevention Trial conducted from 1973 to 1983 were the first to conclude that lowering low-density-lipoprotein (LDL) cholesterol and total blood cholesterol can reduce the incidence of CAD and heart attacks in men at high risk [4]. The birth of the first commercial statin (lovastatin) in 1987 paved the way for a number of successful statins to come to market since then. However, these very first drugs were not tested (at least for bioavailability) using pharmacokinetic (PK) / pharmacodynamics (PD) approaches [11]. Model-based approaches have entered the scene later to study in more detail the mechanisms of cholesterol-lowering drugs using mathematical modelling. The following subsections give a brief introduction to the work based on this approach.
2.1. PK/PD Models of Statins: Characterizing Drug Behavior
Our own search has revealed relatively few papers dealing directly with the mathematical modelling of statins’ mechanisms. The work of Faltaos et al. [12], already in 2006 appears as one of the oldest ones, quite far down the road if we take the late 1980s/early 1990s as the period in which the first statins came to market. Other search terms, involving simulation or numerical modelling for example, do not really yield results older than this. In their paper, Faltaos et al. [12] develop a relatively simple model to dynamically describe LDL-Cholesterol (LDL-C) after treatment with three different statins: atorvastatin, simvastatin and fluvastatin. The model was a based on a physiologically indirect-response model, describing the effect of statins as the inhibition of LDL-C production and additionally increasing LDL-C clearance in a dose-dependent pattern. Assuming the statins inhibit HMG-CoA in hepatocytes, two compartments were proposed in this model, one representing the hepatocyte and another one to model the effect on the circulating LDL-C. The main observation from this study was that the three molecules had a similar inhibitory effect on LDL-C synthesis, however the authors could find some differences on how these drugs affect LDL-C clearance when using an Emax (maximum effect) model, showing a dependency of the administered drug dose. Additional analyses showed no effect of gender, body weight, body surface area, age, calories/day, sugar/day, lipids/day, hyperlipidaemia types and waist/hip circumference, on the pharmacodynamic parameters.
Only a few years later, a more sophisticated approach was proposed by Kim et al. [13], using a population PK/PD approach to study simvastatin based on the prediction of dose-response relationship of the drug in patients with hyperlipidaemia. The model was developed using data from twenty-seven healthy volunteers with a 40 mg daily dose of simvastatin for 14 days. The PKPD model was proposed for simvastatin and its active metabolite (simvastatin acid) and a turnover model was used to describe the change in LDL-C levels (Fig. 2). The model was validated comparing the simulated dose-response relationship of simvastatin with similar data reported in the literature. A similar approach was used by Oh et al. [14], this time to develop a model of the effect of atorvastatin on the time-course of LDL-C profiles, where an indirect response model consisting of production in hepatocyte and elimination from plasma stimulated by atorvastatin was calibrated and validated in a population of dyslipidemic patients and non-patient volunteers. Around the same period, Yang et al. [15] used population PD analyses to describe the dose-response relationship of rosuvastatin with mean values of LDL-C reduction (%) from dose-ranging trials. The interesting angle of this work is that baseline LDL-C and race were analyzed as the potential covariates but also, model robustness was evaluated using the bootstrap and data-splitting methods, and Monte Carlo simulations were performed to assess the predictive performance of the PPD model with the mean effects from the one-dose trials.
2.2. PK/PD Models for other Cholesterol-Lowering drugs: Supporting Drug Design During Phase 2 Dose Selection
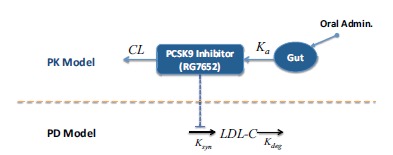
An interesting application of modelling in drug design in this context is presented in 2015 by Budha et al. [16]. They developed a PK/PD model for RG7652 (see Fig. 3), a fully humanized monoclonal antibody targeting human PCSK9. RG7652 prevents degradation of the hepatic LDL-C receptors by blocking PCSK9 binding and thereby resulting in efficient LDL-C uptake by hepatocytes.
