

Abstract
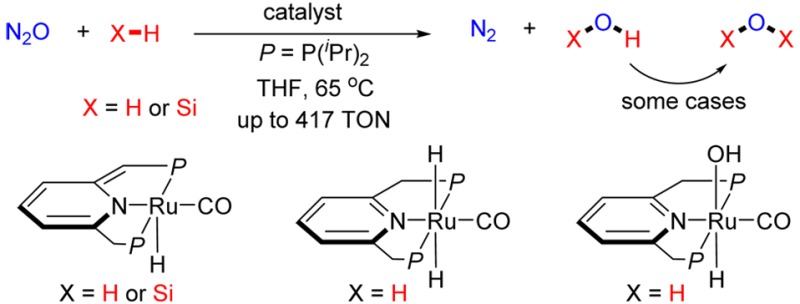
Due to its significant contribution to stratospheric ozone depletion and its potent greenhouse effect, nitrous oxide has stimulated much research interest regarding its reactivity modes and its transformations, which can lead to its abatement. We report the homogeneously catalyzed reaction of nitrous oxide (N2O) with H2. The reaction is catalyzed by a PNP pincer ruthenium complex, generating efficiently only dinitrogen and water, under mild conditions, thus providing a green, mild methodology for removal of nitrous oxide. The reaction proceeds through a sequence of dihydrogen activation, “O”-atom transfer, and dehydration, in which metal–ligand cooperation plays a central role. This approach was further developed to catalytic O-transfer from N2O to Si–H bonds.
Nitrous oxide (N2O), emitted due to agriculture activities, industrial processes, combustion of fossil fuels and biomass, is a potent greenhouse gas and regulator of atmospheric ozone concentrations.1,2 Although accounting for only 6% of all greenhouse gas emissions from human activities, nitrous oxide shows ca. 300 times greater warming potential than CO2.3 Due to its highly destructive environmental effects, the degradation, reduction, and/or application of nitrous oxide have drawn much attention.4 Hydrogenation of nitrous oxide using dihydrogen, which is driven by release of dinitrogen and water, is considered an attractive reductive process. Although there are reports on catalytic hydrogenation reactions of N2O by heterogeneous systems, (metal surfaces, zeolites),5homogeneously catalyzed reactions by metal complexes are highly desirable, as they may be more amenable to catalytic design by catalyst structural modifications, and might occur under mild, selective conditions. Such systems may also shed light on mechanistic steps of importance regarding N2O activation.
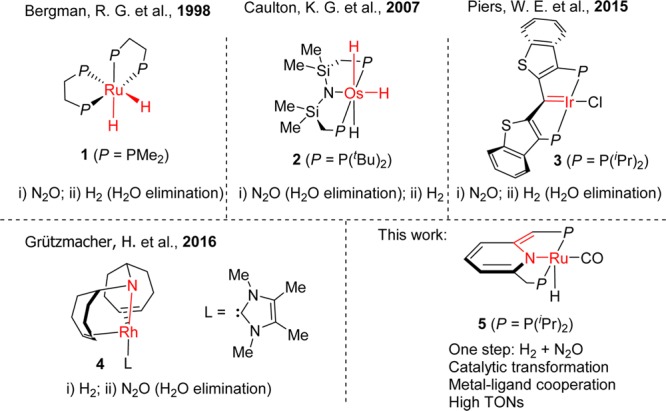
Stochiometric hydrogenation of N2O involving metal complexes was reported (Scheme 1).6 In seminal work by Bergman in 1998,6b the reaction of (DMPE)2Ru(H)21 (DMPE = 1,2-bis(dimethylphosphino)ethane) with 1 equiv N2O afforded the hydroxoruthenium complex (DMPE)2Ru(H)(OH), which further reacted with hydrogen gas to regenerate complex 1 and release a water molecule, representing stepwise stoichiometric hydrogenation of N2O.7 Using excess N2O, the dihydroxoruthenium complex (DMPE)2Ru(OH)2 was formed, preventing water formation and thus prohibiting catalytic hydrogenation. In 2007, Caulton reported that the reaction of (PNP)Os(H)3 (2) (PNP = N(SiMe2CH2PtBu2)2) with 1 atm of N2O resulted in formation of H2O and (PNP)OsH(N2). A separate reaction of the later with hydrogen slowly formed the complex (PNP)Os(H)3 (2).6c No catalysis was reported. In 2015, Piers reported a formal stochiometric hydrogenation of N2O using a PCsp2P iridium pincer carbene complex 3,6d which reacts with N2O with loss of N2 to form an iridiaepoxide complex; reaction of the latter with H2 followed by heating resulted in release of H2O. However, no catalytic turnover was observed. In 2016, Grützmacher reported the Rh-catalyzed dehydrogenative coupling of alcohols using N2O as a hydrogen acceptor, which proceeds by metal–ligand cooperation.6e Mechanistic studies of this system showed that reaction of N2O with H2 in the presence of complex 4 generated N2, and catalysis was mentioned, but catalytic data (TON, conversion, or yield) was not reported. To the best of our knowledge, there is currently no detailed report on homogeneously catalyzed hydrogenation of nitrous oxide.
