

Abstract
Background
The BRAF protein kinase is widely studied as a cancer driver and therapeutic target. However, the regulation of its expression is not completely understood.
Results
Taking advantage of the RNA-seq data of more than 4800 patients belonging to 9 different cancer types, we show that BRAF mRNA exists as a pool of 3 isoforms (reference BRAF, BRAF-X1, and BRAF-X2) that differ in the last part of their coding sequences, as well as in the length (BRAF-ref: 76 nt; BRAF-X1 and BRAF-X2: up to 7 kb) and in the sequence of their 3’UTRs. The expression levels of BRAF-ref and BRAF-X1/X2 are inversely correlated, while the most prevalent among the three isoforms varies from cancer type to cancer type. In melanoma cells, the X1 isoform is expressed at the highest level in both therapy-naïve cells and cells with acquired resistance to vemurafenib driven by BRAF gene amplification or expression of the Δ[3–10] splicing variant. In addition to the BRAF-ref protein, the BRAF-X1 protein (the full length as well as the Δ[3–10] variant) is also translated. The expression levels of the BRAF-ref and BRAF-X1 proteins are similar, and together they account for BRAF functional activities. In contrast, the endogenous BRAF-X2 protein is hard to detect because the C-terminal domain is selectively recognized by the ubiquitin-proteasome pathway and targeted for degradation.
Conclusions
By shedding light on the repertoire of BRAF mRNA and protein variants, and on the complex regulation of their expression, our work paves the way to a deeper understanding of a crucially important player in human cancer and to a more informed development of new therapeutic strategies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-017-0645-4) contains supplementary material, which is available to authorized users.
Keywords: BRAF, RNA-sequencing, Exon-spanning reads, Melanoma, Transcript variants, Protein variants
Background
BRAF is a Ser-Thr protein kinase that belongs to the highly oncogenic RAS/RAF/MEK/ERK signaling pathway. A direct effector of RAS, which induces its activation by dimerization, BRAF phosphorylates and activates MEK, which in turn phosphorylates and activates ERK [1]. Downstream of ERK, the known effectors of BRAF are mainly cytoplasmic proteins and transcription factors that promote cell survival, proliferation, and motility while inhibiting differentiation. Recently, non-coding effectors such as microRNAs and long non-coding RNAs have also been discovered [2–5].
BRAF plays a crucial role in human cancer. About 7% of all cancer cases carry a BRAF mutation, including 100% of hairy cell leukemia (HCL) cases, 50–60% of melanomas, 30–50% of papillary thyroid carcinomas, 10–20% of colorectal cancers, and 3–5% of non-small cell lung cancers. The most common mutation (accounting for up to 98% of all BRAF mutations) is a nucleotide substitution that transforms the Val at position 600 into Glu (V600E, 98% of cases), Lys (V600K, 5–10% of cases), or other amino acids (V600A/D/G/L/M/Q/R, up to 5% of cases). This mutation renders BRAF independent of RAS activation and constitutively active as a monomer [1, 6].
Furthermore, the causal link between mutant BRAFV600E and cancer has been shown in animal models of melanoma [7], colorectal cancer [8], lung cancer [9], and thyroid cancer [10].
Finally, thanks to the development of first- and second-generation selective inhibitors (BRAFi), mutant BRAFV600E has become a valuable therapeutic target in melanoma [11–13], and it holds promise for lung adenocarcinoma [14] and HCL [15].
The regulation of BRAF gene expression remains a rather unexplored field of investigation. This knowledge can contribute to a deeper understanding of the functioning and deregulation of such an important gene, in addition to more effective forms of targeted therapy.
Prompted by our recent study, in which we showed that BRAF mRNA exists in at least two transcript variants that differ in the very last part of their coding sequence (CDS) and in their 3’UTRs [16, 17], we undertook a comprehensive analysis of all the BRAF transcript variants that are expressed in 9 human cancer types. In our results, we confirm that BRAF mRNA is indeed a pool of 3 transcript variants, including the two on which we previously reported. We establish the existence of distinct BRAF protein variants that differ in their C-terminal domains. Finally, we provide insights into the mechanisms at the basis of the different expression levels displayed by the BRAF transcript and protein variants, and into their functional activities.
