

SUMMARY
The Reelin signaling pathway plays a crucial role in regulating neocortical development. However, little is known about how Reelin controls the cytoskeleton during neuronal migration. Here, we identify CLASP2 as a key cytoskeletal effector in the Reelin signaling pathway. We demonstrate that CLASP2 has distinct roles during neocortical development regulating neuron production and controlling neuron migration, polarity, and morphogenesis. We found down-regulation of CLASP2 in migrating neurons leads to mislocalized cells in deeper cortical layers, abnormal positioning of the centrosome-Golgi complex, and aberrant length/orientation of the leading process. We discovered Reelin regulates several phosphorylation sites within the positively-charged serine/arginine rich region that constitute consensus GSK3β phosphorylation motifs of CLASP2. Furthermore, phosphorylation of CLASP2 regulates its interaction with the Reelin adaptor Dab1 and this association is required for CLASP2 effects on neurite extension and motility. Together, our data reveal CLASP2 is an essential Reelin effector orchestrating cytoskeleton dynamics during brain development.
INTRODUCTION
The complex architecture of the brain requires precise control over the timing of neurogenesis, neuron migration, and differentiation. These three developmental processes are exquisitely controlled during the expansion of the mammalian neocortex. The six morphologically distinct layers of the neocortex form in an “inside-out” pattern with early-born neurons forming deeper layers and later-born neurons migrating past them to form superficial layers of the cortical plate (Rakic 1974). The Reelin signaling pathway plays a crucial role in cortical lamination. Reelin is a secreted glycoprotein that exerts its function by binding to the lipoprotein receptors ApoER2 and VLDLR and inducing tyrosine phosphorylation of the intracellular adaptor protein Disabled (Dab1) (Howell et al., 2000; Bock and Herz, 2003). Phosphorylated Dab1 then recruits downstream signaling molecules to promote cytoskeletal changes necessary for neuronal migration, final positioning, and morphology (D’Arcangelo, 2005). Mutations of Reelin, the dual ApoER2/VLDLR receptor, or Dab1 lead to an inversion of the normal inside-out pattern of cortex development (D’Arcangelo et al., 1995; Howell et al., 1997; Trommsdorff et al., 1999). In addition, a number of mutations in cytoskeletal-encoded genes produce deficits in neuron migration and cortical lamination phenotypically similar to Reelin mutants, firmly establishing a mechanistic and developmentally critical connection between Reelin and the cytoskeleton. For example, human mutations in lissencephaly-1, doublecortin, and tubulin, integral components of the microtubule cytoskeleton, cause severe cortical lamination defects with later born neurons failing to migrate past previously born neurons (Reiner et al., 1993; Gleeson et al., 1998; Romaniello et al., 2015). The culmination of these genetic studies indicates that several signaling pathways, including the Reelin pathway, converge on downstream cytoskeletal proteins to affect proper neuronal migration and brain development. However, the molecular effectors of these pathways have not been fully characterized.
CLASPs (cytoplasmic linker associated proteins) belong to a heterogeneous family of plus-end tracking proteins (+TIPs) that specifically accumulate at the growth cone. This localization strategically places them in a position to control neurite growth, directionality, and the crosstalk between microtubules and the actin cytoskeleton (Akhmanova and Hoogenraad, 2005, Basu and Chang, 2007 Akhmanova and Steinmetz, 2008). Previous evidence showed CLASPs accumulate asymmetrically towards the leading edge of migrating fibroblasts indicating a role for CLASPs in cell polarity and movement (Akhmanova et al., 2001; Wittmann and Waterman-Storer, 2005). We found that CLASP2 protein levels steadily increase throughout neuronal development and are specifically enriched at the growth cones of extending neurites. In particular, short-hairpin RNA (shRNA) mediated knockdown of CLASP2 in primary mouse neurons decreases neurite length, whereas overexpression of human CLASP2 causes the formation of multiple axons, enhanced dendritic branching, and Golgi condensation (Beffert et al., 2012). These results implicate a role for CLASP2 in neuronal morphogenesis and polarization; however, the function of CLASP2 during brain development is unknown.
Here we demonstrate CLASP2 is a modifier of the Reelin signaling pathway during cortical development. In vivo knockdown experiments demonstrate CLASP2 plays significant roles in neural precursor proliferation, neuronal migration, and morphogenesis. In addition, we show that GSK3β-mediated phosphorylation of CLASP2 controls its binding to the Reelin adaptor protein Dab1, a required molecular step governing CLASP2’s regulatory effects on neuron morphology and movement.
RESULTS
CLASP2 Expression is Functionally Associated With the Reelin Signaling Pathway
To identify novel genes downstream of Reelin signaling, we examined the expression of mRNA transcripts by microarray between adult brain cortices from mice deficient in either Reelin, the double ApoER2/VLDLR receptor mutant, or Dab1, and compared Affymetrix gene expression profiles against age-matched, wild-type mice. Importantly, each of these mutant mouse models present a similar phenotype which includes severe neuronal migration defects (D’Arcangelo et al., 1995; Howell et al., 1997; Trommsdorff et al., 1999). We defined a large network of genes perturbed above a threshold of 1.5-fold in response to deficient Reelin signaling, identifying 832 up-regulated and 628 down-regulated genes which were common to all three mouse models (Figure 1A). Ingenuity Pathway Analysis revealed a network of genes that is functionally related to cytoskeleton organization, microtubule dynamics, neurogenesis, and migration of cells (Figure 1B). Of the few cytoskeletal candidate genes identified, CLASP2 was the only microtubule +TIP. Specifically, CLASP2 mRNA expression was increased in all three Reelin mutant phenotypes while CLASP1 mRNA expression remained unchanged (Figure 1B). Consistent with the microarray data, CLASP2 protein levels were ~2.8-fold higher in Dab1 knockout mice (Figure 1C). These findings suggest Reelin signaling controls CLASP2 expression and establishes the first molecular link between a plus-end, microtubule binding protein downstream of extracellular Reelin signaling.
Figure 1. CLASP2 is a Key Cytoskeletal Effector in the Reelin Signaling Pathway.
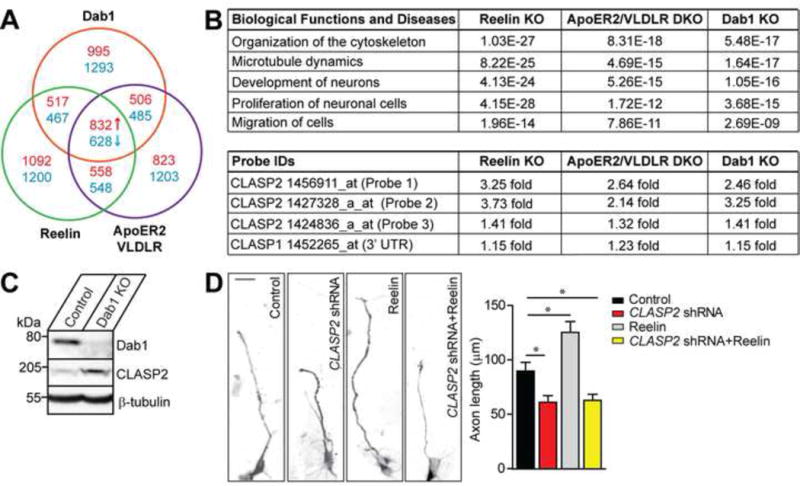
(A) Chart representing expression of mRNA transcripts between brain cortices from mice deficient in Reelin (green), the double ApoER2/VLDLR receptor mutant (purple) or Dab1 (red) compared Affymetrix gene expression profiles against age-matched wild type mice by microarray. The screen identified 832 up-regulated (red) and 628 down-regulated (blue) genes that were common to all three mouse models above a threshold of 1.5 fold.
(B) Ingenuity Pathway Analysis identified several gene networks impaired across Reelin, double ApoER2/VLDLR receptor and Dab1 knockout (KO) mouse models. CLASP2 but not CLASP1 mRNA expression was increased in all three Reelin mutants (data presented are in p-values).
(C) Consistent with the microarray data, CLASP2 protein levels were 2.8-fold higher in Dab1 knockout mice.
(D) Representative images of primary dissociated mouse wild type neuron cultures co-infected with scrambled control, CLASP2 shRNA, Reelin treatment or CLASP2 shRNA with Reelin and immunostained against tau axonal marker at 2 days in vitro. CLASP2 shRNA caused a decrease in axon length when compared to control. Reelin increased axon length; however, Reelin treatment with CLASP2 shRNA was unable to rescue the axonal effects of CLASP2 knockdown (control, n = 35; CLASP2 shRNA, n = 39; Reelin alone, n = 41; CLASP2 shRNA plus Reelin, n = 39 cells). Data are means ± SEM and statistical significance was assessed using one-way ANOVA (*p < 0.05).
To directly test whether CLASP2 functions downstream of Reelin signaling, we evaluated axon length following Reelin treatment with or without CLASP2 in primary hippocampal neurons (Figure 1D). Treatment of neurons with CLASP2 shRNA reduced axon length by 32%. Reelin treatment alone caused a 39% increase in axon length. Interestingly, Reelin treatment had no effect in neurons following CLASP2 knockdown indicating that CLASP2 lies downstream of Reelin signaling and is necessary for the effects of Reelin on axon extension.
CLASP2 Expression is Developmentally Regulated
Having established a functional link between CLASP2 and Reelin signaling, we examined CLASP2 expression in the developing neocortex. CLASP2 is predominantly expressed in the nervous system in two distinct isoforms, CLASP2α and CLASP2γ (Akhmanova et al., 2001). These isoforms differ only at the N-terminus where the longer CLASP2α contains a unique dis1/TOG1 domain previously shown to regulate mitotic spindle assembly, actin binding, and tubulin polymerization (Figure 2A) (Tsvetkov et al., 2007; Patel et al., 2012; Al-Bassam et al., 2010). To determine whether both CLASP2 isoforms are altered in Reelin pathway knockouts, we examine the expression of CLASP2α and CLASP2γ isoforms in wild type and Reelin knockout brains by quantitative PCR. We found CLASP2α and CLASP2 γ isoforms are differentially regulated in Reelin knockout brains. CLASP2α mRNA levels were elevated while CLASP2γ mRNA levels were decreased in Reelin knockout brains (Figure 2B). We next examined the developmental expression profile of CLASP2α and CLASP2γ on mouse cortical brain lysates across a range of developmental time points. We found elevated CLASP2α mRNA at embryonic stages as early as embryonic day 12.5 (E12.5) while CLASP2γ mRNA remained relatively stable throughout brain development (Figure 2C). In addition, in situ hybridization analysis using antisense probes specific to individual CLASP2 isoforms revealed that CLASP2α was preferentially expressed in the ventricular and subventricular zone (VZ/SVZ) at E16.5 and postnatal day 0 (P0) which are enriched with proliferative, neural precursors (the first 200 μm from the ventricular surface, Figure 2D and S1). An antisense probe specific to CLASP2γ mRNA revealed that CLASP2γ was also highly expressed in the VZ/SVZ at E16.5; however, by P0, CLASP2γ mRNA expression was evenly distributed across the cortical wall (Figure 2D and S1). The early expression pattern of CLASP2α and CLASP2γ isoforms during brain development suggests that CLASP2 may play a role in progenitor proliferation and/or neuronal migration.
Figure 2. CLASP2α and γ Isoforms are Differentially Regulated During Brain Development.
