

ABSTRACT
The pathogen-associated 16S rRNA methyltransferase NpmA catalyzes m1A1408 modification to block the action of structurally diverse aminoglycoside antibiotics. Here, we describe the development of a fluorescence polarization binding assay and its use, together with complementary functional assays, to dissect the mechanism of NpmA substrate recognition. These studies reveal that electrostatic interactions made by the NpmA β2/3 linker collectively are critical for docking of NpmA on a conserved 16S rRNA tertiary surface. In contrast, other NpmA regions (β5/β6 and β6/β7 linkers) contain several residues critical for optimal positioning of A1408 but are largely dispensable for 30S binding. Our data support a model for NpmA action in which 30S binding and adoption of a catalytically competent state are distinct: docking on 16S rRNA via the β2/3 linker necessarily precedes functionally critical 30S substrate-driven conformational changes elsewhere in NpmA. This model is also consistent with catalysis being completely positional in nature, as the most significant effects on activity arise from changes that impact binding or stabilization of the flipped A1408 conformation. Our results provide a molecular framework for aminoglycoside resistance methyltransferase action that may serve as a functional paradigm for related enzymes and a starting point for development of inhibitors of these resistance determinants.
KEYWORDS: RNA binding proteins, aminoglycosides, antibiotic resistance, methyltransferase, rRNA modification, ribosomes
INTRODUCTION
Aminoglycosides are potent antimicrobial agents used for clinical treatment of life-threatening infections of both Gram-positive and Gram-negative bacteria and are also used routinely for both veterinary and growth promotion applications in agricultural settings (1, 2). Most aminoglycosides bind 16S rRNA helix 44 (h44) to induce conformational changes in the universally conserved nucleotides A1492 and A1493 in the ribosome decoding center. As a result, the bacterial ribosome is rendered unable to accurately discern cognate mRNA-tRNA pairing, thus impairing translational fidelity (3–9). More recent evidence has also suggested an additional 23S rRNA binding site for some aminoglycosides which disrupts intersubunit bridge B2, impacting a ribosomal conformational change required during elongation (10).
Both aminoglycoside-producing and human-pathogenic bacteria can achieve high levels of resistance to aminoglycosides by reducing drug permeability or increasing efflux from the cell, enzymatic chemical modification of the drug, or mutation or chemical modification of the aminoglycoside binding site (4, 11, 12). In particular, S-adenosyl-l-methionine (SAM)-dependent 16S rRNA methyltransferases are the predominant resistance mechanism found in aminoglycoside-producing bacteria and are an increasing clinical concern with their continued emergence in major human pathogens (2, 13). These enzymes site specifically methylate 16S rRNA in the ribosome decoding center to prevent aminoglycoside binding and thus confer exceptionally high levels of resistance to this class of drug (3, 13–15). Genes encoding aminoglycoside resistance 16S rRNA methyltransferases appear to be globally disseminated and are often located on plasmids or within other mobile genetic elements, frequently in conjunction with other antimicrobial resistance genes (2, 16). Together, the conserved nature of the ribosomal target of these methyltransferase enzymes as well as the apparent transmissibility of their activity to both Gram-positive and pathogenic Gram-negative bacteria underscore the urgent need to develop the means to counteract these resistance determinants (2, 17).
The structure of the pathogen-associated m1A1408 aminoglycoside resistance methyltransferase NpmA bound to the 30S ribosomal subunit provided a first snapshot of this enzyme-substrate complex in a precatalytic state (18). Comparison of free and 30S-bound NpmA revealed structural differences that suggested substrate recognition and methyltransferase activity are controlled by a combination of rigid docking of complementary surfaces and binding-induced conformational changes in both enzyme and substrate. However, the molecular mechanisms and relative contributions of these distinct processes to precise substrate recognition and modification cannot be discerned from the structures alone. Here, we describe the development of a fluorescence-based binding assay for 30S-NpmA interaction and its application in defining the mechanism of 30S substrate recognition and modification by the pathogen-associated aminoglycoside resistance methyltransferase NpmA.
RESULTS
A fluorescence assay to probe 30S-NpmA interaction.
To examine the 30S-NpmA interaction, we sought to develop a quantitative, fluorescence polarization (FP)-based assay using a single fluorescent probe incorporated site specifically into NpmA. We reasoned that this labeled NpmA probe should retain both 30S binding affinity and catalytic activity in this in vitro assay, i.e., exhibit methylation of A1408 in the presence of SAM and subsequent release from the 30S subunit. As a starting point, we used four previously created single-cysteine-substituted NpmA proteins (S89C, K131C, E184C, and E188C), which each exhibited high modification efficiency with other Cys-reactive reagents and retained the ability to confer resistance to kanamycin (Kan; MIC of >1,000 μg/ml) (18). These substitutions are distributed across the solvent-exposed surface of the 30S-bound NpmA (Fig. 1A and B).
FIG 1.

NpmA structure and development of a 30S-NpmA binding assay. (A) View of NpmA (purple) bound to the 30S subunit. Locations of unique Cys residues in NpmA incorporated for site-specific fluorescein labeling are shown as yellow spheres. Ribosomal proteins are shown in green and 16S rRNA in white, except helix 44 (h44), which is highlighted in tan. 30S features are labeled as head (h), platform (p), base (b), and stalk (s). (B) NpmA structure in three orthogonal views (top orientation is viewed from the 30S subunit, i.e., ∼180° rotation around the y axis from panel A). Sites of label incorporation are shown as described for panel A, and the NpmA β2/3 (cyan), β5/6 (slate), and β6/7 (purple) linkers are also highlighted. (C) Pilot analyses comparing FP signal for labeled NpmA variants before and after addition of SAM (noted by the dotted vertical line), either in the presence of 30S or alone (colored black for all three proteins). Only NpmA E184C* (green; bottom plot) exhibits increased FP in the presence of 30S and decreased FP after SAM addition, indicative of initial binding and subsequent dissociation of the enzyme following catalysis of methyl transfer, respectively.
Each of the four fluorescein-labeled NpmA variants was assessed in pilot FP experiments to determine which dye location provided the optimal probe of 30S-NpmA interaction. Labeling of NpmA at residue 131 (NpmA-K131C*) appeared to block 30S-NpmA interaction, as no difference in FP was observed in the presence of the 30S subunit compared to the free protein (Fig. 1C, top). In contrast, both NpmA-S89C* and NpmA-E188C* bound to 30S, as indicated by an increase in FP in the presence of 30S (Fig. 1C, center, and data not shown). However, both labeled proteins failed to dissociate upon addition of SAM, suggesting these labeled proteins were either defective in catalysis, unable to release the methylated 30S, or bound with comparable affinity to another site(s) on the 30S subunit. Finally, NpmA-E184C* also bound the 30S subunit and, critically, dissociated from the substrate following addition of SAM (Fig. 1C, bottom). Additionally, we found that NpmA-E184C* bound only 30S with unmethylated A1408 and not premodified m1A1408 subunits, NpmA-E184C* could be competed with unlabeled wild-type NpmA, and FP from untreated 30S-NpmA-E184C* remained stable over the full time course of these experiments (see Fig. S1 in the supplemental material). Thus, NpmA-E184C* fulfils the criteria to be a useful probe of 30S-NpmA interaction, and this protein was selected for use in all subsequent experiments.
