

Abstract
Sustained Ca2+/calmodulin dependent kinase II (CaMKII) activation plays a central role in the pathogenesis of a variety of cardiac diseases. Emerging evidence suggests CaMKII evoked programmed cell death, including apoptosis and necroptosis, is one of the key underlying mechanisms for the detrimental effect of sustained CaMKII activation. CaMKII integrates β-adrenergic, Gq coupled receptor, reactive oxygen species (ROS), hyperglycemia, and pro-death cytokine signaling to elicit myocardial apoptosis by intrinsic and extrinsic pathways. New evidence demonstrates CaMKII is also a key mediator of receptor interacting serine/threonine kinase 3 (RIP3) induced myocardial necroptosis. The role of CaMKII in cell death is dependent upon subcellular localization and varies across isoforms and splice variants. While CaMKII is now an extensively validated nodal signal for promoting cardiac myocyte death, the upstream and downstream pathways and targets remain incompletely understood, demanding further investigation.
Keywords: CaMKII, programmed cell death, apoptosis, necroptosis, oxidized CaMKII, RIP3, ROS, mitochondria, mitochondrial permeability transition pore
Introduction
Apoptosis, a process of programmed cell death, is considered a vital component of cardiovascular development [1]. However, inappropriate apoptosis or accelerated apoptosis is one of the major factors contributing to cardiovascular disease [2, 3]. Because of this, prevention of apoptosis has been viewed as a promising therapeutic target. Apoptosis is defined, in part, by distinct morphological characteristics and energy-dependent biochemical mechanisms. Apoptosis yields almost no residual cellular components and is unattended by inflammatory reaction. An alternative to apoptotic cell death is necrosis, which is considered to be a toxic process where the cell is a passive agent and follows an energy-independent mode of death. However, this paradigm has evolved over the past decade. Emerging evidence has strongly supported a concept that a subset of necrosis is regulated and follows defined signaling mechanisms. This programmed necrosis, often referred as necroptosis, has been recognized to play an important role in variety of cardiovascular diseases and is now considered be a candidate target for therapeutic intervention [4, 5]. Necroptosis appears to play no role in cardiac development. Instead, suppression of necroptosis is important for normal development and tissue homeostasis [6–8]. The essential mechanistic feature of necroptosis is the opening of mitochondrial permeability transition pore (mPTP) [5], which dissipates the inner mitochondrial membrane potential (ΔΨm), disrupts mitochondrial respiratory function and results in metabolic shutdown. Thus, in stark contrast to apoptosis, which requires mitochondrial derived ATP to fuel the multiple processes aiming to dissemble the cell, necroptosis is a consequence of ATP deficiency. Necroptosis yields cellular residua that trigger innate immune inflammatory responses that contribute to the pathology of tissue injury.
The Ca2+ and calmodulin dependent protein kinase II (CaMKII) is a pleiotropic signal that regulates cardiomyocyte Ca2+ cycling, contractility, inflammation, metabolism, gene expression, and cell survival [9–11]. Although CaMKII plays an important physiological role, such as augmenting the “fight or flight response”, [12] sustained CaMKII activation is now recognized to promote heart failure [13, 14], arrhythmia [15] and sudden cardiac death [16]. Excessive CaMKII activation promotes cardiac myocyte death under conditions of neurohumoral agonist signaling [17, 18], oxidant stress [19–22], hyperglycemia [23, 24], ischemic injury [25–29], doxorubicin toxicity [26, 30], acidosis induced injury and other adverse stimuli associated with increased intracellular [Ca2+] (e.g. thapsigargin, ionomycin, high extracellular [K+]) [31]. Thus, improved understanding of the pathophysiology of CaMKII mediated cell death is an important goal for cardiovascular biology with the potential to provide new targets for treating disease.
Molecular physiology of CaMKII
CaMKII is a serine-threonine kinase that is abundant in myocardium and other excitable tissues. CaMKII exists as four distinct isoforms (α, β, γ, δ) that are encoded by four separate genes. Each of these genes resides on a different chromosome, in humans and in mice [19]. The CaMKII isoforms are homologous, consist of the same domains (i.e. association, regulatory and catalytic), operate by shared activation mechanisms, and may assemble heterologously into holoenzymes. CaMKIIδ is the major myocardial isoform and CaMKIIγ is a minor component [32]. CaMKIIα and CaMKIIβ are absent or scantily represented in heart. CaMKIIδ is most clearly linked to myocardial disease because CaMKIIδ knock out mice are resistant to transaortic banding surgery, [13, 14] while transgenic mice with myocardial CaMKIIδ over-expression develop severe heart failure, arrhythmias and premature death. [33] Although CaMKII is widely distributed throughout cellular compartments, CaMKIIδ exists in a splice variant (CaMKIIδB) where a nuclear localization sequence is part of the hypervariable region (between the regulatory and association domains). CaMKIIδB is relatively, but not exclusively, enriched in the nucleus. [34]
Under basal conditions, CaMKII is biased toward inactivation because the catalytic domain is constrained by the pseudosubstrate region of the regulatory domain (Fig 1). When intracellular Ca2+ rises it calcifies calmodulin (Ca2+/CaM), a Ca2+ sensing protein, to engage the calmodulin binding region of the regulatory domain. Ca2+/CaM binding allosterically displaces the catalytic domain from the inhibition of the regulatory domain, leading to CaMKII activation. Once active, the pseudosubstrate region of the regulatory domain is available for various post-translational modifications that convert CaMKII from a Ca2+/CaM activated enzyme to a Ca2+/CaM autonomous enzyme. These Ca2+/CaM independent forms of CaMKII, due to threonine 287 ‘autophosphorylation’, [35, 36] methionine 281/282 oxidation [20], and serine 280 O-GlcNAclyation [37] are linked to myocardial disease, including increased myocardial death, heart failure and arrhythmias. [11] S-nitrosylation of Cysteine 290 in the regulatory domain also induces autonomous activation of CaMKII [38, 39]. This S-nitrosylation dependent activation of CaMKII takes part in β-adrenergic stimulation evoked Ca2+ sparks [39]. In neuron, S-nitrosylation induced CaMKII activation is involved in cell death [38]. However, whether this C290 nitrosylation induced CaMKII activation is also involved in β-adrenergic stimulation evoked myocyte death remains unclear. A recent study revealed that, besides cysteine 290, cysteine 273 can also be S-nitrosylated [40]. In contrast to C290 nitrosylation, C273 nitrosylation does not activate CaMKII, but inhibits Ca2+/calmodulin dependent activation of CaMKII. Therefore, nitric oxide plays a dual role in CaMKII signaling. It was suggested in this study that nitrosylation of CaMKII activates or inhibits the kinase activity depending on the timing of CaMKII exposure to nitric oxide relative to the timing of Ca2+/calmodulin binding. Exposure of CaMKII to nitric oxide prior to Ca2+/calmodulin binding, results in inhibition of CaMKII by nitrosylation of C273. Conversely, after Ca2+/calmodulin binding to CaMKII, S-nitrosylation of CaMKII preferentially occurs at the C290 site, leading to activation of CaMKII. Our view is that in vivo studies will be required to more completely elucidate the role of nitric oxide regulation of CaMKII in cardiac pathophysiology. Cysteine 290 can also be oxidized by hydrogen peroxide, resulting in Ca2+/calmodulin autonomous CaMKII activation [40]. Further research will be necessary to illuminate how the post-translational modifications in the regulatory domain interact with each other.