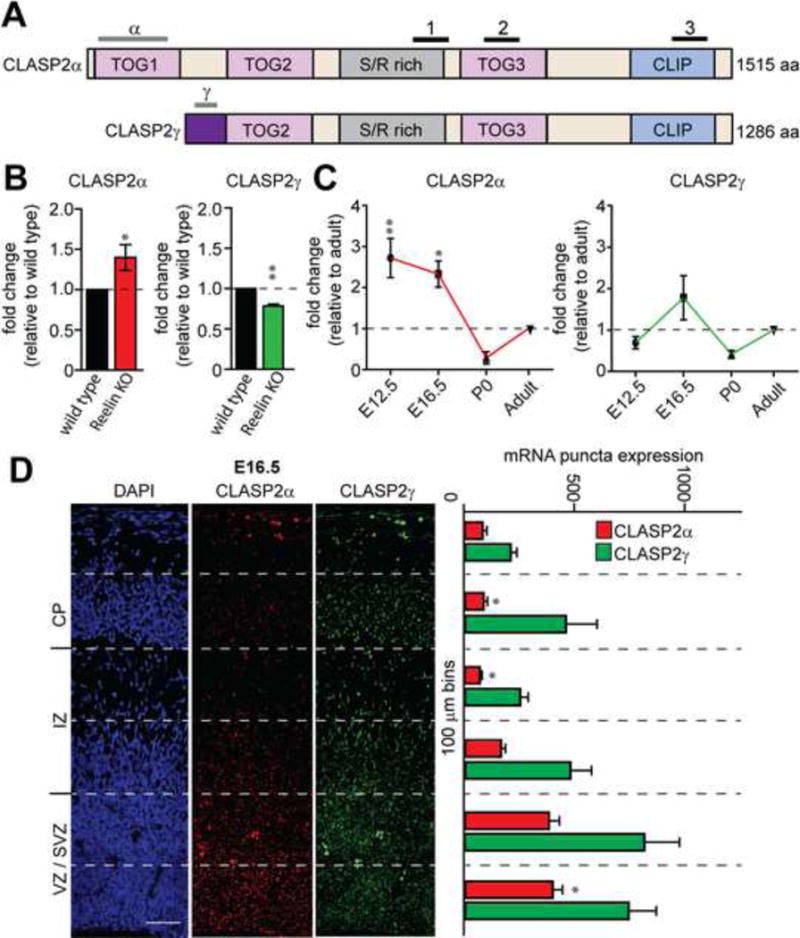
(A) Human CLASP2α and CLASP2γ domain organization (TOG = N-terminal tumor overexpressed gene; S/R rich = repetitive region rich in serine and arginine residues; CLIP = CLIP-170). Lines above indicate the location of probes used for qPCR, in situ hybridization (gray) and the three probe IDs for microarray (black).
(B) Fold change of CLASP2α and CLASP2γ mRNA expression relative to wild type measured by qPCR from cortical lysates collected from wild type and Reelin knockout adult mice (n = 3 brains for each group).
(C) Fold change of CLASP2α and CLASP2γ mRNA expression relative to adult measured by qPCR from dissociated wild type cortical lysates collected at E12.5, E16.5, P0 and adult (n = 5 brains for each time point).
(D) Representative images showed in situ hybridization of cortical sections for CLASP2α and CLASP2γ at E16.5. mRNA fluorescence was measured using imageJ by counting the number of puncta in 100 μm bins from the ventricle to the pial surface (VZ = ventricular zone, SVZ = subventricular zone, IZ = intermediate zone, CP = cortical plate). CLASP2α expression was the highest in the first 200 μm which represents the proliferative VZ/SVZ zones (n = 6 brain sections for each group). Scale bar represents 50 μm. For additional data, see Figure S1.
Data are means ± SEM and statistical significance was assessed using unpaired t test (*p < 0.05, **p<0.001 for B) and one-way ANOVA (*p < 0.05, **p<0.001 for C and D).
CLASP2 Regulates Cell Cycle Exit in Neuronal Progenitors
Because we found elevated CLASP2α mRNA levels in the developing mouse brain during periods of active neurogenesis (Figure 2C), we examined whether CLASP2 expression affected the numbers and/or cell cycle dynamics of neuronal stem and progenitor cells. To test this, we previously identified two CLASP2 shRNA constructs capable of knocking down expression of endogenous CLASP2 in mouse primary neurons (Beffert et al., 2012). Individual and combined CLASP2 shRNA knockdowns efficiently downregulated endogenous CLASP2 but not CLASP1 protein expression in mouse primary neurons (Figure S2). We electroporated scrambled control or combined CLASP2 shRNA constructs, which also contained CAG-GFP reporter cassettes, into E14.5 mouse embryos and labeled cells in the mitotic phase of the cell cycle with an antibody against phospho-Histone H3 (PH3) at E16.5. We found CLASP2 knockdown resulted in a greater percentage of GFP-expressing electroporated cells positive for PH3, consistent with an increase in the percentage of actively dividing cells (Figure 3A).
Figure 3. CLASP2 is Required for Neural Progenitor Differentiation and Controls Terminal Division of Neural Progenitor Cells.
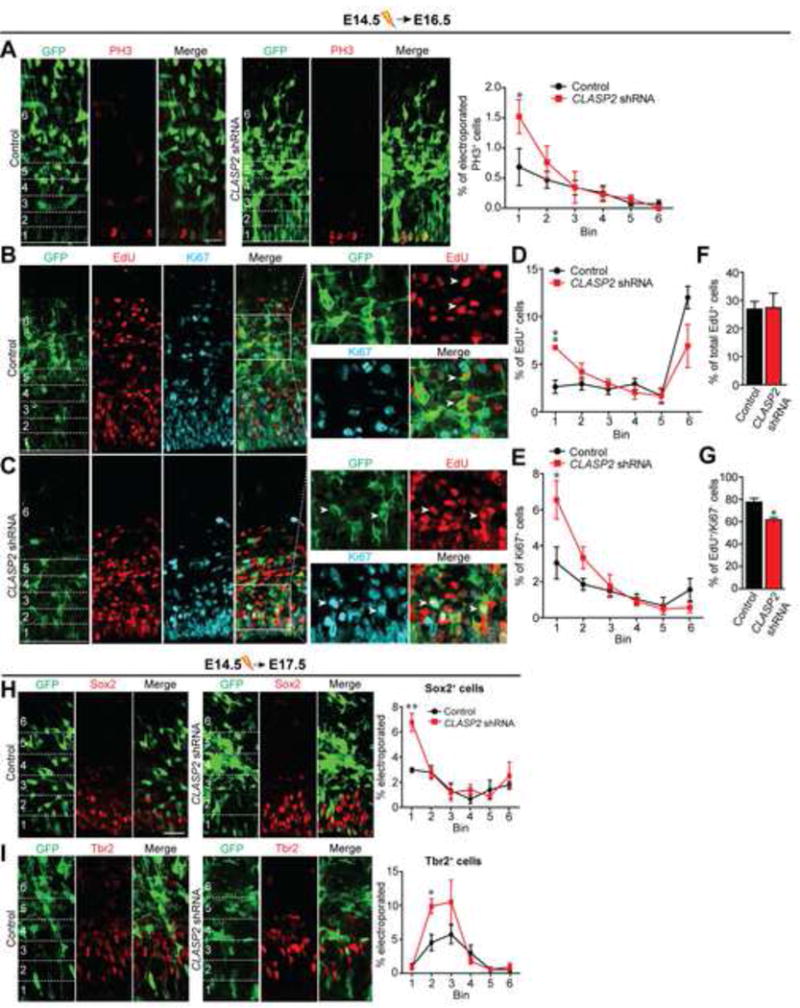
(A) Mouse embryos were electroporated in utero with GFP-tagged scrambled control or CLASP2 shRNA plasmids at E14.5 and analyzed at E16.5. Immunostaining for mitotic marker PH3 (red) shows more dividing cells at the ventricular zone in CLASP2 knockdown cells within the first 20 μm bin (control, n = 4 brains; CLASP2 shRNA, n = 4 brains). For additional data, see Figure S2.
(B–G) Mouse embryos were electroporated in utero with GFP-tagged scrambled control (B) or CLASP2 shRNAs (C) at E14.5. The pregnant dams were injected intraperitoneally with EdU 24 hours following electroporation and embryos were analyzed at E16.5. Coronal brain sections immunostained for EdU (red) and Ki67 (blue) showed an increased in the percentage of electroporated cells which were positive for Edu (D) and Ki67 (E) located in the VZ (bin area 1, within the first 20 μm) in CLASP2 knockdown cells. There was no effect of CLASP2 shRNA on the total number of EdU-positive cells (F); however, there was a significant decrease in the number of EdU-positive/Ki67-negative cells exiting the cell cycle (G). Arrowheads for (B) represents GFP-positive cells which is EdU-positive/Ki67-negative in control brains. Arrowheads for (C) represents GFP-positive cells which is EdU/Ki67-positive in CLASP2 shRNA brains. Control, n = 5 brains and CLASP2 shRNA, n = 4 brains analyzed.
(H–I) Mouse embryos were electroporated in utero with GFP-tagged scrambled control or CLASP2 shRNAs at E14.5 and analyzed at E17.5. Apical and basal progenitors were immunolabeled with antibodies against the transcriptional factors Sox2 (H) and Tbr2 (I). The percentage of transfected cells positive for Sox2 or Tbr2 was calculated every 20 μm bins from the ventricle (control, n = 4 brains; CLASP2 shRNAs, n = 4 brains).
Data are means ± SEM and statistical significance was assessed using one-way ANOVA (*p < 0.05, **p<0.001 for A, D, E, H, I) and unpaired t test (*p < 0.05 for F, G). Scale bar represents 20 μm.
To determine whether the effects of CLASP2 on cell division were due to changes in cell cycle dynamics or cell fate, we performed IUE at E14.5, pulse labeled with an injection of EdU at E15.5, and then analyzed embryonic brains at E16.5 by double staining with EdU and Ki67 antibodies (Figure 3B and 3C). Since Ki67 is expressed throughout the cell cycle, EdU/Ki67 double-positive cells remain active in the cell cycle, whereas EdU-positive/Ki67-negative cells are postmitotic neurons. CLASP2 knockdown caused an increase in the percentage of electroporated cells positive for either EdU (Figure 3D) or Ki67 (Figure 3E) located within the VZ (bin 1, specifically within the first 20 μm). In addition, while we did not observe any changes in total EdU-positive cells (Figure 3F), we found that the fraction of EdU-positive and Ki67-negative cells was significantly lower following CLASP2 knockout (Figure 3G) demonstrating that CLASP2 knockdown is decreasing neuronal output.
During mid-neurogenesis, excitatory neurons are being generated by a heterogeneous pool of cycling cells. Namely, radial glial cells (RGCs), the primordial stem cell of the neocortex, give rise to neurons directly via asymmetric cell division or to two populations of intermediate progenitor cells, apical intermediate progenitor cells (aIPCs) and basal intermediate progenitor cells (bIPCs) (Malatesta et al., 2003; Anthony et al., 2004; Gal et al., 2006; Stancik et al., 2010; Tyler and Haydar, 2013). These progenitor cell types can be distinguished by the expression of Sox2 (in RGCs and aIPCs) and Tbr2 (in bIPCs) (Haubensak et al., 2004; Miyata et al., 2001; Noctor et al., 2004). Therefore, we performed IUE at E14.5 to determine whether CLASP2 regulates the neuronal output globally or specifically affects RGCs or bIPCs. At E17.5, 72 hours post IUE, the number and position of GFP-positive cells expressing Sox2 or Tbr2 were quantified. We found a higher percentage of CLASP2 knockdown cells positive for Sox2 (Figure 3H) and Tbr2 (Figure 3I) compared to control indicating a general increase in the progenitor cell population retained in the VZ/SVZ. Together, this data indicates CLASP2 regulates neural stem cell division and neuronal output.
