

Abstract
Our purpose was to identify causative mutations and characterize the phenotype associated with the genotype in 10 unrelated families with autosomal recessive retinal degeneration. Ophthalmic evaluation and DNA isolation were carried out in 10 pedigrees with inherited retinal degenerations (IRD). Exomes of probands from eight pedigrees were captured using Nimblegen V2/V3 or Agilent V5+UTR kits, and sequencing was performed on Illumina HiSeq. The DHDDS gene was screened for mutations in the remaining two pedigrees with Ashkenazi Jewish ancestry. Exome variants were filtered to detect candidate causal variants using exomeSuite software. Segregation and ethnicity-matched control sample analysis were performed by dideoxy sequencing. Retinal histology of a patient with DHDDS mutation was studied by microscopy. Genetic analysis identified six known mutations in ABCA4 (p.Gly1961Glu, p.Ala1773Val, c.5461–10T>C), RPE65 (p.Tyr249Cys, p.Gly484Asp), PDE6B (p.Lys706Ter) and DHDDS (p.Lys42Glu) and ten novel potentially pathogenic variants in CERKL (p.Met323Val fsX20), RPE65 (p.Phe252Ser, Thr454Leu fsX31), ARL6 (p.Arg121His), USH2A (p.Gly3142Ter, p.Cys3294Trp), PDE6B (p.Gln652Ter), and DHDDS (p.Thr206Ala) genes. Among these, variants/mutations in two separate genes were observed to segregate with IRD in two pedigrees. Retinal histopathology of a patient with a DHDDS mutation showed severe degeneration of retinal layers with relative preservation of the retinal pigment epithelium. Analysis of exome variants in ten pedigrees revealed nine novel potential disease-causing variants and nine previously reported homozygous or compound heterozygous mutations in the CERKL, ABCA4, RPE65, ARL6, USH2A, PDE6B, and DHDDS genes. Mutations that could be sufficient to cause pathology were observed in more than one gene in one pedigree.
Keywords: exome sequencing, genetics, phenotype-genotype, retinal degeneration, IRD
inherited retinal degenerations (IRD) are a heterogeneous group of diseases characterized by the progressive loss of photoreceptors leading to vision loss (8, 14, 17). Some of the common features of retinal degeneration include night blindness, progressive loss of peripheral vision, central vision loss, changes in the retinal pigment epithelium (RPE), and abnormal electroretinogram (ERG) responses (14). Significant heterogeneity has been reported in the phenotype of IRD patients with a wide variation in the age of onset, rate of progression, and severity of disease and clinical symptoms (14, 36). Considerable overlap has been observed in the phenotype of various IRD (13). More than 250 genes have been implicated in retinal degenerations (https://sph.uth.edu/retnet/sum-dis.htm) (8, 11, 13, 23). These genes code for proteins that are structural components of photoreceptors or extracellular matrix. They also can play a critical role in phototransduction, ciliogenesis, oxidative stress, and other biological processes. Mutations in some of these genes are associated with multiple phenotypes including nonsyndromic and syndromic forms of IRD (8, 17, 32).
Although a large number of genes associated with IRD are known, the identification of the molecular cause of disease in many patients remains a challenge because of genetic and phenotypic heterogeneity and overlap in the symptoms of IRD. Currently available next-generation sequencing methodologies offer an opportunity to screen the entire genome, exome, or all the genes that have been reported to cause IRD (26). Here we describe the phenotype of 10 pedigrees with recessive IRD and the identification of the underlying mutations by sequencing the exomes.
METHODS
Patients.
Research procedures were performed in accordance with the Declaration of Helsinki and with the approval of the UC San Diego institutional review board. Blood samples and medical and family history were collected, and detailed ophthalmic evaluation was carried out on all affected members and available relatives.
Clinical examination.
Ophthalmic evaluation including measurement of best-corrected visual acuity (BCVA), Goldmann kinetic perimetry, full-field electroretinography (ERG), and color vision testing were carried out as described earlier (10). Retinal changes were also evaluated by fluorescein angiography and spectral-domain optical coherence tomography (SDOCT) in selected patients. Medical records were reviewed for the affected patients (Table 1).
Table 1.
Clinical features of patients with IRD
| Visual Acuity | Refractive Error | Color Vision Farnsworth D-15 | Color Vision Lanthony Desaturated 15-Hue | Goldmann Visual Field (V4e) Horizontal Diameter, ° | Goldmann Visual Field (I4e) Horizontal Diameter, ° | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pedigree | Patient ID Sex | Age at Visit, yr | Age at Onset, yr | Age at Diagnosis, yr | Gene | OD OS | OD OS | TCDS, CCI OD OS | TES, CCI OD OS | OD OS | OD OS | Full-Field ERG |
| RF.GU.0113 | II-I/M | 31 | 22 | 22 | CERKL | 20/40–2 | Plano | 307.1, 2.62 | N/A | 25° central scotoma OD | 30 | not measurable to dim scotopic; bright scotopic reduced to 9% of average OU |
| 20/63–1 | −0.25+0.25x105 | 278.6, 2.38 | 8° central scotoma OS | 15 | cone responses reduced to 29% of average OU | |||||||
| RF.GU.0113 | II-II/M | 28 | 17 | 17 | CERKL | 20/50 | −0.75+2.00x092 | 288.8, 2.47 | N/A | full OU | 35–40° central scotoma | not measurable to dim scotopic; bright scotopic reduced to 9% of average OU |
| 20/40 | Plano+1.50x090 | 351.3, 3.00 | unable to see I4e OU | cone responses reduced to 18% of average OU | ||||||||
| RF.RA.0812 | III-I/M | 34 | 2 | 34 | ABCA4 | 20/30- | −9.75+2.50x113 | 272.4, 2.33 | 282 (56), 2.99; abnormal | full OU | 55 | N/A |
| RPE65 | 20/40+ | −9.75+2.75x057 | 266.7, 2.28 | 270 (56), 2.90; abnormal | 35 | N/A | ||||||