Fig 1. Activation pathways and subunit structure of CaMKII.
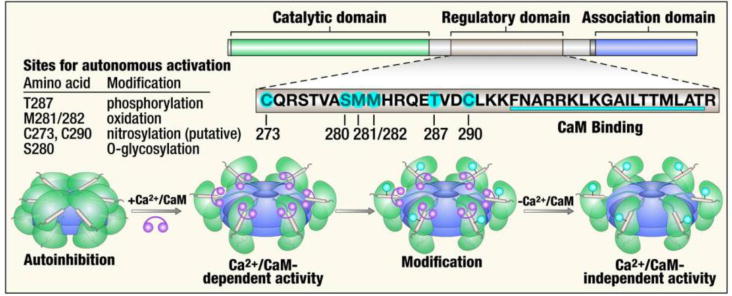
(top right and bottom) CaMKII monomers consist of an N-terminal catalytic domain, a central regulatory domain and a C-terminal association domain. The regulatory domain is bound to the catalytic domain under resting conditions, locking the kinase in the inactive state. The regulatory domain contains a calmodulin binding site, and a rise in intracellular Ca2+ promotes binding of Ca2+/calmodulin to this region, releasing the catalytic domain and activating the enzyme, (bottom right) Ca2+/calmodulin autonomous activation of CaMKII occurs by post-translational modifications. There are multiple post translational modification sites in the regulatory domain (table).
The role of CaMKII in myocardial apoptosis
Apoptosis is orchestrated by a series of evolutionarily conserved caspase cysteine proteases [2]. There are two categories of apoptosis pathways, intrinsic and extrinsic. Mitochondria are at the center of the intrinsic pathway. The activation of Bax and Bak proteins, the family members of Bcl2, permeabilize the outer membrane of mitochondria, leading to the release of mitochondrial apoptogenic proteins, including cytochrome C, to the cytosol. Cytosolic cytochrome C activates the execution pathway, consisting of caspase-3, caspase-6, and caspase-7. Extrinsic apoptosis is initiated by ligand-mediated activation of death receptors, including tumor necrosis factor-α (TNF-α), CD95 (Fas/APO-1) and TNF-related apoptosis-inducing ligand (TRAIL) receptor-1. Ligand binding stimulates the assembly of the death-inducing signaling complex (DISC, mainly consists of Fas-associated protein with death domain (FADD) and FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP)), which induces caspase-8 activation. After Caspase 8 activation, the extrinsic apoptosis pathway converges with the intrinsic apoptosis pathway at the level of executioner caspases-3, -6, and -7. [2]
CaMKII, Ca2+ overload and mitochondria-mediated intrinsic apoptotic pathway
Cytosolic Ca2+ overload is one of the hallmarks of heart failure and ischemic heart disease. Augmented L-type Ca2+ channel (LTCC) influx by overexpression of LTCC (CaV1.2, beta2a subunit) induced sarcoplasmic reticulum (SR) Ca2+ overload, which elicited the mitochondria dependent intrinsic apoptosis pathway [41]. Sustained, excessive CaMKII activation is an upstream signaling event for promoting SR Ca2+ triggered arrhythmias [42, 43] and increased LTCC opening probability, [23, 44] which contribute to excitation-contraction coupling dysfunction, myocardial hypertrophy, heart failure, arrhythmia and sudden cardiac death [10]. CaMKII induced loss of cellular Ca2+ homeostasis also appears to play a crucial role in cardiac myocyte apoptosis [17]. CaMKII inhibition in vivo, by transgenic expression of a myocardial targeted inhibitory peptide (AC3-I), protected against myocardial apoptosis induced by myocardial infarction or isoproterenol administration [18]. This protection appeared to involve reduced SR Ca2+ content in AC3-I mice. Interbreeding AC3-I mice with phospholamban knockout (Pln−/−) mice restores SR Ca2+ content to WT levels. The protection of AC3-I expression against myocardial apoptosis due to isoproterenol injection is reduced, and entirely lost after myocardial infarction in AC3-I transgenic mice interbred into a Pln−/− background. These findings highlight the SR Ca2+ dependence of CaMKII induced myocardial apoptosis. In contrast, interbreeding mice with transgenic myocardial CaMKIIδ over-expression with Pln−/− mice caused increased mitochondrial Ca2+, augmented cardiomyocyte death, worsened heart failure, and increased mortality [45]. Mutant ryanodine receptor (RyR2) knock-in mice, lacking a validated serine 2814 phosphorylation site (S2814A), were resistant to apoptosis and displayed improved cardiac function after myocardial infarction [46]. In addition, overexpression of mutant voltage-gated Ca(2+) channel (Ca(V)1.2) β subunits, which are resistant to CaMKII binding (L493A) and phosphorylation (T498A), delayed rapid-pacing induced cardiomyocyte death [47]. The final execution pathway for this CaMKII mediated, Ca2+ dependent apoptosis appears to be the mitochondria-mediated intrinsic pathway. Overexpression of CaMKIIδ in cultured cardiomyocytes led to increased apoptosis in the condition of oxidative stress, accompanied by elevated cytosolic Ca2+ and enhanced mitochondrial cytochrome c release [31], which is consistent with LTCC overexpression induced mitochondria dependent apoptosis [41]. However, a clear and comprehensive picture of CaMKII target proteins required for this process has yet to emerge.
