

Abstract
Cigarette smoke usage is prevalent in human immunodeficiency virus (HIV)-positive patients, and, despite highly active antiretroviral therapy, these individuals develop an accelerated form of chronic obstructive pulmonary disease (COPD). Studies investigating the mechanisms of COPD development in HIV have been limited by the lack of suitable mouse models. Here we describe a model of HIV-induced COPD in wild-type mice using EcoHIV, a chimeric HIV capable of establishing chronic infection in immunocompetent mice. A/J mice were infected with EcoHIV and subjected to whole body cigarette smoke exposure. EcoHIV was detected in alveolar macrophages of mice. Compared with uninfected mice, concomitant EcoHIV infection significantly reduced forced expiratory flow 50%/forced vital capacity and enhanced distal airspace enlargement following cigarette smoke exposure. Lung IL-6, granulocyte-macrophage colony-stimulating factor, neutrophil elastase, cathepsin G, and matrix metalloproteinase-9 expression was significantly enhanced in smoke-exposed EcoHIV-infected mice. These changes coincided with enhanced IκBα, ERK1/2, p38, and STAT3 phosphorylation and lung cell apoptosis. Thus, the EcoHIV smoke exposure mouse model reproduces several of the pathophysiological features of HIV-related COPD in humans, indicating that this murine model can be used to determine key parameters of HIV-related COPD and to test future therapies for this disorder.
Keywords: cigarette smoke, human immunodeficiency virus, animal model
chronic obstructive pulmonary disease (COPD) is a major public health concern for human immunodeficiency virus (HIV)-positive (+) subjects (10, 13, 14, 21). Several factors have been implicated in the enhanced susceptibility to COPD in this population, including high smoking rates (43), altered lung immunity, chronic airway infection, and even direct toxicity from the long-term effects of highly active antiretroviral therapy (HAART). HIV and COPD are both major causes of morbidity and mortality worldwide. Indeed, COPD is now the third leading cause of death in the United States (47), and global estimates indicate that ~33 million people are HIV+ (70). Over 20% of HIV+ smokers display signs of COPD development compared with ~2% of age- and smoke-matched HIV-negative (−) control subjects (13). Several factors promote this increased susceptibility to lung injury. For one, HIV infection alters the cellular profile of the alveolar space of the lung (65). In addition, both COPD (3) and HIV infection (46) compromise host immune responses, and microbial infections of the airways are frequently observed in both populations.
HIV+COPD+ subjects have reduced lung numbers of CD4+ cells that have diminished antimicrobial function (53). Recently, alveolar macrophages were shown to harbor proviral HIV DNA in HIV+ healthy subjects with undetectable plasma viral loads (9). Therefore, the persistence of viral DNA in the lung could contribute to the unique phenotypes observed in the HIV+ population. Because of the relatively slow development of COPD, the scarce number of human studies on combined HIV and COPD, and limited animal models, very little is known about the biological players in the acceleration of pulmonary obstruction in the HIV+ population.
In recent years, new models to investigate the systemic effects of HIV infection, such as the humanized mouse models, nonhuman primate immunogenetics, and recombinant challenge viruses, have been developed. Equally, our group has established a systemic infection model in mice (55). Because HIV is ineffective at binding and entering murine cells, the coding region of gp120 in HIV-1/NDK was replaced with that of gp80 from ecotropic murine leukemia virus (MLV) to generate a retrovirus that only infects rodents (55). Modifying the HIV virus to infect murine cells enables investigators to infect healthy immunocompetent mice with HIV-1 in a safe manner. EcoHIV causes chronic infection in mice targeting typical HIV host cells but also elicits host immune responses controlling virus replication, creating an animal model of HIV infection that physiologically resembles chronically infected HIV+ patients with immune or antiretroviral therapy (ART) control of HIV replication (36, 55). The model has been applied to study of HIV neuropathogenesis (29, 36), testing antiretroviral drugs (27), testing HIV vaccines (61), testing HIV-mediated gut disease (66), and studying the effect of HIV infection on platelet-mediated dysregulation of the blood-brain barrier (34). Because many murine genetic models are widely available and EcoHIV is unable to infect human cells, the EcoHIV infection model is well suited to study respiratory diseases like COPD. Therefore, in this study, the systemic effects of EcoHIV infection (55) and the whole body cigarette smoke exposure model (16, 20) were used to examine disease parameters of COPD. Combined stimuli of smoke inhalation and EcoHIV infection directly affected lung function and the development of pulmonary obstructive disease. These findings indicate that EcoHIV infection in combination with cigarette smoke exposure can be used to study disease parameters of COPD progression in HIV in small animals in a safe and convenient manner.
MATERIALS AND METHODS
Virus construction and preparation.
EcoHIV was constructed as previously described (55). Virus stocks were prepared by transfection of plasmid DNA into 293T cells, and p24 content was determined using the HIV Ag kit (Beckman Coulter, Indianapolis, IN) to quantify vital titer. Chimeric viruses were washed and suspended in saline for injection.
Animal exposure model.
Male A/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in a specific pathogen-free facility at Mount Sinai Medical Center. Mice (8 wk old) were used at the initiation point for all experiments, and each experimental parameter had 10 animals/group. Mice were anesthetized by intraperitoneal injection of a mixture of ketamine and xylazine (100:10 mg/kg ip). Animals were tail vein inoculated with 105 pg of EcoHIV in PBS. Controls were injected with PBS alone. Later (1 mo), mice were exposed to cigarette smoke in a chamber (Teague Enterprises, Davis, CA) for 4 h/day 5 days/wk at a total particulate matter concentration of 80 mg/m3 as per standard laboratory protocol (20), which equates to a mixture of passive and side-stream smoke from two cigarettes delivered to the smoking chamber every 9 min. Approximately 53 cigarettes were burned over this time point per day. Smoke exposure was continued daily for 2 mo. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and Institutional Animal Care and Use Committee guidelines. Mount Sinai Medical Center’s Institutional Animal Care and Use Committee approved the protocol. All animals were anesthetized by intraperitoneal injection of a mixture of ketamine and xylazine and killed by asphyxiation with carbon dioxide.
Forced oscillation and expiratory measurements.
Mice were anesthetized, tracheostomized, and connected via an endotracheal cannula to the SCIREQ flexiVent system (SCIREQ, Montreal, Canada). After mechanical ventilation was initiated, animals were paralyzed with a 1 mg/kg pancuronium bromide intraperitoneal injection, and forced expiratory flow at 50% of forced vital capacity [FVC (FEF50)] was determined as previously described (63). FEF50/FVC measurements were undertaken before determining airway hyperresponsiveness via methacholine challenge.
Histological analysis.
Bronchoalveolar lavage fluid (BALF) and lung tissue were collected. The lungs underwent pressure fixation as previously described (16) and in accordance with the American Thoracic Society/European Respiratory Society-issued statement on quantitative assessment of lung structure (32). Fixed tissue was stained with hematoxylin and eosin and trichrome (ab150686; Abcam) to investigate airway enlargements and matrix accumulation, respectively. Mean linear intercept (MLI) determination was conducted as previously described (20). Apoptosis was determined on paraffin-embedded tissue by the terminal deoxynucleotidyltransferase dUTP nick end-labeling (TUNEL) in situ cell death detection kit AP (Roche Diagnostics), following the manufacturer’s instructions. For each lung, 10 random sections were obtained at three different depths of tissue sectioning, and a minimum of 1,000 cells/section was visually evaluated after staining. The positively staining cells were expressed as a percentage of total nuclei. Each histological analysis was performed on 10 animals/treatment group. Slides were randomized, read blindly, and scored for each parameter.