The binding affinity of NpmA for the 30S subunit was first estimated by FP measurement using NpmA-E184C* and a range of 30S concentrations. Although complicated by apparent nonspecific binding at the highest 30S concentrations, this analysis yielded a binding affinity (Kd) for 30S-NpmA interaction of 25 nM (Fig. 2A). A competition assay was next established to simplify analysis of numerous NpmA variants; the binding affinities determined are from displacement of labeled NpmA-E184C* from 30S by each variant NpmA (and thus are denoted Ki). First, unlabeled NpmA protein (0.002 to 10 μM) was used to compete off prebound NpmA-E184C* from the 30S subunit (both at 0.05 μM), and a range of NaCl and Mg2+ concentrations was tested to identify conditions that provided the optimal initial FP signal while minimizing nonspecific 30S-NpmA interaction (i.e., increased FP above free NpmA-E184C* at the highest competitor concentration) (Fig. S2). Under the final conditions used, this assay yielded a Ki of 62 nM for unlabeled wild-type NpmA competitor (Fig. 2B) to serve as a benchmark for analyses of variant NpmA protein interactions with the 30S subunit.
FIG 2.

Measurement of 30S-NpmA binding affinity. (A) Direct measurement of NpmA-E184C* binding to the 30S subunit by FP. The arrow denotes the concentration of 30S subunits used in subsequent competition assays with unlabeled wild-type and variant NpmA proteins. (B) Competition FP binding experiment using wild-type NpmA to displace the NpmA-E184C* probe. Analysis of binding (Ki) of all NpmA variants was performed using this competition assay. Error bars represent the standard errors of the means (SEM).
The previously reported 30S-NpmA complex structure (18) revealed the likely predominant sites of interaction between NpmA and its substrate to reside in the regions linking β-strands 2 and 3 (β2/β3 linker), 5 and 6 (β5/β6 linker), and 6 and 7 (β6/β7 linker) of the conserved class I methyltransferase core fold. We therefore used our newly established competition FP assay in combination with complementary functional analyses to define the contributions of these linker regions, and putative key residues within them, to 30S substrate binding and site-specific recognition by NpmA.
The NpmA β2/3 linker drives interaction with the 30S subunit.
In the 30S-NpmA complex structure, the largely α-helical NpmA β2/3 linker directly contacts three 16S rRNA helices (h24, h27, and h45), which form a contiguous RNA surface in the assembled 30S structure (18). Potential interactions with the 16S rRNA phosphate backbone are mediated by four positively charged residues, K66, K67, K70, and K71 (Fig. 3A), suggesting that docking of NpmA onto the 30S subunit is driven by electrostatic interaction between two conformationally rigid surfaces. Consistent with this idea, the process noted above of optimizing conditions for the NpmA binding experiments revealed a marked sensitivity of the 30S-NpmA interaction to the solution ionic strength (Fig. S2).
FIG 3.

β2/3 linker residues are critical for 30S-NpmA interaction. (A) Four Lys residues of the NpmA β2/3 linker (cyan) interact with the phosphate backbone of nucleotides in h24, h27, and h45 of 16S rRNA. (B) Competition FP binding experiments with NpmA-E184C* and unlabeled NpmA proteins with single Lys-to-Glu substitutions in the β2/3 linker. Wild-type NpmA data, shown for comparison, are the same as those shown in Fig. 2B. Error bars represent the SEM. Binding affinity (Ki) for each variant protein derived from these data are shown in Table 1. (C) Same as panel B but for the double (K66E/K67E and K70E/K71E)- and quadruple (4× K→E)-substitution NpmA variants. (D) RT primer extension analysis of m1A1408 modification from E. coli cells expressing the indicated β2/3 linker variant NpmA proteins. The position of the band corresponding to A1408 is marked with an arrowhead, and the values below each lane are average intensities from at least two independent experiments normalized to wild-type NpmA (100%).
To directly examine the contribution of these β2/3 linker lysine residues to the 30S-NpmA interaction, we used single lysine-to-glutamic acid substitutions and measured binding of each NpmA variant in the competition FP assay (Fig. 3B). Each singly substituted variant exhibited a decrease in binding affinity, ranging from approximately 6-fold (K66E, K67E, and K71E) to 25-fold (K70E) reduction compared to wild-type NpmA (Table 1). Binding affinities for each doubly substituted NpmA (K66E/K67E or K70E/K71E) and the quadruple variant (K66E/K67E/K70/K71E) were reduced below the level measureable in this assay, with only a small reduction in FP at the highest competitor concentrations (Fig. 3C and Table 1). It is noteworthy that each double variant and the quadruple variant retained essentially identical affinity for SAM cosubstrate and the reaction by-product S-adenosyl-l-homocysteine (SAH) to wild-type NpmA (Table 1; Fig. S3), indicating that each variant is correctly folded and the lysine substitutions exclusively affect NpmA interaction with the 30S substrate.
TABLE 1.
Summary of kanamycin MICs and 30S and SAM/SAH binding affinities for substituted and linker deletion NpmA proteins
| Region and substitution | Kan MIC (μg/ml) | Kib (μM) 30S |
Kdc (μM) |
|
|---|---|---|---|---|
| SAM | SAH | |||
| Wild type | >1,024a | 0.06 (0.05, 0.08) | 46.3 (34.4, 58.2) | 0.61 (0.52, 0.70) |
| β2/β3 linker | ||||
| K66E | >1,024a | 0.37 (0.22, 0.64) | ND | ND |
| K67E | >1,024a | 0.39 (0.24, 0.64) | ND | ND |
| K70E | >1,024a | 1.5 (0.6, 3.5) | ND | ND |
| K71E | >1,024a | 0.30 (0.21, 0.43) | ND | ND |
| K66E/K67E | 256a | >5 | 19.2 | 0.7 |
| K70E/K71E | 1,024a | >5 | 19.0 | 0.6 |
| K66E/K67E/K70E/K71E | 16a | >5 | 17.2 | 0.8 |
| Linker deletions | ||||
| Δβ5/β6 linker | 8 | 0.02 (0.01, 0.04) | ∼2,900 | 24.5 |
| Δβ6/β7 linker | 16 | 0.15 (0.08, 0.27) | 375 | 3.8 |
| β5/β6 linker | ||||
| A148P | >1,024 | ND | ND | ND |
| E149P | >1,024 | ND | ND | ND |
| R153E | 512a | 0.34 (0.22, 0.53) | ND | ND |
| β6/β7 linker | ||||
| V190P | >1,024 | ND | 48 | 0.5 |
| L196Δ | 1,024 | ND | 84 | 8.3 |
| L196G | >1,024 | ND | 47.2 | 0.71 |
| R200E | 1,024a | 0.11 (0.07, 0.18) | ND | ND |
| L201P | 16 | 0.02 (0.01, 0.04) | 255 | 13 |
| R207E | 4–16a | 0.07 (0.04, 0.11) | 47 | 2.3 |
MICs for these NpmA variants were previously reported (18).