| RF.RA.0812 | III-II/M | 30 | 3 | 19 | ABCA4 | 20/25 | −3.50+4.00x095 | 117.0, 1.00 | 194 (56), 2.37; abnormal | full OU | 35 | not measurable to dim scotopic; bright scotopic reduced to 5% of average OU |
| RPE65 | 20/25 | −5.00+4.25x075 | 117.0, 1.00 | 210 (56), 2.48; abnormal | 40 | cone responses reduced to 7% of average OU | ||||||
| RF.H.0909 | III-I/F | 11 | 10 | N/A | ABCA4 | 20/200 | −0.50+0.75x102 | N/A | N/A | N/A | 5° central scotoma | decreased photopic a and b wave amplitudes |
| 20/200 | −0.25+0.75x073 | N/A | N/A | N/A | 5° central scotoma | decreased photopic a and b wave amplitudes; Scotopic amplitudes below normal in the left eye | ||||||
| RF.H.0909 | III-II/M | 9 | 7 | N/A | ABCA4 | 20/400 | N/A | N/A | N/A | N/A | peripheral island | photopic ERG was barely recordable while the rod ERG was 30% of normal |
| 20/400 | N/A | N/A | N/A | N/A | constricted to central 35° | photopic ERG was barely recordable while the rod ERG was 15% of normal | ||||||
| RF.H.0909 | III-III/M | 7 | 7 | N/A | ABCA4 | 20/100 | N/A | N/A | N/A | N/A | N/A | photopic ERG was 50% of normal; scoptic ERG was abnormal with right eye showing larger signals than left |
| 20/200 | N/A | N/A | N/A | N/A | N/A | photopic ERG was 50% of normal; scoptic ERG was abnormal with right eye showing larger signals than left | ||||||
| RF.T.1111 | II-I/M | 35 | 6 mo | 8 | RPE65 | 20/200 | −7.50+2.00x095 | tritanomaly | N/A | constricted to central 7° | unable to see I4e OU | not measurable to all stimuli OU |
| 20/200 | −7.00+2.25x070 | tritanomaly | N/A | constricted to central 5° | unable to see I4e OU | |||||||
| RF.T.1111 | II-V/F | 35 | 2 mo | N/A | RPE65 | 20/400 | −3.50 DS | N/A | N/A | N/A | unable to see I4e OU | N/A |
| 20/400 | −4.00 DS | N/A | N/A | N/A | unable to see I4e OU | |||||||
| RF.S.0711 | II-I/M | 39 | 5 | 22 | ARL6 | LP | −9.75+0.75x165 | N/A | N/A | N/A | N/A | not measurable to all stimuli OU |
| ATP7B | LP | −9.50+0.25x030 | ||||||||||
| RF.SH.0008 | II-I/F | N/A | N/A | N/A | USH2A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| N/A | N/A | N/A | N/A | N/A | N/A | N/A | ||||||
| RF.SH.0008 | II-III/F | 63 | 45 | 54 | USH2A | 20/40–2+2 | RE:+0.25 + 2.00 X 080 | N/A | N/A | constricted to central 18° | constricted to central 6° | not measurable to dim scotopic flash OU; bright scotopic responses severely reduced |
| 20/40–2 | LE:-0.75 + 1.25 X 094 | N/A | N/A | constricted to central 20° | constricted to central 8° | cone responses severely reduced OU | ||||||
| RF.M.0711 | II-I/F | 42 | 2 | 30 | PDE6B | 20/200 | −7.50 DS | N/A | N/A | N/A | N/A | not measurable to all stimuli OU |
| CF | −7.50 DS | N/A | N/A | N/A | N/A | |||||||
| RF.TE.0512 | II-I/F | 22 | 17 | 22 | DHDDS | 20/25 | N/A | N/A | 44 (78); 1.26; normal | 125° horizontal diameter | 90° horizontal diameter (II4e) | severely reduced a-wave and b-wave amplitudes to 0 dB scotopic stimulus, no measurable responses to photopic stimuli |
| 20/25 | N/A | N/A | 68 (78), 1.40; normal | 140° horizontal diameter | 90° horizontal diameter (II4e) | severely reduced a-wave and b-wave amplitudes to 0 dB scotopic stimulus, no measurable responses to photopic stimuli | ||||||
| RF.TE.0512 | II-II/M | 28 | 26 | 22 | DHDDS | 20/16 | N/A | N/A | N/A | |||
| RF.AA.1104 | II-I/M | 22 | 22 | 22 | DHDDS | 20/16 | N/A | N/A | 134 (78), 1.79; abnormal | 150° horizontal diameter | N/A | moderately reduced a-wave and b-wave amplitudes to 0 dB scotopic and 30 Hz flicker stimuli |
| 20/16 | N/A | N/A | 95 (78), 1.56; abnormal | 145° horizontal diameter | N/A | moderately reduced a-wave and b-wave amplitudes to 0 dB scotopic and 30 Hz flicker stimuli | ||||||
| RF.PO.1109 | II-I/M | 50 | 18 | N/A | DHDDS | LP | N/A | N/A | N/A | N/A | N/A | N/A |
IRD, inherited retinal degeneration; ERG, electroretinogram; OD, right eye; OS, left eye; OU, oculus uterque (both eyes); CCI, color confusion index; TCDS, total color difference score; N/A, not available.
Genetic analysis.
DNA was isolated from blood samples. Exomes of 10 members of eight pedigrees were captured using NimbleGen SeqCap EZ Human Exome Library V2/V3 kit or Agilent V5-UTR probes and sequenced on Illumina HiSeq to screen for mutations in known IRD genes. Unique paired-end (2 × 100 base) DNA sequence reads that passed quality control were mapped against hg19 with samTools (22, 25), and variants were called and annotated as described earlier (9, 27). The detected variants in individuals were filtered by exomeSuite software to identify rare or novel potentially pathogenic changes in genes reported to be associated with retinal degeneration (25). The frequencies of novel variants were determined using ExAC database and our own data set of whole exome variants from 300 individuals. Patients from two additional pedigrees were analyzed by targeted sequencing of the DHDDS gene because of their Ashkenazi Jewish ancestry. Segregation analysis on all available family members and screening of ethnicity-matched control samples were carried out by PCR amplification of relevant exons followed by dideoxy sequencing (24).
RESULTS
Ten unrelated families with nonsyndromic autosomal recessive IRD were studied. One family each was of Indian, Pakistani, Iraqi, and Brazilian origins, while the remaining were of European ancestry and from North America. Among these, three pedigrees were of Ashkenazi Jewish ancestry.
Exomes of selected individuals were sequenced at a minimum of 60x depth. The total number of variants including single nucleotide polymorphisms and indels detected in each of these patients ranged from 23,620 to 108,502. In each pedigree, a number of variants ranging from 240 to 747 were detected in known retinal disease genes by filtering for variants consistent with recessive inheritance. Among these, rare and low-frequency variants, where the allele frequency is <0.005, were selected for further analysis (Table 2).
Table 2.