EcoHIV detection.
Lungs of EcoHIV-infected mice were excised and homogenized using a mechanical homogenizer (Kinematica). RNA was isolated with RNeasy kits (Qiagen). EcoHIV RNA present in lung tissue was determined as previously described (55). Murine β-actin was amplified in parallel to standardize RNA input.
BALF analysis.
Macrophage, neutrophil, and lymphocyte numbers were evaluated by microscopic examination of the BALF cells after Diff-Quick staining. Lactate dehydrogenase (LDH) released into BALF was determined using a commercially available assay (Sigma Aldrich). Matrix metalloproteinase (MMP)-9 (EMD MILLIPLEX MAP MMP Magnetic Bead Panels; EMD Millipore, Billerica, MA), granulocyte-macrophage colony-stimulating factor (G-CSF), and IL-6 (Bio-Rad Cytokines Magnetic Bead Panel) were measured in BALF using a bead assay with the Bio-Rad Bio-Plex 200 system (Bio-Rad, Hercules, CA). The limits of detection for MMP-9, G-CSF, and IL-6 were 12.2, 4.2, and 2.1 pg/ml, respectively. BALF gelatinase activity was determined by gelatin zymography (17). Collagenase units were calculated as the number of micromoles of leucine liberated per milligram of enzyme by a colorimetric ninhydrin method (40, 50). Neutrophil elastase activity was determined using 50 μM fluorogenic substrate N- (methoxysuccinyl)-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin (Enzo Life Sciences) in 0.1 M HEPES, 0.5 M NaCl, pH 7.5, by excitation at 360 nm and emission at 460 nm. Experiments were performed ± neutrophil elastase inhibitor [N-(methoxysuccinyl)-Ala-Ala-Pro-Val-chloromethyl ketone, 1 mM]. Cathepsin G activity was determined in 50 μl of BALF with a colorimetric cathepsin G activity assay kit (ab126780; Abcam, Cambridge, MA), as per the manufacturer’s instructions.
Intracellular signaling.
Lung tissue was homogenized in radioimmunoprecipitation assay buffer and centrifuged at 13,000 g for 10 min, and supernatants were collected. Immunoblots on tissue lysate protein were performed for phospho-IκBα, IκBα, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), ERK1/2, phospho-p38 MAPK (Thr180/Tyr182), p38, phospho-STAT3 (Tyr705), STAT3, caspase-3, and β-actin (Cell Signaling, Danvers, MA). Chemiluminescence was detected and quantified with a Molecular Imager ChemiDoc XRS+ (Bio-Rad). Densitometry was performed on each target and represented as a ratio of pixel intensity compared with total ERK, p38, STAT3, or β-actin using Bio-Rad Laboratories Image Laboratory software (version 4.0, build 16).
Cell line.
Murine alveolar macrophages from the MH-S cell line [CRL-2019; American Type Culture Collection (ATCC), Manassas, VA] were maintained in RPMI 1640 medium (ATCC) supplemented with 10% fetal bovine serum and 1% streptomycin-penicillin-glutamate solution (Life Technologies). Cells were grown at 37°C in a humidified 5% CO2 incubator, and 1 × 106 cells/ml were seeded in 12-well culture clusters 12 h before EcoHIV infection. Cells were exposed to 5% cigarette smoke extract (CSE) 24 h after infection for a further 24 h. CSE was prepared as previously described (19).
Statistical analyses.
Data are expressed as dot blots with the means ± SE highlighted. Differences between two groups were compared by Student’s t-test (2 tailed). Experiments with more than two groups were analyzed by two-way ANOVA with Tukey’s post hoc test analysis. P values for significance were set at >0.05, and all significant changes were noted. All analyses were performed using GraphPad Prism Software (version 6.0h for Mac OS X).
RESULTS
EcoHIV readily infects alveolar macrophages in mice.
A/J mice were inoculated with EcoHIV 1 mo before smoke exposure. Following 2 mo of daily smoke exposure, animals were killed, and lungs were collected (as depicted in Fig. 1A). Because EcoHIV efficiently depletes CD4+ T cells and infects peritoneal macrophages in mice (55), we tested whether the virus can establish infection in whole lung tissue or alveolar macrophages. To detect viral RNA, we quantified viral RNA for the MLV envelope gene gp80 in EcoHIV (hence called EcoHIV envelope) and HIV-1 viral infectivity factor (Vif). We tested vif RNA because it encodes HIV-1 Vif protein that is essential for infectious virus production (68), and vif RNA is a single-spliced viral RNA that is not present in virions. Vif must be synthesized de novo during virus replication. Therefore, detection of vif expression demonstrates new viral infection. A low level of EcoHIV envelope expression was detected in the lungs compared with the alveolar macrophages of mice injected with EcoHIV (Fig. 1B). EcoHIV envelope expression was also examined following smoke exposure in EcoHIV infection. In humans, smoke exposure has been reported to either increase (1) or adjust HIV viral loads (53). We found that smoke exposure did not alter the level of EcoHIV envelope in whole lung (Fig. 1B) or alveolar macrophages (Fig. 1C). Smoke exposure also did not alter vif RNA expression in alveolar macrophages (Fig. 1C). Therefore, alveolar macrophages contain a significant pool of the EcoHIV virus similar to HIV+ subjects (65), but smoke exposure did not enhance proliferation of the virus in these cells.
Fig. 1.

A/J mice are susceptible to Eco-human immunodeficiency virus (HIV) infection within the lungs. A: A/J mice were inoculated one time with EcoHIV or PBS. Later (1 mo), animals began daily exposure to cigarette smoke. The mice were euthanized after 2 mo of smoke exposure. B: relative quantification (RQ) was determined from alveolar macrophages and mouse lungs by RT-qPCR, amplifying a region in the EcoHIV envelope. RQ levels were also determined for EcoHIV envelope expression in whole lung from mice exposed to room air (RS) and smoke. C: RQ was further determined for EcoHIV envelope and viral infectivity factor (vif) gene RNA in alveolar macrophages from EcoHIV-infected animals following room air or smoke exposure. Dot blots are represented as means ± SE, where each measurement was performed on at least 6 animals/group. Student t-tests analyzed differences between groups.
Cigarette smoke exposure decreases lung CD4+ T cell numbers in EcoHIV-infected mice.
The absolute number of CD4+ T cells in the lung is decreased in HIV+ COPD subjects compared with control subjects (53), and HAART rescues peripheral but not lung CD4+ cell proliferation in HIV+ smokers (54). Therefore, to test whether EcoHIV infection and smoke exposure affected lung CD4+ cell depletion during smoke exposure, flow cytometry was used to quantify the number of CD4+ T cells in the lungs of each group of animals. CD4+ T cells were significantly reduced in the lungs of EcoHIV-infected animals but were further depleted in smoke-exposed animals (Fig. 2A). EcoHIV infection did not deplete CD4+ T cells in the spleen (Fig. 2A), suggesting this phenotype may be lung specific both in humans and the EcoHIV model. Interestingly, smoke exposure enhanced BALF immune cell infiltration, but this inflammatory response was not altered by EcoHIV infection (Fig. 2B). Smoke exposure elevated macrophages and neutrophil BALF numbers in the mice (Fig. 2C) as it does in both HIV− and HIV+ human subjects (35). Similar to HIV in COPD subjects (53), EcoHIV infection alters lung CD4 counts without increased lung HIV viral load. Together, these data demonstrate that EcoHIV infection in smoke-exposed mice produces immune cell responses similar to those observed in the lungs of HIV+ smokers.
