

Abstract
A major cause of cancer death is its metastasis to the vital organs. Few effective therapies are available for metastatic castration‐resistant prostate cancer (PCa), and progressive metastatic lesions such as lymph nodes and bones cause mortality. We recently identified AES as a metastasis suppressor for colon cancer. Here, we have studied the roles of AES in PCa progression. We analyzed the relationship between AES expression and PCa stages of progression by immunohistochemistry of human needle biopsy samples. We then performed overexpression and knockdown of AES in human PCa cell lines LNCaP, DU145 and PC3, and determined the effects on proliferation, invasion and metastasis in culture and in a xenograft model. We also compared the PCa phenotypes of Aes/Pten compound knockout mice with those of Pten simple knockout mice. Expression levels of AES were inversely correlated with clinical stages of human PCa. Exogenous expression of AES suppressed the growth of LNCaP cells, whereas the AES knockdown promoted it. We also found that AES suppressed transcriptional activities of androgen receptor and Notch signaling. Notably, AES overexpression in AR‐defective DU145 and PC3 cells reduced invasion and metastasis to lymph nodes and bones without affecting proliferation in culture. Consistently, prostate epithelium‐specific inactivation of Aes in Pten flox/flox mice increased expression of Snail and MMP9, and accelerated growth, invasion and lymph node metastasis of the mouse prostate tumor. These results suggest that AES plays an important role in controlling tumor growth and metastasis of PCa by regulating both AR and Notch signaling pathways.
Keywords: Androgen receptors, neoplasm invasiveness, neoplasm metastasis, prostatic neoplasms, transcription factors
In many types of cancer, the major cause of cancer death is metastasis to the vital organs. Prostate cancer (PCa) is the most common male malignancy and the second leading cause of cancer death of men in the Western world.1, 2 Currently, advanced PCa is treated by androgen ablation therapy. While it is usually effective in the beginning, most PCa patients develop resistance to the therapy sooner or later.3 Once the disease reaches the castration‐resistant stage, there are few effective therapies, and the uncontrolled metastatic lesions cause mortality, including those to regional lymph nodes and distant organs such as bones. Unlike other epithelial tumors that metastasize to the bone only occasionally, metastatic PCa almost invariably targets the bone, and displays characteristic osteoplastic rather than osteolytic lesions.4
In both normal prostate physiology and PCa, androgen receptor (AR) mediates key signaling as a nuclear hormone receptor.5 AR is expressed in most PCa cases, and dysregulation of its downstream growth control mechanism plays a significant role in tumorigenesis and metastasis of PCa.6, 7
In the normal growth and development of the prostate, Notch signaling plays a critical role.8 Several lines of evidence indicate that abnormal expression of Notch receptor and/or ligands is often associated with the progression and metastasis of PCa9, 10, 11, 12 including frequent metastasis to the bone.13
Amino‐terminal enhancer of split (AES), also called Grg5 in mice, is a distinct member of the Groucho/Transducin‐Like Enhancer of split (Gro/TLE) gene family, with its Q/GP domains showing similarities to those in TLEs.14, 15 We demonstrated earlier that AES suppresses colon cancer metastasis through Notch signaling inhibition.16 It was also reported that AES regulates AR transcriptional activity. For example, it disrupts the interaction between AR N‐ and C‐terminal domains, inhibiting AR–DNA interaction.17, 18 But the role of AES in PCa has not been investigated fully.
Alterations in the PTEN/PI3K/AKT pathway are implicated in PCa development, with approximately 70% of late‐stage tumors showing loss of PTEN or activation of phosphoinositide 3‐kinase (PI3K).19 Pten‐deficient mice develop PCa through prostatic intraepithelial neoplasia (PIN), and are thought to mimic human PCa.20 In the present study, we have investigated the roles of AES in PCa in both humans and mice.
Materials and Methods
Characteristics of PCa patients
Formalin‐fixed, paraffin‐embedded (FFPE) sections of primary tumors were obtained from 82 men who underwent prostate needle biopsy with informed consents signed as approved by the Kyoto University Ethics Committee (G52), and consequently were diagnosed to have PCa at Kyoto University Hospital. Patient characteristics are shown in Table 1. Decalcified FFPE sections of bone metastasis lesions were obtained from nine PCa patients who underwent a palliative surgery (mostly decompression laminectomy) for vertebral bone metastasis. All nine patients were on androgen deprivation therapy (ADT) and had developed castration‐resistant disease at the time of surgery.
Table 1.
Inverse correlation between AES expression levels and metastatic spread extents in human PCa. AES expression in the primary tumors of 82 patients was evaluated by immunohistochemistry as positive (+) or negative (−)
| Clinicopathological factors | AES expression | P‐value | |
|---|---|---|---|
| + (n = 55) | − (n = 27) | ||
| Age (years) | 71.5 ± 1.2 | 73.0 ± 1.5 | 0.95* |
| PSA (ng/mL) | 424 ± 244 | 310 ± 80 | 0.66* |
| Lymph node involvement | |||
| Positive | 12 | 12 | 0.042† |
| Negative | 43 | 15 | |
| Bone metastasis | |||
| Positive | 18 | 14 | 0.097† |
| Negative | 37 | 13 | |
| Stage | |||
| Localized | 32 | 9 | 0.03† |
| Metastatic | 23 | 18 | |
*Mann–Whitney U‐test, †Fisher's exact test.
Mouse strains
Mice with the following genotypes were used: a conditional allele for Pten (Pten f/f in C57Bl/6 background) with loxP sites flanking exon 5,21 a conditional allele for Aes with loxP sites flanking Exon2 of Aes,16 a Probasin‐Cre allele (TgPbsn Cre in C57Bl/6 background) having Cre recombinase gene placed under the control of Probasin promoter,22 obtained from NCI.
Histological, immunohistochemical and immunofluorescent analyses
Tissues were fixed in 4% paraformaldehyde, embedded and sectioned at a thickness of 4 μm for histological, immunohistochemical (IHC), and immunofluorescent analyses. These sections were stained with hematoxylin and eosin (H&E) or processed further for immunostaining as described previously.23 For immunofluorescence staining, Alexa Fluor 594‐conjugated goat anti‐mouse IgG or 488‐conjugated goat anti‐rabbit IgG (Molecular Probes, Invitrogen, Carlsbad, CA) was used as the secondary antibody. Bright‐field and fluorescence images were captured with Olympus DP73 (Olympus, Tokyo, Japan) or Leica CTR6000 fluorescent microscope (Leica, Wetzler, Germany) and analyzed using Adobe Photoshop software (Adobe, San Jose, CA, USA).