Homozygous and compound heterozygous variants observed in known RD genes
| Pedigree | Chr | rsID | ExAC | Gene | Zygosity | cDNA Change | AA Change | Polyphen | Status of Variants (Ref) |
|---|---|---|---|---|---|---|---|---|---|
| RF.GU.0113 | 2 | rs750151209 | 0.00001647 | CERKL | Hom | c.967_968delAT | p.Met323Val fsX20 | N/A | novel |
| RF.RA.0812 | 1 | rs1800553 | 0.0051 | ABCA4 | Hom | c.5882G>A | p.Gly1961Glu | 1 | reported(1, 6, 12) |
| 1 | rs373652862 | 0.00004 | RPE65 | Het | c.746A>G | p.Tyr249Cys | 1 | reported(15) | |
| 1 | rs62653015 | 0.00001647 | RPE65 | Het | c.1451G>A | p.Gly484Asp | 1 | reported(33) | |
| RF.H.0909 | 1 | rs760549861 | 0.0001 | ABCA4 | Het | c.5318C>T | p.Ala1773Val | 0.998 | reported(30) |
| 1 | rs1800728 | 0.0002 | ABCA4 | Het | c.5461–10T>C | intronic | N/A | reported(20) | |
| RF.T.1111 | 1 | NL | N/A | RPE65 | Het | c.755T>C | p.Phe252Ser | 1 | novel |
| 1 | NL | N/A | RPE65 | Het | c.1360delA | p.Thr454Leu fsX31 | 0.782 | novel | |
| RF.S.0711 | 3 | rs765715798 | 0.00006591 | ARL6 | Hom | c.362G>A | p.Arg121His | 1 | novel |
| 13 | NL | N/A | ATP7B | Hom | c.2966A>C | p.Leu989Arg | 1 | novel | |
| RF.SH.0008 | 1 | rs397518048 | 0.00003295 | USH2A | Het | c.9424G>T | p.Gly3142Ter | N/A | novel |
| 1 | rs749228276 | 0.00001647 | USH2A | Het | c.9882C>G | p.Cys3294Trp | 1 | novel | |
| RF.M.0711 | 4 | rs373037737 | 0.00003299 | PDE6B | Het | c.1954C>T | p.Gln652Ter | N/A | novel |
| 4 | rs746552548 | 0.00001648 | PDE6B | Het | c.2116A>T | p.Lys706Ter | N/A | reported(28) | |
| RF.TE.0512 | 1 | rs147394623 | 0.0001 | DHDDS | Hom | c.124A>G | p.Lys42Glu | 0.786 | reported(35) |
| RF.AA.1104 | 1 | rs147394623 | 0.0001 | DHDDS | Het | c.124A>G | p.Lys42Glu | 0.786 | reported(35) |
| 1 | NL | N/A | DHDDS | Het | c.616A>G | p.Thr206Ala | 0.998 | novel | |
| RF.PO.1109 | 1 | rs147394623 | 0.0001 | DHDDS | Hom | c.124A>G | p.Lys42Glu | 0.786 | reported(35) |
NL, not listed; N/A, not applicable.
Pedigree 1 (RF.GU.0113).
A family of Indian Punjabi descent had two male siblings affected with rod-cone degeneration and central scotomas (Fig. 1A). No additional family members had vision loss, and there was no known history of consanguinity. Both siblings reported difficulty with night vision, abnormal color vision, and central vision loss beginning in their late teens to early 20s when visual acuity was corrected to 20/25- in each eye. There was bilateral perifoveal RPE atrophy, and diffuse RPE pigmentary mottling extended throughout the fundus in each eye of both siblings at ages 31 and 28 yr (Fig. 1, B1 and C1). Full-field ERG testing at ages 31 and 28 yr for subjects II-I and II-II, respectively, revealed severely reduced but measurable dim scotopic responses. Mixed scotopic responses reduced to 15% of average with delayed timing, and photopic responses reduced to 30% of average with timing delayed by several milliseconds in each eye are consistent with rod-cone IRD (Table 1).
Fig. 1.
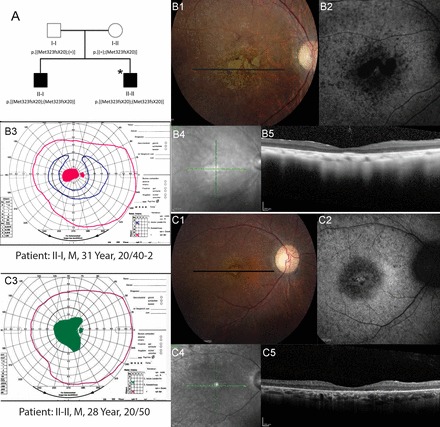
Pedigree and phenotype of 2 siblings with CERKL mutations. A: pedigree with homozygous 2 base pair deletion (c.967_968delAT, p.Met323Val fsX20) in CERKL gene. *Individuals who underwent whole exome sequencing. Clinical features of affected members with CERKL mutations. B1 and C1: color fundus photos; B2 and C2: fundus autofluorescence; B3 and C3: Goldmann visual fields; pink lines indicate the V4e isopter, green lines show the II4e isopter in a patient who was unable to perceive the I4e target, and blue lines show the I4e isopter. Black lines on color photos indicate location of spectral-domain optical coherence tomography (SD-OCT) scans. Homozygotes show heterogeneous, increased autofluorescence (AF) around the fovea; SD-OCT shows severe outer retinal layer loss; visual fields demonstrate central scotomas. B4 and C4: infrared fundus photos; B5 and C5: optical coherence tomography scans.
Central scotomata and color vision abnormalities with a mixed axis of confusion were observed in both siblings by ages 31 and 28 yr (Table 1; Fig. 1, B3 and C3). SDOCT images showed severe loss of the outer retinal layers throughout the central macula in both patients, with a small island of preserved outer retinal structure near the fovea in patient II-I only (Fig. 1, B5 and C5), consistent with a clinical diagnosis of rod-cone IRD with central involvement.
Sequencing the exome of patient II-II and analysis of variants detected three candidate variants. Among these, only one novel, homozygous, two-base pair deletion, c.967_968delAT; p.Met323Val fsX20 (rs750151209) in exon 7 of the CERKL gene (NM_201548.4), was predicted to be causative. This frame shift mutation was predicted to result in premature truncation of the protein at amino acid position 343 or to cause nonsense-mediated decay of the transcript.
Pedigree 2 (RF.RA.0812).
A consanguineous family of Pakistani descent included two affected male siblings (Fig. 2A) whose maternal grandfather was reported to have vision abnormalities since age 4 yr that progressed to complete blindness by age 25. The affected male III-I (Fig. 2A) had early night vision loss beginning around age 2. Fundus examination at age 34 revealed mild nonproliferative diabetic retinopathy without clinically significant macular edema and focal areas of RPE atrophy consistent with focal laser treatment in the left eye only (Fig. 2B1). Goldmann visual fields revealed constriction to the I4e isopter in each eye with full peripheral fields to the V4e isopter (Fig. 2B2). Patient III:II also developed abnormal night vision and dark adaptation beginning in childhood. At age 19, white spots were reported in the fundus. In both siblings, RPE pigmentation was irregular in the macula extending along the temporal arcades and anteriorly but was preserved in the central 10 degrees (Fig. 2C1 shows III-II). No flecks or white spots were observed on examination at ages 30 and 34, respectively. SDOCT scans through the central 20 degrees of the macula showed atrophy of the outer retinal layers near the optic nerve and loss of the outer retinal layers beginning ~5–10 degrees from the fovea (Fig. 2, B3 and C3). Full-field ERG testing in III-II revealed unmeasurable responses to dim scotopic stimuli, while bright scotopic amplitudes were severely reduced to 5%, and photopic amplitudes were reduced to 7% of the lower limit of normal, consistent with rod-cone degeneration.
Fig. 2.