Scheme 1. Homogeneous Hydrogenation of N2O.
To enable homogeneously catalyzed hydrogenation of nitrous oxide, several challenges have to be met: first, high selectivity in the sequential reaction of N2O and H2 in the catalytic cycle is required because nitrous oxide and dihydrogen are in large excess relative to the catalyst. In addition, over-reduction by hydrogen or overoxidation by nitrous oxide can inhibit the catalytic efficiency of the reaction. Third, the catalyst should be active in the presence of excess amount of the generated water.
Here we report the development of the homogeneously catalyzed hydrogenation of N2O with H2. The reaction is catalyzed by ruthenium pincer complexes and it very likely involves a unique mechanism based on metal–ligand cooperation (MLC). The reaction proceeds smoothly in high TON (TON = turnover number) under very mild conditions, thus providing a highly efficient method for hydrogenation of nitrous oxide. Moreover, the reaction was extended to catalytic O-transfer from N2O into Si–H bonds.
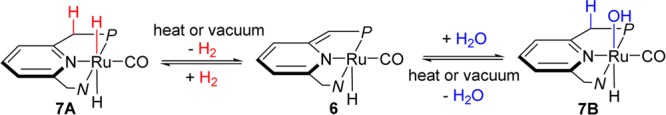
In recent years, our group has developed a series of transition metal complexes with pyridine- and acridine-based LNL′-type (L = N or P) pincer ligands, capable of facile activation of various X–H bonds, including H–H, O–H, N–H, C–H, B–H, and S–H bonds via metal–ligand cooperation.8 As a typical example, the pyridine-based PNN-ruthenium complex 6, first reported by us in 2005,9 can undergo a reversible hydrogenation/dehydrogenation sequence via metal–ligand cooperation (MLC) (Scheme 2).9 Similarly, O–H bond activation of H2O with dearomatized complex 6 takes place smoothly and reversibly at room temperature.10 Both reactions proceed with no change in the formal metal oxidation state. These two reversible reactions, and the stability of the complex under excess of water, encouraged us to explore the catalytic hydrogenation of nitrous oxide by pyridine-based pincer Ru complexes, based on metal–ligand cooperation.
Scheme 2. Reversible Activation of H2 and H2O by 6.
Initially, the hydrogenation of N2O was examined using 0.01 mmol of the PNN-ruthenium complex 6 (0.08 mol % catalyst) and ca. 13 mmol of N2O11 and 50 psi of H2 in a 90 mL of Fisher-Porter tube (50 psi of H2 corresponds to 13 mmol at rt). After heating in 5 mL THF at 65 °C for 36 h, 0.13 mmol of H2O was detected by 1H NMR of the reaction mixture using mesitylene as internal standard with calibration. A control experiment without the catalyst failed to afford products. Thus, the homogeneously catalyzed reaction (13 TON, 1% conversion for N2O) has been realized. The relatively low turnover number is probably due to decomposition of the catalyst since the free PNN ligand and the corresponding phosphine oxide were observed by 31P{1H} NMR of the reaction mixture. Various pyridine-based dearomatized ruthenium pincer complexes were then examined (Table 1). Replacing the diethylaminomethylene group (Et2NCH2−) of the pincer ligand by a 2-pyridinyl group resulted in a significant yield increase. Thus, using the PN(Py) complexes 8 (P = P(tBu)2)12a and 9 (P = P(iPr)2) resulted in an increase in the TONs to 53 (4% conversion for N2O) and 28 (2% conversion for N2O), respectively. However, complete decomposition of the catalysts was observed by 31P{1H} NMR spectroscopy. Compared to the PNN or PN(Py) complexes, the PNP ruthenium complex 10 (P = P(tBu)2)12b exhibited higher stability and led to a higher TON (110 TONs/37 h, 8% conversion for N2O). The phosphine group of the PNP ligands were then modified; whereas use of the PNP complex 12 (P = PPh2)12c resulted in a lower TON (10 TONs/36 h, <1% conversion of N2O), complex 5 (P = P(iPr)2) achieved the best result, namely 2.2 mmol of water (220 TONs, 17% conversion for N2O) were formed in 36 h. Significantly, the NMR spectra of the reaction solution showed that most of the starting ruthenium complex was converted to the complex (PNP)RuH(CO)(OH) 14 (spectrum iv in Figure 1; 1H NMR for Ru–H bond, δ = −14.7 ppm (t, 2JPH = 18.0 Hz); 31P{1H} NMR, δ = 74.0 ppm (s); ESI analysis, MS-ESI for C20H36NOP2Ru (MW = 470, (M–OH)+; for full mass data, see SI). Complex 14 was independently synthesized from 5 and H2O (spectrum v in Figure 1). Moreover, the corresponding reaction using 14 as catalyst led to an even better result (307 TONs/37 h, 24% conversion for N2O), indicating that complex 14 is very likely involved in the catalytic cycle, and is the resting state of the catalytic cycle. Replacing the CO ligand by N2 decreased the yield significantly (110 TON for 10 vs 61 TON for 11(12d)). The acridine-based PNP ruthenium complex 13(12e) was less efficient than complex 5, affording 94 TON (7% conversion for N2O) in 36 h.