Results and discussion
Reference BRAF has a short 3’UTR
The sequences of human BRAF were retrieved from Ensembl Genome Browser (http://www.ensembl.org/index.html) and NCBI (http://www.ncbi.nlm.nih.gov/nucleotide/). As shown in Additional file 1: Figure S1, in the fall of 2015 Ensembl reported 5 BRAF transcript variants, the reference (BRAF-001) and 4 more (BRAF-002 to 005). Analogously, NCBI (GRCh38.p1) reported 10 BRAF transcript variants, the reference (NM_004333.4) and 9 more (BRAF-X1 to X9). BRAF-001 and NM_004333.4, the two reference sequences, are indistinguishable except for the length of their 18th and last exon. In NM_004333.4, the 18th exon (from now on E18.1) is 759 bp long. In BRAF-001, the 18th exon (from now on E18.4) has the same starting point as E18.1, but is shorter (292 bp). This implies that NM_004333.4 and BRAF-001 transcripts differ in the length of their 3’UTRs (590 and 121 nt, respectively). Compared to the two reference sequences, the remaining variants display different alterations such as truncations (BRAF-003, 004, 005), the presence of additional exons (E19 and/or new exons 1–6 (NE1-6) in BRAF-002, 003, 004, 005, X1, X2, X3, X4, X5, X6, X8, X9), and the absence of reference exons (BRAF-X2, X5, X7, X8, X9).
To establish the actual length of the 3’UTR of reference BRAF (BRAF-ref), we considered all five variants of exon 18 retrieved from Ensembl and NCBI. We named them E18.1-5 and noticed that, though they all share the same starting point, they have different end points and thus different lengths: E18.1, reported in NM_004333.4, is 759 bp long; E18.2, reported in BRAF-003 and BRAF-X1, is 154 bp long; E18.3, reported in BRAF-005, BRAF-X3, BRAF-X4, BRAF-X6, BRAF-X7, BRAF-X8, is 250 bp long; E18.4, reported in BRAF-001, is 292 bp long; E18.5, reported in BRAF-002, is 174 bp long (Additional file 2: Table S1 for exon coordinates and length). The reads mapped to each E18 variant were counted on RNA-sequencing (RNA-seq) raw reads, which were downloaded from The Cancer Genome Atlas (TCGA). These reads belong to 4807 samples and 9 cancer types, including those in which BRAF mutations are frequently observed (melanoma, colon cancer, lung adenocarcinoma, and thyroid carcinoma) and others in which BRAF mutations are rare (breast cancer, head and neck cancer, lung squamous cell carcinoma (SCC), acute myeloid leukemia (AML), and diffuse large B-cell lymphoma (DLBCL)) (Additional file 2: Table S2). As shown in Fig. 1a for melanoma and in Additional file 1: Figure S2 for the other 8 cancer types, we observed a net drop of mapped reads at the end of E18.3. Therefore, we concluded that the last exon of BRAF-ref is in fact E18.3. This means that its 3’UTR is as short as 76 nt and rarely reaches the ~120 nt reported in BRAF-001, or the ~600 nt reported in NM_004333.4.
Fig. 1.
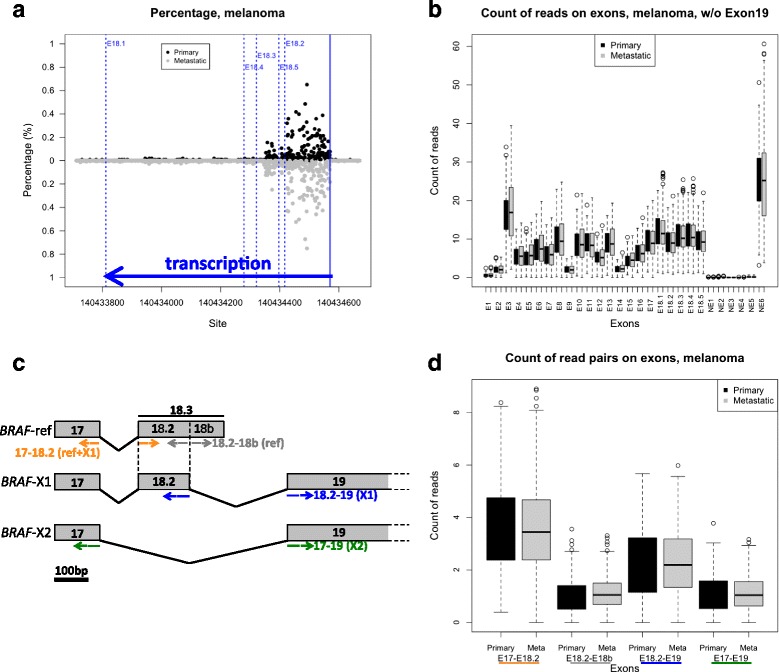
Expression of BRAF transcript variants in melanoma. a Analysis of the length of BRAF 3’UTR by counting the reads mapped to E18.1,2,3,4, and 5. b Count of the reads mapped to all BRAF exons, except E19. c Cartoon depicting the strategy used to measure the relative expression levels of BRAF-ref, BRAF-X1, and BRAF-X2. Paired reads spanning: exon 17 and exon 18.2 were counted as a measure of the cumulative levels of BRAF-ref and BRAF-X1 (yellow); exon 18.2 and exon 18b were counted as a measure of BRAF-ref levels (grey); exon 18.2 and exon 19 were counted as a measure of BRAF-X1 levels (blue); and exon 17 and exon 19 were counted as measure of BRAF-X2 (green). d Box plot of the reads that span E17-E18.2, E18.2-E18b, E18.2-E19, and E17-E19 in primary (black boxes) and metastatic (grey boxes) melanoma samples
The relative abundance of BRAF-ref, BRAF-X1, and BRAF-X2 varies from cancer type to cancer type
To identify transcribed variants other than BRAF-ref, we mapped raw reads on BRAF transcripts and counted mapped reads on each exon in all nine cancer types. As expected, reads that mapped to the reference exons (E1-18) were retrieved in all the cancer types we analyzed, though E1 (which contains the ATG) and E2 are mapped much less compared to the other exons (Figs. 1b and 2, left panels and Additional file 1: Figures S3 and S4). This is possibly due to a sequencing artifact. RNA-seq data obtained from polyA libraries are known to be biased against the 5’end exons [18].