Binding affinities (Ki) for 30S-NpmA were determined by FP, with lower and upper limits of the 95% confidence interval (CI) shown in parentheses. ND, not determined.
NpmA-SAM/SAH affinities (Kd) were determined by ITC. The 95% CI of the means for 10 replicate experiments is shown in parentheses for wild-type NpmA; for other NpmA variants, a difference of <2-fold was not considered significant (see Materials and Methods). ND, not determined.
Previous analysis of resistance to kanamycin conferred by each NpmA β2/3 linker variant in Escherichia coli revealed no detectable effect for any single substitution (18), whereas both of the double variants had a reproducible but modest impact and the quadruple-substituted NpmA conferred essentially no resistance (Table 1). To more directly correlate 30S binding and NpmA activity, we next performed an analysis of 30S methylation in E. coli cells expressing each NpmA β2/3 linker variant (Fig. 3D). Consistent with the in vitro binding data, each single β2/3 linker substitution only modestly reduced the extent of A1408 methylation (∼66 to 89%). These results indicate that complete modification of all subunits is not required for a level of resistance (MIC) indistinguishable from that conferred by the wild-type enzyme (within the limits of the MIC assay with a maximum kanamycin concentration of 1,024 μg/ml). In contrast, cells expressing either doubly substituted or quadruple-substituted variants were more significantly reduced in their extent of A1408 methylation (to ∼11 to 14% and 4%, respectively; Fig. 3D, bottom), consistent with the greater impact on 30S binding and correlating with the observed MICs.
Collectively, these analyses reveal that the impact on NpmA activity of increasing charge reversal (K to E) substitutions in the β2/3 linker correlates well with both 30S-NpmA binding affinity and the enzyme's ability to incorporate the aminoglycoside resistance m1A1408 modification, within the limits of each assay. These results demonstrate the collective importance of the β2/3 linker residues in docking onto the rigid 16S rRNA tertiary surface of the 30S subunit and the subsequent capacity of NpmA to incorporate the m1A1408 modification and confer aminoglycoside resistance. Additionally, there appears to be sufficient redundancy in this set of electrostatic interactions that considerable loss of affinity may be tolerated in the bacterial cell (see Discussion).
Contributions of the NpmA β5/6 and β6/7 linkers to 30S-NpmA binding affinity.
NpmA variants with a complete deletion of either the β5/6 linker (amino acids 145 to 155) or β6/7 linker (amino acids 187 to 207) were created to examine the overall contribution of each region to 30S-NpmA binding affinity. Despite the size of each deletion, both proteins were soluble and appeared well folded from their elution as a symmetrical peak at the expected volume from a gel filtration column (Fig. S4). These deletions may be structurally tolerated given their location on the protein surface and the observed structural variability observed in these linker regions in NpmA and related enzymes (19–21).
Deletion of either linker fully ablated the ability of the enzyme to confer resistance in the MIC assay (Table 1), confirming the expected overall importance of these regions for NpmA activity. Additionally, in contrast to the case for all β2/3 linker variants tested, deletion of the NpmA Δβ5/6 and Δβ6/7 linkers also significantly impacted binding of SAM and SAH, with affinities reduced for both ligands ∼100- and ∼10-fold, respectively (Table 1; Fig. S5). Remarkably, however, the impact of each linker deletion on interaction with the 30S substrate was much less dramatic and also distinct for each variant (Fig. 4, Table 1). While NpmA-Δβ6/7 had slightly reduced 30S-NpmA binding affinity (∼2.5-fold), NpmA lacking the Δβ5/6 linker bound 30S with modestly (∼3-fold) higher affinity than the wild-type enzyme. Thus, despite making essential contributions to NpmA activity, these regions contribute minimally to the overall 30S substrate binding affinity.
FIG 4.

Deletion of the NpmA β5/6 or β6/7 linker has opposite impact on 30S-NpmA affinity. (A) Wild-type NpmA structure shown in two orthogonal views (top) and the remaining structure after deletion of the β5/6 linker (bottom left) or β6/7 linker (bottom right) regions. Color coding of the NpmA linkers and the right-side view of wild-type NpmA are the same as those shown in Fig. 1B. (B) Competition FP binding experiments with NpmA-E184C* and unlabeled NpmA linker deletion variants, NpmA-Δβ5/6 and NpmA-Δβ6/7. The wild-type NpmA data, shown for comparison, are the same as those shown in Fig. 2. Error bars represent the SEM. Binding affinities (Ki) for each variant protein derived from these data are shown in Table 1.
Role of the β5/6 linker in 30S substrate recognition.
In its 30S-bound state, the NpmA β5/6 linker forms an α-helical structure that contacts both strands of 16S rRNA across the h44 major groove. The N-terminal end of the β5/6 linker α-helix packs against the distorted 16S rRNA backbone between C1409 and the flipped A1408 target nucleotide and undergoes the most pronounced conformational reorganization between the free and 30S-bound states of NpmA (18). The opposite end of the β5/6 linker α-helix is positioned via an electrostatic interaction between R153 and the phosphate group of C1484 (Fig. 5A). As this α-helical β5/6 linker structure appears to be unique to NpmA among the known m1A1408 methyltransferase structures (19–22), we individually replaced two central residues, A148 and E149, with proline to test whether disrupting the α-helix would impact NpmA activity. However, both variants conferred wild-type MICs (Table 1), suggesting that the precise structure formed by the β5/6 linker is not critical for NpmA activity.
FIG 5.

R153 contributes to 30S-NpmA binding affinity. (A) View of NpmA β5/6 linker residue R153 interaction with the phosphate group bridging 16S rRNA nucleotides A1483 and C1484. (B) Competition FP binding experiments with NpmA-E184C* and unlabeled charge reversal substitution of NpmA residue 153 (R153E). The wild-type NpmA and NpmA-Δβ5/6 data, shown for comparison, are the same as those shown in Fig. 2 and 4, respectively. Error bars represent the SEM. Binding affinity (Ki) for NpmA-R153E derived from these data are shown in Table 1.