Table 1. Catalyst Screening for Hydrogenation of N2Oa,b,c.
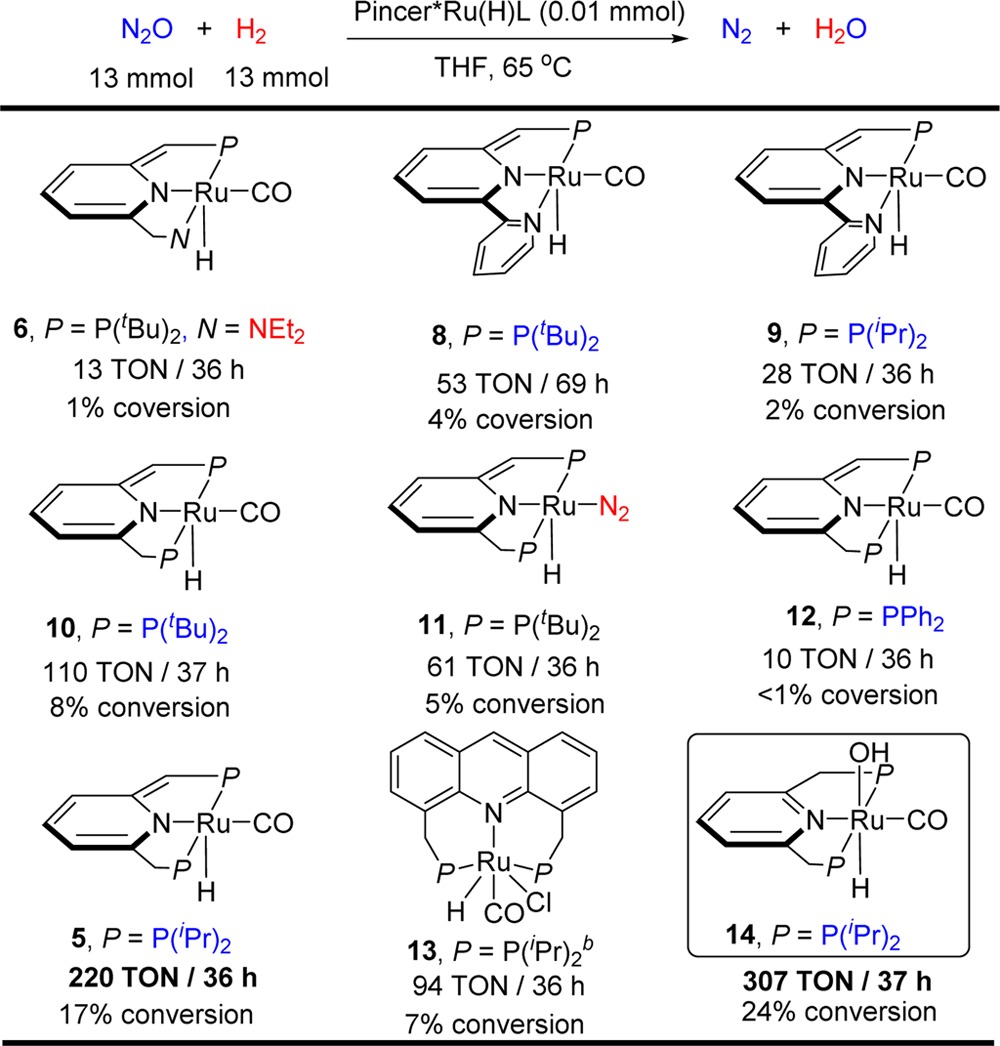
All the reactions were conducted in a 90 mL Fisher-Porter tube using 13 mmol of H2 and N2O in 5 mL THF.