Microinvasion of the mouse prostate tumor cells were defined as broken basement membrane by tumor cells infiltrated into the interstitial space. Microinvasion was counted in three randomly selected light microscopy fields at 100× magnification for each section. Cells positive for nuclear Ki67 immunostaining were counted, and the proportion of Ki67‐positive cells in at least 200 cells was determined for the positive cell frequency. The extent of AES immunohistochemical staining was validated using the intestinal polyp tissue from Apc Δ716 polyposis mice16 as positive control, whereas the prostate tumor tissue from TgPbsnCre;Pten f/f;Aes f/f mice as negative control. It was scored as none (negative control), weak (positive but weaker than positive control), moderate (as strong as positive control) or strong (stronger than positive control) as shown in the representative images (Fig. S1).
Cell culture, and proliferation and invasion assays
PCa cell lines LNCaP, PC3, and DU145 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI 1640 with 10% fetal bovine serum (FBS). Cell proliferation in culture was evaluated using Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan).
For Matrigel invasion assays, cells were serum starved for 6 h and suspended in serum‐free RPMI 1680. The cell suspension (5 × 104 cells) was then added to the Matrigel‐coated 8.0‐μm pore polyethylene terephthalate filter insert of a 24‐well cell culture chamber (BD Falcon, Franklin Lakes, NJ, USA) and was incubated for 24 h with the medium containing 10% FBS in the bottom chamber. Residual cells on the upper side of chambers were removed by scraping with cotton swabs and the cells that attached to the lower side of the membrane were fixed with 70% ethanol and stained with H&E. Invaded cells were counted under light microscopy.
Animal experiments
All experiments using animals were performed according to the protocols approved by Animal Research Committee of Kyoto University. Mice were housed in a specific pathogen‐free room.
To assess the influence of Aes on prostate tumor development, male TgPbsn Cre Pten +/+ Aes +/+ (wild‐type), TgPbsn Cre Pten f/f Aes +/+ (Pten), TgPbsn Cre Pten f/f Aes f/f (Aes/Pten) mice (n = 5 for each) were sacrificed at 3, 6, 12, and 18 months of age and the genitourinary complex (bladder, urethra, prostate glands, and seminal vesicles) was excised and photographed. Then, each prostatic lobe was dissected, weighed, and subjected to further experiments. Samples were snap‐frozen in liquid nitrogen for western blot analyses and isolation of RNA or genomic DNA. At dissection, the intraperitoneal cavity was also carefully inspected for any metastatic lesions. The lungs and liver were excised and subjected to histopathological examinations.
For in vivo transplantation models, we generated PCa cells that stably expressed firefly luciferase, and Aes or none (control). Cells were collected by trypsinization and centrifugation at 1000 g, resuspended in PBS and kept on ice until injection. In the cardiac injection model, 6‐week‐old male nude mice (Clea, Japan) were anesthetized and 5 × 105 cells/mouse were injected into the left ventricle as described previously.24 Correct injection into the systemic circulation was confirmed by the lack of photon flux from the injection site detected by an in vivo imaging system (IVIS, Xenogen, Alameda, CA, USA) 2 min after each injection. In the orthotopic inoculation model, 6‐week‐old male NOD/SCID mice (Clea, Japan) were anesthetized and 1 × 106 cells were injected into the anterior prostate as described previously.25
In the femur implantation model, 6‐week‐old male nude mice (Clea, Japan) were anesthetized and 1 × 106 cells were injected into the left femur body as described previously.26 Following injections, mice were monitored daily for their general condition, and the extent of bone metastasis was determined on post‐injection days 14, 28, and 42 by injecting luciferin intraperitoneally followed by photon flux determination using IVIS. The mice were euthanized, and bones, prostates and other organs with tumor involvement detected by IVIS were collected, fixed in formalin, and embedded in paraffin. Metastatic tumors were identified in sections under a light microscope.
Western blotting
For Western blots, tissues or cells were lysed in the NP‐40 lysis buffer containing 20 mM Tris‐HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Nonidet P‐40, 10 mM sodium fluoride, 1 mM sodium pyrophosphate, 1 mM sodium orthovanadate, and Cømplete protease inhibitor (Roche Applied Science). Lysates containing 20 or 40 μg of protein were separated by SDS/PAGE and transferred to Immobilon P membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% nonfat dry milk (in Tris‐buffered saline, 0.05% Tween‐20) for 1 h at room temperature. They were then probed with the primary antibodies for at 4°C 8 h. After incubation with horseradish peroxidase‐conjugated secondary antibodies (anti‐mouse IgG, Pierce, Rockford, IL; anti‐rabbit IgG, GE Healthcare Amersham, Buckinghamshire, UK), bound proteins were detected by incubation with Immobilon chemiluminescent substrate (Millipore).
Antibodies
Anti‐Aes antibody was described previously.16 Goat polyclonal anti‐prostate‐specific antigen (PSA) was purchased from Santa Cruz Biochemistry (Santa Cruz, CA, USA), mouse polyclonal β‐actin from SIGMA (A5316), mouse polyclonal Cyclyn D1 from BD (556470), mouse monoclonal anti‐Cyclyn E1 from Santa Cruz (sc‐248), goat polyclonal anti‐Probasin from Santa Cruz (sc‐17126), rat monoclonal anti‐Hes1 from MBL (D134‐3), rabbit monoclonal anti‐Snail from CST (C15D3), rabbit polyclonal anti‐EZH2 from CST (4905S), and rabbit polyclonal anti‐MMP9 from Abcam (ab38898).
Quantitative RT‐PCR (q‐RT‐PCR)
q‐RT‐PCR was performed using an ABI StepOnePlus (Applied Biosystems, Foster City, CA, USA). We used the following primer sets and probes: human AES primers (F, forward, 5′‐ TCCCTTTTCTTTGACAGATGGG ‐3′; and R, reverse, 5′‐ AGGAGTCCGAGGTGGTGAATT ‐3′) and probe (5′‐ CTCCTCGCACCTACCCCAGCAACTC ‐3′), mouse Aes primers (F, 5′‐ GGCGGAAATTGTGAAGAGGCT ‐3′; R, 5′‐ CTTGGCTCTCTCGATGGCTC ‐3′) and probe (5′‐ TTTGCGCCCAGGTTCTGCCC ‐3′).