CLASP2 is Necessary for Proper Cortical Positioning
To determine whether CLASP2 plays a role in neuronal migration, we electroporated mouse embyros at E14.5 and analyzed the distribution of GFP positive cells within the cortical wall at E16.5 and P0. At E16.5, GFP-positive cells in control brains were found equally distributed between the VZ/SVZ and intermediate zone (IZ) (Figure 4A). However, there were obvious differences in spatial distribution in CLASP2 knockdown cells with more GFP-positive cells in the VZ/SVZ and fewer in the IZ (Figure 4A). At P0, while most of the control GFP-positive neurons migrated into the cortical layers (CL); CLASP2 knockdown neurons remained in the IZ with fewer neurons in the upper cortical layers (upCL) (Figure 4B). Interestingly, when we electroporated E14.5 embryonic brains with CLASP2 shRNAs and analyzed the brains at P14, many CLASP2 knockdown neurons were found at deeper positions within the lower cortical wall (loCL) suggesting that CLASP2 knockdown caused permanent defects in cortical lamination (Figure 4C). To distinguish whether this defect was due to altered cell cycle dynamics and neuronal output from progenitors or to a direct effect on neuronal migration, we sought to specifically knockdown CLASP2 expression in postmitotic neurons. To accomplish this, we co-electroporated conditional Cre-dependent CLASP2 shRNA plasmids with murine DCX-Cre-IRES-mCherry as previously described (Franco et al., 2011). We found knockdown of CLASP2 restricted to postmitotic neurons also resulted in a significant number of neurons mispositioned in deeper layers at P0 (Figure S3).
Figure 4. CLASP2 is Necessary for Radial Migration of Cortical Projection Neurons in the Mammalian Brain.
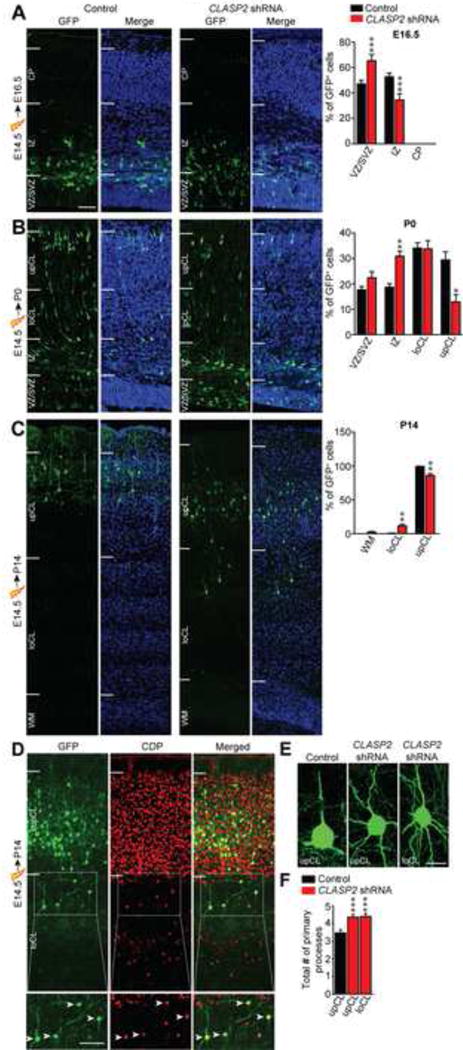
(A–C) Mouse embryos were electroporated in utero with GFP-tagged CLASP2 shRNAs or scrambled control at E14.5 and analyzed at E16.5 (A, control, n = 6 brains; CLASP2 shRNA, n = 6 brains), P0 (B, control, n = 3 brains; CLASP2 shRNA, n = 6 brains) and P14 (C, control, n = 4 brains; CLASP2 shRNA, n = 6 brains). Coronal sections of the cortex were visualized for transfected GFP-positive neurons (green) and cell nuclei (Hoeschst 33342, blue). White lines indicate the demarcations for different cortical regions (VZ = ventricular zone, SVZ = subventricular zone, IZ = intermediate zone, CP = cortical plate, loCL = lower cortical layer, upCL = upper cortical layer, WM = white matter). For additional data, see Figure S3.
(D) Coronal brain sections from E14.5 CLASP2 shRNA electroporation and analyzed at P14 were immunostained with layers II/III marker CDP (red).
(E–F) Representative images of morphological defects of CLASP2 shRNA neurons in the upCL and loCL. CLASP2 knockdown caused an increase in the number of primary neurites independent of cortical layer (control, n = 53 cells; CLASP2 shRNA upCL, n = 60 cells; CLASP2 shRNA loCL, n = 39 cells were analyzed). For additional data, see Figure S4.
Data are means ± SEM and statistical significance was assessed using one-way ANOVA (*p < 0.05, **p<0.001, ***p<0.0001). Scale bar represents 50 μm (A–D) and 10 μm (E).
We next analyzed the molecular and morphological profiles of electroporated neurons at P14, a stage at which migration is complete. As expected, we found the majority of control GFP-positive neurons electroporated at E14.5 were located in layers II and III and expressed the characteristic layer II/III marker CCAAT Displacement Protein (CDP; data not shown). Interestingly, we found that mislocalized neurons electroporated with CLASP2 shRNA also expressed the CDP marker even though they were permanently arrested at deeper cortical layers (Figure 4D). This data demonstrates that although CLASP2 knockdown permanently alters the location of some neurons in the neocortex, it does not change their intrinsic transcriptional identity.
We next wanted to determine the effect of CLASP2 on the postmigratory differentiation of cortical neurons in vivo. In cortical layers II and III, pyramidal neurons primarily exhibit a polarized morphology with a single long axon crossing the corpus callosum with connectivity to the contralateral cortex, a thick apical dendrite, and smaller basal dendrites. We found CLASP2 knockdown neurons had more primary processes originating from the cell soma when compared to layer-matched controls at P14 (Figure 4E, 4F). In addition, CLASP2 knockdown caused the loss of a clear apical dendrite suggesting that these neurons may have lost their ability to correctly polarize or respond to extracellular cues. The effects of CLASP2 knockdown on the number of primary processes was independent of final cortical layer indicating the effects of CLASP2 on neuron polarization and morphology were cell autonomous and not the result of mislocalization within the cortical plate.
Having determined that CLASP2 knockdown leads to developmental defects, we tested the effects of CLASP2 overexpression on neuronal lamination. Recent human genetic evidence listed CLASP2 as a top neuronal gene with the greatest intolerance to dosage changes from copy number variants (Ruderfer et al., 2016). Intolerance to genetic variation, such as copy number, implies higher functional importance of a gene (Lek et al., 2016). Therefore, we performed IUE at E14.5 with control GFP, GFP-CLASP2α or GFP-CLASP2γ full-length expression vectors and analyzed the distribution of GFP-positive cells at P0. Overexpression of CLASP2α or CLASP2γ caused a robust increase in the number of GFP-positive neurons in the IZ and fewer neurons in the upCL compared to control (Figure S4), a phenotype similar to that seen following CLASP2 knockdown. We therefore conclude that CLASP2 expression must be tightly regulated during cortical development to enable proper cortical lamination.
CLASP2 is Required for the Extension and Orientation of the Leading Process of Migrating Neurons
Migrating neurons are highly polarized in the direction of their movement, which is achieved through the correct orientation and remodeling of a leading process that acts to generate traction forces and guide cell movement. Therefore, we asked whether the lamination defects observed in response to CLASP2 knockdown were due to a direct affect on the polarization of migrating neurons. We reasoned that this may be the case since CLASP2 has been shown to regulate neurite length and dendritic arborization in mouse primary neurons (Beffert et al., 2012) in a Reelin responsive manner (Figure 1D). To test this, we performed IUE at E14.5 and measured the length and orientation of the leading process in GFP-positive neurons of control and CLASP2 knockdown neurons as they migrated through the lower cortical layers at P0. Our results demonstrate that CLASP2 knockdown caused a 48% decrease in the length of the leading process when compared to controls (Figure 5A). In addition, we examined the orientation of the thickest leading process in GFP-positive neurons to determine the likely migratory path of each cell and calculated this as an angle in relation to the pial surface. CLASP2 knockdown resulted in an increase in the angle of the leading process (Figure 5B). In particular, 85% of migrating GFP-positive control neurons had a leading process oriented between 0–10 degrees to the pia (Figure 5C); however, only 13% of CLASP2 knockdown neurons kept this normal, apical orientation. Instead, we found nearly 65% of CLASP2 knockdown neurons had a leading process oriented between 11–90 degrees and that 22% had their leading process inverted between 91–180 degrees. The morphological alterations of the leading process in CLASP2 knockdown neurons were not due to disruptions in the morphology of radial glial fibers (Figure 5D), suggesting CLASP2 plays an intrinsic role in regulating both the length and orientation of the leading process in migrating cortical projection neurons, regardless of the orientation of the glial guides.
Figure 5. CLASP2 is Required for Neuronal Polarity.
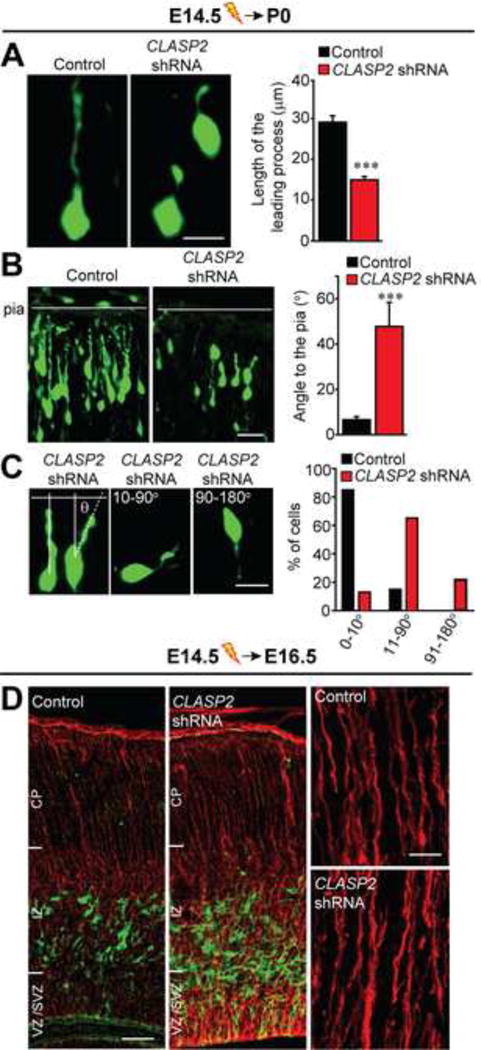
Mouse embryos were electroporated in utero with GFP-tagged CLASP2 shRNAs or scrambled control at E14.5 and analyzed at P0.
(A) Morphological and quantitative analysis of migrating GFP-positive neurons showing the length of the leading process (control, n = 44 cells; CLASP2 shRNA, n = 34 cells).
(B–C) Representative images of the angle of the leading proess in relation to the pial surface. The angle was measured by using the closest connecting line from the cell soma to the pia (control, n = 20 cells; CLASP2 shRNA, n = 23 cells). The percentage of cells with a leading process oriented between 0–10°, 11–90° and 91–180° to the pia were quantitated.
(D) Immunohistochemical analysis of ascending radial glial fibers immunostained against Nestin (red) showed normal stereotypical radial fibers spanning the entire cortical wall in control and CLASP2 shRNA cortex, oriented from the VZ towards the pial surface (VZ = ventricular zone, SVZ = subventricular zone, IZ = intermediate zone, CP = cortical plate).
Data are means ± SEM (A–C) and statistical significance was assessed using unpaired t test (***p<0.0001). Scale bar represents 10 μm (A, C), 20 μm (B), 25 and 50 μm for (D).