All the complexes except 13 were freshly prepared from the corresponding aromatized (pincer)RuH(Cl)(L) with 1 equiv tBuOK in THF and used directly in the reactions. Complex 13 was obtained similarly using KHMDS as base.
The TONs are based on the generated H2O as measured by 1H NMR of the reaction mixture using mesitylene as internal standard (see SI for details).
Figure 1.
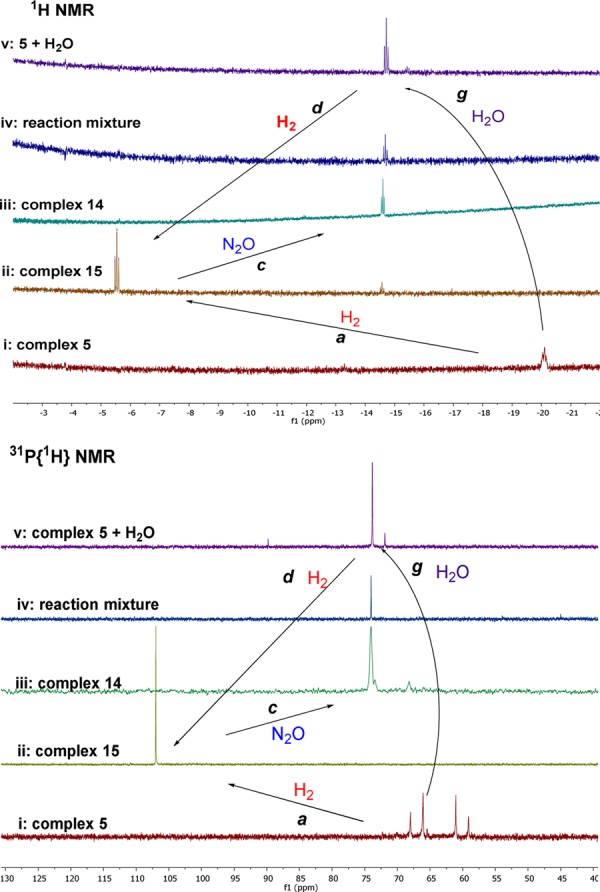
1H NMR of Ru–H bonds and 31P{1H} NMR spectra of complexes 5, 14, and 15 in THF and the corresponding reactions.
To have a better understanding of this catalytic transformation and to further develop it, the reaction mechanism was explored by studying individual steps that might be involved in the catalytic cycle (Scheme 3, Table S1 (see SI), and Figure 1). First, the reaction of the dearomatized ruthenium complex 5 (spectrum i in Figure 1) with dihydrogen is known to occur smoothly to afford the ruthenium trans-dihydride complex 15 (spectrum ii in Figure 1, pathway a in Scheme 3).13 On the other hand, in the absence of H2, N2O was found to decompose complex 5, resulting in a complicated mixture (pathway e), which failed to convert to the ruthenium hydroxo complex 14 in the presence of excess H2 (pathway f). Importantly, the competitive experiment of 5 in the presence of both N2O and H2 (1:1 mixture) resulted in formation of the ruthenium trans-dihydride complex 15 as the only product. Thus, the much faster reaction of 5 with H2 than with N2O inhibits the decomposition of 5 and enables the whole catalytic cycle. Because the hydrogenation of 5 is a reversible reaction, via metal–ligand cooperation (pathway b), catalyst 5 can be regenerated from 15, as observed under vacuum or upon heating.13
Scheme 3. Mechanistic Studies of Individual Steps.