Levels of human ribosomal S18 RNA, human ACTB, and mouse Gapdh were determined using Taqman Ribosomal RNA, human β‐actin and rodent GAPDH Control Reagents (Applied Biosystems). Reactions were prepared using Taqman Universal PCR Master Mix (Applied Biosystems). Relative transcript levels were calculated by the comparative CT method (ABI User Bulletin #2). Primers used were as follows, HES1 (111 bp); F, 5′‐ TCAACACGACACCGGATAAA ‐3′ and R, 5′‐ TCAGCTGGCTCAGACTTTCA ‐3′, PSA (74 bp); F, 5′‐ GGAAATGACCAGGCCAAGAC ‐3′ and R, 5′‐ CAACCCTGGACCTCACACCTA ‐3′.
Reporter activity assays
Notch1 transcription activity was evaluated using Notch reporter assay. Cultured cells were transfected with pGa981‐6 luciferase reporter27 simultaneously with Aes or control (Mock) expression vector with or without RAMIC.16 Twenty‐four hours after transfection, luciferase activities were determined using Dual‐Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol with a MITHORAS LB 940 luminometer (Berthold Biolumat, Bad Wildbad, Germany). Firefly luciferase activities were normalized against those of Renilla luciferase.
Statistical analyses
Each experimental series was performed at least in triplicate, and the results are presented as means ±SEM. Student's t‐test, Fisher's exact test and a logistic regression test were employed using commercially available software (SPSSII, SPSS, Tokyo, Japan Inc.). All tests were two‐sided and P‐values <0.05 were considered statistically significant.
Results
AES levels in primary PCa are inversely correlated with extents of progression and metastasis
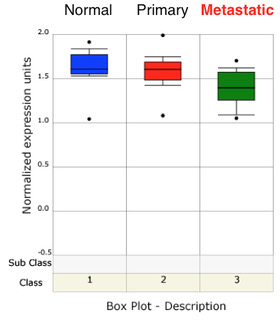
We have recently found that AES suppresses colon cancer metastasis through inhibition of Notch signaling.16 PCa data extracted from a gene expression database show that AES expression is down‐regulated in PCa in a stage‐dependent manner (Fig. S2),19 suggesting that AES is a candidate tumor and/or metastasis suppressor in PCa.
To test the possibility, we examined human PCa tissues for AES expression by IHC, and asked whether its expression is associated with clinical aggressiveness of PCa. We analyzed needle biopsy specimens of the prostate obtained from 82 untreated PCa patients, and found that AES expression levels were inversely correlated with lymph node involvement and clinical stage of PCa (Table 1). In this patient population, AES‐negative PCa had twice higher frequency of lymph node involvement, which was statistically significant (44% vs 22%, P = 0.042). Additionally, AES‐negative PCa was more likely to have bone metastasis than AES‐expressing PCa, although the difference was not statistically significant (52% vs 33%, P = 0.097). Overall, 67% of patients with PCa lacking AES expression had metastasis, whereas only 42% of those expressing AES did (P = 0.03). On the other hand, AES expression was not significantly associated with the PSA level or Gleason grade (data not shown) suggesting that AES expression is an independent biomarker that predicts metastatic spread of PCa. Indeed, after adjusting to the Gleason score, expression of AES was still significantly associated with lymph node involvement (odds ratio (OR) 2.79, 95% confidence interval (CI) 1.079–7.191, P = 0.0343, logistic regression test) as well as to metastasis (OR 2.87, 95% CI: 1.037–7.918, P = 0.0423), except bone metastasis (OR 2.26, 95% CI: 0.894–5.723, P = 0.0848).
Furthermore, AES expression was abrogated in 78% (n = 7) of nine bone metastatic lesions (Fig. S3), whereas it was negative only in 44% (n = 14) of primary prostatic lesions of PCa with bone metastasis.
AES suppresses prostate tumor growth in culture and in vivo through androgen receptor inhibition
To investigate the effects of AES expression on androgen activity, we first studied the relationship between tumor growth and AES expression using human PCa cells in culture. Exogenous expression of Aes inhibited the proliferation by approximately 50% in androgen‐dependent LNCaP cells (Fig. 1a), whereas it did not affect the proliferation of AR‐independent PC3 (Fig. 1b) or DU145 (Fig. 1c) cells. Consistently, silencing of AES by shRNA promoted the proliferation rate twice in LNCaP cells (Fig. 1d). These data suggest that AES reduces the proliferation of PCa cells through inhibition of AR activity.
Figure 1.