CLASP2 Controls the Spatial Positioning of the Centrosome and Golgi Complex
The positioning of the centrosome and Golgi apparatus is a key step in selecting migratory direction, and their translocation into the leading neurite precedes cell movement during neuronal migration (Yanagida et al., 2012; Sakakibara et al., 2013). Because CLASP2 is enriched at both the Golgi and centrosome, two organelles that control cell polarity (Miller et al., 2009; Beffert et al., 2012), we examined whether CLASP2 expression influenced the position and shape of the centrosome/Golgi complex during neuronal migration. We performed IUE at E14.5 with GFP-CLASP2 shRNAs and scrambled GFP-control plasmids. At P0, brains were dissected and stained separately for the centrosome (pericentrin) and golgi complex (GM130). Previous studies demonstrate that nucleus-centrosome coupling is an important mechanism controlling neuron polarization during radial migration (Xie et al., 2003; Solecki et al., 2004). We therefore measured the distance between the nucleus and centrosome in migrating neurons within the cortical wall. At P0, our results show that CLASP2 knockdown caused a reduction in the distance between the nucleus and centrosome when compared to controls (Figure 6A). Similar to previous reports, we found co-localization of Golgi and centrosome markers in both control and CLASP2 shRNA electroporated cells (Yanagida et al., 2012). The Golgi and centrosome complex was localized to the leading process in ~80% of GFP-positive control cells; however, following CLASP2 knockdown, there was a mislocalization of this complex with a higher percentage of cells with Golgi and centrosome staining adjacent to the nucleus (Figure 6B and 6C).
Figure 6. CLASP2 is Necessary for Centrosome-Golgi Localization.
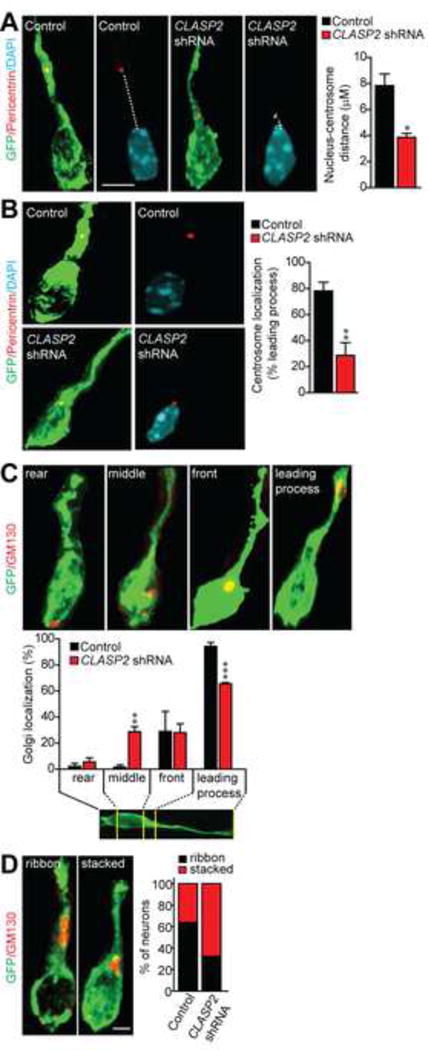
(A–B) Immunostaining for Pericentrin (red) shows centrosome localization and demonstrates a shorter distance between the nucleus and centrosome in CLASP2 knockdown neurons (control, n = 43 cells; CLASP2 shRNA, n = 53 cells). Nuclei were stained with DAPI (blue).
(C–D) Immunostaining for GM130 shows Golgi localization and percentage of neurons exhibiting a ribbon or stacked Golgi phenotype (control, n = 51 cells; CLASP2 shRNA, n = 36 cells). For additional data, see Figure S5.
Data are means ± SEM and statistical significance was assessed using unpaired t test and one-way ANOVA (*p < 0.05, **p<0.001, ***p<0.0001). Scale bar represents 5 μm.
The shape of the Golgi has previously been shown to be critical for directed cargo delivery to the leading edge of motile cells (Miller et al., 2009) and nucleation of microtubules at the Golgi is dependent on CLASP2 (Efimov et al., 2007; Miller et al., 2009). Therefore, we qualitatively assessed the shape of the Golgi in GFP-positive neurons following electroporation with control or CLASP2 shRNA plasmids. Our results indicate that CLASP2 knockdown caused a decrease in the frequency of Golgi ribbons within migrating neurons (Figure 6D). Therefore, CLASP2 expression controls Golgi/centrosome dynamics in the leading process which are necessary for nucleokinesis and directed cell movement during neuron migration. In addition, these data support the conclusion that CLASP2 performs a distinct function in migrating neurons apart from its role in proliferating neural progenitors.
We next questioned whether the delayed migration of CLASP2 knockdown neurons was due to leading process defects and/or to reduced speed of migration caused by alterations in centrosome-nucleus coupling. To address this, we performed IUE at E14.5 with GFP-CLASP2 shRNAs and scrambled GFP-control plasmids and then measured the migration rate of GFP-positive neurons in cortical slices prepared on E17.5 using multiphoton time-lapse imaging. We found that CLASP2 knockdown did not reduce the speed of migration (Figure S5, movie S1 and movie S2), suggesting the delayed migration of CLASP2 knockdown neurons is likely due to alterations in the morphology and polarity of migrating neurons during the final phase of migration.
GSK3β Phosphorylation of CLASP2 Regulates Binding to Dab1
To investigate the molecular mechanism by which Reelin regulates CLASP2 expression and function, we examined the intersection between CLASP2 and known components of the Reelin signaling pathway. Dab1, the intracellular adaptor protein that mediates the Reelin signal in cells, interacts with target proteins through a phosphotyrosine-binding domain that preferentially binds to NPXY motifs (Trommsdorff et al., 1998; Howell et al., 1999). Since CLASP2 contains several putative NPXY motifs (see red asterisks; Figure 7A), we hypothesized that CLASP2-mediated effects on neuronal development may be dependent on an interaction between Dab1 and CLASP2 and/or activated through Dab1-dependent phosphorylation signaling pathways. To examine these possibilities, we co-transfected Dab1 with GFP-CLASP1 or GFP-CLASP2α in HEK293T cells and performed immunoprecipitation assays (Figure 7B). Both CLASP1 (lane 3) and CLASP2 (lane 2 and 4) co-immunoprecipitated with Dab1. In particular, the N-terminal portion of CLASP2 (1-820 amino acids – lane 5) selectively associated with Dab1. In addition, we generated five GFP-CLASP2 fragments that consisted of the first TOG1 domain (1-270 amino acids), TOG2 domain (271-573 amino acids), positively-charged serine/arginine rich region (S/R rich, 574-820 amino acids), TOG3 domain (821-1200 amino acids) or the C-terminal CLIP domain (1200-1515 amino acids) and used them to map the domain responsible for the CLASP2/Dab1 interaction (Figure 7A). We found the S/R rich domain (574-820 amino acids) of CLASP2 specifically co-immunoprecipitated with Dab1 (lane 4 in Figure 7C).
Figure 7. GSK3β-Mediated Phosphorylation of CLASP2 Affects Dab1 Binding.
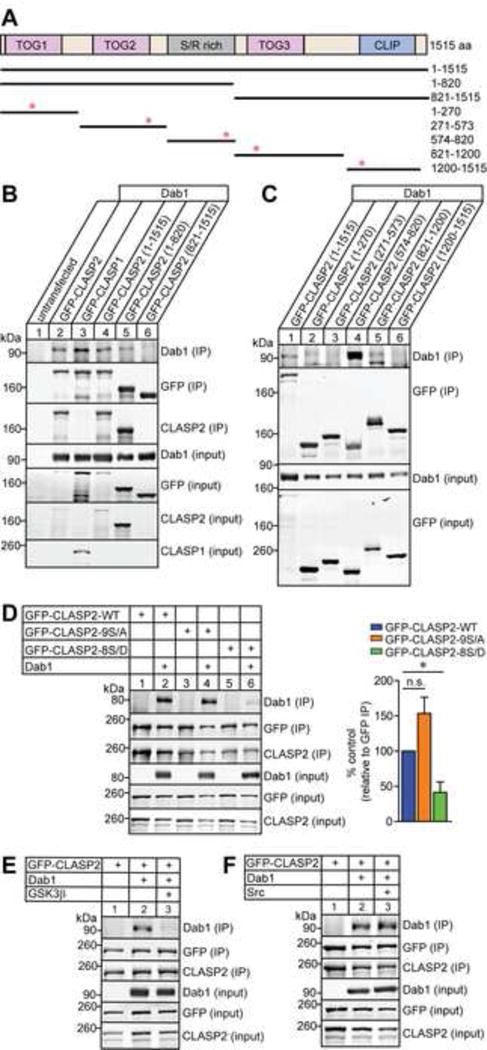
(A) Schematic of human CLASP2α domain organization and GFP-tagged CLASP2 constructs used for immunoprecipitation with corresponding amino acid (aa) residues on the right. Red asterisks indicate the position of the putative NPxY motifs in CLASP2.
(B–C) HEK293T cells were co-transfected with Dab1 and GFP-CLASP2 plasmids. Cell lysates were collected 48 h post-transfection and immunoprecipitated with GFP antibody and immunoblotted for Dab1, GFP and CLASP2. Both CLASP1 (lane 3) and CLASP2 (lane 2 and 4) bind to Dab1 and the binding region for Dab1 is within the first 820 amino acids (lane 5) of CLASP2 (B). CLASP2 specifically binds to Dab1 at the S/R domain (lane 4) between 574-820 amino acids (C).
(D) HEK293T cells were co-transfected with Dab1 and GFP-CLASP2 wild type (WT) or GFP-CLASP2-9S/A or -8S/D phospho-mutants. Cell lysates were collected 48 h post-transfection and immunoprecipitated with GFP antibody and immunoblotted for Dab1, GFP and CLASP2. The phospho-mimetic CLASP2-8S/D mutant (lane 6) showed less Dab1 binding compared to CLASP2-WT (lane 2) or the phospho-resistant CLASP2-9S/A (lane 4) mutant. These results were quantified as relative to the immunoprecipitated level of GFP (n = 3 independent experiments).
(E) The addition of constitutively active GSK3β (lane 3) abolished CLASP2 binding to Dab1 (n = 2 independent experiments).
(F) Phosphorylation of Dab1 with constitutively active Src (lane 3) did not affect CLASP2 binding to Dab1 (n = 3 independent experiments).
Data are means ± SEM and statistical significance was assessed using one-way ANOVA (*p < 0.05). n.s. represents not significant.
Interestingly, the S/R rich region contains nine serine residues that constitute consensus motifs for GSK3β phosphorylation, which disrupt the electrostatic bonds formed between CLASP2 and microtubules thus preventing plus-end tracking (Kumar et al., 2009; Watanabe et al., 2009; Kumar and Wittmann, 2012). To determine whether GSK3β-mediated phosphorylation of CLASP2 affects Dab1 binding, we examined GFP-CLASP2 phospho-mutants which mimic a constitutively active phosphorylated state in which eight serine resides in the S/R domain of CLASP2 were replaced with aspartate (GFP-CLASP2-8S/D) or a phospho-resistant CLASP2 mutant (GFP-CLASP2-9S/A) where each of the nine serine residues were replaced with alanine (see Figure S6 for a list of mutated sites). When HEK293T cells were co-transfected with Dab1 and GFP-CLASP2 phospho-mutants, we found both wild type (lane 2 in Figure 7D) and phospho-resistant GFP-CLASP2-9S/A (lane 4) co-immunoprecipated with Dab1. In contrast, phospho-mimetic GFP-CLASP2-8S/D (lane 6 in Figure 7D) exhibited greatly reduced binding to Dab1 indicating that mimicking the phosphorylation of CLASP2 decreases Dab1 binding affinity. Next, we co-transfected HEK293T cells with plasmids encoding GFP-CLASP2, Dab1 and consitutively active GSK3β to determine whether GSK3β is the kinase responsible for modulating the CLASP2-Dab1 interaction. GSK3β over-expression completely abolished the CLASP2-Dab1 interaction (lane 3 in Figure 7E).