Fig. 2.

Animals infected with EcoHIV have altered lung immune cell profile following cigarette smoke exposure. A/J mice were inoculated one time with EcoHIV or PBS. Later (1 mo), the animals began daily exposures to cigarette smoke. Mice were euthanized at 2 mo. A: absolute lung CD4+ cell numbers were quantified by flow cytometery. The percentage of spleen CD4+ T cells was also quantified in animals infected with EcoHIV for 3 mo. B and C: total bronchoalveolar lavage fluid (BALF) cells (B) and macrophages, neutrophils, and lymphocytes (C) were quantified in each animal group by Quik Diff analysis of BALF cells following cytospin. Graphs are represented as means ± SE of 3 separate measurements on independent days. *P <0.05 when comparing both treatments connected by a line, determined by 2-way ANOVA with Tukey's post hoc test; n = 10 experiments/group.
EcoHIV-infected mice have enhanced pulmonary obstructive disease following cigarette smoke exposure.
Airway remodeling plays a central role in the development of COPD (26, 44, 67). Lower CD4+ T cell counts in HIV+ subjects have been linked to decrements in lung function, as measured by diffusing capacity of the lungs for carbon monoxide (DLCO) (59). HIV+ subjects have a higher prevalence of cough symptoms and sputum production and a lower absolute and percent-predicted DLCO than HIV− subjects (59). We have also observed that the presence of another virus, respiratory syncytial virus, in combination with smoke exposure enhances lung damage (16). Therefore, we performed studies to examine lung tissue remodeling and pulmonary function changes in mice infected with EcoHIV and exposed to smoke. EcoHIV-infected mice exposed to smoke developed severe emphysematous changes (Fig. 3). Indeed, morphometric quantification demonstrated that combined EcoHIV infection and smoke exposure increased MLI compared with cigarette smoke alone (69.6 ± 8.4 EcoHIV infection and smoke exposed vs. 55.8 ± 3.4 smoke exposed; P < 0.01) (Fig. 3A). Forced oscillation and expiratory measurements also determined that smoke exposure induced changes in FEF50/FVC (Fig. 3B). Despite the same cigarette smoke exposure, EcoHIV-infected and smoke-exposed animals demonstrated significantly reduced FEF50/FVC compared with control smoke-exposed animals (Fig. 3B), suggesting the presence of heightened obstructive lung disease (58).
Fig. 3.

Concurrent EcoHIV infection and cigarette smoke exposure exacerbates lung remodeling. A/J mice were inoculated one time with EcoHIV or PBS and exposed to 2 mo of cigarette smoke. A: lung morphology was determined in mice inoculated with EcoHIV or PBS and exposed to cigarette smoke for 2 mo. Mean linear intercepts (MLI) and representative images (scale bar = 50 µM) of mice lungs are presented here of the smoke-exposed groups. B: negative pressure-driven forced expiratory (NPFE) and forced oscillation technique (FOT) maneuvers were performed to determine changes in pulmonary mechanics in all animal groups. Forced expiratory flow at 50% of forced vital capacity (FVC) (FEF50) was determined in each animal. Graphs are represented as means ± SE of at least 3 separate measurements. *P < 0.05 when comparing both treatments connected by a line, determined by 2-way ANOVA with Tukey's post hoc test; n = 10/group.
EcoHIV-infected mice display elevated lung inflammation following exposure to cigarette smoke.
Exposure to cigarette smoke leads to phosphorylation of ERK (45), p38 (37), and IκBα (2). HIV uses the p38 and ERK pathways to aid in virion production and CD4+ T cell depletion (18). Each of these signaling factors is linked to pathogenesis of HIV-linked diseases (33, 57, 64). Therefore, immunoblots examined the phosphorylation of these targets in each mouse group (Fig. 4). The combination of EcoHIV infection and cigarette smoke exposure significantly enhanced phosphorylation of ERK, p38, and IκBα (Fig. 4, A and B). Enhanced STAT3 phosphorylation was also observed in this experimental group and coincided with heightened IL-6 secretion (Fig. 4, A–C). Importantly, IL-6 levels are associated with mortality rates in COPD subjects (15). Expression of p38 can also regulate G-CSF expression (11), and G-CSF levels were significantly elevated in the EcoHIV and smoke exposure cohort (Fig. 4D). Thus, smoke exposure enhanced MAPK activation, IκBα degradation, STAT3 phosphorylation, and IL-6 and G-CSF production in EcoHIV-infected animals.
Fig. 4.
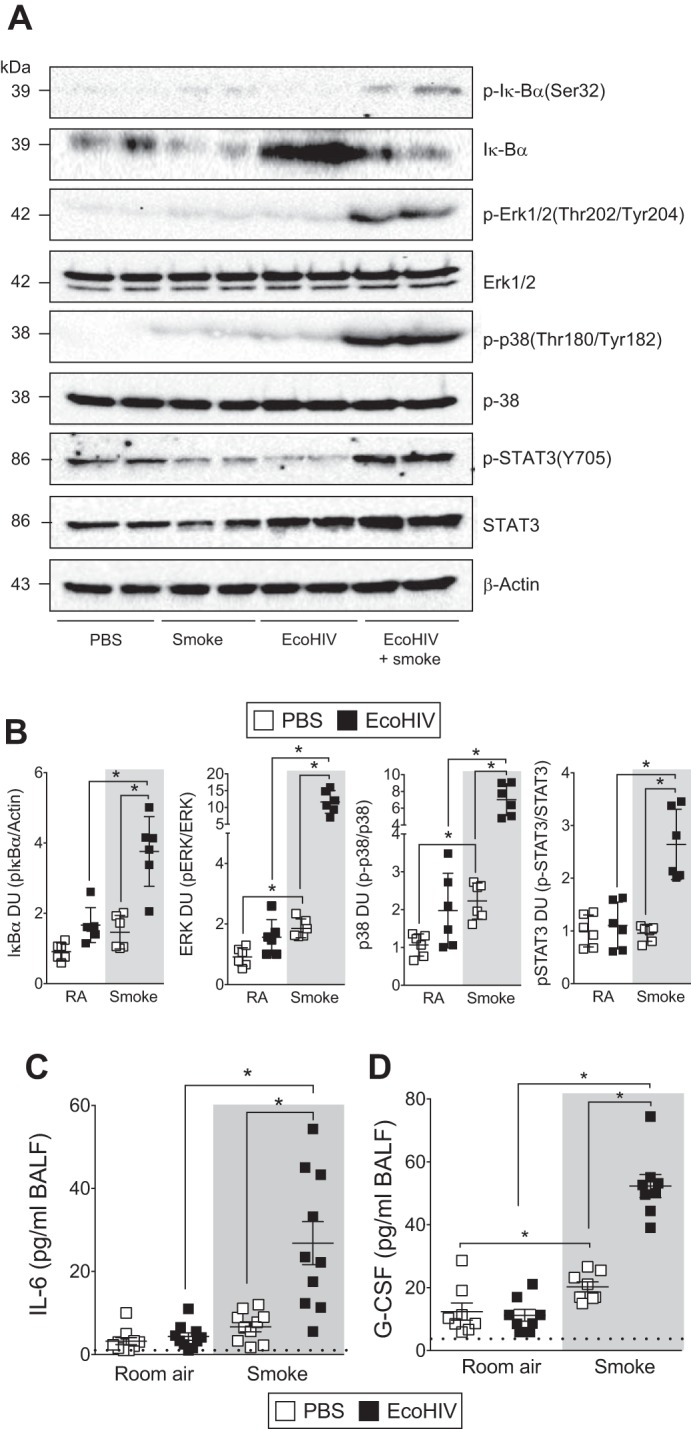
Enhanced inflammation is observed in the lungs of EcoHIV-infected animals also exposed to smoke. A/J mice were inoculated one time with EcoHIV or PBS and exposed to 2 mo of cigarette smoke. A: immunoblots on lung homogenates determined enhanced phosphorylation of p38, ERK IκBα, and STAT3 in mice infected with EcoHIV and smoke exposed. Representative blots are shown here with 2 individual mice/group. B: densitometry was performed on immunoblots. Graphs are represented as densitometry units (DU) of pixel intensity expressed as a ratio to β-actin or total ERK, p38, or STAT3. C and D: BALF was analyzed for IL-6 (C) and granulocyte-macrophage colony-stimulating factor (G-CSF, D) levels in each mouse group. Dotted horizontal line denotes limit of detection of assay. Graphs are represented as mean ± SE of 3 separate measurements on independent days. *P < 0.05 when comparing both treatments connected by a line, determined by 2-way ANOVA with Tukey's post hoc test; n ≥ 6/group.