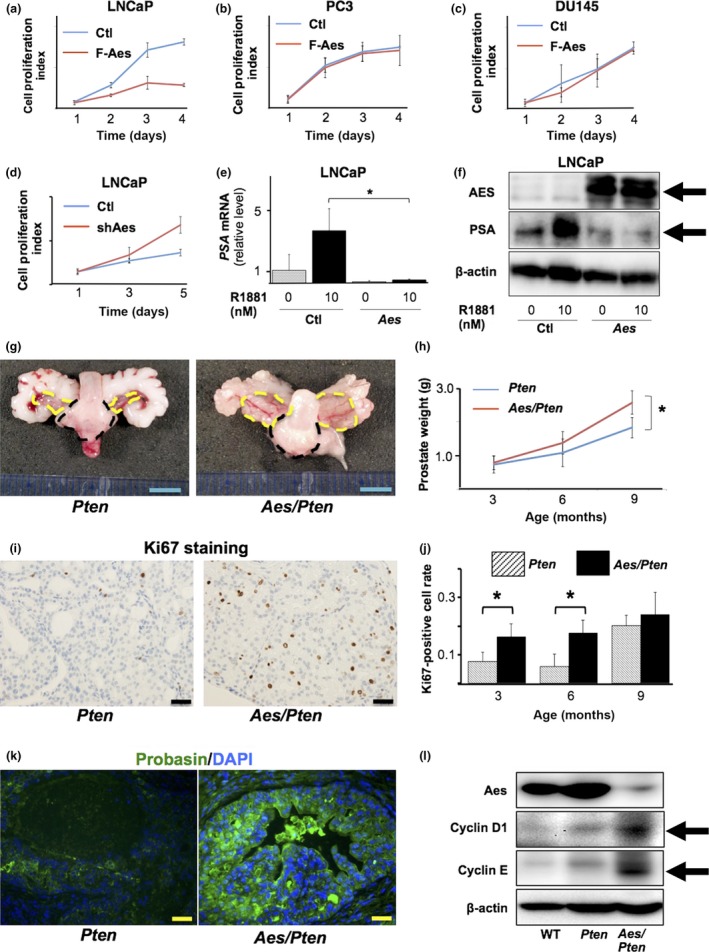
Aes suppresses growth of PCa through androgen receptor inhibition. (a–c) Effects of Aes overexpression on the growth of PCa cell lines determined by MTT assay; (a) LNCaP, (b) PC3, (c) DU145. F‐Aes, flag‐tagged Aes; Ctl, no‐Aes control vector. Note that LNCaP expresses AR whereas either PC3 or DU145 does not. (d) Effects of Aes knockdown by shRNA against AES mRNA (shAes) on the growth of LNCaP determined by MTT assay. (e) Expression levels of PSA mRNA in LNCaP determined by q‐RT‐PCR, in the absence (0 nM) or presence (10 nM) of synthetic androgen R1881 with exogenous expression of Aes (Aes) or none (Ctl). (f) Western analysis of PSA protein in LNCaP cells upon expression of exogenous Aes. Arrows indicate the positions of the authentic proteins. (g) Dissection micrographs of prostates from Pten f/f PbCre mice (Pten) and Aes f/f Pten f/f PbCre mice (Aes/Pten) at 6 months of age. Yellow circles indicate anterior prostate, whereas black ones show ventral and lateral prostate. Ruler is in mm. (h) Prostate weights of Pten and Aes/Pten mice at 3, 6 and 9 months of age. (n = 5, for each age group) (i) Immunohistochemical staining for Ki67 in Pten and Aes/Pten mouse prostates at 6 months of age. Scale bar 20 μm. (j) Quantification Ki67‐ positive cells for PIN in Pten and Aes/Pten mice at 3, 6 and 9 months of age. (k) Immunofluorescence staining for probasin (green) and nuclear DAPI (blue) in Pten and Aes/Pten mouse prostates. Scale bar 20 μm. (l) Western analysis of cyclin D1 and cyclin E in wild‐type (WT) and mutant mouse prostates. β‐actin was used as the loading control. Arrows indicate the positions of the authentic proteins. Asterisks (*) show the statistical significance (P < 0.05).
Because it was reported that AES could inhibit the AR transcriptional activity,17, 18 we next tested whether AES blocked the AR activity by determining the expression levels of an AR target gene PSA upon androgen stimulation. As anticipated, q‐RT‐PCR (Fig. 1e) and Western blotting (Fig. 1f) demonstrated significant reductions in both mRNA and protein levels of PSA upon AES expression.
To investigate the inhibitory function of AES on prostate tumor growth in vivo, we crossed mice carrying a floxed allele of Aes 16 (Aes f/f) with PIN model mice with prostate‐specific deletion of a tumor suppressor Pten under the control of probasin promoter (TgPbsn Cre Pten f/f). The latter model develops PIN in an androgen‐dependent manner.20, 28 The weight of the prostate gland was 1.5–2 times heavier in Aes/Pten compound knock‐out mice than that in Pten single knock‐out mice (Fig. 1g,h) (P < 0.05).
The enlarged prostate size in Aes/Pten mice was attributable to an increased growth of prostate epithelium as shown by larger area of PIN lesion in histology, and as evident by approximately twice higher density of Ki67‐expressing cells in the prostatic epithelium of Aes/Pten mice compared with that of Pten littermates (Fig. 1i,j), suggesting accelerated proliferation of prostatic epithelial cells in Aes/Pten mice.
It has been reported that AR transcriptional activity is reduced by Pten loss‐of‐function mutation in the prostatic epithelial cells.29 Consistently, we found that probasin was scarcely expressed in the prostatic epithelium of Pten mice by immunofluorescence analysis (Fig. 1k, left). Notably, expression of probasin was remarkably restored in that of Aes/Pten mice (Fig. 1k, right), suggesting that Aes suppressed AR transcriptional activity in the Pten‐deficient prostatic epithelium. It is known that activation of the AR promotes cell‐cycle progression in the prostate epithelium and cancer cells.7 Consistent with the AR activation by Aes loss‐of‐function mutation, cyclin family proteins were expressed more abundantly in the prostate of Aes/Pten mice than that of Pten mice (Fig. 1l). Taken together, these results suggest that AES inhibits the proliferation of prostate tumor cells through AR inhibition.
AES blocks PCa cell invasion through Notch signaling inhibition
We previously reported that AES inhibits Notch signaling, and subsequently suppresses the invasion and metastasis of mouse colorectal cancer cells.16 Western blotting revealed that the AES expression levels were inversely correlated with the metastatic ability30 among three PCa cell lines (Fig. 2a,b). We also confirmed that all of PC3, DU145, and LNCaP cell lines express Notch1 and Notch2 receptors, and their ligands Jagged1 (data not shown) as reported previously.31 We then assessed the effects of Aes overexpression on Matrigel invasion activity. Exogenous expression of Aes decreased the number of invading cells by 50%–70% in both PC3 (Fig. 2c) and DU145 (Fig. 2d) cell lines without affecting proliferation. In LNCaP cells, reduced invasion may be partly attributable to the inhibition of proliferation, which was reduced by ~50% upon exogenous expression of Aes (Fig. 1a).
Figure 2.