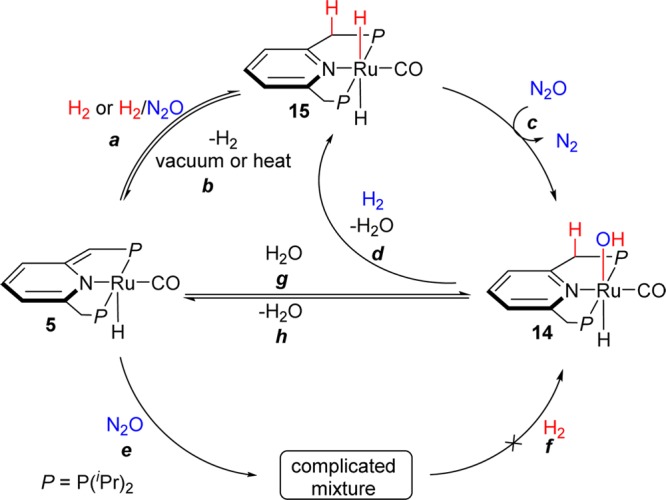
Moreover, even in the presence of 2 equiv N2O, only mono “O”-atom transfer took place upon reaction with 15, highly efficiently forming the hydrido hydroxo complex 14 (spectrum iii in Figure 1, pathway c in Scheme 3).14 This is important because double insertion to give the dihydroxo complex (as observed by Bergman6a,6b) would hinder subsequent water elimination. Reversible water elimination from complex 14 is facilitated by the trans effect of Ru–H bond, affording the dearomatized PNP pincer catalyst 5 under vacuum (pathway h). Furthermore, the ruthenium trans-dihydride complex 15 was regenerated directly by the reaction of ruthenium hydroxide 14 with H2, combining water release and hydrogen addition via metal–ligand cooperation (pathway d).
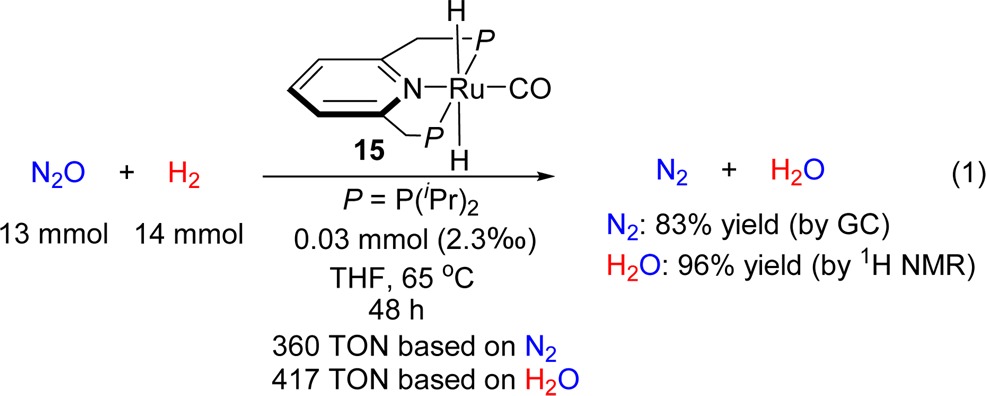
Complex 15 was found to be the best catalyst in the hydrogenation of N2O. Full conversion of nitrous oxide and the highest TON were achieved by generating 15in situ from complex 5 (0.03 mmol, 0.23% of catalyst) and increasing the reaction time to 48 h (eq 1). The produced 83% yield of dinitrogen (360 TON based on N2) was determined carefully by GC, and 96% yield of H2O (417 TON based on H2O) was measured by 1H NMR with calibration.11
 |
1 |
On the basis of these results, we propose that the mechanism of hydrogenation of nitrous oxide follows the reaction sequence of pathway a-c-h, involving hydrogenation of the dearomatized PNP pincer complex 5 by metal–ligand cooperation (MLC) to afford the ruthenium trans-dihydride compound 15, followed by (likely rate determining) selective mono oxygen transfer from nitrous oxide to give complex 14, and water release by MLC to regenerate complex 5, thus completing an efficient, selective catalytic cycle. Essentially, each one of the complexes 5, 14 and 15, which are involved in the catalytic cycle, can serve as a catalyst, as we indeed observed. However, due to the potential decomposition of 5 if N2O is added prior to H2, complex 15 is practically the best catalyst. To be mentioned, due to the reversible hydration/dehydration sequence via metal–ligand cooperation, it is difficult to isolate complex 14 and it was characterized in situ.
Silicon–oxygen bond formation of silane with nitrous oxide is the key process for deposition of nonstoichiometric silicon oxide or semi-insulating polysilicon (SIPOS), which is used as a substitute for silicon dioxide as passivation material in high voltage power devices.15 Thus, homogeneous “O”-atom transfer of nitrous oxide into silane was then examined by using catalyst 5 under N2O atmosphere (50 psi) (Table 2): the reaction of PhMe2SiH 16a with nitrous oxide took place smoothly using 1 mol % of catalyst, affording the desired silanol 17a together with disilyl ether 18a in 32% and 64% yields, respectively. H2 was detected by GC, indicating the disilyl ether is formed from the direct dehydrogenative coupling reaction of formed silanol and residual silane. Ph2MeSiH 16b, which contains a bulkier substituent (Ph over Me group), exhibited lower reactivity and afforded 30% of silanol 17b together with 46% of disilyl ether 18b using 2 mol % of catalyst and increasing reaction time to 3 days. Substrate 16c with tert-butyl group afforded only silanol 17c in 46% yield together with 54% of recovered starting material. No disilyl ether 18c was formed because of the steric hindrance of the bulky substituents.