CaMKII in p53 mediated apoptosis
p53 is a multi-functional protein, and a pro-apoptotic transcription factor with connections to intrinsic and extrinsic apoptotic pathways. Nuclear p53 accumulation and activation with excessive apoptosis appeared to be essential mechanisms promoting heart failure progression in dilated cardiomyopathy due to mutations in genes encoding cytoskeletal or myofilament proteins [48]. The mutant myofilament often led to decreased Ca2+ sensitivity. In apparent compensation for this deficiency, β-adrenergic signaling and Ca2+ transients are increased, which favors sustained CaMKII activation. Inhibition of CaMKII, by transgenic expression of AC3-I or small molecule inhibitors, in a mouse model of dilated cardiomyopthy due to overexpression of a mutated cardiac α-actin (R312H), attenuated p53 activation, reduced apoptosis, and improved cardiac function. It remains to be determined how CaMKII activates p53.
CaMKII in ER stress mediated apoptosis
Endoplasmic reticulum (ER) mediated synthesis and proper folding of multiple proteins, some posttranslational modifications, trafficking of newly synthesized proteins to the Golgi apparatus, lipid biosynthesis, and intracellular Ca2+ homeostasis are crucial for normal cellular functioning. However, defects in ER activities (i.e. ER stress) can trigger cell death [5]. The precise molecular mechanisms governing the switch from physiological ER functions to ER stress, and the crosstalk between stressed ER and death machinery remains incompletely understood. ER stress evoked increases in cytosolic Ca2+, and subsequent sustained CaMKII activation was a key mechanism of ER stress induced apoptosis in cholesterol loaded macrophages [22]. Under these conditions, CaMKII was activated by Ca2+ overload and oxidation, and contributed to 3 pro-apoptotic pathways. In the first pathway CaMKII directly activated the C-Jun N-terminal kinase (JNK) pathway, resulting in the upregulation of FAS ligand and the activation of the extrinsic apoptotic pathway. The second pathway was facilitated mitochondrial Ca2+ uptake by CaMKII that resulted in activation of the intrinsic apoptotic pathway with loss of ΔΨm and release of apoptogens, including cytochrome C. In cholesterol loaded macrophages, oxidized CaMKIIγ appeared to selectively partition into mitochondria, and CaMKIIγ−/− mice were protected from apoptosis by this pathway. The third independent pathway involved the signal transducer and activator of transcription 1 (STAT1) activation, a proapoptotic signal. This CaMKII mediated ER stress elicited cell death is also observed in other tissues and organs, including myocardium. CaMKII inhibition prevents ER stress induced cardiac dysfunction in a tunicamycin induced cardiomyopathy model, although whether the pathways described in cholesterol loaded macrophages are implicated in this model remains to be elucidated [49]. CaMKII appears to link excessive oxidant stress with ER stress and mitochondrial dysfunction in a mouse model of polycystic kidney disease, [50] suggesting this pathway may contribute to multiple diseases. Taken together, CaMKII activation either by Ca2+ overload or oxidation, in the setting of ER stress, appears to be a key event driving the transition from ER adaptation to ER stress triggered apoptosis, and CaMKII activation is one of the crucial mediators in the crosstalk between stressed ER and mitochondria.
CaMKII, inflammation and apoptosis
Excessive inflammation following ischemia-reperfusion injury or myocardial infarction exacerbates myocyte death and worsens cardiac function. Several lines of evidence consistently showed CaMKII augments myocardial inflammatory responses after myocardial infarction and ischemia-reperfusion injury [21, 25]. We performed a gene expression profiling study on hearts isolated from mice with myocardial specific transgenic expression of a CaMKII inhibitory peptide (AC3-I) subjected to MI [21]. Interestingly, CaMKII inhibition substantially reduced the myocardial infarction-triggered expression of a constellation of proinflammatory genes. The activity of NF-κB, a master regulator of inflammation, is augmented after myocardial infarction and suppressed by CaMKII inhibition. Similarly, nuclear NF-κB mediated CaMKII activation augmented myocardial ischemia-reperfusion injury [25]. Activated CaMKII in cardiomyocytes following ischemia-reperfusion injury led to IκB kinase phosphorylation and concomitant increases in nuclear p65, heightened inflammation, marked by increased monocyte infiltration and elevated monocyte chemoattractant protein-1 (MCP-1), and apoptosis [25]. NF-κB activation, increased inflammatory response and apoptosis following MI are blunted in CaMKIIδ knockout mice (Camk2−/−) and in mice with myocardial transgenic expression of AC3-I or CaMKIIN. [21, 51] Increased inflammatory response by CaMKII in I/R injury was also observed in another independent study [28]. However, this study showed that CaMKII mediated inflammation played little role in the first five days following I/R injury, but was essential in chronic ventricular remodeling after I/R injury. In this study, Camk2−/− mice showed a similar increase in cell death in the acute phase, but displayed improved cardiac function 5 weeks after I/R injury. This improvement was associated with an attenuated inflammatory response, evidenced by decreased leukocyte infiltration and reduced expression of members of the chemokine (C-C motif) ligand family, in particular CCL3 (macrophage inflammatory protein-1α, MIP-1α). Overall, CaMKII appears to be an important mediator for integrating neurohormonal signaling and inflammatory responses in MI or I/R injury. This CaMKII mediated inflammatory response is, at least in part, cardiac myocyte dependent, as cardiac specific CaMKII knockout or myocardial transgenic overexpression of CaMKII inhibitory peptide were employed in the studies discussed here.