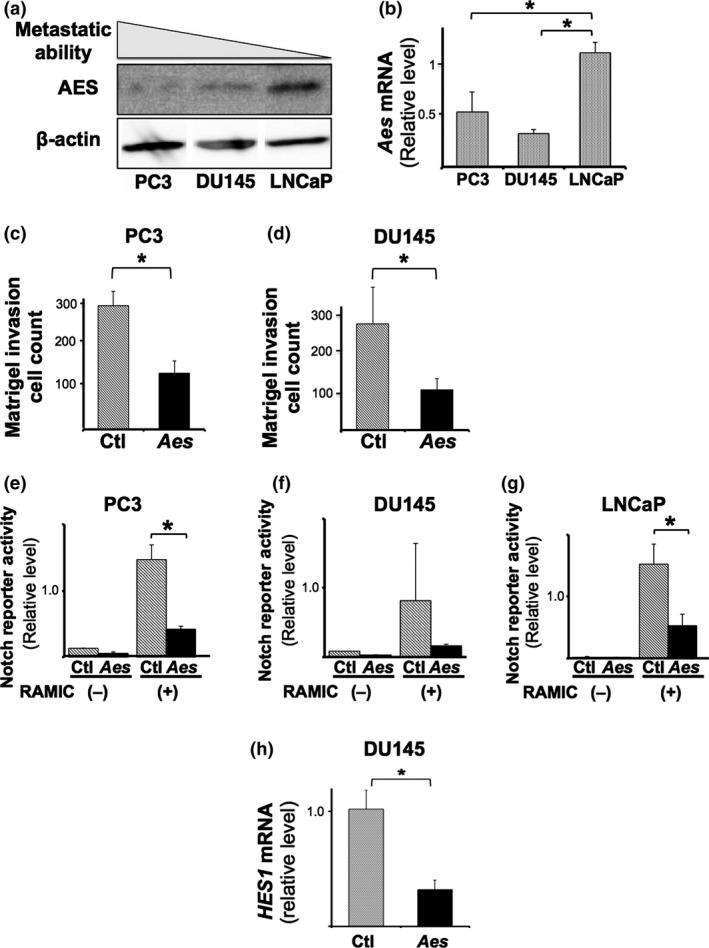
AES suppresses PCa cell invasion through Notch signaling inhibition. (a) Expression of AES protein in PCa cell lines, PC3, DU145 and LNCaP, analyzed by western blotting. (b) Expression levels of AES mRNA in PC3, DU145 and LNCaP determined by q‐RT‐PCR. (c, d) Effects of Aes overexpression on Matrigel invasion in PC3 (c) and DU145 (d) PCa cell lines (per x200 field). (e–f) Effects of Aes overexpression on Notch reporter activity in PC3 (e), DU145 (f) and LNCaP (g) cell lines, either in the absence (–) or presence (RAMIC) of the recombinant form of activated Notch receptor. Ctl: no‐Aes control vector. (h) Expression levels of HES1 mRNA in DU145 determined by q‐RT‐PCR upon exogenous expression of Aes (Aes) or none (Ctl). In (b–h), asterisks (*) show the statistical significance (P < 0.05).
We next determined the effects of Aes expression on Notch signaling transcription using a luciferase‐reporter assay. Forced expression of Aes suppressed the Notch reporter activity by 70%–90% in all three PCa cell lines either in the absence or presence of RAMIC, a recombinant form of activated Notch receptor (Fig. 2e–g). Moreover, exogenous expression of Aes caused approximately 70% reduction in the mRNA levels for HES1, one of the representative transcriptional targets of Notch signaling (Fig. 2h). Collectively, these results suggest that AES reduces Notch signaling transcription and cellular invasion also in PCa cells as in CRC cells.
AES suppresses metastatic spread of human PCa cell grafts in mice
To further investigate the role of AES in PCa metastasis in vivo, we next evaluated the effect of AES expression using two transplantation‐metastasis mouse models. When injected into the cardiac left ventricle, PC3 and MDA‐PCa2b cells can metastasize to bones such as the mandible, humerus and femur of nude mice. Notably, expression of Aes in PC3 cells significantly reduced (by ~50 times) the metastatic spread to the bone after cardiac injection when assessed with an in vivo photon‐count imaging system (Fig. 3a–c).
Figure 3.
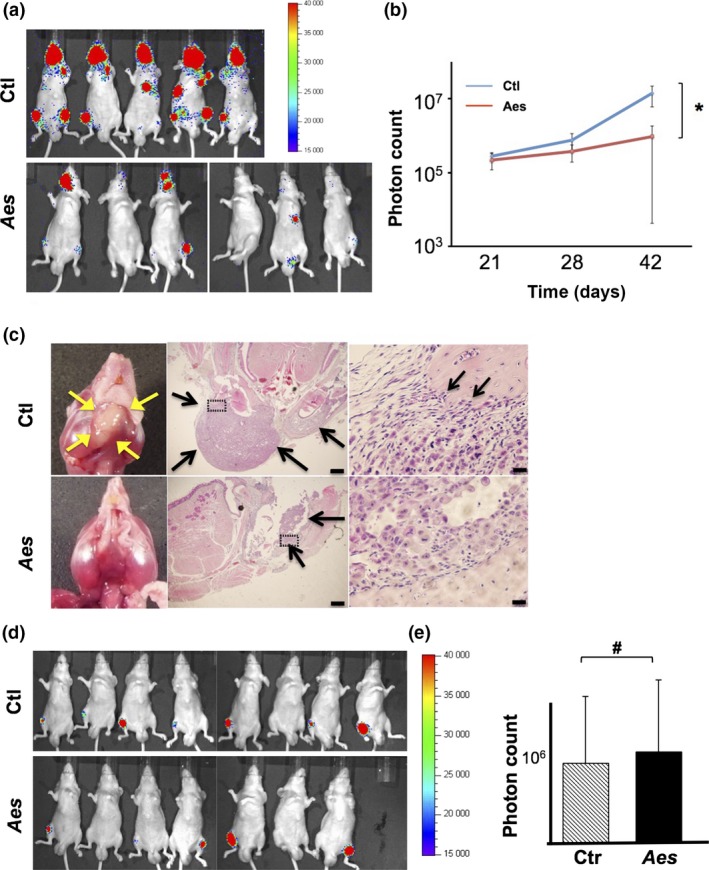
Aes suppresses metastatic spread of human PCa cell grafts to the bone in mice. (a) In vivo bioluminescence images of mice injected with luciferase‐expressing PC3 cells into the left cardiac ventricle. (b) Quantification of the bone metastatic lesions (photon counts) in mice injected with luciferase‐expressing PC3 cells as shown in (a). Aes, Aes‐expressing PC3 cells; Ctl, no‐Aes control cells. (c) Microphotographs of bone metastatic lesions in mice injected with luciferase‐expressing PC3 cells into the left ventricle. Arrows indicate metastatic lesions. Framed regions (dotted rectangles) are enlarged on the right. Scale bar; middle 500 μm, right 20 μm. (d) In vivo bioluminescence images of mice injected with luciferase‐expressing PC3 cells into femurs directly. (e) Quantification of the bone tumor lesions (photon counts) in mice injected with luciferase‐expressing PC3 cells as shown in (d). Asterisk (*) in (b) indicates statistically significant difference (P < 0.05), whereas pound (#) in (e) indicates no statistical significance.