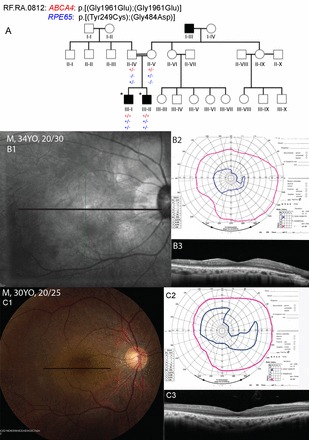
Pedigree and phenotype of 2 siblings with ABCA4 and RPE65 mutations. A: Segregation of the homozygous mutation (c.5882G>A; p.Gly1961Glu) in the ABCA4 gene and compound heterozygous mutation (c.746A>G; p.Tyr249Cys and c.1451G>A; p.Gly484Asp) in RPE65 gene. *Individuals who underwent whole exome sequencing. Clinical features of patient III-I at 34 yr and patient III-II at 30 yr. B1 and C1: fundus photos; B2 and C2: Goldmann visual fields; pink lines indicate the V4e isopter and blue lines show the I4e isopter. B3 and C3: SD-OCT. Black lines on color and infrared fundus photos indicate location of SD-OCT scans. Patients show heterogeneous, irregular retinal pigment epithelial pigmentation in the macula with relative preservation around the fovea; SD-OCT shows normal outer retinal layers at the fovea and in the central 10 degrees; visual fields demonstrate relative constriction of the I4e isopter with preserved peripheral visual fields to the V4e target.
Exome sequencing and analysis of variants identified a previously reported homozygous mutation, c.5882G>A; p.Gly1961Glu (rs1800553) in exon 42 of the ABCA4 (NM_000350) gene, in both affected siblings (Table 2) (1, 6, 12). In addition, two previously reported mutations in the RPE65, c.746A>G; p.Tyr249Cys (rs373652862) and c.1451G>A; p.Gly484Asp (rs62653015), were observed in the compound heterozygous state in both affected siblings (Table 2) (15, 33). Both the ABCA4 and the RPE65 mutations segregated with disease in this pedigree (Fig. 2A).
Pedigree 3. (RF.H.0909).
Pedigree RF.H.0909 (Fig. 3A) is a Caucasian family with no prior history of retinal degeneration in which the three offspring of nonconsanguineous parents developed visual abnormalities within the first decade of life. All three children had visual acuities of 20/100 OU (oculus uterque/both eyes) or worse when examined at the ages of 11, 9, and 7 yr, respectively. Fundus examination revealed bull’s eye maculopathy and pigment clumps surrounded by atrophy in the equatorial region OU with a flat, darkly pigmented choroidal lesion in the temporal macula OD (oculus dextrus/right eye) of III:I (Fig. 4A1), while III:II exhibited foveal atrophy with subretinal hypopigmented lesions anterior to the arcades and optic disk pallor (Fig. 4A2); the subretinal lesions and macular atrophy were hyperfluorescent, but there was a dark choroid around the optic nerve on fluorescein angiography (Fig. 4A3). Patient III:III exhibited foveal atrophy OU and faint pigment clumps surrounded by RPE atrophy anterior to the arcades, with a superotemporal flat pigmented choroidal lesion OS (oculus sinister/left eye) (Fig. 4A4) similar to that seen in III:I. Visual field testing revealed central scotomata OU in III:I, but III:II showed a peripheral island OD and constriction to the central 35 degrees OS; visual field testing was unreliable in III:III. Electroretinography demonstrated a cone-rod pattern of dysfunction in all three patients.
Fig. 3.
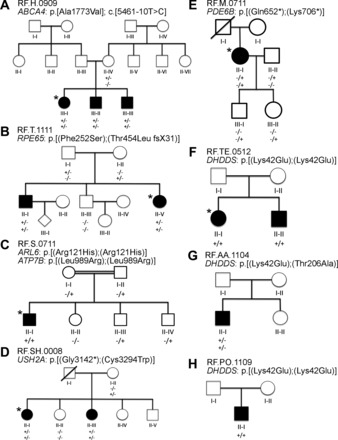
Eight different unrelated pedigrees involved in this study. The enrolled members for this study are indicated by the presence of genotype below the icon. +/+, The presence of a homozygous mutation; −/−, the homozygous wild-type allele; +/−, heterozygous allele. *Individuals who underwent whole exome sequencing. The pedigrees are RF.H.0909 (A), RF.T.1111 (B), RF.S.0711 (C), RF.SH.0008 (D), RF.M.0711 (E), RF.TE.0512 (F), RF.AA.1104 (G), RF.PO.1109 (H).
Fig. 4.
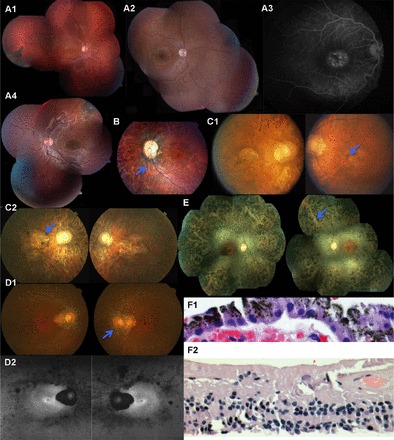
A1: fundus examination of RF.H.0909: III-I at age 11 yr. A2 and A3: the fundus image and fluorescein angiography of RF.H.0909: III-II. A4: the fundus image of patient RF.H.0909: III-III. B: fundus photos of patient RF.T.1111: II-V showing optic disk pallor with an optic pit temporally retinal vascular attenuation and bone spicule pigmentary change. C1 and C2: the color fundus photos of the patient RF.S.0711: II-I at age 36 and 50 yr. D1: color fundus photos at age 59 yr; D2: autofluorescence (AF) fundus photos of patient RF.SH.0008: II-III at age 63 yr demonstrate mottled areas of reduced AF most prominent along arcades and nasal midperiphery, a large patch of complete lack of AF associated with peripapillary chorioretinal atrophy, and small perifoveal hyper-AF ring oculus uterque (OU). E: fundus examination of RF.M.0711: II-I showed mild optic nerve pallor, attenuation of retinal blood vessels. F1 and F2: hematoxylin-and-eosin stain of retinal biopsy of RF.PO.1109: II-I reveals retinal degeneration of all layers including retinal ganglion cell, inner nuclear layer, and outer nuclear layer and intact retinal pigment epithelial (RPE) monolayer above choriocapillaris.
Exome sequencing of proband and subsequent analysis of variants identified two previously described compound heterozygous mutations in the ABCA4 (NM_000350.2), a missense change c.5318C>T; p.Ala1773Val (rs760549861) (30) and a an intronic variant c.5461–10T>C (rs1800728) associated with RD (20). Dideoxy sequencing revealed segregation of these variants with the phenotype.
Pedigree 4 (RF.T.1111).