The role of CaMKII in myocardial necroptosis
Necrosis has been regarded as a passive and unregulated process. However, this concept has been challenged by a growing body of work, strongly suggesting that a subset of necrosis is also regulated or programmed. The recognition of programmed necrosis shifted the focus on cardiac myocyte death in cardiovascular disease, and emerging evidence indicates that programmed necrosis or necroptosis also plays a critical role in the pathogenesis of common cardiovascular diseases [4].
The best characterized necroptosis molecular pathway is partially overlapping with apoptosis, and shares death-domain containing receptors including TNF-α, Fas receptor, and the TRAIL receptor [52, 53]. After the engagement of a pro-necroptotic ligand to its death receptor, the receptor recruits multiple proteins to form a complex mainly consisting of cellular inhibitor of apoptosis proteins (cIAPs), TNFR-associated death domain (TRADD), receptor-interacting protein 1 (RIP1) [54] and TNFR-associated factor 2 (TRAF2), which is different from DISC (Death Inducing Signaling Complex) [55]. This protein complex evolves to the necroptosome after disassociation from the death receptor, and association of proteins, including, RIP1 and RIP3 [52, 53, 56, 57]. Necrostatin, a RIP1 inhibitor, has been shown to protect against myocardial ischemia-reperfusion injury [58, 59]. The downstream signaling pathways of RIP1/RIP3 complex are less understood, but are increasingly studied. Interestingly, CaMKII was recently found to be one of the targets of RIP3 kinase, and RIP3 deletion was broadly protective against acute and chronic ischemia-reperfusion injury, and doxorubicin induced cardiomyopathy. The RIP3 knock out mice (Ripk3−/−) displayed attenuated myocyte death, including necrosis and apoptosis, and improved cardiac function and survival after ischemia-reperfusion injury or doxorubicin challenge [26]. CaMKII phosphorylation, oxidation and activity were blunted in Ripk3−/− mice subjected to ischemia-reperfusion injury or doxorubicin. In cultured neonatal myocytes, CaMKII phosphorylation was increased with overexpression of RIP3, and CaMKII inhibition either by dominant-negative CaMKII or KN-93, a small molecule CaMKII inhibitor, decreased cell death in the setting of RIP3 over-expression. Intriguingly, RIP3 directly binds and phosphorylates CaMKII. RIP3 co-localized with CaMKII in myocytes and a co-immunoprecipitation study showed the binding of RIP3 and CaMKII was enhanced in ischemia-reperfusion injury or after doxorubicin challenge. Importantly, in a cell-free recombinant protein system, RIP3 kinase directly phosphorylated recombinant CaMKII protein at the site of T287, the autophosphorylation site [26]. This is the first evidence, to our knowledge, that a kinase, other than CaMKII itself, is able to directly phosphorylate CaMKII. Thus, RIP3 contributes to myocardial necroptosis by acting as a CaMKII kinase.
A growing body of evidence suggests that the opening of mPTP is part of a final pathway in programmed necrosis. siRNA mediated knockdown of Cyclophilin D, a protein that increases the opening probability of the mPTP, markedly protected against RIP3 induced cardiomyocyte necrosis. Overexpression of RIP3 led to depolarization of ΔΨm in a Cyclophilin D-dependent manner, and RIP3 deficiency protected the heart from ischemia-reperfusion injury and doxarubicin-induced loss of ΔΨm. Inhibition of CaMKII blocked RIP3-induced ΔΨm depolarization. Thus, this line of evidence indicated mPTP is a critical downstream effector of the RIP3-CaMKII necroptosis pathway. While this and other studies [29] indicate that CaMKII increases mPTP opening, the identity of the CaMKII targets remains unknown. Taken together, these findings suggest CaMKII importantly contributes to cardiovascular disease by RIP3 activation and that inhibition of the RIP3-CaMKII pathway inhibition may be an attractive candidate therapeutic target.
The role of oxidized CaMKII in myocardial apoptosis
Both calcium and reactive oxygen species (ROS) play a crucial role in pathogenesis of cardiac diseases. CaMKII, a kinase originally identified by Ca2+ and calmodulin dependent activation, is persistently activated through the oxidation of a pair of methionines in its regulatory domain (at positions M280/281), independent of Ca2+ and calmodulin binding [20]. Oxidative activation of CaMKII (ox-CaMKII) occurs via a variety of upstream ROS resources, including NAPDH oxidases (NOXs) and mitochondria. Angiotensin II [20, 60] and aldosterone [61]are each capable of oxidizing CaMKII by activation of Nox2. This AngII/Nox2/ox-CaMKII pathway is essential to angiotensin II induced cardiomyocyte apoptosis. CaMKII inhibition, over-expression of methionine sulfoxide reductase A (MsrA), a reductase capable of reversing the first oxidation step for methionines (i.e. to a sulfoxide) and loss of p47, a key component of Nox2 in myocardium, all protected against angiotensin II induced apoptosis. MsrA knockout mice (Msra−/−) infused with angiotensin II exhibit elevated ox-CaMKII, extensive apoptosis, rapidly accelerated cardiac dysfunction and increased mortality. The same pattern is observed in Msra−/− mice subjected to myocardial infarction. [20] This angiotensin II/Nox2/ox-CaMKII pathway can contribute to apoptosis of cardiac pacemaker cells, leading to sinus node dysfunction. [60] Our group developed knockin mice where CaMKIIδ M280/281 were replaced by valines (MMVV) in order to directly determine the impact of ox-CaMKII on cardiovascular disease in vivo. Patients with diabetes who suffer myocardial infarction showed increased myocardial ox-CaMKII compared to non-diabetic patients after myocardial infarction. Mice with streptozotocin (STZ)-induced diabetes and subjected to myocardial infarction, displayed increased ox-CaMKII, profound bradycardia secondary to sinoatrial nodal pacemaker cell apoptosis, loss of normal heart rate variability, and markedly increased mortality. STZ-treated, ox-CaMKII resistant MMVV mice showed substantially improved pacemaker cell survival, preserved heart rate and elimination of diabetes attributable increases in mortality after myocardial infarction. [24] Interestingly, in this STZ induced diabetes model [62], and in heart failure model with Gαq protein overexpression [63], CaMKII was also a crucial mediator of ROS production, suggesting the possibility that ox-CaMKII contributes to a ROS induced ROS positive feedback loop. Importantly, pro-oxidant environments can enhance the sensitivity of CaMKII activation by Ca2+ and calmodulin, so that CaMKII activation occurs at resting or ambient intracellular Ca2+ levels [64].