Next we injected PC3 cells with or without exogenous Aes expression directly into the mouse femur to interrogate tumor growth between Aes‐expressing and ‐nonexpressing PC3 cells in the bone. Notably, we found no significant difference between PC3 cells with and without exogenous Aes expression by this experimental design (Fig. 3d,e), suggesting that the less metastatic spread with Aes‐expressing cells is attributable to other features of PCa cells than the proliferative properties at the metastatic sites, and that Aes expression is critical for the processes before metastatic colonization.
Taken together, these results with the in vivo transplantation and metastasis models suggest that Aes suppresses the ability of PCa cells to progress through the metastatic steps such as invasion, intravasation and extravasation without affecting tumor growth at the primary or metastatic sites.
Loss of Aes accelerates local microinvasion and lymph node metastasis of Pten‐deficient mouse PCa
Next we investigated whether loss of Aes affects local invasion and distant metastasis of PCa in mutant knock‐out mice along the course of progression. At 3 and 6 months of age, we found neither local microinvasion from the primary PIN or cancer to mesenchymal stroma, nor distant metastasis in Pten or Aes/Pten mice. However, at 12 months, local microinvasion was detectable and it was more frequent in Aes/Pten than in Pten mice (Fig. 4a,b). Notably, at 18 months of age, we observed lymph node metastasis in 70% (7/10) of Aes/Pten mice whereas such metastasis was detected only in 14% (1/7) of Pten simple mutant mice of the same age (P < 0.05, Fig. 4c,d).
Figure 4.
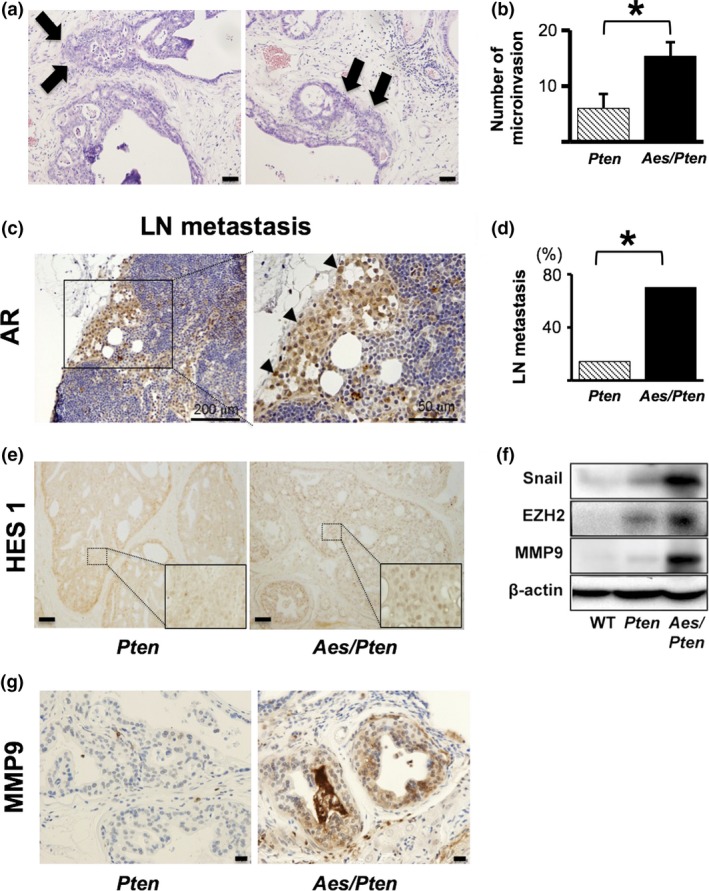
Homozygous Aes gene knockout in Pten −/−mice promotes PCa invasion. (a) Tumor microinvasions (arrows) from PIN lesions into stroma in Aes/Pten mice at 12 months of age. (b) Quantification of microinvasion in Pten (n = 3) and Aes/Pten (as shown in A; n = 3) mice at 12 months of age. (c) Microphotographs of lymph node metastasis from PCa in Aes/Pten mice at 18 months of age detected by immunohistochemistry with anti‐AR antibody. (d) Quantification of lymph node metastasis in Pten (n = 7) and Aes/Pten (as shown in C; n = 10) mice at 18 months of age. (e) Immunohistochemical staining for Hes1 protein in Pten and Aes/Pten mouse prostate at 6 months of age. (f) Expression of Snail, EZH2 and MMP9 in the prostates of wild‐type (WT) and mutant mice analyzed by western blotting. β‐actin was used as the loading control. (g) Immunohistochemical staining for MMP9 in Pten and Aes/Pten mouse prostates at 6 months of age. Scale bars; (a), 50 μm; (c), 200 μm (left) and 50 μm (right); (e), 20 μm; (g), 20 μm. Asterisks (*) in (b) and (d) indicate statistically significant differences (P < 0.05).
As our cell culture analysis showed that AES inhibited Notch signaling, we analyzed Hes1 expression in mouse prostates by IHC. Nuclear Hes1 expression was more prevalent in Aes/Pten than in Pten mice (Fig. 4e), indicating that Aes deletion in Pten mice activated Notch signaling.
Snail, EZH2 and MMP9 were reported to be involved in PCa invasion through epithelial‐mesenchymal transition (EMT) 32. Expression levels of these proteins were increased in Aes/Pten mouse prostates compared with those in Pten prostates (Fig. 4f). Additionally, we observed more abundant expression of MMP9 in the prostate of Aes/Pten than that of Pten mice (Fig. 4g), suggesting more active invasive phenotype of the former mice.
Discussion
We previously reported that AES suppresses invasion and metastasis of colon cancer through Notch signaling inhibition.16 In this study, we have shown that the same is true with PCa. The present results suggest that loss of AES promotes invasion and metastasis of PCa through Notch activation both in humans and mice. Indeed, we have shown inverse correlation between metastatic spread of PCa and expression of human AES that was markedly suppressed in bone metastasis lesions. Interestingly, Notch pathways were not associated with proliferation of prostate cancer cells as evident by little effect of exogenous AES expression on the proliferation of AR‐deficient prostate cancer cells PC3 or DU145.
We have also demonstrated that human AES regulates AR transcriptional activity in PCa cells as reported in the previous studies.17, 18 Additionally, our data suggests that AES inhibits proliferation and growth of prostate tumors through AR inhibition in vivo in a widely used mouse model of PCa caused by prostate‐specific Pten deletion. Taken together, AES acts as a tumor suppressor targeting both Notch and AR signaling pathways.