Reelin exerts its function by binding to the lipoprotein receptors ApoER2 and VLDLR and inducing tyrosine phosphorylation of the intracellular adaptor protein Dab1 by Src family kinases (Howell et al., 2000; Bock and Herz, 2003) therefore, we set out to determine whether Src-mediated phosphorylation of Dab1 affects the interaction with CLASP2. Surprisingly, the addition of active Src kinase had no effect on CLASP2-Dab1 binding indicating that the phosphorylation state of Dab1 does not control CLASP2 binding (lane 3 in Figure 7F). Altogether, we conclude that GSK3β-mediated phosphorylation of CLASP2 regulates its interaction with Dab1.
GSK3β Phosphorylation of CLASP2 Controls Cell Motility and Neurite Extension
To determine whether Reelin regulates phosphorylation of CLASP2, we used tandem mass tagging spectrometry to identify and quantitate specific phosphorylation sites on CLASP2 in mouse primary cortical neurons in response to Reelin stimulation. Our analysis identified several phosphorylation sites within the positively-charged S/R rich region that were specifically altered in response to Reelin, including three of the same sites known to be directly regulated by GSK3β (Figure S6). These findings indicate that Reelin signaling alters the status of specific phosphorylation sites on CLASP2 that likely leads to functional changes (Figure 8A and S7).
Figure 8. GSK3β Phosphorylation of CLASP2 Regulates Cell Motility and Neurite Extension .
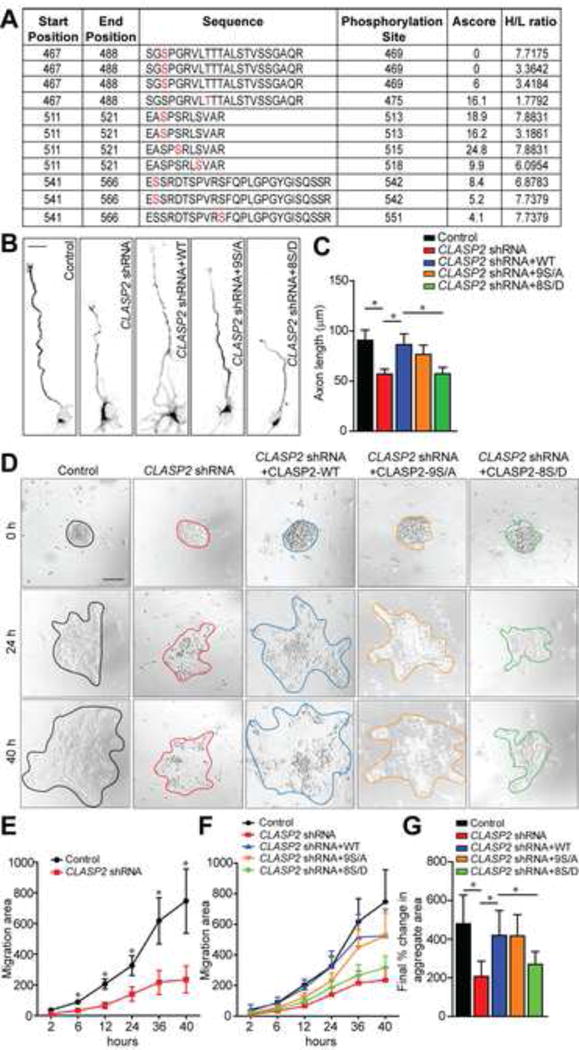
(A) Table lists several CLASP2 phosphopeptides identified by tandem mass spectrometry showing changes following Reelin treatment of mouse primary neurons. The first two columns indicate the start and end position of the peptides within the mouse CLASP2γ sequence. The amino acid sequence is indicated with the putative phosphorylation site (marked in red) followed by the position of the residue within CLASP2γ. Ascore represents the probability of correct phosphorylation site localization based on the presence and intensity of site-determining ions from the MS/MS spectra. H/L represents the ratio of signal of the specific phosphopeptide in Reelin (H) or control (L) treated neurons. For additional data, see Figure S6 and S7.
(B) Representative images of primary dissociated mouse wild type neuron cultures co-infected with scrambled control, CLASP2 shRNAs or CLASP2 shRNAs with CLASP2-wild type (WT), CLASP2-9S/A or -8S/D phospho-mutants and immunostained against tau axonal marker at 2 days in vitro.
(C) CLASP2 shRNA caused a decrease in axon length when compared to control. Both CLASP2-WT and phospho-resistant CLASP2-9S/A rescued the CLASP2 shRNA phenotype whereas phospho-mimetic CLASP2-8S/D was unable to rescue the axonal effects of CLASP2 knockdown (control, n = 19; CLASP2 shRNA, n = 28; CLASP2-WT, n = 21; CLASP2-9S/A, n = 17; CLASP2-8S/D, n = 33 cells).
(D) Primary mouse wild type neuron cultures were co-infected with scrambled control, CLASP2 shRNAs or CLASP2 shRNAs with CLASP2-wild type (WT), CLASP2-9S/A or -8S/D phospho-mutants and cell migration was assessed by measuring the area of migration away from the initial aggregates over a 48 h period
(E) CLASP2 shRNA caused a decrease in migration area compared to control (control, n = 8; CLASP2 shRNA, n = 8 aggregates).
(E–F) Phospho-mimetic CLASP2-8S/D was unable to rescue the deleterious effects of CLASP2 shRNA on cell motility compared to CLASP2 WT and CLASP2-9S/A phosphomutant (control, n = 8; CLASP2 shRNA, n = 8; CLASP2-WT, n = 5; CLASP2-9S/A, n = 9; CLASP2-8S/D, n = 13 aggregates).
Data are means ± SEM and statistical significance was assessed using one-way ANOVA (*p < 0.05). Scale bar represents 10 μm (B) and 50 μm (D).
We next evaluated whether GSK3β phosphorylation of CLASP2 affects neurite extension. We compared mouse primary hippocampal neurons infected with control shRNA, CLASP2 shRNAs, CLASP2 shRNAs rescued with human CLASP2α wild type, phospho-resistant CLASP2-9S/A, or phospho-mimetic CLASP2-8S/D lentivirus by immunofluorescence. At 2 days in vitro, quantitative analysis showed CLASP2 knockdown resulted in a 27% decrease in axon length (Figure 8B and 8C). Both CLASP2α wild type and phospho-resistant CLASP2-9S/A rescued the CLASP2 shRNA phenotype whereas phospho-mimetic CLASP2-8S/D was unable to rescue the axonal deficits following CLASP2 knockdown. These results showed GSK3β phosphorylation regulates the effects of CLASP2 on neurite extension.
To determine whether GSK3β phosphorylation is associated with CLASP2’s effects on cell motility, we used an in vitro neuronal migration assay to compare the effects of control shRNA, CLASP2 shRNAs, CLASP2 shRNAs rescued with human CLASP2 wild type, phospho-resistant CLASP2-9S/A, or phospho-mimetic CLASP2-8S/D mutants. Cortical neurons isolated from newborn pups were dissociated and allowed to form aggregates by incubating in a polypropylene tube before plating on matrigel-coated coverglass. Cell migration was assessed by measuring the area of migration away from the inital aggregates at day of plating using live imaging over a 48 hour period (Figure 8D). The shRNA-mediated knockdown of CLASP2 decreased cell migration compared to control (Figure 8E and 8G). Both CLASP2 wild type and phospho-resistant CLASP2-9S/A rescued the CLASP2 shRNA phenotype whereas the phospho-mimetic CLASP2-8S/D was unable to rescue the inhibitory effects of CLASP2 knockdown on cell migration. These results indicate GSK3β-mediated CLASP2 phosphorylation alters cell motility (Figure 8F and 8G). These results provide strong evidence in neurons that phosphorylation of CLASP2 by GSK3β, can control neurite extension and subsequent migration.
DISCUSSION
Here, we investigated how microtubule dynamics and organization are coupled to extracellular signaling during brain development. We used a systems analysis approach to identify genes perturbed in the Reelin signaling pathway and identified the microtubule plus-end binding protein CLASP2 as a key cytoskeletal effector of Reelin mutant phenotypes. We show down-regulation of CLASP2 in migrating neurons within the developing cortex leads to the mis-localization of neurons to deeper cortical layers, in part due to abnormal positioning of the centrosome-Golgi complex, and aberrant length/orientation of the leading process during migration. Mechanistically, we demonstrate GSK3β-mediated phosphorylation of CLASP2 controls binding to the intracellular adaptor protein Dab1 and is essential for regulating CLASP2’s effects on neurite extension and cell motility. Together, these results represent the first demonstration of CLASP2 function in the developing mammalian brain and firmly establish CLASP2 is a downstream cytoskeletal effector of Reelin signaling pathway in reshaping the cytoskeleton necessary for neurite growth and motility.
Reelin Controls CLASP2 Function
Our initial genome-wide screen discovered CLASP2 as the most highly regulated gene common to three independent mouse models with deficits in Reelin signaling and provides a strong in vivo functional connection between CLASP2 and the Reelin pathway (Figure 1). In addition, our data demonstrate that Reelin signaling leads to specific post-translational modifications on CLASP2 which affect its function. The unbiased experiment using quantitative mass spectrometry on CLASP2 demonstrates the ability of Reelin to regulate CLASP2 by altering the number of functionally important phosphorylation sites, most of which are GSK3β substrates in primary mouse neurons (Figure 8). Moreover, we demonstrate that specific mutations of CLASP2-GSK3β phosphorylation sites regulate the interaction between CLASP2 and Dab1, a finding consistent with a recent Drosophila screen which showed the fly homolog of Dab interacts with CLASP (Long et al., 2013). We further show GSK3β-mediated phosphorylation of CLASP2 is crucial for its function in regulating neurite extension and cell motility. Indeed, earlier supporting studies have demonstrated the importance of the multi-site phosphorylation by GSK3β where it was found to disrupt the electrostatic bonds formed between CLASP2 and the acidic C-terminal tail of tubulin, preventing the association of CLASP2 with microtubules (Watanabe et al., 2009; Kumar and Wittmann, 2012).
There is significant experimental evidence to support a direct role of GSK3β in both regulating downstream effects of Reelin signaling as well as that of CLASP2. First, we identified a number of strongly regulated CLASP2 phosphorylation sites following Reelin treatment, most of which were GSK3β substrate sites. In addition, others have confirmed a role for GSK3β in mediating the downstream effects of Reelin (Beffert et al., 2002). Second, a role for GSK3β directly regulating CLASP2 function including growth cone regulation in development strongly support a functional role for phospho-specific regulation of CLASP2 in neuronal development (Hur et al., 2011). Third, activated GSK3β has been shown to cause cortical migration defects, abnormal centrosome positioning, and destabilization of microtubules at the leading process (Asada and Sanada, 2010; Morgan-Smith et al., 2014), phenotypes which are similar to those resulting from mutations in Reelin signaling and those presented in the current study following CLASP2 knockdown. Therefore, we propose a model whereby the localization of CLASP2 during radial migration is regulated by GSK3β through Reelin signaling and CLASP2 enrichment at the leading edge is necessary to stabilize the leading process for directed movement.