We next assessed the contribution of β5/6 linker residue R153 to 30S binding affinity using a variant which was previously found to result in a modestly reduced MIC (18): NpmA-R153E bound ∼6- and ∼17-fold more weakly than the wild-type enzyme and NpmA-Δβ5/6 variant, respectively (Fig. 5B, Table 1). Thus, while the reduction in binding affinity is comparable to equivalent changes made in the β2/3 linker, the R153E substitution results in a more significant functional deficit in the MIC assay. These observations suggest an important, specific contribution of R153 to NpmA activity beyond its contribution to overall enzyme-substrate binding affinity. Further, the role of R153 and the finding that NpmA lacking the entire β5/6 linker (including R153) binds with higher rather than lower affinity can be reconciled in a mechanism of substrate recognition in which 30S-driven conformational changes in the β5/6 linker contribute primarily to stabilization of the catalytically competent state of the enzyme rather than enzyme-substrate affinity (see Discussion).
Role of the β6/7 linker in SAM binding and 30S substrate recognition.
The NpmA β6/7 linker forms two α-helices connected by a short loop (Fig. 6A and B), the second of which contains several residues important for enzyme activity (18). These residues include R200 and R207, which make electrostatic interactions with the 16S rRNA, and Trp197, which stacks on the flipped A1408 base. Additionally, the β6/7 linker forms part of the SAM binding pocket, and 30S binding appears to induce a localized change in the NpmA backbone of L196 to create an additional interaction with the cosubstrate that is not observed in the free NpmA-SAM complex (18). Thus, the β6/7 linker potentially contributes to NpmA activity by influencing cosubstrate and substrate binding, as well as precisely orienting the flipped A1408 target base in the enzyme active site for modification.
FIG 6.

Role of NpmA β6/7 linker structure and critical residues in substrate binding affinity and NpmA activity. (A) View of the NpmA β6/7 linker and its electrostatic interactions with 16S rRNA via residues R200 and R207. W197 was previously shown to be critical for positioning the A1408 target base in the NpmA active site, and the backbone carbonyl of L196 is positioned within hydrogen bonding distance from the bound SAM analog sinefungin (SFG) (18). (B) Comparison of the NpmA β6/7 linker in its 30S bound (purple) and free (semitransparent gray) forms, revealing a binding-induced, local conformational change centered on L196 (the view shown on the left is the same as that in panel A). Two residues, V190 and L201, replaced with proline, are also highlighted as a sphere (Cα). (C) RT primer extension analysis of m1A1408 modification from E. coli cells expressing the indicated β6/7 linker variant NpmA proteins. (D) Competition FP binding experiments with NpmA-E184C* and unlabeled NpmA with L201P or R207E substitutions. (E) Same as panel D but for the NpmA-R200E variant. In panels D and E, the wild-type NpmA and NpmA-Δβ6/7 data, shown for comparison, are the same as those shown in Fig. 2 and 4, respectively. Error bars represent the SEM. Binding affinities (Ki) derived from these data are shown in Table 1.
We first tested whether the secondary structure of the NpmA β6/7 linker is critical for activity by replacing a single residue in the center of each short α-helix with proline (Fig. 6B). Disrupting the first α-helix of the β6/7 linker (residues 186 to 193) with a V190P substitution had no impact on resistance to kanamycin or SAM/SAH binding affinity (Table 1; Fig. S5). In contrast, placement of a proline residue in the second β6/7 linker α-helix (residues 198 to 203) with an L201P substitution fully ablated the ability of NpmA to methylate 16S rRNA and confer antibiotic resistance (Fig. 6C and Table 1). Interestingly, however, the L201P substitution also resulted in a modest (3-fold) increase in 30S-NpmA binding affinity and differentially impacted NpmA interaction with SAM and SAH (Kd reduced 6- and 30-fold, respectively) (Table 1). These observations suggest that precise structure of only the second α-helix of the β6/7 linker is critical for NpmA activity through its optimal positioning of key residues (including W197, R200, R207, and the backbone of L196).
We next created two variants with changes at L196 to test the prediction that this residue contributes to regulating NpmA interaction with SAM, or the reaction by-product SAH, following methylation (18). An L196G substitution was found to have no impact on either SAM or SAH binding affinity or the ability of the NpmA protein to confer resistance (Table 1), consistent with the observed 30S-binding-induced interaction between SAM and L196 being mediated by the protein backbone. However, while an L196 deletion (L196Δ) had a modest impact on the kanamycin MIC, a strongly differential impact was observed on SAM and SAH binding affinity (Kd reduced 2- and 20-fold, respectively), comparable to the effect of the L201P substitution. These observations point to a role for the β6/7 linker in regulating NpmA activity through its interaction with cosubstrate and the reaction by-product (see Discussion).
Finally, we assessed the β6/7 linker contribution to 30S substrate binding affinity using two variants, R200E and R207E, previously found to impact NpmA activity (18) (Table 1). NpmA-R200E bound 30S with a slightly reduced affinity (∼2-fold), comparable to the deficit in substrate binding upon complete β6/7 linker deletion (NpmA-Δβ6/7, ∼2.5-fold reduced Ki). Thus, the small decrease in 30S affinity in both NpmA variant proteins likely can be attributed to the loss of a single favorable electrostatic interaction mediated by R200 (Fig. 6E and Table 1). RT analyses additionally revealed that m1A1408 methylation was reduced in E. coli cells expressing NpmA-R200E (Fig. 6C). This result is consistent with the small reduction observed in the kanamycin MIC (Table 1), and the impact of R200E substitution was comparable to that of the two β2/3 linker double variants on both MIC and extent of methylation. However, the greater effect of R200E on activity relative to loss of binding affinity compared to the β2/3 linker variants likely further reflects its importance in precisely positioning the β6/7 linker and, thus, other critical residues within this region. In contrast to NpmA-R200E, while NpmA-R207E bound 30S with the same affinity as the wild-type enzyme, RT analyses showed almost complete loss of methylation, consistent with the prior observation that the NpmA-R207E variant is unable to confer resistance (18) (Fig. 6C and D and Table 1). Thus, R207 has no role in 30S-NpmA binding but is instead critical exclusively for stabilization of the flipped conformation of A1408.