These findings have implications in clinical management of castration‐resistant PCa (CRPC). Incomplete response to androgen deprivation therapy (ADT) of PCa is attributed largely to insufficient blockade of AR signaling pathway3, 33 as demonstrated by the complementary effect of recently developed second‐generation androgen‐blocking agents.34, 35, 36, 37 Stimulating AES may facilitate AR blockade and further strengthen ADT because AES affects AR activity by binding to the N‐terminus of AR,17, 18 which is distinct from the mode of action by already existing AR‐targeting agents. It is conceivable that AES inhibits not only full‐length AR but also C‐terminus‐deficient AR truncate variants. The resistance to the second‐generation AR antagonist is often caused by ARV7, one of the commonly expressed variants lacking the ligand‐binding and C‐terminus domains.38 Further study is needed regarding detailed molecular mechanisms for interaction between ARV7 and AES.
Additionally, our results open the therapeutic possibility of enforcing the AES function to solve current clinical dilemma in PCa treatment. There have been a number of reports showing that ADT induces EMT in PCa cells, and consequently facilitates invasion and metastasis.39 The present results show that AES suppresses EMT and invasion of PCa cells through simultaneous blockade of Notch and AR signaling pathways. Taken together, enforcing the AES function in combination with existing agents for ADT may help overcome drug resistance and treatment‐induced metastatic progression of PCa.
Accumulating evidence suggests tumor‐promoting functions of Notch signaling pathway in PCa. Several reports suggest that aberrant activation of Notch signaling promotes both invasion and proliferation of PCa through activation of NF‐κB or AKT pathway.10, 11 Consistently, examinations of clinical samples show that Notch1 and Jagged1 are overexpressed in metastatic lesions of PCa.9, 12 However, cytoplasmic molecular mechanisms that activate Notch signaling have not been fully investigated in prostate cancer. The present study has demonstrated that AES functions as an upstream regulator of the Notch signaling also in PCa.
Like in other cancers, EMT is implicated in PCa metastasis. Among a number of EMT markers, only Notch1 is significantly more abundantly expressed in bone metastases than in primary sites of PCa,40 suggesting that Notch1 might be involved directly in PCa bone metastasis. Another study shows that Snail1 facilitates PCa cell migration and invasion without E‐cadherin suppression.41 Furthermore, Snail1 and EZH2 also up‐regulate MMPs, which facilitates the invasion of various human cancers including PCa.42, 43 Consistently, Snail and MMP9 are more abundantly expressed in Aes/Pten than Pten mouse prostates, suggesting that decreased AES levels and downstream Notch activation affect EMT of PCa cells (Fig. 4f,g).
Only limited experimental models are available for investigation of PCa bone metastases; for example, intracardiac or intratibial injection of highly transformed tumor cells to induce metastases.24, 44 The results of the present study indicate that AES affects the multistep metastatic cascade after intravasation of PCa cells to the blood stream, but before dissemination to the bone.45 The late occurrence of detectable LN metastasis in Pten mouse in the present study may be attributable to the strain difference or breeding environment. Indeed, various aggressiveness and progression rates were reported.5, 20, 28, 46, 47 In this study, we did not observe metastasis in Pten mouse until 18 months of age, which is consistent with a previous report showing that prostate tumors of Pten‐deficient mouse of C57BL/6 background was slow‐growing and showed no metastasis.48 Analyzing more detail role of Aes in PCa initiation and progression are expected with multiple other models.
In conclusion, the results of the present study suggest that AES plays roles as tumor and metastasis suppressors in PCa regulating activation of both AR and Notch signaling pathways.
Disclosure Statement
The authors have no conflicts of interest to declare.
Supporting information
Fig. S1. Immunohistochemical evaluation of AES expression in human PCa specimens.
{kind=link}
Fig. S2. AES expression in normal prostate, and primary and metastatic PCa tissues.
{kind=link}
Fig. S3. Immunohistochemistry of AES in bone metastatic lesions of human PCa.
{kind=link}
Acknowledgements
This work was supported by Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Y. O. and M. M. T.). The authors thank Teruaki Fujishita, Koji Aoki, Masahiro Aoki, Satoshi Yamashita, Naoki Terada, Noboru Shibasaki, Takeshi Yoshikawa, Takayuki Goto, Yu Miyazaki, Junya Toguchida and Tak W. Mak.
Cancer Sci 108 (2017) 744–752
Funding Information
Japan Society for the Promotion of Science, Grant / Award Number: ‘Kiban A/25253022′.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29. [DOI] [PubMed] [Google Scholar]
- 2. Grönberg H. Prostate cancer epidemiology. Lancet 2003; 361: 859–64. [DOI] [PubMed] [Google Scholar]
- 3. Feldman B, Feldman D. The development of androgen‐independent prostate cancer. Nat Rev Cancer 2001; 1: 34–45. [DOI] [PubMed] [Google Scholar]
- 4. Logothetis C, Lin S‐H. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer 2005; 5: 21–8. [DOI] [PubMed] [Google Scholar]
- 5. Shen M, Abate‐Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Gene Dev 2010; 24: 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone‐related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2014; 33: 3225–34. [DOI] [PubMed] [Google Scholar]
- 7. Nantermet PV, Xu J, Yu Y et al Identification of genetic pathways activated by the androgen receptor during the induction of proliferation in the ventral prostate gland. J Biol Chem 2004; 279: 1310–22. [DOI] [PubMed] [Google Scholar]
- 8. Leong KG, Gao WQ. The Notch pathway in prostate development and cancer. Differentiation 2008; 76: 699–716. [DOI] [PubMed] [Google Scholar]
- 9. Santagata S, Demichelis F, Riva A et al JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res 2004; 64: 6854–7. [DOI] [PubMed] [Google Scholar]
- 10. Bin Hafeez B, Adhami V, Asim M et al Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase‐9 and urokinase plasminogen activator. Clin Cancer Res 2009; 15: 452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Z, Li Y, Banerjee S et al Down‐regulation of Notch‐1 and Jagged‐1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF‐kappaB signaling pathways. J Cell Biochem 2010; 109: 726–36. [DOI] [PubMed] [Google Scholar]
- 12. Zhu H, Zhou X, Redfield S, Lewin J, Miele L. Elevated Jagged‐1 and Notch‐1 expression in high grade and metastatic prostate cancers. Am J Transl Res 2013; 5: 368–78. [PMC free article] [PubMed] [Google Scholar]