Combined smoke exposure and EcoHIV infection enhance lung apoptosis in mice.
Increased structural and immune cell apoptosis is observed in COPD lungs (30). EcoHIV-infected animals had enhanced lung cell apoptosis following smoke exposure, observed by TUNEL, caspase-3 cleavage, and LDH release analyses (Fig. 5). Combined EcoHIV infection and smoke exposure caused a significant increase in the number of TUNEL-positive cells (Fig. 5, A and B). As we have previously observed (16), smoke alone triggers a modest induction of TUNEL-positive cells within the lung (Fig. 5, A and B). However, this induction was significantly augmented by concomitant infection with EcoHIV (Fig. 5, A and B). Additionally, elevated levels of LDH were observed in the BALF of EcoHIV-infected mice exposed to smoke compared with the other mouse groups (Fig. 5C), which indicates enhanced cell membrane damage in the lung. Likewise, enhanced caspase-3 cleavage was observed in the mice infected with EcoHIV and exposed to smoke (Fig. 5D). Therefore, enhanced apoptosis in the lungs could contribute to the destructive lung remodeling changes that occurred with combined EcoHIV infection and cigarette smoke exposure.
Fig. 5.
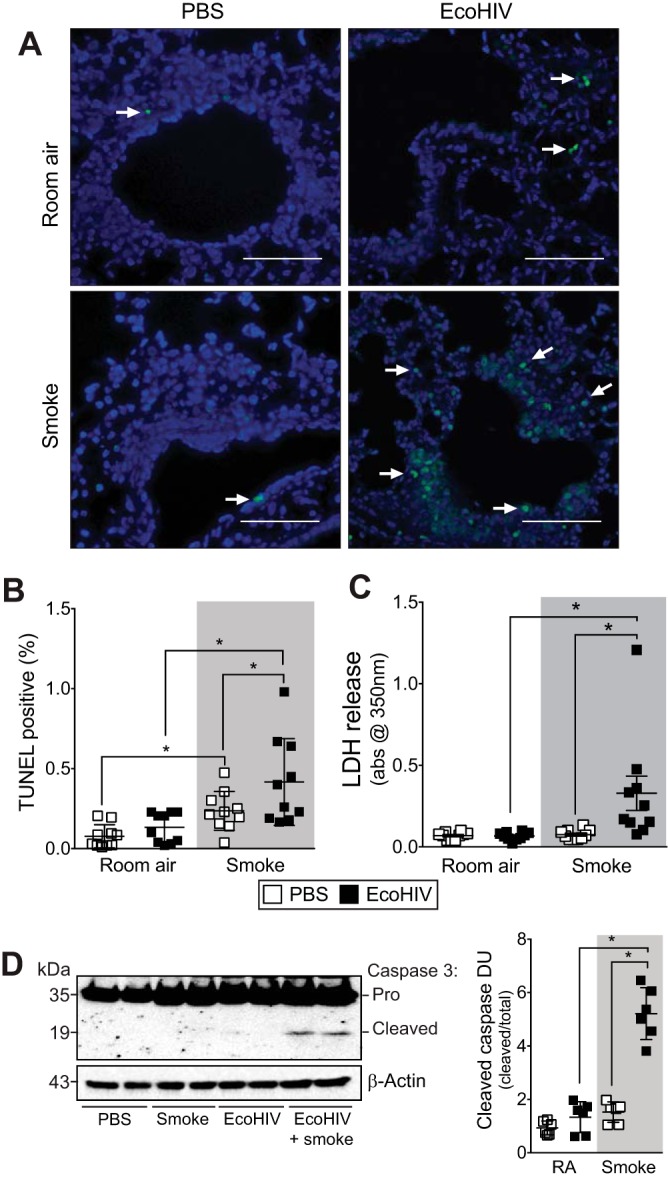
EcoHIV infection enhances cigarette smoke-induced fibrosis and apoptosis. A/J mice were inoculated one time with EcoHIV or PBS and exposed to 2 mo of cigarette smoke. A and B: representative images (A) and scoring of terminal deoxynucleotidyltransferase dUTP nick end-labeling (TUNEL) analysis (B) was performed on lung tissue from each mouse group. C and D: lactate dehydrogenase (LDH) release (C)- and caspase-3 cleavage (D)-determined enhanced activation of caspase-3 coincided with higher cell death in lungs of mice infected with EcoHIV and smoke exposed. Representative blots are shown here with 2 individual mice/group. Densitometry was performed on immunoblots. Graph is represented as DU of pixel intensity expressed as a ratio to total caspase-3. Graphs are represented as means ± SE of 3 separate measurements on independent days. *P < 0.05 when comparing both treatments connected by a line, determined by 2-way ANOVA with Tukey's post hoc test; n ≥ 6/group.
EcoHIV-infected mice display elevated lung protease following exposure to cigarette smoke.
Proteases, especially neutrophil derived, are frequently implicated in the pathogenesis of COPD (31). In humans, several proteases are selectively upregulated in the lungs of HIV+ COPD subjects, and this induction is associated with the enhanced loss of lung function (35). Protease production and release can also be mediated by activation of mitogen-activated protein kinases (MAPK), such as ERK (38) and p38 (48). Therefore, we investigated several proteases known to play a role in disease progression. Here, we show that mice infected with EcoHIV and exposed to cigarette smoke have increased airway collagenase, neutrophil elastase, and cathepsin G activity in their BALF (Fig. 6A). Neutrophil elastase is elevated in the sputum of smokers and correlates with the severity of airflow obstruction (52). Also, it has been reported that inhibition of cathepsin G is an effective means of preventing pulmonary inflammation (41). The influence of EcoHIV infection and smoke exposure on MMP expression was also investigated via gelatin zymography and multiplex analysis (Fig. 6, B and C). EcoHIV infection and smoke exposure increased BALF gelatinase activity, particularly for MMP-9 (Fig. 6B), and markedly elevated MMP-9 protein levels in the BALF fluid of the mice (Fig. 6C). These data suggest that enhanced protease activation may contribute to the development of COPD in HIV+ subjects.