Many questions arise with the finding of ox-CaMKII induced cell death. Does the ox-CaMKII induced cell death share the same upstream and downstream pathways of cell death linked to autophosphorylation? How do upstream signals, such as ROS, O-GlcNAcylation, and Ca2+, interact to activate CaMKII? Are these activation pathways hierarchical, and is the fidelity of regulatory pathways (e.g. MsrA for ROS, RIP3 for autophosphorylation and OGA for O-GlcNAcylation) preserved in the setting of multiple CaMKII activating post-translational modifications? Do the various post-translationally modified forms of CaMKII have the same subcellular domain localization and protein targets?
ox-CaMKII in inflammation and apoptosis
Oxidative stress and ROS production are associated with inflammation and in the context of disease with excessive inflammatory response. Toll-like receptor (TLR) activation promotes NF-κB-dependent inflammatory transcription and oxidative injury in myocardium after myocardial infarction. Our group demonstrated that ox-CaMKII is a mediator of TLR-4 signaling induced myocardial injury following myocardial infarction [51]. TLR-4 activation by lipopolysaccharide, or endotoxin, induces a ROS burst followed by elevated ox-CaMKII in cardiomyocytes. The myeloid differentiation protein 88 (MyD88) is an adapter protein critical for TLR-4 signaling. MyD88 knockout mice subjected to myocardial infarction or lipopolysaccharide injection showed decreased ox-CaMKII, attenuated pro-inflammatory gene expression, reduced myocyte apoptosis, reduced infarction size and improved cardiac function. Thus, ox-CaMKII may be increased by a diverse array of upstream signals associated with oxidant stress, and contribute to inflammation and apoptosis.
CaMKII activation pathways in myocardial necroptosis
ROS is a critical determinant of necroptosis. TNFα treatment induced necroptosis is associated with increased ROS production and treatment with the ROS scavengers, butylated hydroxnisole (BHA) or N-acetylcysteine (NAC), delays TNFα-induced necroptosis in several cell types [65]. Inhibition of mitochondrial complexes I and II reduce ROS and protect L929 cells against TNF-induced necroptosis [66]. In addition to mitochondrial ROS, RIP1 and RIP3 also can directly activate NAPDH oxidases, which is involved in the execution of necroptosis [67]. The production of ROS following TNFα treatment is decreased by RIP1 or RIP3 ablation [56]. RIP3 knock out mice subjected to ischemia-reperfusion injury or exposed to doxorubicin were protected from increases in ROS, ox-CaMKII and autophosphorylated CaMKII [26]. In cultured neonatal cardiomyocytes, Nox2 knockdown or Tiron (a ROS scavenger) treatment, ameliorated RIP3 overexpression induced cell necroptosis [26]. CaMKIIδ overexpression augmented RIP3 induced necroptosis, but CaMKII mutants resistant to autophosphorylation (T287A) or oxidation (MMVV) attenuated RIP3 induced necroptosis [26]. Cells expressing CaMKII lacking the oxidation and autophosphorylation sites were completely protected against RIP3 mediated necroptosis [26]. Thus, both CaMKII phosphorylation and oxidative activation of CaMKII seem to contribute to RIP3 mediated necroptosis.
Selective participation of subcellular CaMKII compartments in programmed cell death
Under physiological conditions, mitochondrial Ca2+ uptake is part of a metabolic response pathway for increasing activity of the tricarboxcylic acid cycle to increase reducing equivalents, mostly in the form of NADPH, and thereby enhance oxidative phosphorylation and ATP production. However, excessive mitochondrial Ca2+ triggers apoptosis or programmed necrosis through mPTP opening. We identified CaMKII as a component of the mitochondrial matrix, and generated transgenic mice with myocardial and mitochondrial targeted expression of CaMKIIN (mtCaMKIIN) to determine if mitochondrial CaMKII activity could participate in myocardial injury responses [29]. Hearts from mtCaMKIIN mice were resistant to ischemia reperfusion injury, myocardial infarction, and isoproterenol toxicity. These mtCaMKIIN hearts displayed reduced myocardial infarction size and increased myocardial performance after ischemia-reperfusion injury. Isolated cardiomyocytes from mtCaMKIIN mice were protected from mPTP opening and loss of ΔΨm in response to Ca2+ challenge. We found that mitochondrial CaMKII bound to the mitochondrial Ca2+ uniporter (MCU) and identified 2 CaMKII catalyzed phosphorylation sites of CaMKII on MCU (serine 57 and 92). We developed evidence that CaMKII increased MCU current. However, this last finding is controversial and we are continuing to evaluate mitochondrial targets that explain CaMKII actions. Two recent studies employing cardiac specific MCU knockout mice (Mcu−/−) showed MCU deletion prevented mPTP opening and ischemia-reperfusion injury in vivo. [68, 69] The Mcu−/− mice had deficient exercise responses, consistent with the known metabolic benefits of physiological increases in mitochondrial matrix Ca2+. In contrast, mice with constitutive, global loss of MCU showed mild decreases in exercise capacity and strength but no protection from ischemia-reperfusion injury [70]. We developed mice with loss of myocardial MCU activity by α-myosin heavy chain promoter driven expression of a dominant-negative MCU, due to charge reversal mutations in the pore forming domain [71]. These dominant negative MCU mice showed reduced heart rate adaptation to stress [72], increased oxygen consumption and diminished ATP production in response to physiological stress, confirming that MCU mediated mitochondrial Ca2+ uptake is essential to metabolic ‘fight or flight’ responses. Similar to findings in mice with constitutive loss of MCU, the dominant negative MCU transgenic hearts were not protected from ischemia-reperfusion injury despite preservation of the ΔΨm and reduced ROS production. The dominant negative MCU mice had elevations in cytosolic Ca2+ that was repaired by cell dialysis of ATP, suggesting that metabolic deficiency, rather than loss of mitochondrial Ca2+ buffering was primarily responsible for defective Ca2+ homeostasis and impaired mechanical performance. Chronic loss of MCU led to widespread alterations in gene transcription in dominant negative MCU hearts. Notably, expression of Bax was significantly increased, potentially contributing to the lack of improved survival after myocardial ischemia. Taken together, these findings suggest that mitochondrial CaMKII contributes to cell death and mitochondrial CaMKII inhibition protects against clinically relevant models of myocardial injury and death by incompletely understood mechanisms. Timing and duration appear to be critical variables determining the consequences of loss of MCU current.