After evaluating the pharmacokinetics RG7652 and data fitting from a Phase I study (healthy subjects) where both PK and LDL-C profiles for this population were successfully described, the model simulation results provided useful insights and quantitative understanding for the selection of Phase 2 study doses in patients with coronary heart disease. This paper describes the successful use of modelling and simulation as tools for drug dosage but perhaps more interesting, the capacity of models to provide a better grasp of the mechanisms behind a certain behavior, condition or effect.
2.3. Simulation of Combined Therapies: Dose-Response Models and Quantitative Decision-Making
Mandema et al. [17] used a dose-response model to evaluate the product profile of gemcabene, a lipid-altering agent (which incidentally, never made it to market) alone and in combination with a statin. The model estimates the lipid effects (LDL-C and HDL-C); adverse effects, such as persistent alanine aminotransferase elevation and myalgia; and for tolerability issues, such as headache, for 5 of the currently marketed statins, ezetimibe, gemcabene, and the combination of ezetimibe or gemcabene with a statin. After evaluation, authors found out that gemcabene had almost no effect when combined with atorvastatin. The authors also evaluated the impact of treatment with a combination of a statin with gemcabene or ezetimibe on coronary artery disease, using additional analyses to predict the risk reduction relative to placebo or compared with other statin treatments on the basis of the lipid effects. This work is a prime example of quantitative decision-making as it facilitated a quick decision to stop development of the drug, saving millions of US dolars to the Company in R&D and expensive clinical trials.
Work from Vargo et al. [18] used a PKPD model derived from a literature-based meta-analyses to evaluate a fixed-dose combination (FDC) of ezetimibe and atorvastatin for the treatment of dyslipidemia, under a combined dose range. Their results encourage the use of computational modeling to estimate dose-response relationships and predicting their clinical equivalence, which could have a beneficial impact on lowering cost/timelines through effective management of resources/studies, and predicting the effectiveness of new dosage formulations allowing to reduce the number of additional clinical efficacy trials in regulatory settings.
2.4. Using PK/PD Models to Improve/Understand Treatment: Investigating the Effect of Compliance and Chronological Dosing
Aoyama et al. [19] developed a study defining the PK/PD model of rosuvastatin in order to predict its response, using simulated plasma mevalonic acid (MVA) concentration in various dosage regimes (or ‘scenarios’) such as poor compliance and morning and evening dosage. They calculated the MVA concentration-time curves for 24 h in the steady state and they simulated the reduction ratio of baseline levels from the 0.33-3.0-fold value of each PK parameter estimate. They found out that chronological dosing had an effect and suggested that the parameters of the PK/PD model could be used to determine the effective rosuvastatin dosage, providing useful clinical information in practice.
The model from Wright et al. [20] (Fig. 4) explored the influence of simvastatin dosing time, variable compliance and circadian cholesterol production on the reduction of low-density lipoprotein (LDL). This study used a modified version of the model of Kim et al. [13] previously described (Fig. 1) where a model for circadian LDL production was incorporated into the PKPD model. By using stochastic simulation techniques, they reduced LDL from baseline at different dose levels daily for 30 days, also varying dosing time (8.00 a.m.), evening (22.00 p.m.), a combination of time-administration and failure to fully adhere (evening with reduced compliance) and evening administration for a hypothetical bioequivalent generic. The final conclusion of this work was that differences were overall negligible between day/night dosage, whatever small differences were negated when having variable compliance and there was no significant difference with the bioequivalent generic.
Fig. (4).
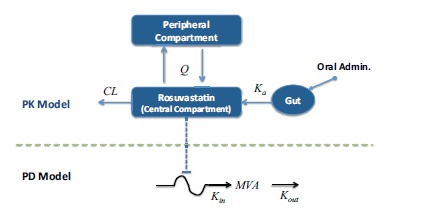
PKPD Model of Statin: Evaluating the effect of chronological time in dosing and compliance on MVA (mevalonic acid) levels in plasma [19]. Note that the arrow associated with Kin is not straight as this model considers that the rate of production of MVA is variable (expressed as a function of chronological time).