Two of three offspring developed night blindness in the first decade of life in a Caucasian family with no prior history of IRD (Fig. 3B). Patients II-I and II-V were noted to have nystagmus and abnormal vision at ages 6 and 2 mo, respectively; II-I was diagnosed with rod-cone dystrophy at age 8 yr. Both affected siblings had subtle, very-low-frequency nystagmus; acuity was reduced to 20/200 OU in the affected male at the age of 39 yr and 20/400 OU in the affected female at 35 yr of age. Moderate myopia (−3.50 Dsph OD and −4.00 Dsph OS) was noted in the affected female, while myopia was more severe in the affected male (−7.50 Dsph+ 2.00 Dcyl x 095 OD and −7.00 Dsph + 2.25 DCyl x 070 OS). Both patients had bone spicules, thin retinal vessels, and optic disk pallor; II-V had an optic pit temporally OS (Fig. 4B), but neither patient had cataracts (14, 19). Patient II-III did not have retinitis pigmentosa (RP) but had keratoconus with classic signs of the disease including Vogt’s striae and Fleischer rings OU.
Analysis of exome variants identified novel compound heterozygous variants in the RPE65 gene: c.755T>C; p.Phe252Ser (NM_000329.2), predicted to be damaging, and an additional novel heterozygous frame shift variant, (NM_000329.2) c.1360delA; p.Thr454Leu fsX31, predicted to cause a premature stop codon.
Pedigree 5 (RF.S.0711).
A 39 yr old male patient from a consanguineous Iraqi family (Fig. 3C) was reported to have vision problems since age 5 yr, photosensitivity at 12 yr, and nyctalopia with peripheral vision loss at age 30 yr. His visual acuity was 20/400 OU with manifest refraction of −9.75 Dsph +0.75 Dcyl x165 OD and −9.50 Dsph +0.25 Dcyl x 30 OS, intraocular pressures (IOP) 18 mmHg OU with a tilted disk and increased cup-to-disk ratio, and severely reduced color vision (Ishihara plates 0/12 OU) at age 22 yr. Anterior segment was normal, and attenuated blood vessels and bone spicules were observed on ophthalmoscopy OU. The patient was diagnosed with Wilson disease at age 10 yr and had liver transplant and umbilical hernia repair at age 36 yr but was otherwise healthy. ERG testing under both photopic and scotopic stimulation elicited no detectable responses at age 22 yr, based on the reduced visual acuity and color vision abnormalities, findings consistent with cone-rod dystrophy OU. At age 50 yr, vision was light perception OU, IOP was 23 mmHg OU with an enlarged cup-to-disk ratio OU, attenuated retinal blood vessels, bone spicules encroaching upon the posterior pole, and macular atrophy, which expanded after age 36 yr (Fig. 4, C1 and C2, shows fundus images at ages 36 and 50 yr, respectively).
Exome variants analysis identified two novel homozygous variants, one in ATP7B on chromosome 13 and ARL6 on chromosome 3.
A novel homozygous missense variant, c.362G>A; p.Arg121His (rs765715798) in the gene ARL6 (NM_001278293), identified in the affected male was predicted to be probably damaging by PolyPhen2 and damaging by SIFT. The second homozygous variant found in the affected proband in ATP7B (NM_000053), c.2966A>C; p.Leu989Arg, was predicted to be damaging by PolyPhen2 and SIFT. Segregation analysis by dideoxy sequencing showed that both variants found in ARL6 as well as in ATP7B segregated with the disease in this family (Fig. 3C).
Pedigree 6 (RF.SH.0008).
Pedigree RF.SH.0008 (Fig. 3D) is a Caucasian family with no prior history of IRD in which two of five offspring of nonconsanguineous parents developed vision loss and also reported ringing in ears but not hearing loss. Patient II-III noticed night vision abnormalities in her mid-40s followed by decreasing peripheral vision ~10 yr later. Fundus exam showed waxy pallor and peripapillary RPE atrophy extending toward the macula, severely attenuated retinal blood vessels with sclerosis and RPE mottling with a few bone spicules (Fig. 4D1); fundus autofluorescence (AF) images demonstrated mottled areas of reduced AF most prominent along the arcades and in the nasal midperiphery, a large patch of complete lack of AF associated with peripapillary chorioretinal atrophy, and a perifoveal hyper-AF ring in the macula OU (Fig. 4D2). Kinetic visual field testing demonstrated a central island that was severely constricted to an approximate diameter of 20 degrees OS and 18 degrees OD with the V4e test target, with a large superior/temporal/inferior crescent of peripheral field in each eye, and a large absolute complete ring scotoma in the right eye and an incomplete ring scotoma that was connected by a narrow isthmus of field superiorly to the central field in the left eye.
Analysis of sequence variants in II-III revealed a novel nonsense variant c.9424G>T; p.Gly3142Ter (rs397518048) and a novel missense c.9882C>G; p.Cys3294Trp (rs749228276) variant identified in the USH2A (NM_206933.2) gene. Subsequent dideoxy sequencing confirmed segregation of these variants with IRD.
Pedigree 7 (RF.M.0711).
A 42 yr old female (Fig. 3E) had BCVA reduced to 20/200 in the right eye and counting fingers in the left eye. She was reported to have vision problems since age 2 yr and became legally blind by her third decade. Anterior segment examination was normal. Fundus examination showed mild optic nerve pallor, attenuation of retinal blood vessels, and RPE mottling in the macula with prominent bone spicules in the retinal periphery approaching the arcades (Fig. 4E).
Analysis of exome variants identified a novel nonsense variant, c.1954C>T; p.Gln652Ter (rs373037737), and a previously reported nonsense mutation (28), c.2116A>T; p.Lys706Ter (rs746552548) in the PDE6B gene (NM_000283.3), in the heterozygous state. Segregation analysis suggested that the two detected PDE6B variants exist in trans configuration and segregate with IRD in this family.
Pedigree 8 (RF.TE.0512).
Review of the clinical and family history of two siblings of Ashkenazi Jewish origin revealed retinal degeneration (Fig. 3F). The older female sibling (II-I) developed loss of night vision at age 17 yr. At age 22 yr, visual acuities were 20/25 with mild posterior subcapsular cataracts, moderate optic nerve pallor, retinal vascular attenuation, and RPE pigmentary change with white spots in the periphery OU (Fig. 5, A and C). At this age, visual field testing revealed mild constriction to the V4e and II4e isopters (Fig. 5F), and full-field ERG showed severely reduced responses to scotopic and photopic stimuli with similar reduction of a- and b-wave amplitudes (Fig. 5, D and E). An audiogram revealed normal hearing function bilaterally. Visual acuity declined over time to 20/40 by age 32 yr, then 20/50 by age 33 yr, then 20/70 in the right eye, and declined to 20/36 in the left eye, by age 40 yr. Visual fields showed progressive constriction over time; by age 31 yr, visual fields were constricted to less than 20 degrees diameter in each eye (Fig. 5G).
Fig. 5.