Nuclear CaMKII plays a critical role in transcriptional regulation in response to pathological stress. Nuclear CaMKII activation phosphorylates and exports class IIa HDACs [73]. The inhibition of Class IIa HDACs by nuclear CaMKII contributes to hypertrophic gene activation and fetal gene reprogramming in cardiac hypertrophy and heart failure. CaMKIIδ has more than 10 splicing variants [74, 75]. Among them, CaMKIIδB and CaMKIIδC are present in cardiac myocyte and have been extensively studied. CaMKIIδB and CaMKIIδC possess similar catalytic activity and Ca2+ and calmodulin binding affinity. CaMKIIδB contains a nuclear localization sequence that is absent in CaMKIIδC. Overexpression of CaMKIIδB and CaMKIIδC with GFP-Tag Showed that CaMKIIδB is predominantly localized in the nucleus whereas CaMKIIδC is localized mainly in cytosol [34]. Although CaMKIIδB and CaMKIIδC have distinct distribution in myocytes, it should be kept in mind that the localization of both subtypes is not exclusively restricted to nuclear or cytosol. The lack of high fidelity nuclear targeting by CaMKIIdB may be because the CaMKII NLS has multiple serines that when phosphorylated prevent nuclear targeting. [76]
CaMKIIδB and CaMKIIδC have opposite impact on cardiomyocyte death. Overexpression of CaMKIIδB protected against doxorubicin induced cardiomyocyte apoptosis and doxorubicin induced cardiomyopathy. CaMKIIδB induced cardioprotection appears to be related to promotion of GATA4 binding to the promoter of Bcl2, an antiapoptotic gene, thus increasing the expression of BCL2 protein [77]. This CaMKIIδB induced prosurvival signaling pathway may contribute to cardiac progenitor cell survival, commitment and differentiation [78]. CaMKIIδB can also protect cardiac myocytes against oxidative stress, angiotensin II or hypoxia induced apoptosis and myocardial ischemia/reperfusion injury [79]. The underlying mechanism of this protection appears to be mediated by promotion of inducible heat shock protein 70 (HSP70) expression through phosphorylation and activation of heat shock factor 1 (HSF1) [79]. Increased HSF1/HSP70 was not observed in CaMKIIδC overexpression. However, CaMKIIδB over-expression also induces modest myocardial hypertrophy, so is not entirely beneficial [80]. In contrast to CaMKIIδB over expression models, CaMKIIδC over expression promotes cell death and sensitizes myocardium to a variety of stress conditions [31].-Despite these opposing actions, it seems clear that the total deletion of CaMKIIδ is beneficial in pathological disease, at least in mouse models [13, 14].
CaMKII isoforms may heteromultimerize to form the holoenzyme, and the subcellular distribution of CaMKIIδ holoenzymes depends on the ratio of the B and C splice variants [34]. Importantly, the spatial and functional specificity of CaMKIIδ appears to be determined by the selective mobilization of subcellular Ca2+ stores [34]. For example, phenylepherine stimulation induced HDAC4 nuclear exportation by activation of CaMKIIδC, the cytosolic isoform of CaMKIIδ. Caffeine evoked Ca2+ SR release was able to activate CaMKIIδB, the nuclear isoform of CaMKIIδ, and subsequently phosphorylated phospholamban. It will be important to better understand the function of potential subcellular pools of CaMKII. However, there is now broad agreement that CaMKII inhibition, without subcellular targeting, is protective against cardiac injury. Thus, it seems unlikely to us that subcellular targeting of CaMKII inhibition will be necessary to evolve new CaMKII inhibitor therapies.
In summary, emerging evidence shows CaMKII is a nodal signal promoting cardiac myocyte death. CaMKII plays a central role, in part, because it is a convergence point for β-adrenergic receptor signaling, ROS, hyperglycemia, Gαq protein coupled signaling (e.g. endothelin-1, angiotensin II), mineralocorticoid receptor signaling, TLR signaling and signaling by pro-death cytokines. Many questions remain to be answered regarding the upstream and downstream pathways and subcellular domains participating in CaMKII induced cell death.
Figure 2. Molecular mechanisms of CaMKII mediated myocardial necroptosis.
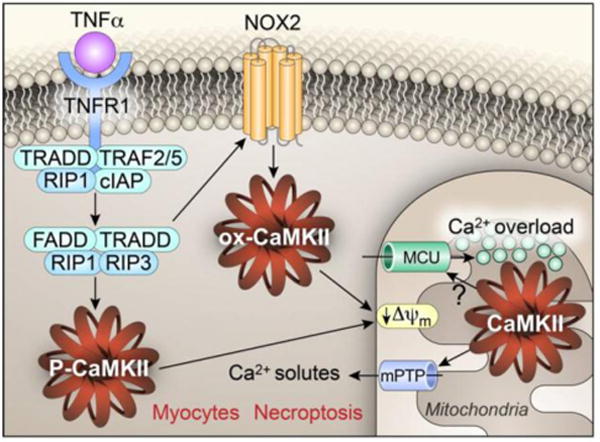
The engagement of tumor necrosis factor alpha (TNFα) with its receptor leads to the formation of necroptosome. mainly consisting of receptor-interacting serine/threonine-protein kinase 1 and 3 (RIP1 and RIP3). RIP3 activation induces CaMKII activation through direct phosphorylation of CaMKII at threonine 287 (P-CaMKII, CaMKIId numbering) or oxidation of CaMKII via NOX2 stimulation (ox-CaMKII) at methionines 281 and282. Both P-CaMKII and ox-CaMKII contribute to myocardial necroptosis by triggering mitochondrial permeability transition pore (mPTP) opening, which leads to the collapse of the inner mitochondrial membrane potential (ΔΨm). The mechanism of CaMKII biased mPTP opening is unclear, but CaMKII induced mitochondrial Ca2+ uniporter (MCU) hyperactivity and Ca2+ overload may contribute.