Fig. 6.

Neutrophil-associated proteases are enhanced in the lungs of EcoHIV-infected animals also exposed to smoke. A/J mice were inoculated one time with EcoHIV or PBS and exposed to 2 mo of cigarette smoke. A: total collagenase, neutrophil elastase, and cathepsin G activity levels were determined in BALF from each animal group. Total collagenase and cathepsin G activity levels were determined colormetrically and neutrophil elastase fluorescently. FI, fluorescent intensity. B: gelatin zymography was performed to determine BALF gelatinase activity. Bands at 92 and 82 kDa are representative of latent and active matrix metalloproteinase (MMP)-9, respectively. Bands at 72 and 66 kDa are representative of latent and active MMP-2, respectively. Representative gels are shown here with 3 individual mice/group. Densitometry was performed on zymograms. Graph is represented as DU of pixel intensity for MMP-9. C: BALF MMP-9 levels were determined by multiplex analysis. Dotted horizontal line denotes limit of detection of assay. Graphs are represented as means ± SE of 3 separate measurements on independent days. *P < 0.05 when comparing both treatments connected by a line, determined by 2-way ANOVA with Tukey's post hoc test; n ≥ 6/group.
Because mouse alveolar macrophages are infected with EcoHIV in our model and are capable of producing proteases that degrade extracellular matrix components, we infected an alveolar macrophage cell line, MH-S cells, with EcoHIV and exposed these cells to CSE before examining protease expression. CSE exposure had no effect on EcoHIV infectivity in MH-S cells, as determined by EcoHIV envelope or HIV-1 vif expression (Fig. 7A). Alveolar macrophages from HIV+ smokers with early emphysema produce more MMP-1, −7, −9, and −12 compared with HIV− smokers with early emphysema (35). Therefore, we examined several macrophage-specific proteases. Gene expression of cathepsin S (CatS) and MMP-2, −7, −9, −10, −12, and −13 was examined by qPCR. EcoHIV alone significantly enhanced expression of CatS and MMP-13, and smoke alone induces expression of MMP-9 and MMP-12 (Fig. 7, B and C). In combination with CSE, EcoHIV infection enhanced expression of CatS and MMP-9, −10, and −13 in MH-S cells (Fig. 7, B and C). Therefore, EcoHIV infection of alveolar macrophages stimulates the expression of proteolytic enzymes that can contribute to smoke-driven lung damage.
Fig. 7.

EcoHIV infection enhances cigarette smoke extract-induced protease expression in alveolar macrophages. MH-S cells were inoculated one time with EcoHIV or PBS and 24 h later were exposed to PBS or cigarette smoke extract (CSE) for a further 24 h. RQ was determined for EcoHIV envelope and vif genes in EcoHIV-infected cells (A) and cathepsin S (CatS, B) and MMP-2, −7, −9, −10, −12, and −13 (C) in each cell group. Graphs are represented as means ± SE of 3 separate measurements on independent days. *P < 0.05 when comparing both treatments connected by a line, determined by 2-way ANOVA with Tukey's post hoc test; n = 10/group.
DISCUSSION
The emerging new clinical manifestations in the HIV+ populations on ART, which are generally characterized by chronic HIV infection with stably suppressed HIV replication and functioning immune system, require novel research models to investigate these important phenotypes. The mouse model of EcoHIV infection and smoke exposure here allows the inoculation of conventional mice with chimeric HIV-1 without systemic immunosuppression (36) and subsequent exposure of these animals to long-term cigarette smoke. This EcoHIV infection spreads to multiple organs and cells (55), including alveolar macrophages, and subsequently alters lung responses to smoke exposure. Following exposure to smoke, EcoHIV-infected animals display enhanced loss of CD4+ T cells in the lung, reduced lung function, exaggerated protease responses, apoptosis, and heightened inflammation markers that are all linked to human COPD progression. Because EcoHIV cannot enter human cells, this infectious HIV derivative represents a safer means to study HIV in COPD and allows the use of numerous already established mouse knockout and transgenic animals. Therefore, using EcoHIV in combination with smoke exposure is a useful tool in murine models to study multiple aspects of COPD pathogenesis in the HIV population.
Elevated proteases may influence lung remodeling in HIV, since alveolar macrophages from HIV+ COPD subjects express much higher levels of MMP-1, −7, −9, and −12 (35). Here, the EcoHIV smoke-exposed mice exhibited significantly higher MMP-9 expression and gelatinolytic activity and enhanced cathepsin G and neutrophil elastase activity. These proteases have been clearly linked to COPD and likely contribute to the emphysema (25) that develops in HIV1+ COPD subjects. Knockout mice that lack neutrophil serine proteases, proteinase 3, cathepsin G, and neutrophil elastase, are substantially protected against lung tissue destruction after long-term exposure to cigarette smoke (25). Combined smoke and EcoHIV infection upregulates a broad spectrum of proteases that target several extracellular matrix substrates, plasma proteins, cell surface receptors, cytokines, protease inhibitors, and proteases (23). This suggests that many matrix components could undergo additional degradation and prompt an exaggerated phenotype. MMP-9 is constitutively present in neutrophil granules (4, 5) and can be induced in alveolar macrophages (60). MMP-9 can promote COPD development by targeting elastin, gelatin, and other key lung matrix components. Our group recently observed that MMP-9 has antiviral properties, but increases in its expression significantly contribute to lung cell apoptosis (11). Viral suppression reduces Fas receptor and programmed death-1 expression in lung CD4+ T cells (54), which may be mediated by protease activity (42). In addition, it plays an important role in lung inflammation by modulating the activity cytokines like IL-1β (62) that modulate neutrophil influx in the lung (24). Cigarette smoke induces neutrophil degranulation in the lung (8), which increases elastase, cathepsin G, and MMP-9 levels within the airways. Inhibiting cathepsin G prevents pulmonary inflammation in asthma and cigarette smoke exposure animal models (41). Cathepsin G also increases monocyte/macrophage susceptibility to acute HIV-1 infection (51). Equally, despite no difference in neutrophil numbers between smoke-exposed infected and noninfected animals, our study demonstrates significant neutrophil-associated signaling. Additionally, the ability of immune cells to phagocytose apoptotic neutrophils is impaired in HIV+ individuals (69). This may result in heightened inflammation. Neutrophils produce CCL2 and IL-10 in response to the HIV stimulation and enhance replication of the virus in monocyte-derived macrophages (71). These responses may have contributed to the persistence of EcoHIV in alveolar macrophages in this study. Together, these findings suggest that neutrophils and proteases play a significant role in the accelerated disease formation that occurs in HIV+ smokers.