2.5. Improving PK/PD Models: Developing PBPK Models
2.5.1. Understanding the Pleiotropic Effect of Statins
The use of PBPK (Physiologically Based Pharmacokinetic) models is continuously increasing to support drug development and regulatory submissions due to the possibility of using this approach to predict drug concentrations in blood and specific tissues. Given that PBPK models are an extension of (simpler) PK models, it is probable that using PBPK approaches provides a better understanding of the PD of a drug and patient variability due to PD aspects or drug disposition to the site of action.
As an example of the use of PBPK models for statins, Rose et al. [21] modelled the liver concentrations of rosuvastatin (see Fig. 5). This model in particular, studied the effect of the uptake (normally dependent on different genotypes) by OATP1B1 (organic anion transporting polypeptide 1B1) transporters on the PD response. Given the additional physiological mechanisms added to this model, one of the main outcomes from this was the possibility to give an explanation to the disconnection between different polymorphisms of OATP1B1 and their effect on plasma concentrations of rosuvastain and cholesterol synthesis.
2.5.2. PBPK Models for Safety Assessment of Statins
Another PBPK model of statins was proposed by Lippert et al. [22]. Their aim was to provide a modeling platform where physiological and pharmacological knowledge combined with pre-clinical and clinical data would allow prediction of adverse events related to drug exposure. This modeling approach seems particularly relevant to evaluate different dosing scenarios, test new compounds or evaluate the effect on high-risk populations. They applied the approach to model simvastatin and pravastatin for a special population having a particular genotype leading to an increased risk of developing myopathy. Fig. (6) shows a simplified diagram of the PBPK model used in this study and the workflow for model-based safety assessment.
Fig. (6).
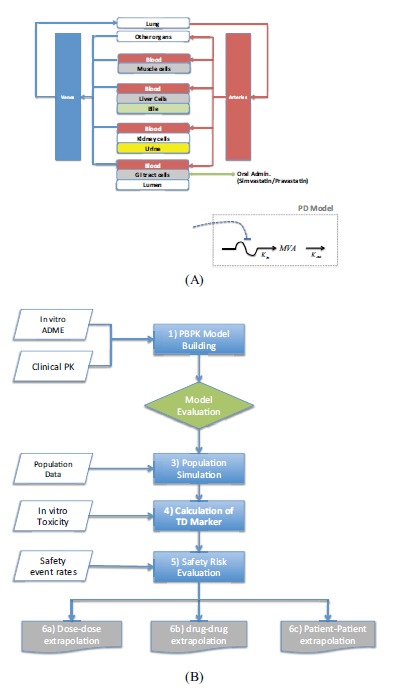
(A) Simplified diagram of the PBPK Model for Safety Assessment of Statins. (B) Model-based Workflow for Safety Assessment [22].
2.5.3. A Discussion: from PBPK to QSP
PBPK has become an established methodology within the Pharmaceutical industry together with PK/PD models and over the past decade, a significant number of publications have appeared in the literature [23]. One clear advantage of PBPK with respect to PK/PD, is that although PBPK models are built using a similar mathematical framework to PK/PD, they are parameterized using known physiology and consist of a larger number of compartments, which correspond to the different organs or tissues in the body. These compartments are connected by flow rates that parallel the circulating blood system. PBPK models, like the more empirical models, provide estimates of common PK parameters, e.g., clearance, volume of distribution, and effective half-life [24]. However, because of their own nature and description, being more physiologically orientated, they provide a quantitative mechanistic framework by which scaled drug-specific parameters can be used to predict the plasma and, importantly, tissue concentration-time profiles of new drugs, following i.v. or oral administration [24].
This been said, mathematical modelling within the pharmaceutical industry has been advancing at increasing speed and a newly-emerging field, ‘Quantitative Systems Pharmacology’ (QSP) is on the rise, with fundamental differences to its predecessors, which make-up for some of their shortcomings. Although PBPK is mechanistic in nature, it is still reliant on concepts familiar to PK/PD in many cases with no direct biological or physiological meaning and an extra-layer of interpretation is necessary. Systems Pharmacology’s attempt is to open-up these models to characterize through quantitative analysis and following fundamental biology principles “the dynamic interactions between drug(s) and a biological system to understand the behaviour of the system as a whole, as
opposed to the behaviour of its individual constituents; thus, it has become the interface between PKPD and systems biology. It applies the concepts of Systems Engineering, Systems Biology, and PKPD to the study of complex biological systems through iteration between computational and/or mathematical modelling and experimentation” [25].