DISCUSSION
The continued emergence of increasingly resistant bacterial pathogens necessitates a deeper understanding of resistance mechanisms and identification of new antimicrobial targets to revive existing drugs or develop novel antimicrobial compounds. In the present study, we set out to dissect the molecular mechanism of action of the pathogen-associated aminoglycoside resistance m1A1408 16S rRNA methyltransferase NpmA as a platform for future development of effective inhibitors of such resistance determinants. Our findings reveal that docking of NpmA on the 30S subunit is driven almost exclusively by electrostatic interactions with 16S rRNA made by a group of lysine residues located in a single region of NpmA, the β2/3 linker. In contrast, the β5/β6 and β6/β7 linkers, which contain the major augmentations to the core class I methyltransferase fold, contribute minimally to overall 30S-NpmA binding affinity but nonetheless are critical for NpmA activity via SAM cosubstrate binding and/or stabilization of A1408 in a flipped conformation for modification. Overall, our data support a model for NpmA action in which 30S binding and adoption of a catalytically competent state on the substrate are distinct events (Fig. 7): docking on the 16S rRNA surface via the β2/3 linker necessarily precedes catalysis of methyl transfer, whereas alterations in the NpmA β5/β6 or β6/β7 linker that ablate activity do not impact 30S binding. This model is also consistent with catalysis of methyl transfer being completely positional in nature, as the most significant impact on NpmA activity occurs upon introduction of defects that impact binding or stabilization of the flipped conformation.
FIG 7.
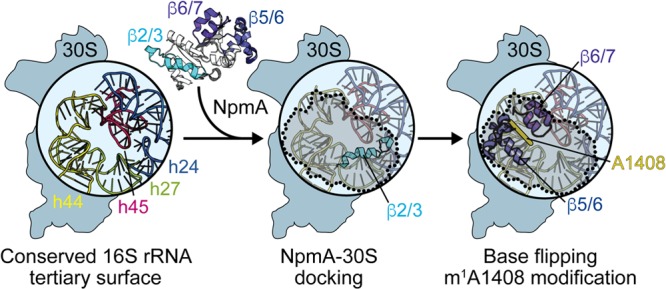
Model for NpmA action. (Left) The structurally conserved 16S rRNA tertiary surface, comprising helices 24, 27, 44, and 45, that is bound by aminoglycoside resistance methyltransferase enzymes. NpmA docking on the 30S subunit is driven by interactions made by the β2/3 linker (center), while conformational changes and specific residues within the β5/6 and β6/7 linkers control base flipping and methyltransferase activity (right). 30S-bound NpmA is denoted by the shaded area outlined with dotted lines.
Other m1A1408 methyltransferases each possess a subset of the equivalent positively charged residues within their β2/3 linker, with conservation highest at residues equivalent to NpmA Lys71 (always Lys) and Lys67 (always Lys or Arg). The partial conservation of these positively charged surface residues suggests that docking on the 16S rRNA surface mediated by electrostatic interactions of the β2/3 linker is a conserved feature of these enzymes. In contrast, for the m7G1405 aminoglycoside resistance methyltransferases, the β2/3 linker is partially surface exposed but also surrounded by the extended α-helical N-terminal domain which is implicated in substrate recognition (23, 24). Thus, whether these or other 16S rRNA modification enzymes exploit a similar strategy for docking on the conserved 16S rRNA tertiary surface surrounding the 30S subunit A site remains to be determined by further high-resolution structural studies of 30S-enzyme complexes.
Using our fluorescence-based binding assay, we determined that wild-type NpmA binds its 30S substrate with high affinity (Kd of ∼25 nM). During the preparation of the manuscript, a report was published of isothermal titration calorimetry (ITC) analysis of 30S-NpmA interaction which suggested a much weaker binding affinity of 5 to 12.5 μM (25). However, these ITC experiments appear unlikely to accurately reflect the true NpmA-30S affinity given the extremely low signal compared to the background, and, most strikingly, the observation that a similar Kd could be measured in the presence of either SAH or SAM. In the latter case, mixing of NpmA, 30S, and SAM would be expected to result in methyl transfer in the ITC cell. In contrast, our carefully controlled assay allowed for more complete validation of the authenticity of specific NpmA-30S binding (Fig. 1; also see Fig. S1 in the supplemental material) and analysis of multiple variants. While several characterized rRNA-modifying enzymes do appear to have substantially lower substrate affinities (26–29), there is also precedence for very-high-affinity 16S rRNA methyltransferase-substrate binding in the case of RsmA (KsgA), which binds pre-30S as part of a subunit assembly quality control mechanism (30, 31).
For NpmA, a high 30S affinity might be important for its ability to compete for its substrate with the other abundant binding partners in the bacterial cell, such as translation initiation factors or the 50S subunit. However, we also found that although individual substitution of each NpmA β2/3 linker lysine residue impacted binding affinity, there was no measurable effect on the ability of the variant enzyme to confer resistance in E. coli. Thus, significant redundancy also appears to exist in the 16S rRNA-NpmA β2/3 linker interface. Why NpmA might be overevolved in its ability to bind E. coli 30S is unclear. However, the origin of NpmA is unknown, and its transfer among diverse bacterial species with subtle alterations of the 16S rRNA binding surface could have promoted accumulation of points of interaction with 30S. Whatever its origin, this inherent redundancy makes NpmA capable of conferring exceptionally high-level aminoglycoside resistance to a broad range of bacterial species, even where alterations to the 16S rRNA docking surface might otherwise have reduced its ability to bind and methylate A1408. Overevolution of 30S binding affinity and the likely positional nature of catalysis by NpmA may also underpin our previous unexpected finding that, among all m1A1408 16S rRNA methyltransferases characterized, NpmA alone is able to similarly modify G1408 mutant 30S ribosome subunits (32). Thus, while the fundamental mechanism of 30S substrate docking via the β2/3 linker likely is conserved in this enzyme family, the overevolution of this interaction may be unique to the only currently identified pathogen-associated member.
Our results support a role for conformational changes in the NpmA β5/6 linker as an essential functional switch that controls substrate specificity. Comparison of the free and 30S-bound NpmA structures reveals that the β5/6 linker must reorganize upon 30S binding to avoid steric clashes and pack closely against h44, stabilizing the distorted backbone between C1409 and A1408. The β5/6 linker conformational change also repositions residue E146 to play its proposed role in supporting R207 of the β6/7 linker, which contacts the phosphate group of A1408 (18). Although NpmA R153 contributes to 30S binding affinity, the overall impact of the β5/6 linker on 30S binding revealed by our analyses is neutral: deletion of the entire linker (including R153) increased 30S binding affinity to the same extent that R153E substitution decreased affinity. Thus, an energetic cost appears to be associated with driving the functionally critical binding-induced conformational change in the β5/6 linker that is balanced by the favorable interaction of R153 with 16S rRNA. Upon β5/6 linker deletion, the cost of binding-induced conformational changes in NpmA is removed, thus increasing 30S binding affinity, but the enzyme cannot confer resistance due to its reduced ability to sufficiently bind or use SAM for methyl transfer and the loss of its contribution to stabilizing the flipped A1408 target nucleotide. Whether a similar mechanism underpins the activity of other m1A1408 methyltransferases in unclear, but the specific molecular details are likely to differ given the structural and sequence diversity of the β5/6 linker.