The major limitations of this study merit discussion. gp120 is not expressed in EcoHIV, and gp120 has several functional roles in HIV pathogenicity outside of viral entrance in host cells. One key function of gp120 is to promote the activation of STAT3 and the subsequent induction of IL-6 (12). Despite lacking gp120, EcoHIV infection promoted a potent STAT3/IL-6 response in the lung. Elevated systemic IL-6 was also observed in a recent study on EcoHIV-induced gut disease in mice, although the mechanism of this induction was not elaborated (66). Similarly, gp120 also influences MMP-9 expression (39) and apoptosis (22) yet EcoHIV potently upregulated lung MMP-9 expression and enhanced smoke-induced apoptosis. Upregulation of inflammatory cytokine endothelial monocyte activating polypeptide II (EMAP II) is linked to lung endothelial cell apoptosis (7), which is induced by gp120 (22) signaling through the CXCR4 receptor and activation of p38 MAPK. HIV tat protein also selectively enhances CXCR4 signaling (49), and MMP-2 negatively regulates EMAP II-mediated apoptosis (6). Therefore, gp120 alone is not solely responsible for elevated IL-6 and MMP-9 expression and elevated apoptosis. Further complimentary in vitro and ex vivo studies are required to determine the role of gp120 on the pathogenicity of COPD. However, the lung phenotype of EcoHIV-infected mice exposed to cigarette smoke mimics the human disease and represents a robust animal model to identify future therapeutic strategies. It should also be noted that the receptor for the ecotropic MLV is a cationic amino acid transporter (CAT-1) that is ubiquitously expressed on multiple murine cells, unlike HIV-1 that primary interacts with CD4 and its coreceptors. Therefore non-CD4+ T cells are susceptible to EcoHIV infection, but we previously observed that CD4+ cells and macrophages are the primary CAT-1-positive cells to become infected with EcoHIV (56). After inoculation in conventional mice, EcoHIV infects CD4+ spleen cells, lymphocytes, and macrophages (55). However, these cells can regulate viral replication in mice (28). Our study did observe a significant but modest reduction in lung CD4+ T cells in smoke-exposed EcoHIV-infected mice. Whether this reduction in lung CD4+ cells is a depletion of cells or reduced infiltration in the lung will be investigated in future studies. Also, alveolar macrophages harbor proviral HIV DNA in HIV+ healthy subjects with undetectable plasma viral loads (9), which suggests that macrophage viral load may play a role in the development of lung disease. As occurs with HIV+ patients, we detected EcoHIV RNA primarily in alveolar macrophages within the lung. Thus, the EcoHIV infection model could be a useful tool to investigate how viral infection alters macrophage biology to promote the development of COPD in humans.
Infection of mice with EcoHIV followed by smoke exposure represents a new small animal model to investigate pulmonary manifestations of HIV. This approach opens up the possibilities of using many genetic mouse models to better understand the development of lung diseases in HIV+ subjects. This model could lead to more effective approaches to counter lung disease development in the HIV+ patient population.
GRANTS
This work was supported by grants made available to P. Geraghty (Flight Attendant Medical Research Institute, YCSA 113380), R. Foronjy [United States National Institutes of Health (NIH) Grant 5R01-HL-098528–05 and Flight Attendant Medical Research Institute Grant CIA 130020)], and D. Volsky (NIH Grants R01-DA-017618–10, R01-DA-037611–03, and R01-MH-104145–04).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.G., E.H., B.-H.K., A.J.D., D.J.V., and R.F. performed experiments; P.G., D.J.V., and R.F. analyzed data; P.G., B.-H.K., D.J.V., and R.F. interpreted results of experiments; P.G. prepared figures; P.G., D.J.V., and R.F. drafted manuscript; P.G., D.J.V., and R.F. edited and revised manuscript; P.G., E.H., B.-H.K., A.J.D., D.J.V., and R.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Pulmonary Division of the State University of New York Downstate Medical Center for support and Dr. Moro Salifu.
REFERENCES
- 1.Ande A, McArthur C, Ayuk L, Awasom C, Achu PN, Njinda A, Sinha N, Rao PS, Agudelo M, Nookala AR, Simon S, Kumar A, Kumar S. Effect of mild-to-moderate smoking on viral load, cytokines, oxidative stress, and cytochrome P450 enzymes in HIV-infected individuals. PLoS One 10: e0122402, 2015. doi: 10.1371/journal.pone.0122402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis 23: 1511–1518, 2002. doi: 10.1093/carcin/23.9.1511. [DOI] [PubMed] [Google Scholar]
- 3.Bauer CM, Morissette MC, Stämpfli MR. The influence of cigarette smoking on viral infections: translating bench science to impact COPD pathogenesis and acute exacerbations of COPD clinically. Chest 143: 196–206, 2013. doi: 10.1378/chest.12-0930. [DOI] [PubMed] [Google Scholar]
- 4.Borregaard N, Sehested M, Nielsen BS, Sengeløv H, Kjeldsen L. Biosynthesis of granule proteins in normal human bone marrow cells. Gelatinase is a marker of terminal neutrophil differentiation. Blood 85: 812–817, 1995. [PubMed] [Google Scholar]
- 5.Chakrabarti S, Zee JM, Patel KD. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: novel pathways for tertiary granule release. J Leukoc Biol 79: 214–222, 2006. doi: 10.1189/jlb.0605353. [DOI] [PubMed] [Google Scholar]
- 6.Chetty C, Bhoopathi P, Lakka SS, Rao JS. MMP-2 siRNA induced Fas/CD95-mediated extrinsic II apoptotic pathway in the A549 lung adenocarcinoma cell line. Oncogene 26: 7675–7683, 2007. doi: 10.1038/sj.onc.1210584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clauss M, Voswinckel R, Rajashekhar G, Sigua NL, Fehrenbach H, Rush NI, Schweitzer KS, Yildirim AO, Kamocki K, Fisher AJ, Gu Y, Safadi B, Nikam S, Hubbard WC, Tuder RM, Twigg HL III, Presson RG, Sethi S, Petrache I. Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. J Clin Invest 121: 2470–2479, 2011. doi: 10.1172/JCI43881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Advances in neutrophil biology: clinical implications. Chest 134: 606–612, 2008. doi: 10.1378/chest.08-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cribbs SK, Lennox J, Caliendo AM, Brown LA, Guidot DM. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retroviruses 31: 64–70, 2015. doi: 10.1089/aid.2014.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, Veterans Aging Cohort 5 Project Team . Increased COPD among HIV-positive compared to HIV-negative veterans. Chest 130: 1326–1333, 2006. doi: 10.1378/chest.130.5.1326. [DOI] [PubMed] [Google Scholar]
- 11.Dabo AJ, Cummins N, Eden E, Geraghty P. Matrix Metalloproteinase 9 Exerts Antiviral Activity against Respiratory Syncytial Virus. PLoS One 10: e0135970, 2015. doi: 10.1371/journal.pone.0135970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Cornò M, Donninelli G, Varano B, Da Sacco L, Masotti A, Gessani S. HIV-1 gp120 activates the STAT3/interleukin-6 axis in primary human monocyte-derived dendritic cells. J Virol 88: 11045–11055, 2014. doi: 10.1128/JVI.00307-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz PT, King MA, Pacht ER, Wewers MD, Gadek JE, Nagaraja HN, Drake J, Clanton TL. Increased susceptibility to pulmonary emphysema among HIV-seropositive smokers. Ann Intern Med 132: 369–372, 2000. doi: 10.7326/0003-4819-132-5-200003070-00006. [DOI] [PubMed] [Google Scholar]