Before moving onto some recent examples, it is important to highlight that this level of complexity is significantly higher when compared to PK/PD or PBPK and one key issue with QSP is that models heavily rely on the modeller’s skill and understanding of basic biological mechanisms and from a purely practical point of view, there are no standard approaches, software or ontologies currently available.
3. QSP MODELS OF CHOLESTEROL-LOWERING DRUGS
3.1. QSP as Modelling Tool to Test the Effect of Combined Therapies on Cholesterol Levels
Gadkar et al. [26] developed a QSP model of the mechanisms of action of statin and anti-PCSK9 therapies predicting changes on LDL-C for different treatment protocols and patient subpopulations using the Simbiology software (Mathworks, Natick, MA) (Fig. 7). Some clinical data was used to validate the proposed mechanistic interactions, which were described in detail, through the simulation of virtual subjects. Starting from the description of a ‘healthy’ or ‘normal’ subject, numerous parameterizations representing different virtual subjects were used to describe ‘virtual populations’. Exploration of alternate parameterizations via virtual subjects is an important aspect of research in mechanistic QSP models [27, 28] as these alternate parametrizations can offer insight into the system and the source of variability. It is important to mention that systems models typically include numerous parameters that are not uniquely identifiable but still, many of these parameters demonstrate inter-subject variability. However, the potential impact of this variability and uncertainty can be addressed by exploring alternate parameterizations that recapitulate higher level behaviors of interest [26]. As the authors very well explain, “virtual population variability addresses not only inter-patient variability, but also alternate mechanistic hypotheses and parameter uncertainty, the virtual population variability does not correspond directly to clinical variability”, which is absolutely at the core of QSP. Models are used as tools to explore a highly uncertain model and parameter space and they can be used as sophisticated tools to test hypotheses and our own understanding of the drug mechanism in interaction with the system, not only for quantification. Finally, as an outcome of their work, their simulations predict a slightly better reduction in LDL-C when a combined therapy of PCSK9 inhibitor and statins is administered compared to the monotherapy.
Fig. (7).
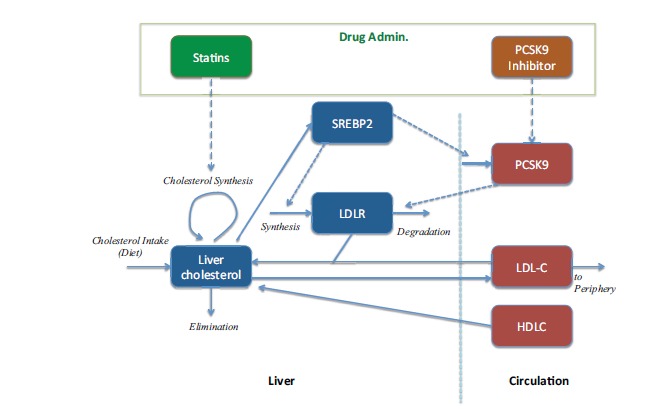
QSP model to evaluate a combined therapy [26].
3.2. QSP and Multiscale Modelling to Understand the Effect on Atherosclerosis Progression: Towards Stratified Medicine
The Association of the British Pharmaceutical Industry defines stratified medicine as “the tailoring of medical treatment to the individual characteristics of each patient. It does not literally mean the creation of medicines or medical devices that are unique to a patient, but rather the ability to classify individuals into stratified subpopulations that differ in their susceptibility to (or severity of) a particular disease or their response to a specific treatment” [29]. This means that preventive or therapeutic interventions can then be concentrated on those who will benefit, sparing expense and side effects for those who will not.