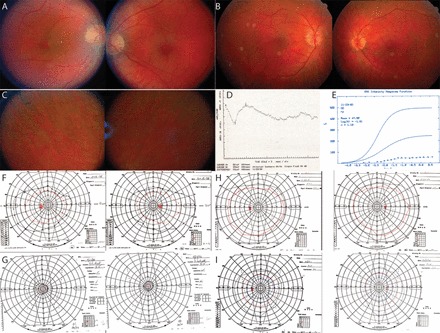
Pedigree RF.TE.0512 with DHDDS c.124A>G; p.Lys42Glu mutations. Top: color fundus photos at age 22 yr for II-I (A) and II-II (B) show mild disk pallor and retinal vascular attenuation. Middle: color fundus photos show peripheral bone spicule pigment and white spots in II-I at age 22 yr (C). Full-field electroretinogram (ERG) shows reduction of a-wave and b-wave responses to a scotopic bright flash (D); an intensity-response curve shows reduced maximum amplitude (Rmax) (E). Bottom: Goldmann visual fields at age 22 yr (F) and age 40 yr (G) in II-I and at age 22 yr (H) and age 32 yr (I) in II-II show progressive constriction.
The younger male sibling (II-II) was examined at age 22 yr when he had no symptoms of vision loss with visual acuity 20/16 in each eye. Fundus examination at ages 26 and 28 yr showed retinal vascular attenuation and mild disk pallor but no bone spicule pigmentary change (Fig. 5B). Visual fields showed progressive constriction over time (Fig. 5, H and I, shows visual fields at ages 22 and 32 yr, respectively). Goldmann visual field testing on three unaffected relatives was normal.
Targeted mutation analysis identified the previously described DHDDS (NM_024887) mutation c.124A>G; p.Lys42Glu (rs147394623) (35) in the homozygous state in both siblings and in the heterozygous state in their parents.
Pedigree 9 (RF.AA.1104).
The exome of a Jewish patient with IRD with no family history of consanguinity or IRD was sequenced (Fig. 3G). Analysis of variants revealed compound heterozygous mutations in the DHDDS gene: the previously reported mutation, c.124A>G; p.Lys42Glu (rs147394623) (35), and a novel pathogenic change, c.616A>G; p.Thr206Ala. Analysis of additional members of this pedigree confirmed segregation of these changes with IRD.
Pedigree 10 (RF.PO.1109).
Analysis of an affected male (Fig. 3H) of Ashkenazi Jewish origin with no family history of retinal degeneration detected the previously described DHDDS change c.124A>G; p.Lys42Glu (rs147394623) in the homozygous state (35). This patient underwent a retinal and choroidal biopsy to rule out viral vs. fungal vs. immunoproliferative disease. No additional clinical characterization was available for this patient. Histology of available retinal biopsy sample of this patient revealed significant degeneration of all retinal layers including ganglion cells (Fig. 4F1). The RPE layer was intact despite significant retinal degeneration and loss of inner and outer segments and nuclei of photoreceptors (Fig. 4F2).
Control analysis.
Analysis of 100 ethnicity-matched appropriate controls did not detect the novel causative variants identified in this study, indicating that these variants are rare in those populations.
DISCUSSION
Exome analysis of the probands of 10 pedigrees identified mutations in the CERKL, ABCA4, RPE65, ARL6, USH2A, PDE6B, and DHDDS genes as the underlying cause of retinal degeneration. Among these, nine were novel, potentially pathogenic variants, while five were previously reported mutations. Although patients from all pedigrees studies had typical symptoms of recessive IRD, variation was observed in clinical features such as the involvement of central retina, RPE atrophy, or lack of RPE abnormalities. Detailed clinical evaluation carried out on these patients provided insight into the phenotype associated with the genotypes observed.
Mutations in ceramide kinase-like (CERKL) gene are associated with autosomal recessive (ar)RP and cone-rod dystrophy (3, 4, 19). Patients with CERKL mutations have been reported with loss of visual acuity, bone spicule pigmentation with white dots in the outer retina, macular RPE atrophy, and central chorioretinal atrophy. The clinical pathology of pedigree RF.GU.0113 and previous reports in literature indicate that the CERKL gene mutations are associated with rod-cone dystrophy with central involvement.
In this study, mutations in the ABCA4 gene were found to be the underlying and/or contributing cause of RD in two pedigrees, RF.RA.0812 and RF.H.0909. Patients from these pedigrees have presented with arRP, bull’s eye maculopathy or macular atrophy phenotype, respectively. Neither of these pedigrees demonstrated lipofuscin deposits or typical flecks seen in patients with STGD1, although the pedigree RF.RA.0812 was reported to have white spots in the fundus at the time of diagnosis before age 20 yr in both individuals. Mutations in ABCA4 are associated with a broad spectrum of recessive retinal degeneration phenotypes including STGD1, cone-rod dystrophy, and arRP (6, 21). The phenotype of the two pedigrees presented here further supports the association of ABCA4 mutations with RP phenotypes and macular atrophy with or without the findings of STGD1. However, the frequency of c.5882G>A; p.Gly1961Glu variant observed in RF.RA.0812 was reported to be high, and the phenotype of some patients carrying this variant was found to be mild (1, 6, 12). In this pedigree, an additional set of previously reported compound heterozygous mutations p.Tyr249Cys and p.Gly484Asp in the RPE65 were observed (Fig. 2). Similar to the reports of patients with p.Gly484Asp mutations in RPE65, the affected individuals in RF.RA.0812 had poor night vision and a history of fine white retinal dots earlier in life that faded over time and were replaced with RPE changes (33). It is likely that the mutations observed in RPE65 are sufficient to explain the RD phenotype in this pedigree. However, the impact of having both the ABCA4 p.Gly1961Asp homozygous variant and the RPE65 compound heterozygous mutations in this pedigree is not known. Co-occurrence of mutations in two genes segregating with disease has been reported in IRD pedigrees previously (16, 19).
The affected male in pedigree RF.S.0711 is of Iraqi origin and has a diagnosis of cone dystrophy and Wilson disease. Three different novel potentially pathogenic homozygous changes in ARL6, CLN3, and ATP7B genes on three different chromosomes were detected in this individual. ARL6 is primarily associated with Bardet-Biedl syndrome (BBS), while one homozygous mutation was reported in a Saudi pedigree with recessive nonsyndromic RP (2). Mutations in the ATP7B gene are associated with Wilson disease. Mutations in ARL6 and ATP7B segregated with disease in this pedigree (Fig. 3C). The novel change in CLN3 is observed in the homozygous state in the affected male and one 47 yr old, unaffected sibling. The clinical evaluation did not reveal signs of ceroid lipofuscinosis except the retinal degeneration, which did not segregate with the novel CLN3 change. In this patient, the c.362G>A; p.Arg121His ARL6 mutation alone may be sufficient to cause nonsyndromic retinal degeneration, while the ATP7B mutation is the underlying cause of Wilson disease. The impact of mutations in CLN3 and ATP7B, if any, on modifying the ARL6-associated phenotype, such as causing nonsyndromic RD instead of BBS, is not known.