- 13. Zayzafoon M, Abdulkadir S, McDonald J. Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines. J Biol Chem 2004; 279: 3662–70. [DOI] [PubMed] [Google Scholar]
- 14. Brantjes H, Roose J, van De Wetering M, Clevers H. All Tcf HMG box transcription factors interact with Groucho‐related co‐repressors. Nucleic Acid Res 2001; 29: 1410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beagle B, Johnson GV. AES/GRG5: more than just a dominant‐negative TLE/GRG family member. Dev Dyn 2010; 239: 2795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sonoshita M, Aoki M, Fuwa H et al Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell 2011; 19: 125–37. [DOI] [PubMed] [Google Scholar]
- 17. Yu X, Li P, Roeder R, Wang Z. Inhibition of androgen receptor‐mediated transcription by amino‐terminal enhancer of split. Mol Cell Biol 2001; 21: 4614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Gao S, Wang Z. Structural and functional analysis of amino‐terminal enhancer of split in androgen‐receptor‐driven transcription. Biochem J 2010; 427: 499–511. [DOI] [PubMed] [Google Scholar]
- 19. Taylor B, Schultz N, Hieronymus H et al Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang S, Gao J, Lei Q et al Prostate‐specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003; 4: 209–21. [DOI] [PubMed] [Google Scholar]
- 21. Lesche R, Groszer M, Gao J et al Cre/loxP‐mediated inactivation of the murine Pten tumor suppressor gene. Genesis 2002; 32: 148–9. [DOI] [PubMed] [Google Scholar]
- 22. Jin C, McKeehan K, Wang F. Transgenic mouse with high Cre recombinase activity in all prostate lobes, seminal vesicle, and ductus deferens. Prostate 2003; 57: 160–4. [DOI] [PubMed] [Google Scholar]
- 23. Oshima H, Taketo MM, Oshima M. Destruction of pancreatic beta‐cells by transgenic induction of prostaglandin E2 in the islets. J Biol Chem 2006; 281: 29330–6. [DOI] [PubMed] [Google Scholar]
- 24. Singh A, Figg W. In vivo models of prostate cancer metastasis to bone. J Urol 2005; 174: 820–6. [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Xue H, Cutz J‐CC et al An orthotopic metastatic prostate cancer model in SCID mice via grafting of a transplantable human prostate tumor line. Lab Invest 2005; 85: 1392–404. [DOI] [PubMed] [Google Scholar]
- 26. Nogawa M, Yuasa T, Kimura S et al Monitoring luciferase‐labeled cancer cell growth and metastasis in different in vivo models. Cancer Lett 2005; 217: 243–53. [DOI] [PubMed] [Google Scholar]
- 27. Kato H, Taniguchi Y, Kurooka H et al Involvement of RBP‐J in biological functions of mouse Notch1 and its derivatives. Development 1997; 124: 4133–41. [DOI] [PubMed] [Google Scholar]
- 28. Susan K. Survey of genetically engineered mouse models for prostate cancer: analyzing the molecular basis of prostate cancer development, progression, and metastasis. J Cell Biochem 2005; 94: 279–97. [DOI] [PubMed] [Google Scholar]
- 29. Carver BS, Chapinski C, Wongvipat J et al Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN‐deficient prostate cancer. Cancer Cell 2011; 19: 575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines–part 1. J Urol 2005; 173: 342–59. [DOI] [PubMed] [Google Scholar]
- 31. Kim SH, Sehrawat A, Sakao K, Hahm ER, Singh SV. Notch activation by phenethyl isothiocyanate attenuates its inhibitory effect on prostate cancer cell migration. PLoS ONE 2011; 6: e26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nelson PS. Targeting the androgen receptor in prostate cancer–a resilient foe. N Engl J Med 2014; 371: 1067–9. [DOI] [PubMed] [Google Scholar]
- 33. Kobayashi T, Inoue T, Kamba T, Ogawa O. Experimental evidence of persistent androgen‐receptor‐dependency in castration‐resistant prostate cancer. Int J Mol Sci 2013; 14: 15615–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scher HI, Fizazi K, Saad F et al Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012; 367: 1187–97. [DOI] [PubMed] [Google Scholar]
- 35. Beer TM, Armstrong AJ, Rathkopf DE et al Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014; 371: 424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Bono JS, Logothetis CJ, Molina A et al Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011; 364: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ryan CJ, Smith MR, de Bono JS et al Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013; 368: 138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Antonarakis ES, Lu C, Wang H et al AR‐V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014; 371: 1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clyne M. Prostate cancer: androgen deprivation causes EMT in the prostate. Nat Rev Urol 2012; 9: 4. [DOI] [PubMed] [Google Scholar]
- 40. Seema S, Jill M, Wei C, Fazlul HS. Molecular signature of epithelial‐mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res 2010; 3: 90–9. [PMC free article] [PubMed] [Google Scholar]
- 41. Barrallo‐Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 2005; 132: 3151–61. [DOI] [PubMed] [Google Scholar]
- 42. Olmeda D, Jordá M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007; 26: 1862–74. [DOI] [PubMed] [Google Scholar]
- 43. Shin YJ, Kim J‐HH. The role of EZH2 in the regulation of the activity of matrix metalloproteinases in prostate cancer cells. PLoS ONE 2012; 7: e30393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corey E, Quinn J, Bladou F et al Establishment and characterization of osseous prostate cancer models: intra‐tibial injection of human prostate cancer cells. Prostate 2002; 52: 20–33. [DOI] [PubMed] [Google Scholar]
- 45. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011; 147: 275–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aytes A, Mitrofanova A, Kinkade CW et al ETV4 promotes metastasis in response to activation of PI3‐kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc Natl Acad Sci U S A 2013; 110: E3506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang J, Kobayashi T, Floc'h N et al B‐Raf activation cooperates with PTEN loss to drive c‐Myc expression in advanced prostate cancer. Cancer Res 2012; 72: 4765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Svensson RU, Haverkamp JM, Thedens DR, Cohen MB, Ratliff TL, Henry MD. Slow disease progression in a C57BL/6 pten‐deficient mouse model of prostate cancer. Am J Pathol 2011; 179: 502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunohistochemical evaluation of AES expression in human PCa specimens.
Fig. S2. AES expression in normal prostate, and primary and metastatic PCa tissues.
Fig. S3. Immunohistochemistry of AES in bone metastatic lesions of human PCa.