Focusing this time on the effect of the drug on atherosclerosis progression and not only on lowering cholesterol levels, a step further in the study of the pharmacology of cholesterol-lowering drugs was proposed by Pichardo-Almarza et al. [30]. This completely novel QSP approach, in-between two different worlds (PK/PD and Systems Biology) used the mathematical model as a vehicle to understand the PK of Simvastatin and its effect on LDL and atherosclerotic plaque evolution and vascular remodeling, which is the clinical endpoint. The multiscale approach adopted describes the most important biological/physiological mechanisms related to atherosclerotic plaque progression combined with the effect of blood flow conditions and how this has an impact on the LDL and Monocytes penetration in the arterial wall. Simulations of a virtual population composed of 1000 patients with different physiological characteristics and simulated during 20 years showed the sensitivity of the plaque growth and statin response to different physiological conditions (e.g. characteristics of the blood flow and the geometry of the artery) and was able to simulate each patient’s trajectory defined by quantifying the effect of the drug and adherence to regime on plaque volume. The model was validated through comparison of the results of the virtual population against well-known clinical trials [31, 32]. This novel approach opens up the way to use QSP for predictive modelling in individual patients, although there is obviously much work to be done. Fig. (8) shows a diagram summarizing the different parts of this QSP model.
Fig. (8).
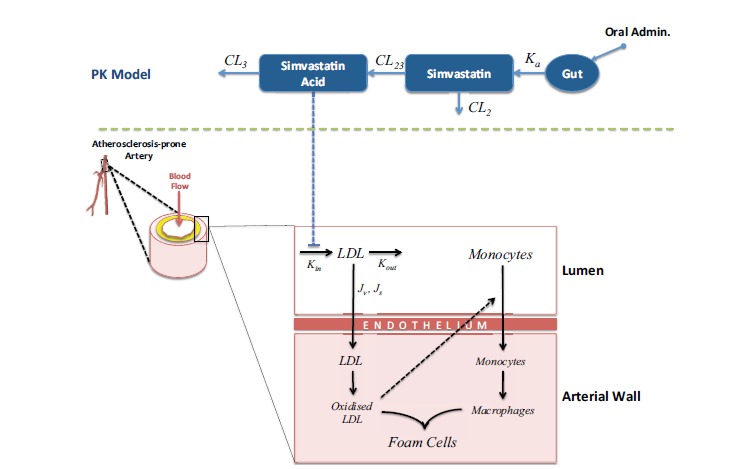
QSP model: Understanding the effect of Statins in LDL and Atherosclerotic Plaque Progression [30].
CONCLUSION
Great and encouraging progress has been made in the modelling and simulation of the pharmacology of cholesterol-lowering drugs, and their effect on cardiovascular diseases. With the emergence of QSP, the combined effort of multidisciplinary teams around the world is making it possible to develop new and improved multiscale approaches that lead to a better understanding of atherosclerosis and its treatment. By focusing on the clinical endpoint, QSP has also the potential introduce a seismic change in our understanding of the relationship between drugs and disease, and not only the relationship of the drug to an organ or surrogate biomarker. We have provided examples of how computational models in QSP have proved to be extremely valuable to understand the pharmacokinetics and immediate pharmacodynamics effect of the drugs whilst additionally showing how they could be extremely useful to understand the behavior of a patient population under specific conditions (i.e. patients in a clinical trial) and the dynamics of the disease progression according to drug administration efficacy and patient behavior.