Table 2. Catalytic Oxygen Transfer to Si–H Bond Using Nitrous Oxidea.
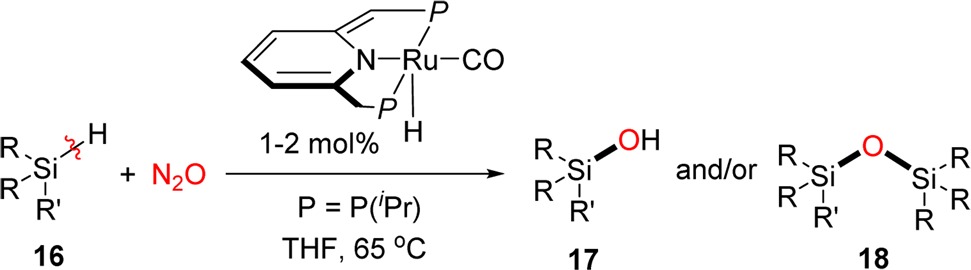
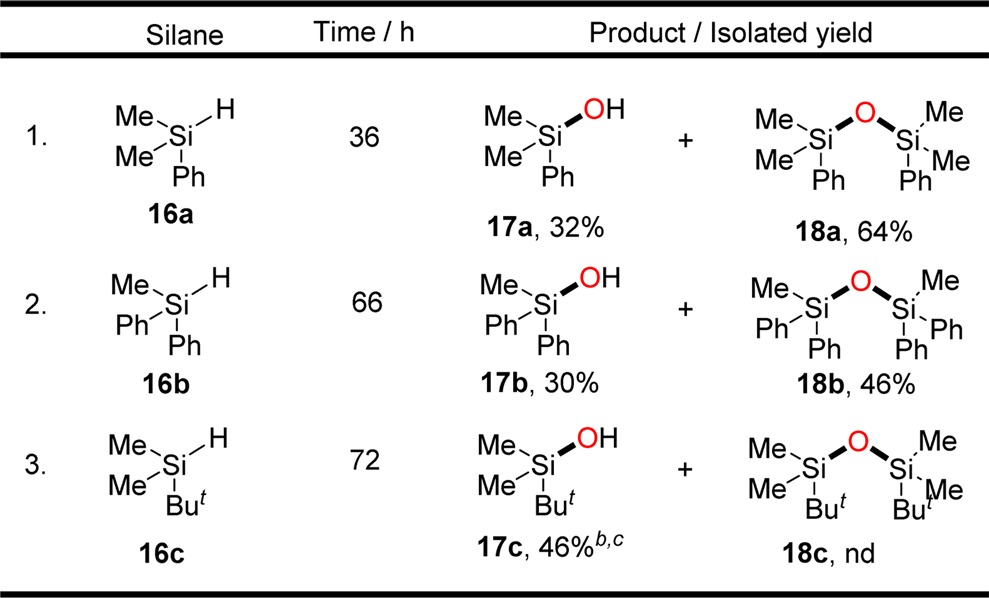
The reactions were conducted using a THF solution containing 0.01 mmol of catalyst 5 and 1.0 mmol (for PhMe2SiH) or 0.5 mmol (for Ph2MeSiH and tBuMe2SiH) of substrate under 50 psi of N2O.
The yields (based on the silane) were determined by GC using standard curve due to the low boiling point of the product.
54% of the starting material was recovered.
In summary, the homogeneously catalyzed hydrogenation of nitrous oxide by a metal complex has been developed. High efficiency and high TON are achieved using the PNP pincer ruthenium complex 15 as the catalyst. Studies of stoichiometric steps indicate that the reaction involves metal–ligand cooperation (MLC). Thus, H2 addition to the dearomatized catalyst 5 takes place with aromatization to form complex 15, followed by mono-oxygen transfer from N2O to a Ru–H bond, and subsequent water release by MLC, regenerating the dearomatized complex. Moreover, catalytic “O”-atom transfer from nitrous oxide to Si–H bonds of silanes, catalyzed by the dearomatized 5, was also demonstrated. Further studies in this area are being carried out in our laboratory.
Acknowledgments
This research was supported by the European Research Council (ERC AdG 692775). D.M. holds the Israel Matz Professorial Chair of Organic Chemistry. R.Z. thanks the Faculty of Chemistry for being awarded a Dean’s Fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02124.