Highlights.
CaMKII is activated and contributes to cardiomyocyte death by β-adrenergic signaling, Gq protein coupled receptor signaling, ROS, intracellular Ca2+, and pro-death cytokines
CaMKII elicits cardiac myocyte apoptosis in both intrinsic and extrinsic apoptotic pathway
CaMKII is an essential mediator of RIP3 induced myocardial necroptosis
Both Ca2+/calmodulin dependent activation and oxidative activation of CaMKII play a critical role in programmed cell death
Acknowledgments
Mr. Shawn Roach produced the artwork for the figures. This work was supported, in part, by National Institutes of Health (NIH) Grants R01-HL079031, R01-HL096652, R01-HL070250, and R01-HL071140 to MEA and K08HL130604 to NF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
MEA is a cofounder of Allosteros Therapeutics, a company aiming to develop CaMKII inhibitor therapeutics.
References
- 1.Fisher SA, Langille BL, Srivastava D. Apoptosis during cardiovascular development. Circ Res. 2000;87(10):856–64. doi: 10.1161/01.res.87.10.856. [DOI] [PubMed] [Google Scholar]
- 2.Kitsis RN, Mann DL. Apoptosis and the heart: a decade of progress. J Mol Cell Cardiol. 2005;38(1):1–2. doi: 10.1016/j.yjmcc.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 4.Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart. Circ Res. 2011;108(8):1017–36. doi: 10.1161/CIRCRESAHA.110.225730. [DOI] [PubMed] [Google Scholar]
- 5.Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32(7):1552–62. doi: 10.1161/ATVBAHA.111.224915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471(7338):368–72. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471(7338):373–6. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rokita AG, Anderson ME. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII) Circulation. 2012;126(17):2125–39. doi: 10.1161/CIRCULATIONAHA.112.124990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ Res. 2013;113(6):690–708. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson ME. Oxidant stress promotes disease by activating CaMKII. J Mol Cell Cardiol. 2015;89(Pt B):160–7. doi: 10.1016/j.yjmcc.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, Hund TJ, Kutschke W, Sarma S, Grumbach IM, Wehrens XH, Mohler PJ, Song LS, Anderson ME. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci U S A. 2009;106(14):5972–7. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106(7):2342–7. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106(10):1288–93. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 16.Wagner S, Maier LS, Bers DM. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res. 2015;116(12):1956–70. doi: 10.1161/CIRCRESAHA.116.304678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111(5):617–25. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP, Anderson ME. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291(6):H3065–75. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 19.Luczak ED, Anderson ME. CaMKII oxidative activation and the pathogenesis of cardiac disease. J Mol Cell Cardiol. 2014;73:112–6. doi: 10.1016/j.yjmcc.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, Weiss RM, Zimmermann K, Yull FE, Blackwell TS, Mohler PJ, Anderson ME. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest. 2009;119(4):986–96. doi: 10.1172/JCI35814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119(10):2925–41. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu W, Tsang S, Browe DM, Woo AY, Huang Y, Xu C, Liu JF, Lv F, Zhang Y, Xiao RP. Interaction of beta1-adrenoceptor with RAGE mediates cardiomyopathy via CaMKII signaling. JCI Insight. 2016;1(1):e84969. doi: 10.1172/jci.insight.84969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123(3):1262–74. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112(6):935–44. doi: 10.1161/CIRCRESAHA.112.276915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22(2):175–82. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 27.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundina-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73(4):689–98. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Weinreuter M, Kreusser MM, Beckendorf J, Schreiter FC, Leuschner F, Lehmann LH, Hofmann KP, Rostosky JS, Diemert N, Xu C, Volz HC, Jungmann A, Nickel A, Sticht C, Gretz N, Maack C, Schneider MD, Grone HJ, Muller OJ, Katus HA, Backs J. CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med. 2014;6(10):1231–45. doi: 10.15252/emmm.201403848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491(7423):269–73. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sag CM, Kohler AC, Anderson ME, Backs J, Maier LS. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J Mol Cell Cardiol. 2011;51(5):749–59. doi: 10.1016/j.yjmcc.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282(14):10833–9. doi: 10.1074/jbc.M611507200. [DOI] [PubMed] [Google Scholar]
- 32.Kreusser MM, Lehmann LH, Keranov S, Hoting MO, Oehl U, Kohlhaas M, Reil JC, Neumann K, Schneider MD, Hill JA, Dobrev D, Maack C, Maier LS, Grone HJ, Katus HA, Olson EN, Backs J. Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation. 2014;130(15):1262–73. doi: 10.1161/CIRCULATIONAHA.114.006185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92(8):912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 34.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res. 2011;109(12):1354–62. doi: 10.1161/CIRCRESAHA.111.248401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schworer CM, Colbran RJ, Soderling TR. Reversible generation of a Ca2+-independent form of Ca2+(calmodulin)-dependent protein kinase II by an autophosphorylation mechanism. J Biol Chem. 1986;261(19):8581–4. [PubMed] [Google Scholar]
- 36.Colbran RJ, Smith MK, Schworer CM, Fong YL, Soderling TR. Regulatory domain of calcium/calmodulin-dependent protein kinase II. Mechanism of inhibition and regulation by phosphorylation. J Biol Chem. 1989;264(9):4800–4. [PubMed] [Google Scholar]
- 37.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502(7471):372–6. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coultrap SJ, Bayer KU. Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) J Biol Chem. 2014;289(28):19458–65. doi: 10.1074/jbc.M114.558254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E. NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc Res. 2013;100(3):392–401. doi: 10.1093/cvr/cvt201. [DOI] [PubMed] [Google Scholar]
- 40.Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-Nitrosylation Induces Both Autonomous Activation and Inhibition of Calcium/Calmodulin-dependent Protein Kinase II delta. J Biol Chem. 2015;290(42):25646–56. doi: 10.1074/jbc.M115.650234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res. 2005;97(10):1009–17. doi: 10.1161/01.RES.0000189270.72915.D1. [DOI] [PubMed] [Google Scholar]