Moreover, with an ageing population and a global pandemic of overweight and obesity, cardiovascular diseases are and will still be a major cause of mortality and morbidity for years to come; the end to this situation is not really in sight and the stakes are as high as ever. With new knowledge, increaseing access to high quality data and computing power, QSP for cardiovascular diseases looks like an excellent target area in which to develop stratified medicine right now.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge support by the EPSRC grant “Personalised Medicine Through Learning in the Model Space” (grant number EP/L000296/1) and the Leverhulme Trust Senior Research Fellowship “Exploring the Unknowable Using Simulation: Structural Uncertainty in Multiscale Models” (Fellowship number RF-2015-482).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Leil T.A., Bertz R. Quantitative Systems Pharmacology can reduce attrition and improve productivity in pharmaceutical research and development. Front. Pharmacol. 2014;5:247. doi: 10.3389/fphar.2014.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferket B.S., van Kempen B.J., Heeringa J., et al. Personalized prediction of lifetime benefits with statin therapy for asymptomatic individuals: a modeling study. PLoS Med. 2012;9(12):e1001361. doi: 10.1371/journal.pmed.1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parish E., Bloom T., Godlee F. Statins for people at low risk. BMJ. 2015;351:h3908. doi: 10.1136/bmj.h3908. [DOI] [PubMed] [Google Scholar]
- 4.Januska B.M. Triumph of the heart: The story of statins. Am. J. Health Syst. Pharm. 2010;67(4):321–322. [Google Scholar]
- 5.Matter C.M., Stuber M., Nahrendorf M. Imaging of the unstable plaque: how far have we got? Eur. Heart J. 2009;30(21):2566–2574. doi: 10.1093/eurheartj/ehp419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lusis A.J. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massberg S., Schulz C., Gawaz M. Role of platelets in the pathophysiology of acute coronary syndrome. Semin. Vasc. Med. 2003;3(2):147–162. doi: 10.1055/s-2003-40673. [DOI] [PubMed] [Google Scholar]
- 8.Kostner G.M., Gavish D., Leopold B., Bolzano K., Weintraub M.S., Breslow J.L. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation. 1989;80(5):1313–1319. doi: 10.1161/01.cir.80.5.1313. [DOI] [PubMed] [Google Scholar]
- 9.Reiner Ž. Statins in the primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 2013;10(8):453–464. doi: 10.1038/nrcardio.2013.80. [DOI] [PubMed] [Google Scholar]
- 10.Dadu R.T., Ballantyne C.M. Lipid lowering with PCSK9 inhibitors. Nat. Rev. Cardiol. 2014;11(10):563–575. doi: 10.1038/nrcardio.2014.84. [DOI] [PubMed] [Google Scholar]
- 11.Li J.J. Triumph of the heart: the story of statins. Oxford, New York: Oxford University Press; 2009. p. 201. [Google Scholar]
- 12.Faltaos D.W., Urien S., Carreau V., et al. Use of an indirect effect model to describe the LDL cholesterol-lowering effect by statins in hypercholesterolaemic patients. Fundam. Clin. Pharmacol. 2006;20(3):321–330. doi: 10.1111/j.1472-8206.2006.00404.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim J., Ahn B.J., Chae H.S., et al. A population pharmacokinetic-pharmacodynamic model for simvastatin that predicts low-density lipoprotein-cholesterol reduction in patients with primary hyperlipidaemia: Simvastatin pharmacokinetic-pharmacodynamics. Basic Clin. Pharmacol. Toxicol. 2011;109(3):156–163. doi: 10.1111/j.1742-7843.2011.00700.x. [DOI] [PubMed] [Google Scholar]
- 14.Oh E.S., Lee S.H., Park M.S., Park K., Chung J.Y. Modeling of the LDL cholesterol-lowering effect of atorvastatin in Korean dyslipidemic patients and non-patient volunteers. Int. J. Clin. Pharmacol. Ther. 2012;50(9):647–656. doi: 10.5414/cp201699. [DOI] [PubMed] [Google Scholar]