Experimental procedures; spectral data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Prather M. Science 1998, 279, 1339. 10.1126/science.279.5355.1339. [DOI] [PubMed] [Google Scholar]
- Ravishankara A. R.; Daniel J. S.; Portmann R. W. Science 2009, 326, 123. 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]; Highlight:Dameris M. Angew. Chem., Int. Ed. 2010, 49, 489. 10.1002/anie.200906334. [DOI] [PubMed] [Google Scholar]
- a U.S. Greenhouse Gas Inventory Report 1990–2014, U.S. EPA;.; b Hansen J.; Sato M. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16109. 10.1073/pnas.0406982101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews on N2O chemistry, see:; a Tolman W. B. Angew. Chem., Int. Ed. 2010, 49, 1018. 10.1002/anie.200905364. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Konsolakis M. ACS Catal. 2015, 5, 6397. 10.1021/acscatal.5b01605. [DOI] [Google Scholar]; c Severin K. Chem. Soc. Rev. 2015, 44, 6375. 10.1039/C5CS00339C. [DOI] [PubMed] [Google Scholar]; d Parmon V. N.; Panov G. I.; Uriarte A.; Noskov A. S. Catal. Today 2005, 100, 115. 10.1016/j.cattod.2004.12.012. [DOI] [Google Scholar]; e Pauleta S. R.; Dell’Acqua S.; Moura I. Coord. Chem. Rev. 2013, 257, 332. 10.1016/j.ccr.2012.05.026. [DOI] [Google Scholar]; f Leont’ev A. V.; Fomicheva O. A.; Proskurnina M. V.; Zefirov N. S. Russ. Chem. Rev. 2001, 70, 91. 10.1070/RC2001v070n02ABEH000631. [DOI] [Google Scholar]; g Lee D.-H.; Mondal B.; Karlin K. D.. Nitrogen Monoxide and Nitrous Oxide Binding and Reduction in Activation of Small Molecules; Tolman W. B., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, 2006; pp 43–79. [Google Scholar]
- For selected reports on heterogeneous hydrogenation of nitrous oxide, see Pt:; a Cassel H.; Glückauf E. Z. Physik. Chem. 1932, 19B, 47. 10.1515/zpch-1932-0106. [DOI] [Google Scholar]; b Dixon J. K.; Vance J. E. J. Am. Chem. Soc. 1935, 57, 818. 10.1021/ja01308a009. [DOI] [Google Scholar]; Ag:; c Benton A. F.; Thacker C. M. J. Am. Chem. Soc. 1934, 56, 1300. 10.1021/ja01321a015. [DOI] [Google Scholar]; Alumina:; d Vance J. E.; Dixon J. K. J. Am. Chem. Soc. 1941, 63, 176. 10.1021/ja01846a041. [DOI] [Google Scholar]; Ru, Rh, Ir, or Pt:; e Miyamoto A.; Baba S.; Mori M.; Murakami Y. J. Phys. Chem. 1981, 85, 3117. 10.1021/j150621a022. [DOI] [Google Scholar]; Cu:; f Dandekar A.; Vannice M. A. Appl. Catal., B 1999, 22, 179. 10.1016/S0926-3373(99)00049-1. [DOI] [Google Scholar]; Au:; g Gluhoi A. C.; Dekkers M. A. P.; Nieuwenhuys B. E. J. Catal. 2003, 219, 197. 10.1016/S0021-9517(03)00185-4. [DOI] [Google Scholar]; Fe:; h Delahay G.; Mauvezin M.; Guzmán-Vargas A.; Coq B. Catal. Commun. 2002, 3, 385. 10.1016/S1566-7367(02)00157-7. [DOI] [Google Scholar]; i Nobukawa T.; Yoshida M.; Okumura K.; Tomishige K.; Kunimori K. J. Catal. 2005, 229, 374. 10.1016/j.jcat.2004.11.009. [DOI] [Google Scholar]; Ir(1,1,0):; j Carabineiro S. A.; Nieuwenhuys B. E. Surf. Sci. 2001, 495, 1. 10.1016/S0039-6028(01)01572-2. [DOI] [Google Scholar]; For the relative photo or radiative chemistry, see:; k Zabor J. W.; Noyes W. A. Jr. J. Am. Chem. Soc. 1940, 62, 1975. 10.1021/ja01865a021. [DOI] [Google Scholar]; l Cheek C. H.; Swinnerton J. W. J. Phys. Chem. 1964, 68, 1429. 10.1021/j100788a026. [DOI] [Google Scholar]