RPE65 and PDE6B are widely studied retinal disease genes associated with early-onset retinal degeneration (5, 29). Similarly, mutations in USH2A are associated with nonsyndromic recessive RP with later age of onset (7, 18). The phenotype of patients in our study from families with mutations in RPE65, PDE6B, and USH2A is consistent with the phenotypic features associated with these gene mutations.
A missense mutation, p.Lys42Glu, in DHDDS has been reported as a common cause of recessive RP in patients of Ashkenazi Jewish ancestry (31, 35). We observed a second novel DHDDS mutation, p.Thr206Glu, in one affected individual. DHDDS knockdown induces photoreceptor degeneration in zebrafish, confirming its causative nature (34). The rare opportunity to study retinal histology of a patient with the common p.Lys42Glu DHDDS mutation revealed the presence of relatively preserved RPE cells despite severe degeneration of outer retinal layers. This information will be helpful in designing therapeutic strategies, including cell-based therapies to replace photoreceptors in the presence of relatively preserved RPE cells, in patients with RD due to DHDDS mutations.
Analysis of pedigrees presented in this study revealed that whole exome sequencing could reveal not only the possible causative mutations, but also pathogenic sequence variants in additional genes. Among the 10 pedigrees analyzed, potentially disease-causing variants were observed in more than one gene associated with RD or neurodegeneration in two pedigrees. It is interesting to observe compound heterozygous causative mutations in a consanguineous pedigree (RF.RA.0812). Detection of novel damaging variants in multiple retinal disease genes segregating with disease in a pedigree presents a challenge in determining the underlying cause of phenotype. The additional variants may independently or, together with the primary mutations, contribute to the phenotype. Additional studies are needed to determine the role of novel damaging variants as either the underlying cause of pathology or phenotype modifiers. These studies will assist in developing effective ways to treat IRD when appropriate gene-based therapies become available.
The results of the analysis presented in this study indicate that novel or rare mutations in known IRD genes may contribute to a significant proportion of cases that are yet to be genotyped. Detailed clinical evaluation over a period of several years and the genotype analysis of the pedigrees studied suggest that the phenotype of patients is consistent with clinical features described to be associated with the genotypes. Accumulation of knowledge on the phenotype and genotype of patients will enable investigators to establish the phenotype associated with mutations in specific genotypes and the natural history of these diseases.
Genetics analysis using next-generation sequencing technologies may reveal causative mutations in multiple genes, and this may present a challenge in determining the primary cause of disease, genetic counseling, and selection of gene-based therapies to treat members of these pedigrees.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants EY-002162 (J. L. Duncan), EY-014375 (S. A. Riazuddin), EY-020846 (J. H. Lin), NIH-R01EY-021237; NIH-R01EY-013198 (R. Ayyagari), DK-53505-12 (P. Lee), U54 RR-025204 (P. Lee); Foundation Fighting Blindness (R. Ayyagari, J. L. Duncan, S. A. Riazuddin, J. R. Heckenlively, R. G. Weleber); Research to Prevent Blindness (R. Ayyagari, J. L. Duncan, J. R. Heckenlively); That Man May See, Inc. (J. L. Duncan); The Bernard A. Newcomb Macular Degeneration Research Fund (J. L. Duncan); Hope for Vision (J. L. Duncan); and The Beutler Foundation (P. Lee).
DISCLOSURES
R. G. Weleber serves on Scientific Advisory Boards for the Foundation Fighting Blindness. This relationship has been reviewed and managed by Oregon Health & Science University. No conflicts of interest, financial or otherwise, are declared by the other author(s).
AUTHOR CONTRIBUTIONS
P.B., J.L.D., B.M., I.K., K.B., L.G., J.H.L., G.B., M.N., J.J.S., M.P., C.S., and G.D. performed experiments; P.B., J.L.D., B.M., I.K., K.B., L.G., J.H.L., G.B., M.N., J.J.S., M.P., C.S., R.G.W., J.R.H., G.D., P.L., and R.A. analyzed data; P.B., J.L.D., B.M., I.K., K.B., L.G., J.H.L., M.N., J.J.S., M.P., C.S., R. G.W., J.R.H., G.D., P.L., S.A.R., and R.A. interpreted results of experiments; P.B., J.L.D., K.B., and J.H.L. prepared figures; P.B., J.L.D., C.S., G.D., and R.A. drafted manuscript; P.B., J.L.D., I.K., K.B., L.G., J.H.L., G.B., R. G.W., J.R.H., P.L., S.A.R., and R.A. edited and revised manuscript; J.L.D., I.K., K.B., L.G., J.H.L., G.B., R. G.W., J.R.H., G.D., S.A.R., and R.A. approved final version of manuscript; R.A. conceived and designed research.
ACKNOWLEDGMENTS
R. Ayyagari, the principal investigator and corresponding author, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.Aguirre-Lamban J, González-Aguilera JJ, Riveiro-Alvarez R, Cantalapiedra D, Avila-Fernandez A, Villaverde-Montero C, Corton M, Blanco-Kelly F, Garcia-Sandoval B, Ayuso C. Further associations between mutations and polymorphisms in the ABCA4 gene: clinical implication of allelic variants and their role as protector/risk factors. Invest Ophthalmol Vis Sci 52: 6206–6212, 2011. doi: 10.1167/iovs.10-5743. [DOI] [PubMed] [Google Scholar]
- 2.Aldahmesh MA, Safieh LA, Alkuraya H, Al-Rajhi A, Shamseldin H, Hashem M, Alzahrani F, Khan AO, Alqahtani F, Rahbeeni Z, Alowain M, Khalak H, Al-Hazzaa S, Meyer BF, Alkuraya FS. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis 15: 2464–2469, 2009. [PMC free article] [PubMed] [Google Scholar]
- 3.Aleman TS, Soumittra N, Cideciyan AV, Sumaroka AM, Ramprasad VL, Herrera W, Windsor EA, Schwartz SB, Russell RC, Roman AJ, Inglehearn CF, Kumaramanickavel G, Stone EM, Fishman GA, Jacobson SG. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci 50: 5944–5954, 2009. doi: 10.1167/iovs.09-3982. [DOI] [PubMed] [Google Scholar]
- 4.Ávila-Fernández A, Cantalapiedra D, Aller E, Vallespín E, Aguirre-Lambán J, Blanco-Kelly F, Corton M, Riveiro-Álvarez R, Allikmets R, Trujillo-Tiebas MJ, Millán JM, Cremers FP, Ayuso C. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol Vis 16: 2550–2558, 2010. [PMC free article] [PubMed] [Google Scholar]