- 15.Yang J., Li L., Wang K., et al. Race differences: modeling the pharmacodynamics of rosuvastatin in Western and Asian hypercholesterolemia patients. Acta Pharmacol. Sin. 2011;32(1):116–125. doi: 10.1038/aps.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budha N.R., Leabman M., Jin J.Y., et al. Modeling and simulation to support phase 2 dose selection for RG7652, a fully human monoclonal antibody against proprotein convertase subtilisin/Kexin Type 9. AAPS J. 2015;17(4):881–890. doi: 10.1208/s12248-015-9750-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandema J.W., Hermann D., Wang W., et al. Model-based development of gemcabene, a new lipid-altering agent. AAPS J. 2005;7(3):E513–E522. doi: 10.1208/aapsj070352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vargo R., Adewale A., Behm M.O., Mandema J., Kerbusch T. Prediction of clinical irrelevance of PK differences in atorvastatin using PK/PD models derived from literature-based meta-analyses. Clin. Pharmacol. Ther. 2014;96(1):101–109. doi: 10.1038/clpt.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoyama T., Omori T., Watabe S., et al. Pharmacokinetic/pharmacodynamic modeling and simulation of rosuvastatin using an extension of the indirect response model by incorporating a circadian rhythm. Biol. Pharm. Bull. 2010;33(6):1082–1087. doi: 10.1248/bpb.33.1082. [DOI] [PubMed] [Google Scholar]
- 20.Wright D.F., Pavan Kumar V.V., Al-Sallami H.S., Duffull S.B. The influence of dosing time, variable compliance and circadian low-density lipoprotein production on the effect of simvastatin: simulations from a pharmacokinetic-pharmacodynamic model. Basic Clin. Pharmacol. Toxicol. 2011;109(6):494–498. doi: 10.1111/j.1742-7843.2011.00757.x. [DOI] [PubMed] [Google Scholar]
- 21.Rose R.H., Neuhoff S., Abduljalil K., Chetty M., Rostami-Hodjegan A., Jamei M. Application of a physiologically based pharmacokinetic model to predict OATP1B1-related variability in pharmacodynamics of rosuvastatin. CPT Pharmaco Syst Pharmacol. 2014;3:e124. doi: 10.1038/psp.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lippert J., Brosch M., von Kampen O., et al. A Mechanistic, Model-based approach to safety assessment in clinical development. CPT Pharmaco Syst Pharmacol. 2012;1(11):e13. doi: 10.1038/psp.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowland M., Peck C., Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 24.Jones H., Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmaco Syst Pharmacol. 2013;2(8):e63. doi: 10.1038/psp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Graaf P.H., Benson N. Systems pharmacology: bridging systems biology and pharmacokinetics-pharmacodynamics (PKPD) in drug discovery and development. Pharm. Res. 2011;28(7):1460–1464. doi: 10.1007/s11095-011-0467-9. [DOI] [PubMed] [Google Scholar]
- 26.Gadkar K., Budha N., Baruch A., Davis J.D., Fielder P., Ramanujan S. A mechanistic systems pharmacology model for prediction of ldl cholesterol lowering by PCSK9 antagonism in human dyslipidemic populations. CPT Pharmaco Syst Pharmacol. 2014;3(11):e149. doi: 10.1038/psp.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoda L., Kreuwel H., Gadkar K., et al. The Type 1 Diabetes PhysioLab Platform: a validated physiologically based mathematical model of pathogenesis in the non-obese diabetic mouse. Clin. Exp. Immunol. 2010;161(2):250–267. doi: 10.1111/j.1365-2249.2010.04166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt B.J., Casey F.P., Paterson T., Chan J.R. Alternate virtual populations elucidate the type I interferon signature predictive of the response to rituximab in rheumatoid arthritis. BMC Bioinformatics. 2013;14:221. doi: 10.1186/1471-2105-14-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. The stratification of disease for personalised medicines [Internet]. Available from: http: //www.abpi.org.uk/our-work/library/medicaldisease/ Documents/strat_med.pdf .
- 30.Pichardo-Almarza C., Metcalf L., Finkelstein A., Diaz-Zuccarini V. Using a systems pharmacology approach to study the effect of statins on the early stage of atherosclerosis in humans. CPT Pharmacometrics Syst. Pharmacol. 2015;4(1):e00007. doi: 10.1002/psp4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lüscher T.F., Pieper M., Tendera M., et al. A randomized placebo-controlled study on the effect of nifedipine on coronary endothelial function and plaque formation in patients with coronary artery disease: the ENCORE II study. Eur. Heart J. 2009;30(13):1590–1597. doi: 10.1093/eurheartj/ehp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okazaki S., Yokoyama T., Miyauchi K., et al. Early statin treatment in patients with acute coronary syndrome: demonstration of the beneficial effect on atherosclerotic lesions by serial volumetric intravascular ultrasound analysis during half a year after coronary event: the ESTABLISH Study. Circulation. 2004;110(9):1061–1068. doi: 10.1161/01.CIR.0000140261.58966.A4. [DOI] [PubMed] [Google Scholar]