- 14.Drummond MB, Kirk GD. HIV-associated obstructive lung diseases: insights and implications for the clinician. Lancet Respir Med 2: 583–592, 2014. doi: 10.1016/S2213-2600(14)70017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrari R, Tanni SE, Caram LM, Corrêa C, Corrêa CR, Godoy I. Three-year follow-up of Interleukin 6 and C-reactive protein in chronic obstructive pulmonary disease. Respir Res 14: 24, 2013. doi: 10.1186/1465-9921-14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foronjy RF, Dabo AJ, Taggart CC, Weldon S, Geraghty P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PLoS One 9: e90567, 2014. doi: 10.1371/journal.pone.0090567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foronjy RF, Taggart CC, Dabo AJ, Weldon S, Cummins N, Geraghty P. Type-I interferons induce lung protease responses following respiratory syncytial virus infection via RIG-I-like receptors. Mucosal Immunol 8: 161–175, 2015. doi: 10.1038/mi.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furler RL, Uittenbogaart CH. Signaling through the P38 and ERK pathways: a common link between HIV replication and the immune response. Immunol Res 48: 99–109, 2010. doi: 10.1007/s12026-010-8170-1. [DOI] [PubMed] [Google Scholar]
- 19.Geraghty P, Dabo AJ, D’Armiento J. TLR4 protein contributes to cigarette smoke-induced matrix metalloproteinase-1 (MMP-1) expression in chronic obstructive pulmonary disease. J Biol Chem 286: 30211–30218, 2011. doi: 10.1074/jbc.M111.238824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geraghty P, Hardigan A, Foronjy RF. Cigarette smoke activates the proto-oncogene c-src to promote airway inflammation and lung tissue destruction. Am J Respir Cell Mol Biol 50: 559–570, 2014. doi: 10.1165/rcmb.2013-0258OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gingo MR, Morris A, Crothers K. Human immunodeficiency virus-associated obstructive lung diseases. Clin Chest Med 34: 273–282, 2013. doi: 10.1016/j.ccm.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green LA, Yi R, Petrusca D, Wang T, Elghouche A, Gupta SK, Petrache I, Clauss M. HIV envelope protein gp120-induced apoptosis in lung microvascular endothelial cells by concerted upregulation of EMAP II and its receptor, CXCR3. Am J Physiol Lung Cell Mol Physiol 306: L372–L382, 2014. doi: 10.1152/ajplung.00193.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greene CM, McElvaney NG. Proteases and antiproteases in chronic neutrophilic lung disease - relevance to drug discovery. Br J Pharmacol 158: 1048–1058, 2009. doi: 10.1111/j.1476-5381.2009.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum 60: 3642–3650, 2009. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guyot N, Wartelle J, Malleret L, Todorov AA, Devouassoux G, Pacheco Y, Jenne DE, Belaaouaj A. Unopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysema. Am J Pathol 184: 2197–2210, 2014. doi: 10.1016/j.ajpath.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Hackett TL, Shaheen F, Zhou S, Wright JL, Churg A. Fibroblast signal transducer and activator of transcription 4 drives cigarette smoke-induced airway fibrosis. Am J Respir Cell Mol Biol 51: 830–839, 2014. doi: 10.1165/rcmb.2013-0369OC. [DOI] [PubMed] [Google Scholar]
- 27.Hadas E, Borjabad A, Chao W, Saini M, Ichiyama K, Potash MJ, Volsky DJ. Testing antiretroviral drug efficacy in conventional mice infected with chimeric HIV-1. AIDS 21: 905–909, 2007. doi: 10.1097/QAD.0b013e3281574549. [DOI] [PubMed] [Google Scholar]
- 28.Hadas E, Chao W, He H, Saini M, Daley E, Saifuddin M, Bentsman G, Ganz E, Volsky DJ, Potash MJ. Transmission of chimeric HIV by mating in conventional mice: prevention by pre-exposure antiretroviral therapy and reduced susceptibility during estrus. Dis Model Mech 6: 1292–1298, 2013. doi: 10.1242/dmm.012617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He H, Sharer LR, Chao W, Gu CJ, Borjabad A, Hadas E, Kelschenbach J, Ichiyama K, Do M, Potash MJ, Volsky DJ. Enhanced human immunodeficiency virus Type 1 expression and neuropathogenesis in knockout mice lacking Type I interferon responses. J Neuropathol Exp Neurol 73: 59–71, 2014. doi: 10.1097/NEN.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodge S, Hodge G, Holmes M, Reynolds PN. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J 25: 447–454, 2005. doi: 10.1183/09031936.05.00077604. [DOI] [PubMed] [Google Scholar]
- 31.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Paré PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350: 2645–2653, 2004. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 32.Hsia CCW, Hyde DM, Ochs M, Weibel ER; ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure . An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 181: 394–418, 2010. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacqué JM, Mann A, Enslen H, Sharova N, Brichacek B, Davis RJ, Stevenson M. Modulation of HIV-1 infectivity by MAPK, a virion-associated kinase. EMBO J 17: 2607–2618, 1998. doi: 10.1093/emboj/17.9.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones LD, Jackson JW, Maggirwar SB. Modeling HIV-1 Induced Neuroinflammation in Mice: Role of Platelets in Mediating Blood-Brain Barrier Dysfunction. PLoS One 11: e0151702, 2016. doi: 10.1371/journal.pone.0151702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaner RJ, Santiago F, Crystal RG. Up-regulation of alveolar macrophage matrix metalloproteinases in HIV1(+) smokers with early emphysema. J Leukoc Biol 86: 913–922, 2009. doi: 10.1189/jlb.0408240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelschenbach JL, Saini M, Hadas E, Gu CJ, Chao W, Bentsman G, Hong JP, Hanke T, Sharer LR, Potash MJ, Volsky DJ. Mice chronically infected with chimeric HIV resist peripheral and brain superinfection: a model of protective immunity to HIV. J Neuroimmune Pharmacol 7: 380–387, 2012. doi: 10.1007/s11481-011-9316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo WH, Chen JH, Lin HH, Chen BC, Hsu JD, Wang CJ. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem Biol Interact 155: 31–42, 2005. doi: 10.1016/j.cbi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 38.Lakka SS, Jasti SL, Gondi C, Boyd D, Chandrasekar N, Dinh DH, Olivero WC, Gujrati M, Rao JS. Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene 21: 5601–5608, 2002. doi: 10.1038/sj.onc.1205646. [DOI] [PubMed] [Google Scholar]
- 39.Louboutin JP, Reyes BA, Agrawal L, Van Bockstaele EJ, Strayer DS. HIV-1 gp120 upregulates matrix metalloproteinases and their inhibitors in a rat model of HIV encephalopathy. Eur J Neurosci 34: 2015–2023, 2011. doi: 10.1111/j.1460-9568.2011.07908.x. [DOI] [PubMed] [Google Scholar]
- 40.Mandl I, MacLennan JD, Howes EL, DeBellis RH, Sohler A. Isolation and characterization of proteinase and collagenase from Cl. histolyticum. J Clin Invest 32: 1323–1329, 1953. doi: 10.1172/JCI102861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maryanoff BE, de Garavilla L, Greco MN, Haertlein BJ, Wells GI, Andrade-Gordon P, Abraham WM. Dual inhibition of cathepsin G and chymase is effective in animal models of pulmonary inflammation. Am J Respir Crit Care Med 181: 247–253, 2010. doi: 10.1164/rccm.200904-0627OC. [DOI] [PubMed] [Google Scholar]