Although the NpmA β6/7 linker makes only a minor contribution to 30S binding affinity (mediated by R200), our results delineated crucial roles for both the precise structure and specific residues within this region. Substitution of R207 does not impact 30S binding affinity but nonetheless results in an enzyme unable to confer resistance in bacteria, consistent with an exclusive role for R207 in stabilization of the distorted phosphate backbone of the flipped A1408 nucleotide. Disrupting the local secondary structure by insertion of a proline residue in the second α-helix of the β6/7 linker (L201P variant) results in a protein that binds more tightly to 30S but is completely unable to confer resistance. Disruption of the NpmA β6/7 linker structure would, for example, impact the placement of critical residues such as Trp197, which stacks on the flipped A1408 base in the NpmA active site. While each of the m1A1408 methyltransferases possesses a residue equivalent to NpmA Trp197, there is significant variation in the β6/7 linker structure and/or the contributions of positively charged residues in this region. For example, while the KamB β6/7 linker structure is essentially identical to that of NpmA, KamB activity is abrogated by single substitutions of two arginine residues distinct from NpmA R207. More strikingly, the β6/7 linker of Kmr (20) is dynamic while that of CacKam (21) is dramatically reorganized into a structure incompatible with 30S binding and modification. Whether these features reflect fundamental mechanistic differences in A1408 recognition and modification or more minor variations on a common theme will require further studies to tease apart.
Both the β5/6 and β6/7 linker deletions significantly impacted interaction of NpmA with SAM and SAH. The more dramatic (∼100-fold) defect in SAM/SAH binding with the β5/6 linker deletion was unexpected, as this region does not directly contact SAM. While gross structural changes in the β5/6 linker deletion variant are unlikely given its retained ability to bind 30S, loss of the β5/6 linker could have long-range impacts on NpmA structure or conformational dynamics, which in turn impact SAM/SAH affinity. Recent hydrogen-deuterium exchange analysis of NpmA-SAM/SAH complexes suggests the potential for allosteric communication between the β2/3 linker 30S binding surface, the β5/6 linker, and the cosubstrate binding pocket between them (25). NpmA interaction with cosubstrate was also previously speculated to be influenced by 30S substrate binding (20, 22), based in part on the observation that substitution of β6/7 linker residue S195 decreases SAM/SAH affinity by >50-fold, yet the variant enzyme is still capable of conferring wild-type-level resistance. In addition to a direct hydrogen bond made by S195 to the SAM carboxylate group, a local 30S binding-induced conformational change also repositions the L196 backbone to directly interact with SAM. Deletion, but not replacement, of L196 altered binding in an unprecedented manner for NpmA, with SAH affinity being significantly more affected, resulting in a relative reduction in affinity of ∼10-fold. The L201P and R207E substitutions resulted in similar, although less pronounced (∼5-fold), relative reductions in SAH binding affinity, underscoring the importance of the precise β6/7 linker structure and functionally connecting 30S-binding-induced reorganization, interaction with SAM, and control of base flipping.
Finally, our analyses of 16S rRNA methylation in bacterial cells expressing NpmA variants gives initial clues about both the extent 30S must be methylated for resistance as well as an approximate threshold for where methylation is no longer protective. Expression of NpmA variants with single substitutions in the β2/3 linker resulted in >66% methylation and a resistance phenotype indistinguishable from that of wild-type NpmA. In contrast, ∼10 to 25% methylation conferred an intermediate resistance phenotype, and <5% methylation conferred no resistance. Thus, although growth may be substantially slowed, only a small fraction of 30S subunits need to be methylated for bacteria to survive treatment with antibiotic. These initial observations reveal the need for future studies directed at carefully determining the lower limit of methylation for aminoglycoside resistance. This limit would represent a key threshold for inhibition that an inhibitor of the aminoglycoside resistance methyltransferases would need to achieve in order to effectively block resistance.
In summary, our results have provided new mechanistic insights into substrate recognition and modification for the pathogen-associated aminoglycoside resistance 16S rRNA methyltransferase NpmA, which can serve as a platform for mechanism-based inhibitor development exploiting unique facets of the enzyme activity. For example, these studies have localized the critical regions within both enzyme and substrate for their interaction, suggesting that the conserved 16S rRNA surface bound by NpmA could be targeted for development of compounds that block binding without impacting ribosome function and thus promoting rapid development of resistance.
MATERIALS AND METHODS
NpmA mutagenesis, expression, and purification.
Amino-terminally hexahistidine-tagged (6× His) wild-type and variant NpmA proteins were expressed from a previously described pET44a expression construct containing an E. coli codon-optimized NpmA gene obtained by chemical synthesis (18, 33). NpmA amino acid substitutions were generated in the pET44a-NpmA plasmid using the megaprimer whole-plasmid PCR method (34) and confirmed by automated DNA sequencing.
Wild-type and variant NpmA proteins were expressed in E. coli BL21(DE3) cells grown at 37°C in Terrific broth containing 100 μg/ml ampicillin, with induction of protein expression at mid-log phase (optical density at 600 nm [OD600] of 0.6 to 0.8) using 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were grown a further 2.5 h postinduction at 37°C (except for NpmA Δβ6/7, which was grown at 20°C overnight), harvested by centrifugation, and lysed by sonication in 50 mM sodium HEPES (pH 7.5) buffer containing 1 M NaCl, 10 mM imidazole, 10% glycerol, 0.5% Triton X-100, 6 mM β-mercaptoethanol (β-ME), 0.27 U/ml DNase I, 1 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Soluble cell lysate was dialyzed against 50 mM sodium HEPES buffer (pH 7.5) containing 2 M NaCl and 10 mM β-ME and 6×His-NpmA proteins purified by Ni2+ affinity chromatography using a HisTrap Fast Flow column (GE Healthcare). Bound protein was eluted using a linear gradient of imidazole (25 to 500 mM) in 50 mM sodium HEPES buffer (pH 7.5) containing 0.5 M NaCl and 10 mM β-ME. Pooled NpmA-containing fractions were further purified by gel filtration chromatography on a Superdex 75 column equilibrated with 50 mM sodium HEPES buffer (pH 7.5) containing 150 mM NaCl and 6 mM β-ME. Purified proteins were either used in experiments immediately or stored at −80°C following flash freezing.
Kanamycin MIC assays.
MIC assays were conducted in 96-well plate format using E. coli BL21(DE3) harboring plasmids expressing wild-type or variant NpmA grown in lysogeny broth (LB) containing 2-fold dilutions of kanamycin over a range of 2 to 1,024 μg/ml. Each well containing 100 μl LB medium, 5 μM IPTG, and kanamycin was inoculated with 1 ×105 CFU/ml in an additional 100 μl of LB. Plates were incubated at 37°C with shaking for 24 h. The MIC was defined as the lowest concentration of kanamycin that inhibited growth (OD600 of <0.05 above background).
ITC.