- 42.Wu Y, Roden DM, Anderson ME. Calmodulin kinase inhibition prevents development of the arrhythmogenic transient inward current. Circ Res. 1999;84(8):906–12. doi: 10.1161/01.res.84.8.906. [DOI] [PubMed] [Google Scholar]
- 43.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127(8):913–22. doi: 10.1161/CIRCULATIONAHA.12.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol. 1999;276(6 Pt 2):H2168–78. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- 45.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106(2):354–62. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, Palomeque J, Aiello EA, Skapura DG, Rinaldi G, Respress JL, Brown JH, Wehrens XH, Salas MA, Mattiazzi A. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2014;74:274–83. doi: 10.1016/j.yjmcc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010;107(11):4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toko H, Takahashi H, Kayama Y, Oka T, Minamino T, Okada S, Morimoto S, Zhan DY, Terasaki F, Anderson ME, Inoue M, Yao A, Nagai R, Kitaura Y, Sasaguri T, Komuro I. Ca2+/calmodulin-dependent kinase IIdelta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation. 2010;122(9):891–9. doi: 10.1161/CIRCULATIONAHA.109.935296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roe ND, Ren J. Oxidative activation of Ca(2+)/calmodulin-activated kinase II mediates ER stress-induced cardiac dysfunction and apoptosis. Am J Physiol Heart Circ Physiol. 2013;304(6):H828–39. doi: 10.1152/ajpheart.00752.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bracken C, Beauverger P, Duclos O, Russo RJ, Rogers KA, Husson H, Natoli TA, Ledbetter SR, Janiak P, Ibraghimov-Beskrovnaya O, Bukanov NO. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. Am J Physiol Renal Physiol. 2016;310(11):F1414–22. doi: 10.1152/ajprenal.00426.2015. [DOI] [PubMed] [Google Scholar]
- 51.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52(5):1135–44. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 53.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 54.Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–96. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 55.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 56.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 58.Koudstaal S, Oerlemans MI, Van der Spoel TI, Janssen AW, Hoefer IE, Doevendans PA, Sluijter JP, Chamuleau SA. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur J Clin Invest. 2015;45(2):150–9. doi: 10.1111/eci.12391. [DOI] [PubMed] [Google Scholar]
- 59.Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, Sluijter JP. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107(4):270. doi: 10.1007/s00395-012-0270-8. [DOI] [PubMed] [Google Scholar]
- 60.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest. 2011;121(8):3277–88. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ, Anderson ME. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med. 2011;17(12):1610–8. doi: 10.1038/nm.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishio S, Teshima Y, Takahashi N, Thuc LC, Saito S, Fukui A, Kume O, Fukunaga N, Hara M, Nakagawa M, Saikawa T. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J Mol Cell Cardiol. 2012;52(5):1103–11. doi: 10.1016/j.yjmcc.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 63.Westenbrink BD, Ling H, Divakaruni AS, Gray CB, Zambon AC, Dalton ND, Peterson KL, Gu Y, Matkovich SJ, Murphy AN, Miyamoto S, Dorn GW, 2nd, Heller Brown J. Mitochondrial reprogramming induced by CaMKIIdelta mediates hypertrophy decompensation. Circ Res. 2015;116(5):e28–39. doi: 10.1161/CIRCRESAHA.116.304682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, Mattiazzi A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105(12):1204–12. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 65.Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA, Liu ZG. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279(11):10822–8. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 66.Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267(8):5317–23. [PubMed] [Google Scholar]
- 67.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26(5):675–87. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 68.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015;12(1):15–22. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015;12(1):23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–72. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A. 2015;112(29):9129–34. doi: 10.1073/pnas.1504705112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS, Anderson ME. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. doi: 10.1038/ncomms7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116(7):1853–64. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J Biol Chem. 1993;268(19):14443–9. [PubMed] [Google Scholar]
- 75.Edman CF, Schulman H. Identification and characterization of delta B-CaM kinase and delta C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim Biophys Acta. 1994;1221(1):89–101. doi: 10.1016/0167-4889(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 76.Heist EK, Srinivasan M, Schulman H. Phosphorylation at the nuclear localization signal of Ca2+/calmodulin-dependent protein kinase II blocks its nuclear targeting. J Biol Chem. 1998;273(31):19763–71. doi: 10.1074/jbc.273.31.19763. [DOI] [PubMed] [Google Scholar]
- 77.Little GH, Saw A, Bai Y, Dow J, Marjoram P, Simkhovich B, Leeka J, Kedes L, Kloner RA, Poizat C. Critical role of nuclear calcium/calmodulin-dependent protein kinase IIdeltaB in cardiomyocyte survival in cardiomyopathy. J Biol Chem. 2009;284(37):24857–68. doi: 10.1074/jbc.M109.003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quijada P, Hariharan N, Cubillo JD, Bala KM, Emathinger JM, Wang BJ, Ormachea L, Bers DM, Sussman MA, Poizat C. Nuclear Calcium/Calmodulin-dependent Protein Kinase II Signaling Enhances Cardiac Progenitor Cell Survival and Cardiac Lineage Commitment. J Biol Chem. 2015;290(42):25411–26. doi: 10.1074/jbc.M115.657775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peng W, Zhang Y, Zheng M, Cheng H, Zhu W, Cao CM, Xiao RP. Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ Res. 2010;106(1):102–10. doi: 10.1161/CIRCRESAHA.109.210914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J, Jr, Brown JH. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277(2):1261–7. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]