Mechanisms By Which CLASP2 Regulates Progenitor Proliferation and Differentiation
CLASPs have been shown to affect microtubule turnover during both interphase and mitosis (Galjart, 2005). In mitotic cells, CLASPs localize to kinetochores where they promote the turnover of attached microtubules in order to ensure spindle bipolarity and chromosome alignment/segregation (Maiato et al., 2003, 2005; Pereira et al., 2006; Maffini et al., 2009; Logarinho et al., 2012; Maia et al., 2012; Samora et al., 2011). In addition, CLASP knockdown causes depolymerization of astral microtubules, mispositioning of the mitotic spindle, and mitotic delays (Maia et al., 2012; Samora et al., 2011); however, the role of CLASPs during neurogenesis is unknown. Here, we found that CLASP2 affects terminal cell division of neural progenitors in the developing mouse cortex. Multiple lines of evidence support this: first, we found differential expression of CLASP2 isoforms during cortical development. CLASP2α isoform peaks during neurogenesis at E12.5–16.5 in the mouse developing brain where we found a specific increase in CLASP2α mRNA in the proliferative VZ/SVZ. Second, CLASP2 knockdown increased the percentage of both apical Sox2 and basal Tbr2 neural progenitor cell population. Third, there was a greater percentage of CLASP2 knockdown cells co-localized with the cell cycle marker PH3 indicating CLASP2 knockdown could be prolonging the cell cycle or preventing terminal differentiation by maintaining progenitors in a proliferative state. Lastly, we found a decrease in the percentage of CLASP2 knockdown cells which exited from the cell cycle over a 24 hour period following EdU injection. Taken together, these results suggest CLASP2 regulates the timing of neuronal output from progenitor cells in the VZ/SVZ. However, these results are likely independent of Reelin-Dab1 signaling since reeler mice display no obvious problems with proliferative mechanisms (Caviness et al., 2008).
Regulation of Neuronal Migration by CLASP2
Our data suggests that Reelin-induced changes in CLASP2 function are essential for regulating neuronal migration during cortical development and that the pleiotropic functions of CLASP2 in the cortex indicate that its activity must be tightly controlled. We show that misregulation of CLASP2 expression clearly leads to alterations in the cortical lamination pattern of affected neurons, as evidenced by knockdown and overexpression of CLASP2 in vivo. This is further supported by recent human genetic findings which identified both CLASP2 and Reelin as neuronal developmental genes demonstrating the greatest intolerance to dosage changes from copy number variants (Ruderfer et al., 2016).
Previous studies have shown both Reelin and CLASP2 play direct roles in modulating neuronal polarization through the regulation of both Golgi shape and deployment into the leading process (Matsuki et al. 2010; O’Dell et al., 2012; Beffert et al., 2012). In addition, CLASP-dependent nucleation of microtubules at the Golgi is required for directional cargo trafficking (Miller et al., 2009; Efimov et al., 2007). Our results indicate CLASP2 knockdown causes the mislocalization of the Golgi away from the leading process and a decrease in the percentage of cells demonstrating Golgi ribbon morphology. Therefore, the disruption of Golgi shape and position following CLASP2 knockdown would prevent directed cargo trafficking to the leading process and cause a loss of neuron polarization. To support this, we show that CLASP2 knockdown led to an overall increase in the number of primary processes, defect in leading process orientation, and the loss of a clear apical dendrite in differentiated neurons indicating intrinsic defects in neuron polarization independent of final cortical position.
Altogether, our data indicate that CLASP2 plays distinct roles in controlling neuronal production and positioning during early brain development. In particular, while CLASP2 knockdown decreases neuronal output from the germinal zones, the cells that are eventually produced express appropriate laminar identity markers and many of these cells eventually reside in the appropriate layer. However, it is clear that a proportion of the generated neurons migrate abnormally and are located in ectopic (deep) locations regardless of whether CLASP2 is knockdown starting at the precursor stage or after the neurons have been generated. Indeed, the conditional expression of CLASP2 shRNA in neurons demonstrates clearly that this ectopia is due to CLASP2-mediated effects at the postmitotic migrating neuron stage.
In the cerebral cortex, defects in neuronal precursor proliferation, morphogenesis, cell migration, and synaptogenesis can lead to mental retardation, autism spectrum disorders and epilepsy. Therefore, elucidating the pathways which integrate extracellular cues with cytoskeleton reorganization are essential to understanding cortical architecture and connectivity.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Angela Ho (aho1@bu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mouse protocols were approved by the Institutional Animal Care and Use Committee at Boston University and Boston University School of Medicine. Wild type C57BL/6J mice were purchased from The Jackson Laboratory. Timed pregnant CD-1 dams were purchased from Charles River Laboratories. For all mice studies, mice of either sex were used.
Primary Neuronal Cultures and Lentiviral Infection
Primary neuronal cultures were prepared from newborn C57BL/6J mice of either sex and infected with lentiviruses on the day of plating as previously described (Ho et al., 2006). Briefly, neurons were dissociated with trypsin, triturated, and plated onto coverslips coated with Matrigel. Recombinant lentiviruses were produced by transfecting HEK293T cells using FuGENE6 (Roche) with plasmids encoding viral enzymes and envelope proteins essential for packing of viral particles (RSV/REV, MDLg/RRE, and VSVG) with the addition of a shuttle vector encoding the gene of interest. To determine axon length, hippocampal neurons were infected with control scrambled or CLASP2 shRNA plasmids at the day of plating and fixed 72 hours later. Neurons were treated each day (a total of 3 times) with 20 nM of reelin or media control. Axons were stained with Tau antibody and the longest neurite was traced using NIH ImageJ software.
Migration Assay of Primary Mouse Neuronal Cultures
Primary neurons were harvested from the cortex of newborn C57BL/6J mice of either sex and dissociated to form a single cell suspension. Cell suspensions were allowed to stand overnight in polypropylene tubes causing them to re-aggregate before being plated into 24-well dishes coated with Matrigel. Four hours after plating, cell aggregates were adherent to the substrate and initial pictures were taken using a Carl Zeiss MZ6 fluorescence microscope. Cell aggregates were photographed at 8–10 hour intervals using 10x phase-contrast optics. Quantification of the migration area was calculated using ImageJ software. Lines were drawn in freehand connecting the outermost cells recognizable to the initial aggregate. All observations were recorded blind to the treatment group and migration area was calculated as a percentage with respect to the size of the initial aggregate.
Organotypic Slice Culture and Multiphoton Time Lapse Imaging
Seventy two hours post-electroporation, embryos were harvested and freshly dissected in complete HBSS which consisted of 1× Hanks Balanced Salt Solution with 2 mM Hepes, 2 mM D-Glucose, 0.2 mM CaCl2, 0.2 mM MgSO4, and 2 mM NaHCO3. Brains were screened for positive expression of GFP using an Olympus MVX10 stereomacroscope equipped with epifluorescence illumination. The brains were embedded in DMEM/3% low melting point agarose and 350–400 micron organotypic sections were prepared using a VT1000S vibrating blade microtome. Slices were embedded in a collagen matrix on inverted transwell inserts and placed in 35 mm dishes in Neuroimaging medium (NIM) for a 30 min recovery period prior to imaging. NIM consisted of neurobasal medium supplemented with B27, N2 supplement, 10% fetal bovine serum, 1% penicillin-streptomycin, and glutamax.
For imaging, the organotypic slices were imaged using a custom perfusion chamber (BiopTechs) attached to the stage of the Zeiss LSM 710 microscope coupled to a Chameleon Vision-S Ti:Sapphire laser (Coherent). GFP was excited at 995 nm and emitted light was collected with 2 LSM binary GaAsP module photodetectors. Z-stack images (100–200 μm, 2 μm intervals) were collected at 10 min intervals with a 20× with 1.0 numerical aperture water dipping objective for the duration of the experiment, which typically ran overnight for greater than 8–12 h. Individual dividing cells were analyzed further by post-processing using the ZEN software package (Carl Zeiss).
METHOD DETAILS
Molecular Cloning
The shRNA constructs for mouse CLASP2 (shRNA-A GCATCAGTCCTTTCAACAAGT and shRNA-B GAACTTGAAGAGACGTTAAAT) and control scrambled shRNA (CCGCAGGTATGCACGCGT) were subcloned into the pLKO.1 (Addgene plasmid 10879), pSico (Addgene plasmid 11578) and pCGLH vectors for lentivirus and in utero electroporation studies, respectively. Full-length human CLASP2α (National Center for Biotechnology Information reference sequence NM_015097.2) was PCR amplified from a full-length cDNA clone (IMAGE clone 9021646; BC140778) and subcloned into pCAG and pEGFP-C1 (Clontech). In addition, we obtained human full-length cDNA for CLASP2α, CLASP2-9S/A and CLASP2-8S/D (kind gift from Dr. Torsten Wittman, University of California San Francisco) and these were subcloned into the lentiviral vector pFUW and sequence verified. To generate truncated GFP-CLASP2 variants, we used full-length human CLASP2α as template and designed PCR primers at the regions of lowest complexity in order to optimally preserve domain structure, specifically 1–820, 821–1515, 1–270, 271–573, 574–820, 821–1200 and 1200–1515 amino acids were amplified by PCR and subcloned into pEGFP-C3 (Invitrogen).
Microarray and IPA System Analysis
Total RNA from control C57Bl/6, Reeler mice, ApoER2/VLDR double knockout, Dab1 knockout mice was prepared by TRIzol isolation (Invitrogen). A total of three cortex samples were taken at 21 days of age and pooled per condition. Samples were hybridized to the GeneChip Mouse Genome 430 2.0 Array (3′IVT, Affymetrix) and Ingenuity Pathway Analysis (Ingenuity Systems) was used to determine downstream effects most likely associated with knockout of the Reelin signaling pathway. Core analysis was run for each mouse model with a cut-off of 1.5 fold change compared to age-matched controls. A comparison analysis was run between mouse models and relevant disease profiles were pulled out based on p-value rank.
Quantitative Polymerase Chain Reaction
Embryonic and newborn cortex was homogenized with a pestle (Thomas) and total RNA was purified using the RNeasy Mini Kit (Qiagen). Total RNA from the adult cortex samples were processed with Trizol (Invitrogen). At least five independent cortex samples were isolated for each developmental timepoint. cDNA was synthesized using Superscript III Reverse Transcriptase (Invitrogen). Reverse transcription quantitative PCR was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems) using the Power SYBR Green PCR Mastermix (Applied Biosystems) with a two-step cycling protocol and an annealing/extension temperature of 60°C. The experiment was performed in duplicate with at least two biological replicates and three technical replicates each. The relative amount for each target was normalized using GAPDH as a reference gene and the fold change in gene expression was calculated using the ddCt method with the adult cortex samples as control.
In situ Hybridization
In situ hybridization (ISH) was outsourced to Reveal BioSciences, San Diego. Brain samples were harvested from CD1 mice and fixed in 10% Neutral Buffered Formalin (NBF). Fixed samples were processed into paraffin wax, embedded, and sectioned at 4 μm onto positively charged slides. Prior to staining, paraffin was removed by heating slides to 90–95°C for 10 min in aqueous buffered solution before protease digestion. Slides were then fixed in 10% NBF for 5 min before target probe set hybridization at 40°C for 2 hours. Probes were labeled with an alkaline phosphatase-conjugated oligonucleotide for 15 min before addition of fast blue or fast red substrates for fluorescent imaging.
In utero electroporation
In utero electroporation (IUE) was performed on timed pregnant CD-1 dams (Charles River Laboratories) at E14.5 as described previously (Gal et al., 2006). Dams were anesthetized via intraperitoneal injection of a ketamine/xylazine mixture and the uterine horns were exposed by midline laparotomy. One to two microliters of plasmid DNA (final concentration of 3 μg/μl) mixed with 0.1% fast green dye (Sigma Aldrich) was injected intercerebrally using a pulled glass micropipette. For the shRNA experiments, both shRNA-A and shRNA-B were transfected together. For conditional shRNA experiments, both shRNA-A and shRNA-B were subcloned into the pSico plasmid and transfected together with Dcx-Cre-mCherry plasmid. For electroporation, the anode of a Tweezertrode (Harvard Apparatus) was placed over the dorsal telencephalon above the uterine muscle and four 35 volt pulses (50 ms duration separated by a 950 ms interval) were applied with a BTX ECM830 pulse generator (Harvard Apparatus). Following electroporation, the uterine horns were returned to the abdomen and the cavity filled with warm 0.9% saline solution. Incisions were closed with silk sutures and dams were returned to a clean cage and monitored closely during recovery. The procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Boston University School of Medicine. For the EdU experiment, IUE was performed on timed pregnant CD-1 dams at E14.5 and pulse labeled by EdU injection into pregnant dams at E15.5 by intraperitoneal injections of 5-ethynyl-2′-deoxyuridine (50 μg of EdU per gram of mouse body weight). Twenty-four hours following EdU injections, pregnant dams were sacrificed and embryos were harvested for immunohistochemical staining with EdU and Ki67 antibodies.