For ITC, purified NpmA protein (60 to 80 μM) was exhaustively dialyzed against 50 mM sodium HEPES buffer (pH 7.5) containing 150 mM NaCl. Final dialysis buffer was used to prepare solutions of SAM (2 mM) and SAH (0.6 to 0.8 mM). Titration experiments were performed at 25°C using an Auto-iTC200 microcalorimeter (Malvern/MicroCal) with 16 2.4-μl injections of SAM or SAH into each protein. Data were fit using Origin 7 software with a single binding site model to extract the binding affinity (Kd) for each protein-ligand pair. All experiments with variant proteins were performed in parallel with an experiment with wild-type NpmA to control for differences in protein buffers and ligand (SAM/SAH) preparations. All measurements for wild-type NpmA were within ∼2-fold of the values shown in Table 1. We therefore only considered differences of >2-fold compared to the wild type to be significant for each variant protein for which high-quality titrations and fits to the single binding site model were obtained (see Fig. S3 and S5 in the supplemental material).
Fluorescence assay to monitor 30S-NpmA interaction.
Small ribosomal (30S) subunits were purified from E. coli (MRE600) grown to mid-log phase (OD600 of 0.6 to 0.7), essentially as previously described (35). Single-Cys variants of NpmA were fluorescein labeled by incubation of purified protein in the dark overnight at 4°C with a 5-fold excess of fluorescein-5-maleimide (AnaSpec, Inc.). This reagent was selected as fluorescein is well established for fluorescence-based studies of macromolecular interaction, including 30S-rRNA methyltransferase interactions (30), and the maleimide moiety provides a minimal linker between fluorophore and protein to minimize undesirable effects due to linker flexibility. Excess dye reagent was removed using a dye removal column (Thermo Scientific), and fluorescently labeled proteins (denoted with an asterisk) were analyzed by SDS-PAGE with visualization on a Typhoon Trio imaging system. Pilot FP experiments were conducted at 25°C in 100-μl reaction mixtures containing 50 nM fluorescently labeled protein (NpmA-S89C*, NpmA-K131C*, NpmA-E184C*, or NpmA-E188C*) and 50 nM 30S subunits in 20 mM HEPES-KOH buffer (pH 7.0) containing 10 mM NH4Cl, 50 mM KCl, 5 mM Mg(OAc)2, and 3 mM β-ME. Fluorescence polarization was monitored using a BioTek Synergy4 plate reader.
FP experiments to measure 30S-NpmA binding affinity (Kd) were performed under the same solution conditions as those used for the pilot FP experiments and contained 20 nM NpmA-E184C* and 30S subunit over a concentration range of 0 to 256 nM. Binding reaction mixtures were incubated at 25°C for 15 min before FP measurement. Experiments were performed in triplicate and data fit using a one-site total binding equation in GraphPad Prism6 to determine the Kd.
Measurements of Ki for each NpmA variant were made using competition binding assays performed at least in triplicate. Each 100-μl reaction mixture contained 50 mM 30S subunit, 50 nM NpmA-E184C*, and the variant NpmA protein (over a concentration range of 0.002 to 10 μM) in 20 mM HEPES-KOH buffer (pH 7.0) containing 10 mM NH4Cl, 75 mM KCl, 5 mM Mg(OAc)2, and 3 mM β-ME. NpmA-E184C* and 30S subunits were preincubated for 10 min at 25°C before addition of unlabeled competitor protein, and the final reaction mixture was incubated at 25°C for 12 min before measurement of FP as described above. Data were fit in GraphPad Prism6 to determine Ki using the one-binding-site competition binding equation.
RT analysis of A1408 methylation.
Reverse transcription (RT) assays (36, 37) were used to determine the extent of m1A1408 modification by wild-type and variant NpmA in cells expressing the proteins. E. coli BL21(DE3) harboring a plasmid expressing wild-type or variant NpmA were grown to mid-log phase in LB containing 5 μM IPTG, and total RNA was extracted using an RNeasy minikit (Qiagen). To determine the extent of methylation, a 32P-labeled DNA primer complementary to E. coli 16S rRNA nucleotides 1459 to 1479 was used for RT primer extension at 37°C using AMV reverse transcriptase (Promega). Primer extension products were run on denaturing (8 M urea) 10% PAGE sequencing-style gels and visualized on a Typhoon Trio imaging system.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health-National Institute of Allergy and Infectious Diseases (R01-AI088025 to G.L.C.).
Single-cysteine NpmA variants were generated and their initial functional characterizations were performed by Miloje Savic and Pooja M. Desai. We also thank Christine Dunham and members of both the Conn and Dunham laboratories for useful discussions during the course of this work and preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00077-17.
REFERENCES
- 1.Wachino JI, Shibayama K, Kurokawa H, Himura K, Yamane K, Suzuki S, Shibata N, Ike Y, Arakawa Y. 2007. Plasmid-mediated novel m1A1408 methyltransferase NpmA, for 16S rRNA found in clinically isolated Escherichia coli resistant to structurally diverse aminoglycosides. Antimicrob Agents Chemother 51:4401–4409. doi: 10.1128/AAC.00926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wachino JI, Arakawa Y. 2012. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: an update. Drug Resist Updat 15:133–148. doi: 10.1016/j.drup.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Kotra LP, Haddad J, Mobashery S. 2000. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother 44:3249–3256. doi: 10.1128/AAC.44.12.3249-3256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jana S, Deb JK. 2006. Molecular understanding of aminoglycoside action and resistance. Appl Microbiol Biotechnol 70:140–150. doi: 10.1007/s00253-005-0279-0. [DOI] [PubMed] [Google Scholar]
- 5.Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem Rev 105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 6.Pfister P, Hobbie S, Vicens Q, Bottger EC, Westhof E. 2003. The molecular basis for A-site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and x-ray crystal structures. Chembiochem 4:1078–1088. doi: 10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]
- 7.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interaction with antibiotics. Nature 407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 8.Francois B, Russell RJM, Murray JB, Aboul-Ela F, Masquida B, Vicens Q, Westhof E. 2005. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res 33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vicens Q, Westhof E. 2001. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure 9:647–658. doi: 10.1016/S0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 10.Wasserman MR, Pulk A, Zhou Z, Altman RB, Zinder JC, Green KD, Garneau-Tsodikova S, Cate JHD, Blanchard SC. 2015. Chemically related 4,5-linked aminoglycoside antibiotics drive subunit rotation in opposite directions. Nat Commun 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cundliffe E. 1989. How antibiotic-producing organisms avoid suicide. Annu Rev Microbiol 43:207–233. [DOI] [PubMed] [Google Scholar]