- 42.Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, Madtes DK, Shapiro SD, Martin TR. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol 37: 210–221, 2007. doi: 10.1165/rcmb.2006-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mdodo R, Frazier EL, Dube SR, Mattson CL, Sutton MY, Brooks JT, Skarbinski J. Cigarette smoking prevalence among adults with HIV compared with the general adult population in the United States: cross-sectional surveys. Ann Intern Med 162: 335–344, 2015. doi: 10.7326/M14-0954. [DOI] [PubMed] [Google Scholar]
- 44.Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. Thorax 70: 482–489, 2015. doi: 10.1136/thoraxjnl-2014-206084. [DOI] [PubMed] [Google Scholar]
- 45.Mercer BA, Kolesnikova N, Sonett J, D’Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem 279: 17690–17696, 2004. doi: 10.1074/jbc.M313842200. [DOI] [PubMed] [Google Scholar]
- 46.Migueles SA, Connors M. Success and failure of the cellular immune response against HIV-1. Nat Immunol 16: 563–570, 2015. doi: 10.1038/ni.3161. [DOI] [PubMed] [Google Scholar]
- 47.Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. Natl Vital Stat Rep 59: 1–52, 2010. [PubMed] [Google Scholar]
- 48.Mócsai A, Jakus Z, Vántus T, Berton G, Lowell CA, Ligeti E. Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol 164: 4321–4331, 2000. doi: 10.4049/jimmunol.164.8.4321. [DOI] [PubMed] [Google Scholar]
- 49.Mondal D, Williams CA, Ali M, Eilers M, Agrawal KC. The HIV-1 Tat protein selectively enhances CXCR4 and inhibits CCR5 expression in megakaryocytic K562 cells. Exp Biol Med (Maywood) 230: 631–644, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Moore S, Stein WH. Chromatography of amino acids; colorimetric ninhydrin method for analysis of the effluent. Fed Proc 7: 174, 1948. [PubMed] [Google Scholar]
- 51.Moriuchi H, Moriuchi M, Fauci AS. Cathepsin G, a neutrophil-derived serine protease, increases susceptibility of macrophages to acute human immunodeficiency virus type 1 infection. J Virol 74: 6849–6855, 2000. doi: 10.1128/JVI.74.15.6849-6855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paone G, Conti V, Vestri A, Leone A, Puglisi G, Benassi F, Brunetti G, Schmid G, Cammarella I, Terzano C. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis Markers 31: 91–100, 2011. doi: 10.1155/2011/139493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popescu I, Drummond MB, Gama L, Coon T, Merlo CA, Wise RA, Clements JE, Kirk GD, McDyer JF. Activation-induced cell death drives profound lung CD4(+) T-cell depletion in HIV-associated chronic obstructive pulmonary disease. Am J Respir Crit Care Med 190: 744–755, 2014. doi: 10.1164/rccm.201407-1226OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Popescu I, Drummond MB, Gama L, Lambert A, Hoji A, Coon T, Merlo CA, Wise RA, Keruly J, Clements JE, Kirk GD, McDyer JF. HIV Suppression Restores the Lung Mucosal CD4+ T-Cell Viral Immune Response and Resolves CD8+ T-Cell Alveolitis in Patients at Risk for HIV-Associated Chronic Obstructive Pulmonary Disease. J Infect Dis 214: 1520–1530, 2016. doi: 10.1093/infdis/jiw422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Potash MJ, Chao W, Bentsman G, Paris N, Saini M, Nitkiewicz J, Belem P, Sharer L, Brooks AI, Volsky DJ. A mouse model for study of systemic HIV-1 infection, antiviral immune responses, and neuroinvasiveness. Proc Natl Acad Sci USA 102: 3760–3765, 2005. doi: 10.1073/pnas.0500649102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Potash MJ, Hadas E, Volsky DJ. Response to ‘Remarks on the article of Hadas et al.: Transmission of chimeric HIV by mating in conventional mice: prevention by pre-exposure antiretroviral therapy and reduced susceptibility during estrus’. Dis Model Mech 7: 178–179, 2014. doi: 10.1242/dmm.014167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puca A, Fiume G, Palmieri C, Trimboli F, Olimpico F, Scala G, Quinto I. IkappaB-alpha represses the transcriptional activity of the HIV-1 Tat transactivator by promoting its nuclear export. J Biol Chem 282: 37146–37157, 2007. doi: 10.1074/jbc.M705815200. [DOI] [PubMed] [Google Scholar]
- 58.Rodrigues MT, Fiterman-Molinari D, Barreto SS, Fiterman J. The role of the FEF50%/0.5FVC ratio in the diagnosis of obstructive lung diseases. J Bras Pneumol 36: 44–50, 2010. [DOI] [PubMed] [Google Scholar]
- 59.Rosen MJ, Lou Y, Kvale PA, Rao AV, Jordan MC, Miller A, Glassroth J, Reichman LB, Wallace JM, Hopewell PC; Pulmonary Complications of HIV Infection Study Group . Pulmonary function tests in HIV-infected patients without AIDS. Am J Respir Crit Care Med 152: 738–745, 1995. doi: 10.1164/ajrccm.152.2.7633736. [DOI] [PubMed] [Google Scholar]
- 60.Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes PJ. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 26: 602–609, 2002. doi: 10.1165/ajrcmb.26.5.4685. [DOI] [PubMed] [Google Scholar]
- 61.Saini M, Hadas E, Volsky DJ, Potash MJ. Vaccine-induced protection from infection of mice by chimeric human immunodeficiency virus type 1, EcoHIV/NL4-3. Vaccine 25: 8660–8663, 2007. doi: 10.1016/j.vaccine.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol 161: 3340–3346, 1998. [PubMed] [Google Scholar]
- 63.Shalaby KH, Gold LG, Schuessler TF, Martin JG, Robichaud A. Combined forced oscillation and forced expiration measurements in mice for the assessment of airway hyperresponsiveness. Respir Res 11: 82, 2010. doi: 10.1186/1465-9921-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shapiro L, Heidenreich KA, Meintzer MK, Dinarello CA. Role of p38 mitogen-activated protein kinase in HIV type 1 production in vitro. Proc Natl Acad Sci USA 95: 7422–7426, 1998. doi: 10.1073/pnas.95.13.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shellito JE. Failure of host defenses in human immunodeficiency virus. Semin Respir Crit Care Med 25: 73–84, 2004. doi: 10.1055/s-2004-822307. [DOI] [PubMed] [Google Scholar]
- 66.Sindberg GM, Sharma U, Banerjee S, Anand V, Dutta R, Gu CJ, Volsky DJ, Roy S. An infectious murine model for studying the systemic effects of opioids on early HIV pathogenesis in the gut. J Neuroimmune Pharmacol 10: 74–87, 2015. doi: 10.1007/s11481-014-9574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sohal SS, Ward C, Danial W, Wood-Baker R, Walters EH. Recent advances in understanding inflammation and remodeling in the airways in chronic obstructive pulmonary disease. Expert Rev Respir Med 7: 275–288, 2013. doi: 10.1586/ers.13.26. [DOI] [PubMed] [Google Scholar]
- 68.Sova P, Volsky DJ, Wang L, Chao W. Vif is largely absent from human immunodeficiency virus type 1 mature virions and associates mainly with viral particles containing unprocessed gag. J Virol 75: 5504–5517, 2001. doi: 10.1128/JVI.75.12.5504-5517.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torre D, Gennero L, Baccino FM, Speranza F, Biondi G, Pugliese A. Impaired macrophage phagocytosis of apoptotic neutrophils in patients with human immunodeficiency virus type 1 infection. Clin Diagn Lab Immunol 9: 983–986, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.UNAIDS Report on the Global AIDS Epidemic. Geneva, Switzerland: World Health Organization, vol 208, 2013. [Google Scholar]
- 71.Yoshida T, Jones VC, Kobayashi M, Li XD, Pollard RB, Suzuki F. Acceleration of R5 HIV replication by polymorphonuclear neutrophils in cultures of macrophages. Immunol Cell Biol 85: 215–219, 2007. doi: 10.1038/sj.icb.7100024. [DOI] [PubMed] [Google Scholar]