- a Kaplan A. W.; Bergman R. G. Organometallics 1997, 16, 1106. 10.1021/om960991i. [DOI] [Google Scholar]; b Kaplan A. W.; Bergman R. G. Organometallics 1998, 17, 5072. 10.1021/om980295d. [DOI] [Google Scholar]; c Lee J.-H.; Pink M.; Tomaszewski J.; Fan H.; Caulton K. G. J. Am. Chem. Soc. 2007, 129, 8706. 10.1021/ja071452f. [DOI] [PubMed] [Google Scholar]; d Doyle L. E.; Piers W. E.; Borau-Garcia J. J. Am. Chem. Soc. 2015, 137, 2187. 10.1021/ja512602m. [DOI] [PubMed] [Google Scholar]; e Gianetti T. L.; Annen S. P.; Santiso-Quinones G.; Reiher M.; Driess M.; Grützmacher H. Angew. Chem., Int. Ed. 2016, 55, 1854. 10.1002/anie.201509288. [DOI] [PubMed] [Google Scholar]; f Co complex catalyzed O-transfer from N2O to phosphines:Gianetti T. L.; Rodríguez-Lugo R. E.; Harmer J. F.; Trincado M.; Vogt M.; Santiso-Quinones G.; Grützmacher H. Angew. Chem., Int. Ed. 2016, 55, 15323. 10.1002/anie.201609173. [DOI] [PubMed] [Google Scholar]
- The mechanism of O insertion into Ru–H was studied by DFT, see:Yu H.; Jia G.; Lin Z. Organometallics 2008, 27, 3825. 10.1021/om8000845. [DOI] [Google Scholar]
- a Gunanathan C.; Milstein D. Acc. Chem. Res. 2011, 44, 588. 10.1021/ar2000265. [DOI] [PubMed] [Google Scholar]; b Gunanathan C.; Milstein D. Science 2013, 341, 1229712. 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]; c Gunanathan C.; Milstein D. Chem. Rev. 2014, 114, 12024. 10.1021/cr5002782. [DOI] [PubMed] [Google Scholar]; d Khusnutdinova J. R.; Milstein D. Angew. Chem., Int. Ed. 2015, 54, 12236. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]; e Zell T.; Milstein D. Acc. Chem. Res. 2015, 48, 1979. 10.1021/acs.accounts.5b00027. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Leitus G.; Ben-David Y.; Milstein D. J. Am. Chem. Soc. 2005, 127, 10840. 10.1021/ja052862b. [DOI] [PubMed] [Google Scholar]
- Kohl S. W.; Weiner L.; Schwartsburd L.; Konstantinovski L.; Shimon L. J. W.; Ben-David Y.; Iron M. A.; Milstein D. Science 2009, 324, 74. 10.1126/science.1168600. [DOI] [PubMed] [Google Scholar]
- For details regarding calibration of the amount of N2O considering its solubility in THF, see SI.
- a Balaraman E.; Gnanaprakasam B.; Shimon L. J.; Milstein D. J. Am. Chem. Soc. 2010, 132, 16756. 10.1021/ja1080019. [DOI] [PubMed] [Google Scholar]; b Khaskin E.; Iron M. A.; Shimon L. J. W.; Zhang J.; Milstein D. J. Am. Chem. Soc. 2010, 132, 8542. 10.1021/ja103130u. [DOI] [PubMed] [Google Scholar]; c Jia G.; Lee H. M.; Williams I. D.; Lau C. P.; Chen Y. Organometallics 1997, 16, 3941. 10.1021/om970207+. [DOI] [Google Scholar]; d Zhang J.; Gandelman M.; Shimon L. J. W.; Rozenberg H.; Milstein D. Organometallics 2004, 23, 4026. 10.1021/om049716j. [DOI] [Google Scholar]; e Gunanathan C.; Milstein D. Angew. Chem., Int. Ed. 2008, 47, 8661. 10.1002/anie.200803229. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Leitus G.; Ben-David Y.; Milstein D. Angew. Chem., Int. Ed. 2006, 45, 1113. 10.1002/anie.200503771. [DOI] [PubMed] [Google Scholar]
- For “O”-atom transfer into M–H bond of N2O to form M–OH, see: M = Ru: refs (6a), (6b), (7), and (10); for M = Rh, see ref (6e); for M = Hf, see:; a Vaughan G. A.; Rupert P. B.; Hillhouse G. L. J. Am. Chem. Soc. 1987, 109, 5538. 10.1021/ja00252a047. [DOI] [Google Scholar]
- Fayolle F.; Couderc J.-P.; Duverneuil P. Chem. Vap. Deposition 1996, 2, 255.and the references therein. 10.1002/cvde.19960020610. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.