Immunohistochemistry
Electroporated pregnant dams and mice underwent transcardial perfusion with 4% paraformaldehyde in phosphate-buffered saline (PBS). Brains were removed, fixed in 4% paraformaldehyde overnight, and cryoprotected in 30% sucrose in PBS. Brains were frozen in tissue molds with OCT compound and coronal 10 μm sections were cut using a LEICA CM1850 cyrostat (LEICA Biosystems). Sections were immediately mounted on SuperFrost microscope slides (Fisher Scientific) and kept at -80◦C for storage. Prior to immunostaining, sections were rinsed with PBS and treated with 0.3% methanol peroxidase for 10 min to quench endogenous peroxidase activity. Antigen retrieval was performed by microwaving brain sections in10 mM sodium citrate buffer pH 6 at 800 watts for 1 min followed by 80 watts for 10 min. Sections were then blocked in 5% goat serum, 0.2% Triton X-100 in PBS for 1 h followed by incubation with primary antibodies overnight at 4°C. Sections were then washed with PBS and incubated with the appropriate Alexa-conjugated secondary antibodies before being mounted with ProLong-Gold mounting medium containing DAPI (Invitrogen).
Immunoprecipitation
Human Embryonic Kidney (HEK) 293T cells were lysed 48 h following transfection in immunoprecipitation buffer containing: 20 mM Tris HCL, 100 mM NaCl, 1 mM EDTA pH 8.0, and 1% NP-40, supplemented with protease/phosphatase inhibitors (Biotool). Protein extracts were incubated for 2 h with rotation at 4°C with the precipitating antibody (rabbit anti-GFP 1:500) followed by overnight incubation with 25 μl of protein A Ultralink resin (Thermo Scientific). The resin were washed several times with immunoprecipitation buffer and precipitated proteins were eluted by boiling for 5 min in reducing SDS sample buffer and resolved by SDS-PAGE.
CLASP2 Immunoprecipitation for Mass Spectrometry
Primary neuronal cultures were infected with GFP control or GFP-CLASPγ lentivirus at 2 days in vitro (DIV). On 7 DIV, neurons were treated with 284 μM of reelin or media control for 20 min. Neurons were briefly rinsed in PBS and lysed in IP buffer containing 20 mM Tris-HCl, 1% NP40, 100 mM NaCl, 1 mM EDTA with protease and phosphatase inhibitors. Neuronal lysates were centrifuged at 13,000 x g for 5 min and the supernatant were incubated with GFP antibody at 1:250 for 2 h at 4°C followed by the addition of A/G resin beads. The beads were incubated overnight and washed with IP buffer 4–5 times and samples were examined by SDS/PAGE followed by Coomassie staining using a Thermo Scientific Pierce Power Station and Power Stain Solution following the manufacturer’s protocol to identify appropriate bands. Excised gel bands were cut and samples were reduced with 1 mM DTT for 30 min at 60°C and alkylated with 5 mM iodoacetamide for 15 min in the dark at room temperature. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure (Shevchenko et al., 1996). Gel pieces were washed and dehydrated with acetonitrile for 10 min followed by removal of acetonitrile and dried in a speed-vac. Rehydration of the gel pieces was with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega, Madison, WI) at 4°C. Samples were placed in a 37°C room overnight and peptides were extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were then dried in a speed-vac.
Tandem Mass Spectrometry
Tandem mass spectrometry was completed at Harvard Medical School, Boston MA. Samples were re-suspended in 20 μl of 200 mM HEPES buffer along with 6 μl of acetonitrile. 2 μl of TMT0 or TMT131 were added as light and heavy labels to each set of samples. After 1 h, 2 μl of a 5% hydroxylamine was added for 15 min followed by the addition of 10 μl of 20% formic acid. Desalting of the samples was performed with an in-house de-salting tube using reverse phase C18 Empore SPE Disks (3M). On the day of analysis the samples were reconstituted in 10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip (Peng et al., 2001). After equilibrating the column, each sample was loaded via a Famos auto sampler (LC Packings) onto the column. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid). As each peptide was eluted they were subjected to electrospray ionization and then they entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific). Eluting peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern by the software program, Sequest (ThermoFinnigan) (Eng et al., 1994). The static modifications of 224.1525 mass units were set for lysine and the N-terminal of peptides (light label), along with 57.0215 mass units on cysteine (iodoacetamide). Differential modifications of 5.0104 mass units were set for lysine and the N-terminal of peptides (heavy label) along with 79.9663 mass units to serine, threonine, and tyrosine was included in the database searches to determine phosphopeptides. Phosphorylation assignments were determined by the Ascore algorithm. All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimenters are blind to condition during data acquisition and analysis. For imaging experiments, images were captured using a Zeiss LSM 700 scanning confocal microscope. Cortical layers were identified based on cell density using DAPI staining and GFP-positive cells were measured using the Volocity software (Improvision). Sample sizes (n) indicated in the figure legends 2, 3, 4A–D correspond to the number of brains analyzed. To quantify the length of the leading process and angle to the pial surface, images were taken from 2–3 representative slices of each brain. Length of the leading process was calculated as the distance from the middle of the cell body to the end of the longest neurite. To assess Golgi and centrosome position, GFP-positive cells were identified and Z-stack images (6–8 μm) were taken at 300× magnification. For both control and CLASP2 shRNA conditions, an average of 20 cells were analyzed per brain across multiple sections. Sample sizes (n) indicated in the figure legends 1, 4E–F, 5, 6, 8 correspond to the number of cells analyzed.
All statistical parameters are presented as means ± SEM. To assess for statistical significance between groups, we used an unpaired t test to analyze all pairwise datasets and one-way analysis of variance (ANOVA) for parametric analysis of multiple comparisons that was conducted using Prism 6 software (GraphPad Software). Number of experiments and statistical information are reported on the corresponding figure legends. In figures, asterisks denote statistical significance marked by *, p < 0.05; **, p<0.001; ***, p<0.0001.
DATA AND SOFTWARE AVAILABILITY
Gene expression data reported in this paper by microarray from three different Reeler mouse models are available at the GEO repository with assession number GSE94896. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94896
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
- IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
-
○A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
-
○
Please see the sample Table at the end of this document for examples of how reagents should be cited. To see how the typeset table will appear in the PDF and online, please refer to any of the research articles published in Cell in the August 25, 2016 issue and beyond.
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. You do not need to delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CDP M-222 | Santa Cruz | Cat# SC-13024, RRID:AB_2261231 |
| Rat polyclonal anti-CLASP1 | Abnova | Cat# MAB9736, RRID:AB_10754999 |
| Rat polyclonal anti-CLASP2 | Abnova | Cat# MAB9738, RRID:AB_10757498 |
| Rabbit polyclonal anti-Dab1 | Dr. J. Herz from UT Southwestern | N/A |
| Mouse monoclonal anti-GFP | Neuromab | Cat# 75-131, RRID:AB_10671445 |
| Rabbit polyclonal anti-GFP | Synaptic Systems | Cat# 132 002, RRID:AB_887725 |
| Mouse monoclonal anti-GM130 | BD Biosciences | Cat# 610822, RRID:AB_398141 |
| Mouse monoclonal anti-Ki67 | Cell Signaling | Cat# 12202, RRID:AB_2620142 |
| Mouse monoclonal anti-Nestin | BD Biosciences | Cat# 611658, RRID:AB_399176 |
| Mouse monoclonal anti-Pericentrin | BD Biosciences | Cat# 611814, RRID:AB_399294 |
| Rabbit polyclonal anti-PH3 | Cell Signaling | Cat# 9701, RRID:AB_331534 |
| Goat polyclonal anti-Sox2 | Santa Cruz | Cat# SC-17320, RRID:AB_2286684 |
| Mouse monoclonal anti-Tau1 | Millipore | Cat# MAB3420, RRID:AB_94855 |
| Rabbit polyclonal anti-Tbr2 | Abcam | Cat# AB23345, RRID:AB_778267 |
| Biological Samples | ||
| Adult brain tissue from C57Bl/6, Reeler mice, ApoER2/VLDR double knockout and Dab1 knockout mice | Dr. J. Herz from UT Southwestern | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit | ThermoFisher Scientific | Cat# C10425 |
| Deposited Data | ||
| Gene expression data from 3 different Reeler mouse models and controls | GEO | GSE94896, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94896 |
| Experimental Models: Cell Lines | ||
| Human: Human Embryonic Kidney (HEK) 293T cells | ATCC | Cat# CRL-11268, RRID:CVCL_1926 |
| Human: HEK293 stable cell line producing Reelin | Beffert et al., 2002 | |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J6 | The Jackson Laboratory | Stock No: 000664, RRID:IMSR_JAX:00 0664 |
| Mouse: CD-1 IGS | Charles River Laboratories | Cat# CRL:022, RRID:IMSR_CRL:022 |
| Recombinant DNA | ||
| pLKO.1 | Addgene | 10879 |
| pSico | Addgene | 11578 |
| pEGFP-C1 | Clontech | Cat# 632470 |
| pEGFP-C3 | Clontech | Cat# 632482 |
| pCGLH | Gal et al., 2006 | N/A |
| pCAG | Gal et al., 2006 | N/A |
| pFUW | Ho et al., 2006 | N/A |
| RSV/REV | Ho et al., 2006 | N/A |
| MDlg/RRE | Ho et al., 2006 | N/A |
| VSVG | Ho et al., 2006 | N/A |
| pENTR/D-TOPA EGFP-CLASP2α WT | Kumar et al., 2009 | N/A |
| pENTR/D-TOPA EGFP-CLASP2α 9SxA | Kumar et al., 2009 | N/A |
| pENTR/D-TOPA EGFP-CLASP2α 8SxD | Kumar et al., 2009 | N/A |
| DCX-Cre-IRES-mCherry | Franco et al., 2011 | N/A |
| Human CLASP2α | IMAGE clone 9021646 | BC140778 |
| Mouse CLASPγ | This paper | N/A |
| pLKO.1-CLASP2 shRNA-A | This paper | N/A |
| pLKO.1-CLASP2 shRNA-B | This paper | N/A |
| pLKO.1-control scrambled | This paper | N/A |
| pSico-CLASP2 shRNA-A | This paper | N/A |
| pSico-CLASP2 shRNA-B | This paper | N/A |
| pCGLH-CLASP2 shRNA-A | This paper | N/A |
| pCGLH-CLASP2 shRNA-B | This paper | N/A |
| pCGLH-control scrambled | This paper | N/A |
| pCAG-CLASP2α | This paper | N/A |
| pCAG-CLASP2γ | This paper | N/A |
| pEGFP-CLASP2α | This paper | N/A |
| pEGFP-CLASP2γ | This paper | N/A |
| pFUW-GFP-CLASP2α WT | This paper | N/A |
| pFUW-GFP-CLASP2α 9SxA | This paper | N/A |
| pFUW-GFP-CLASP2α 8SxD | This paper | N/A |
| pEGFP-CLASP2α 1-820 | This paper | N/A |
| pEGFP-CLASP2α 821-1515 | This paper | N/A |
| pEGFP-CLASP2α 1-270 | This paper | N/A |
| pEGFP-CLASP2α 271-573 | This paper | N/A |
| pEGFP-CLASP2α 574-820 | This paper | N/A |
| pEGFP-CLASP2α 821-1200 | This paper | N/A |
| pEGFP-CLASP2α 1200-1515 | This paper | N/A |
| Sequence-Based Reagents | ||
| GeneChip Mouse Genome 430 2.0 Array | Affymetrix | |
| shRNA targeting sequences for mouse CLASP2 shRNA-A GCATCAGTCCTTTCAACAAGT shRNA-B GAACTTGAAGAGACGTTAAAT |
Beffert et al., 2012 | N/A |
| shRNA control scrambled CCGCAGGTATGCACGCGT | Beffert et al., 2012 | N/A |
| Software and Algorithms | ||
| ImageJ software | NIH | http://imagej.nih.gov/ij/ RRID:SCR_003070 |
| Image Studio 5.2.5 | LI-COR Biosciences | https://www.licor.com/bio/products/software/image_studio/ RRID:SCR_013430 |
| Ingenuity Pathway Analysis | Ingenuity | http://www.ingenuity.com/products/ipa RRID:SCR_008653 |
| Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ RRID:SCR_002798 |
| Volocity software | Improvision | http://www.perkinelmer.com/lab-solutions/resources/docs/BRO_VolocityBrochure_PerkinElmer.pdf RRID:SCR_002668 |
| ZEN software | Zeiss Microscope | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html RRID:SCR_013672 |
| Other | ||
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC:700294 |
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl013041 5 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Sequence-Based Reagents | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
Supplementary Material
Acknowledgments
We thank Drs. Thomas C. Südhof, Joachim Herz, Santos Franco and Torsten Wittmann for plasmids and antibodies. We thank Alicia Dupre, Elias Fong and Christine Learned for technical support. This work was supported by grants from the National Institute of Health (R21 MH100581 to T.F.H., U.B., and A.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
G.M.D. performed the cell biology, protein chemistry and live imaging experiments for Figure 8D–G; W.A.T. performed the in utero electroporation and live imaging experiments for Figure S5; G.M.D., W.A.T., T.F.H, U.B., and A.H. designed experiments and wrote the paper.