- 12.Conn GL, Savic M, Macmaster R. 2008. DNA and RNA modification enzymes: structure, mechanism, function, and evolution. Landes Bioscience, Austin, TX. [Google Scholar]
- 13.Doi Y, Arakawa Y. 2007. 16S ribosomal RNA methylation: emerging resistance mechanisms against aminoglycosides. Antimicrob Resist 45:88–94. [DOI] [PubMed] [Google Scholar]
- 14.Savic M, Lovric Tomic JTI, Vasiljevic B, Conn GL. 2009. Determination of the target nucleosides for members of two families of 16S rRNA methyltransferases that confer resistance to partially overlapping groups of aminoglycoside antibiotics. Nucleic Acids Res 37:5420–5431. doi: 10.1093/nar/gkp575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galimand M, Courvalin P, Lambert T. 2003. Plasmid-mediated high-level resistance to aminoglycosides in Enterobacteriaceae due to 16S rRNA methylation. Antimicrob Agents Chemother 47:2565–2571. doi: 10.1128/AAC.47.8.2565-2571.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao X, Zhang Z, Shen H, Ning M, Chen J, Wei H, Zhang K. 2014. Genotypic characteristics of multidrug-resistant Escherichia coli isolates associated with urinary tract infections. APMIS 122:1088–1095. [DOI] [PubMed] [Google Scholar]
- 17.Liou GF, Yoshizawa S, Courvalin P, Galimand M. 2006. Aminoglycoside resistance by ArmA-mediated ribosomal 16S methylation in human bacterial pathogens. J Mol Biol 359:358–364. doi: 10.1016/j.jmb.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 18.Dunkle JA, Vinal K, Desai PM, Zelinskaya N, Savic M, West DM, Conn GL, Dunham CM. 2014. Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc Natl Acad Sci U S A 111:6275–6280. doi: 10.1073/pnas.1402789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macmaster R, Zelinskaya N, Savic M, Rankin CR, Conn GL. 2010. Structural insights into the function of aminoglycoside-resistance A1408 16S rRNA methyltransferases from antibiotic-producing and human pathogenic bacteria. Nucleic Acids Res 38:7791–7799. doi: 10.1093/nar/gkq627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savic M, Sunita Zelinskaya SN, Desai PM, Macmaster R, Vinal K, Conn GL. 2015. 30S subunit-dependent activation of the Sorangium cellulosum So ce56 aminoglycoside resistance-conferring 16S rRNA methyltransferase Kmr. Antimicrob Agents Chemother 59:2807–2816. doi: 10.1128/AAC.00056-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witek MA, Conn GL. 2016. Functional dichotomy in the 16S rRNA (m1A1408) methyltransferase family and control of catalytic activity via a novel tryptophan mediated loop reorganization. Nucleic Acids Res 44:342–353. doi: 10.1093/nar/gkv1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Husain N, Obranic S, Koscinski L, Seetharaman J, Babic F, Bujnicki JM, Maravic-Vlahovicek G, Sivaraman J. 2011. Structural basis for the methylation of A1408 in 16S rRNA by a panaminoglycoside resistance methyltransferase NpmA from a clinical isolate and analysis of the NpmA interactions with the 30S ribosomal subunit. Nucleic Acids Res 39:1903–1918. doi: 10.1093/nar/gkq1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Husain N, Tkaczuk KL, Tulsidas SR, Kaminska KH, Cubrilo S, Maravic-Vlahovicek G, Bujnicki JM, Sivaraman J. 2010. Structural basis for the methylation of G1405 in 16S rRNA by aminoglycoside resistance methyltransferase Sgm from an antibiotic producer: a diversity of active sites in m7G methyltransferases. Nucleic Acids Res 38:4120–4132. doi: 10.1093/nar/gkq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitt E, Galimand M, Panvert M, Courvalin P, Mechulam Y. 2009. Structural bases for 16S rRNA methylation catalyzed by ArmA and RmtB methyltransferases. J Mol Biol 388:570–582. doi: 10.1016/j.jmb.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 25.Husain N, Tulsian NK, Chien WL, Suresh S, Anand GS, Sivaraman J. 2016. Ligand-mediated changes in conformational dynamics of NpmA: implications for ribosomal interactions. Sci Rep 6:37061. doi: 10.1038/srep37061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basturea GN, Deutscher MP. 2007. Substrate specificity and properties of the Escherichia coli 16S rRNA methyltransferase, RsmE. RNA 13:1969–1976. doi: 10.1261/rna.700507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hager J, Staker BL, Jakob U. 2004. Substrate binding analysis of the 23S rRNA methyltransferase RrmJ. J Bacteriol 186:6634–6642. doi: 10.1128/JB.186.19.6634-6642.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maravic G, Bunjicki JM, Feder M, Pongor S, Flögel M. 2003. Alanine-scanning mutagenesis of the predicted rRNA-binding domain of ErmC′ redefines the substrate-binding site and suggests a model for protein-RNA interactions. Nucleic Acids Res 31:4941–4949. doi: 10.1093/nar/gkg666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ero R, Leppik M, Liiv A, Remme J. 2010. Specificity and kinetics of 23S rRNA modification enzymes RlmH and RluD. RNA 16:2075–2084. doi: 10.1261/rna.2234310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Farrell HC, Musayev FN, Scarsdale JN, Rife JP. 2012. Control of substrate specificity by a single active site residue of the KsgA methyltransferase. Biochemistry 51:466–474. doi: 10.1021/bi201539j. [DOI] [PubMed] [Google Scholar]
- 31.Connolly K, Rife JP, Culver G. 2008. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol Microbiol 70:1062–1075. doi: 10.1111/j.1365-2958.2008.06485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zelinskaya N, Witek MA, Conn GL. 2015. The pathogen-derived aminoglycoside resistance 16S rRNA methyltransferase NpmA possesses dual m1A1408/m1G1408 specificity. Antimicrob Agents Chemother 59:7862–7865. doi: 10.1128/AAC.01872-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zelinskaya N, Rankin CR, Macmaster R, Savic M, Conn GL. 2011. Expression, purification, and crystallization of adenosine 1408 aminoglycoside-resistance rRNA methyltransferases for structural studies. Protein Expr Purif 75:89–94. doi: 10.1016/j.pep.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyazaki K, Takenouchi M. 2002. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques 33:1033–1034. [DOI] [PubMed] [Google Scholar]
- 35.Moazed D, Stern S, Noller HF. 1986. Rapid chemical probing of conformation in 16S ribosomal and 30S ribosomal subunits using primer extension. J Mol Biol 187:399–416. [DOI] [PubMed] [Google Scholar]
- 36.Witek MA, Conn GL. 2014. Expansion of the aminoglycoside-resistance 16S rRNA (m(1)A1408 methyltransferase family: expression and functional characterization of four hypothetical enzymes of diverse bacterial origin. Biochim Biophys Acta 1844:1648–1655. doi: 10.1016/j.bbapap.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stern S, Moazed D, Noller HF. 1988. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol 164:481–489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.