- 5.Bowne SJ, Humphries MM, Sullivan LS, Kenna PF, Tam LC, Kiang AS, Campbell M, Weinstock GM, Koboldt DC, Ding L, Fulton RS, Sodergren EJ, Allman D, Millington-Ward S, Palfi A, McKee A, Blanton SH, Slifer S, Konidari I, Farrar GJ, Daiger SP, Humphries P. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 19: 1074–1081, 2011. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke TR, Fishman GA, Zernant J, Schubert C, Tsang SH, Smith RT, Ayyagari R, Koenekoop RK, Umfress A, Ciccarelli ML, Baldi A, Iannaccone A, Cremers FP, Klaver CC, Allikmets R. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci 53: 4458–4467, 2012. doi: 10.1167/iovs.11-9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen M, Bitner-Glindzicz M, Luxon L. The changing face of Usher syndrome: clinical implications. Int J Audiol 46: 82–93, 2007. doi: 10.1080/14992020600975279. [DOI] [PubMed] [Google Scholar]
- 8.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet 84: 132–141, 2013. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498, 2011. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan JL, Zhang Y, Gandhi J, Nakanishi C, Othman M, Branham KE, Swaroop A, Roorda A. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci 48: 3283–3291, 2007. doi: 10.1167/iovs.06-1422. [DOI] [PubMed] [Google Scholar]
- 11.Fahim AT, Daiger SP, Weleber RG. Nonsyndromic retinitis pigmentosa overview, in GeneReviews. Edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K. Seattle, WA: University of Washington, Seattle, 1993; 20301590. [Google Scholar]
- 12.Fujinami K, Sergouniotis PI, Davidson AE, Mackay DS, Tsunoda K, Tsubota K, Robson AG, Holder GE, Moore AT, Michaelides M, Webster AR. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology 120: 2324–2331, 2013. doi: 10.1016/j.ophtha.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 368: 1795–1809, 2006. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 14.Heckenlively JR. Retinitis Pigmentosa. Philadelphia: J.B. Lippincott, 1988, p. 269. [Google Scholar]
- 15.Henderson RH, Waseem N, Searle R, van der Spuy J, Russell-Eggitt I, Bhattacharya SS, Thompson DA, Holder GE, Cheetham ME, Webster AR, Moore AT. An assessment of the apex microarray technology in genotyping patients with Leber congenital amaurosis and early-onset severe retinal dystrophy. Invest Ophthalmol Vis Sci 48: 5684–5689, 2007. doi: 10.1167/iovs.07-0207. [DOI] [PubMed] [Google Scholar]
- 16.Huynh N, Jeffrey BG, Turriff A, Sieving PA, Cukras CA. Sorting out co-occurrence of rare monogenic retinopathies: Stargardt disease co-existing with congenital stationary night blindness. Ophthalmic Genet 35: 51–56, 2014. doi: 10.3109/13816810.2013.865762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inglehearn CF. Molecular genetics of human retinal dystrophies. Eye (Lond) 12: 571–579, 1998. doi: 10.1038/eye.1998.147. [DOI] [PubMed] [Google Scholar]
- 18.Keats BJ, Corey DP. The usher syndromes. Am J Med Genet 89: 158–166, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 19.Khan AO, Abu-Safieh L. Rod-cone dystrophy with initially preserved visual acuity despite early macular involvement suggests recessive CERKL mutations. Ophthalmic Genet 36: 369– 372, 2015. doi: 10.3109/13816810.2014.889168. [DOI] [PubMed] [Google Scholar]
- 20.Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol 243: 90–100, 2005. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 21.Lee W, Xie Y, Zernant J, Yuan B, Bearelly S, Tsang SH, Lupski JR, Allikmets R. Complex inheritance of ABCA4 disease: four mutations in a family with multiple macular phenotypes. Hum Genet 135: 9–19, 2016. doi: 10.1007/s00439-015-1605-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup . The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079, 2009. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Zhang Q, Pierce EA. Photoreceptor sensory cilia and inherited retinal degeneration. Adv Exp Med Biol 664: 223–232, 2010. doi: 10.1007/978-1-4419-1399-9_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald IM, Gudiseva HV, Villanueva A, Greve M, Caruso R, Ayyagari R. Phenotype and genotype of patients with autosomal recessive bestrophinopathy. Ophthalmic Genet 33: 123–129, 2012. doi: 10.3109/13816810.2011.592172. [DOI] [PubMed] [Google Scholar]
- 25.Maranhao B, Biswas P, Duncan JL, Branham KE, Silva GA, Naeem MA, Khan SN, Riazuddin S, Hejtmancik JF, Heckenlively JR, Riazuddin SA, Lee PL, Ayyagari R. exomeSuite: whole exome sequence variant filtering tool for rapid identification of putative disease causing SNVs/indels. Genomics 103: 169–176, 2014. doi: 10.1016/j.ygeno.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maranhao B, Biswas P, Gottsch AD, Navani M, Naeem MA, Suk J, Chu J, Khan SN, Poleman R, Akram J, Riazuddin S, Lee P, Riazuddin SA, Hejtmancik JF, Ayyagari R. Investigating the molecular basis of retinal degeneration in a familial cohort of Pakistani decent by exome sequencing. PLoS One 10: e0136561, 2015. doi: 10.1371/journal.pone.0136561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303, 2010. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA 92: 3249–3253, 1995. doi: 10.1073/pnas.92.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci USA 95: 3088–3093, 1998. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenirri S, Alaimo G, Manitto MP, Brancato R, Ferrari M, Cremonesi L. Are microarrays useful in the screening of ABCA4 mutations in Italian patients affected by macular degenerations? Clin Chem Lab Med 46: 1250–1255, 2008. doi: 10.1515/CCLM.2008.248. [DOI] [PubMed] [Google Scholar]
- 31.Venturini G, Koskiniemi-Kuendig H, Harper S, Berson EL, Rivolta C. Two specific mutations are prevalent causes of recessive retinitis pigmentosa in North American patients of Jewish ancestry. Genet Med 17: 285–290, 2015. doi: 10.1038/gim.2014.132. [DOI] [PubMed] [Google Scholar]
- 32.Weleber RG, Carr RE, Murphey WH, Sheffield VC, Stone EM. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol 111: 1531–1542, 1993. doi: 10.1001/archopht.1993.01090110097033. [DOI] [PubMed] [Google Scholar]
- 33.Weleber RG, Michaelides M, Trzupek KM, Stover NB, Stone EM. The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest Ophthalmol Vis Sci 52: 292–302, 2011. doi: 10.1167/iovs.10-6106. [DOI] [PubMed] [Google Scholar]
- 34.Wen R, Dallman JE, Li Y, Züchner SL, Vance JM, Peričak-Vance MA, Lam BL. Knock-down DHDDS expression induces photoreceptor degeneration in zebrafish. Adv Exp Med Biol 801: 543–550, 2014. doi: 10.1007/978-1-4614-3209-8_69. [DOI] [PubMed] [Google Scholar]
- 35.Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, Cideciyan AV, Jacobson SG, Sharon D. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet 88: 207–215, 2011. doi: 10.1016/j.ajhg.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng A, Li Y, Tsang SH. Personalized therapeutic strategies for patients with retinitis pigmentosa. Expert Opin Biol Ther 15: 391–402, 2015. doi: 10.1517/14712598.2015.1006192. [DOI] [PMC free article] [PubMed] [Google Scholar]