The authors declare no competing conflict of interests.
References
- Akhmanova A, Hoogenraad CC. Microtubule plus-end-tracking proteins: mechanisms and functions. Curr Opin Cell Biol. 2005;17:47–54. doi: 10.1016/j.ceb.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, Galjart N. CLASPs are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 2001;104:923–935. doi: 10.1016/s0092-8674(01)00288-4. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008;9:309–322. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- Al-Bassam J, Kim H, Brouhard G, van Oijen A, Harrison SC, Chang F. CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev Cell. 2010;19:245–258. doi: 10.1016/j.devcel.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony TE, Klein C, Fishell G, Heintz N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron. 2004;41:881–890. doi: 10.1016/s0896-6273(04)00140-0. [DOI] [PubMed] [Google Scholar]
- Asada N, Sanada K. LKB1-mediated spatial control of GSK3 and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing nNeocortex. J Neurosci. 2010;30:8852–8865. doi: 10.1523/JNEUROSCI.6140-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Chang F. Shaping the actin cytoskeleton using microtubule tips. Curr Opin Cell Biol. 2007;19:88–94. doi: 10.1016/j.ceb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- Beffert U, Dillon GM, Sullivan JM, Stuart CE, Gilbert JP, Kambouris JA, Ho A. Microtubule plus-end tracking protein CLASP2 regulates neuronal polarity and synaptic function. J Neurosci. 2012;32:13906–13916. doi: 10.1523/JNEUROSCI.2108-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock HH, Herz J. Reelin activates Src family tyrosinekinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/s0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Bhide PG, Nowakowski RS. Histogenetic processes leading to the laminated neocortex: migration is only a part of the story. Dev Neurosci. 2008;30:82–95. doi: 10.1159/000109854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcangelo G. Int Rev Neurobiol (Elsevier BV) 2005. The Reeler Mouse: Anatomy of a Mutant; pp. 383–417. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo GG, Miao G, Chen S-C, Scares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Efimov A, Kharitonov A, Efimova N, Loncarek J, Miller PM, Andreyeva N, Gleeson P, Galjart N, Maia ARR, McLeod IX, et al. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev Cell. 2007;12:917–930. doi: 10.1016/j.devcel.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Martinez-Garay I, Gil-Sanz C, Harkins-Perry SR, Müller U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482–497. doi: 10.1016/j.neuron.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal JS, Morozov YM, Ayoub AE, Chatterjee M, Rakic P, Haydar TF. Molecular and morphological heterogeneity of neural precursors in the mouse neocortical proliferative zones. J Neurosci. 2006;26:1045–1056. doi: 10.1523/JNEUROSCI.4499-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galjart N. CLIPs and CLASPs and cellular dynamics. Nat Rev Mol Cell Biol. 2005;6:487–498. doi: 10.1038/nrm1664. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB. From The Cover: Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: A major site of neurogenesis. Proc Natl Acad Sci USA. 2004;101:3196–3201. doi: 10.1073/pnas.0308600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Liu X, Südhof TC. Deletion of mint proteins decreases amyloid production in transgenic mouse models of alzheimer’s disease. J Neurosci. 2008;28:14392–14400. doi: 10.1523/JNEUROSCI.2481-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–885. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol. 1999;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Lee BD, Kim SJ, Xu WL, Zhou FQ. GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 2011;25:1968–1981. doi: 10.1101/gad.17015911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Lyle KS, Gierke S, Matov A, Danuser G, Wittmann T. GSK3 phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J Cell Biol. 2009;184:895–908. doi: 10.1083/jcb.200901042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wittmann T. +TIPs: SxIPping along microtubule ends. Trends Cell Biol. 2012;22:418–428. doi: 10.1016/j.tcb.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennel T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logarinho E, Maffini S, Barisic M, Marques A, Toso A, Meraldi P, Maiato H. CLASPs prevent irreversible multipolarity by ensuring spindle-pole resistance to traction forces during chromosome alignment. Nat Cell Biol. 2012;14:295–303. doi: 10.1038/ncb2423. [DOI] [PubMed] [Google Scholar]
- Long JB, Bagonis M, Lowery LA, Lee H, Danuser G, Van Vactor D. Multiparametric analysis of CLASP-interacting protein functions during interphase microtubule dynamics. Mol Cell Biol. 2013;33:1528–1545. doi: 10.1128/MCB.01442-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffini S, Maia ARR, Manning AL, Maliga Z, Pereira AL, Junqueira M, Shevchenko A, Hyman A, Yates JR, Galjart N, et al. Motor-independent targeting of CLASPs to kinetochores by CENP-E promotes microtubule turnover and poleward flux. Curr Biol. 2009;19:1566–1572. doi: 10.1016/j.cub.2009.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato H, Khodjakov A, Rieder CL. Drosophila CLASP is required for the incorporation of microtubule subunits into fluxing kinetochore fibres. Nat Cell Biol. 2005;7:42–47. doi: 10.1038/ncb1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato H, Rieder CL, Earnshaw WC, Sunkel CE. How do kinetochores CLASP dynamic microtubules? Cell Cycle. 2003;2:511–514. doi: 10.4161/cc.2.6.576. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Götz M. Neuronal or glial progeny. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Matsuki T, Matthews RT, Cooper JA, van ber Brug MP, Cookson MR, Hardy JA, Olson EC, Howell BW. Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell. 2010;143:826–836. doi: 10.1016/j.cell.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Smith M, Wu Y, Zhu X, Pringle J, Snider WD. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. Elife. 2014;395:4–18. doi: 10.7554/eLife.02663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PM, Folkmann AW, Maia ARR, Efimova N, Efimov A, Kaverina I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat Cell Biol. 2009;11:1069–1080. doi: 10.1038/ncb1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Okano H, Ogawa M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron. 2001;31:727–741. doi: 10.1016/s0896-6273(01)00420-2. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- O’Dell RS, Ustine CJ, Cameron DA, Lawless SM, Williams RM, Zipfel WR, Olson EC. Layer 6 cortical neurons require Reelin-Dab1 signaling for cellular orientation, Golgi deployment, and directed neurite growth into the marginal zone. Neural Dev. 2012;7:7–25. doi: 10.1186/1749-8104-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K, Nogales E, Heald R. Multiple domains of human CLASP contribute to microtubule dynamics and organization in vitro and in Xenopus egg extracts. Cytoskeleton. 2012;69:155–165. doi: 10.1002/cm.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- Pereira AL, Pereira AL, Maia AR, Drabek K, Sayas CL, Hergert PJ, Lince-Faria M, Matos I, Duque C, Stepanova T, Rieder CL, Earnshaw WC, Galjart N, Maiato H. Mammalian CLASP1 and CLASP2 cooperate to ensure mitotic fidelity by regulating spindle and kinetochore function. Mol Biol Cell. 2006;17:4526–4542. doi: 10.1091/mbc.E06-07-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science. 1974;183:425–427. doi: 10.1126/science.183.4123.425. [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller–Dicker lissencephaly gene containing G protein β-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Romaniello R, Arrigoni F, Bassi MT, Borgatti R. Mutations in α- and β-tubulin encoding genes: Implications in brain malformations. Brain Dev. 2015;37:273–280. doi: 10.1016/j.braindev.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Exome Aggregation Consortium. Daly MJ, MacArthur DG, Fromer M, Purcell SM. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet. 2016;48:1107–1111. doi: 10.1038/ng.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara A, Ando R, Sapir T, Tanaka T. Microtubule dynamics in neuronal morphogenesis. Open Biol. 2013;3:130061–130061. doi: 10.1098/rsob.130061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samora CP, Mogessie B, Conway L, Ross JL, Straube A, McAinsh AD. MAP4 and CLASP1 operate as a safety mechanism to maintain a stable spindle position in mitosis. Nat Cell Biol. 2011;13:1040–1050. doi: 10.1038/ncb2297. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Model L, Gaetz J, Kapoor TM, Hatten ME. Par6α signaling controls glial-guided neuronal migration. Nat Neurosci. 2004;7:1195–1203. doi: 10.1038/nn1332. [DOI] [PubMed] [Google Scholar]
- Stancik EK, Navarro-Quiroga I, Sellke R, Haydar TF. Heterogeneity in ventricular zone neural precursors contributes to neuronal fate diversity in the postnatal neocortex. J Neurosci. 2010;30:7028–7036. doi: 10.1523/JNEUROSCI.6131-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Tsvetkov AS, Samsonov A, Akhmanova A, Galjart N, Popov SV. Microtubule-binding proteins CLASP1 and CLASP2 interact with actin filaments. Cell Motil Cytoskeleton. 2007;64:519–530. doi: 10.1002/cm.20201. [DOI] [PubMed] [Google Scholar]
- Tyler WA, Haydar TF. Multiplex genetic fate mapping reveals a novel route of neocortical neurogenesis, which is altered in the Ts65Dn mouse model of Down syndrome. J Neurosci. 2013;33:5106–5109. doi: 10.1523/JNEUROSCI.5380-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, et al. Phosphorylation of CLASP2 by GSK-3 regulates its interaction with IQGAP1, EB1 and microtubules. J Cell Sci. 2009;122:2969–2979. doi: 10.1242/jcs.046649. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Waterman-Storer CM. Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3 in migrating epithelial cells. J Cell Biol. 2005;169:929–939. doi: 10.1083/jcb.200412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell. 2003;114:469–482. doi: 10.1016/s0092-8674(03)00605-6. [DOI] [PubMed] [Google Scholar]
- Yanagida M, Miyoshi R, Toyokuni R, Zhu Y, Murakami F. Dynamics of the leading process, nucleus, and Golgi apparatus of migrating cortical interneurons in living mouse embryos. Proc Natl Acad Sci USA. 2012;109:16737–16742. doi: 10.1073/pnas.1209166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.