Fig. 2.
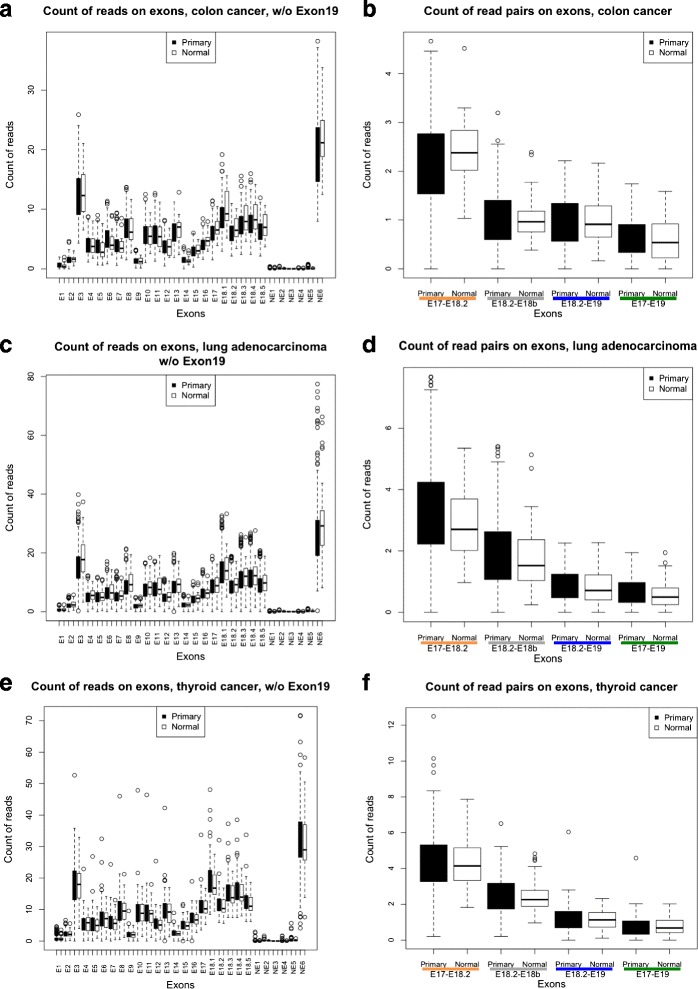
Expression of BRAF exons and transcript variants in colon cancer, lung adenocarcinoma and thyroid cancer. a, c, e Count of the reads mapped to all BRAF exons, except E19. (b, d, f) Box plot of the reads that span E17-E18.2 (BRAF-ref + BRAF-X1, yellow), E18.2-E18b (BRAF-ref, grey), E18.2-E19 (BRAF-X1, blue), and E17-E19 (BRAF-X2, green) in primary (black boxes) and normal (white boxes) samples
For the “non-canonical” exons, we used the absence of mapped reads on NE1, NE2, NE3, and NE4 (Figs. 1b and 2, left panels and Additional file 1: Figures S3 and S4) to rule out the transcription of BRAF-005, and of the BRAF-X3, X4, X5, X6, X8, and X9 variants. Analogously, we used the absence of reads mapping to exon NE5 to rule out the transcription of BRAF-002.
To rule out the transcription of BRAF-X7 (which lacks exon 14 and 15), we used exon-spanning reads. In such reads, one of a read pair is mapped on an exon and the other is mapped on a different exon. Because of the nature of paired-end reads, exon-spanning reads can be used to assess whether two exons are expressed together. The absence of reads that span exon 13 and exon 16 (Additional file 1: Figure S5) indicates that BRAF-X7 is not transcribed and confirms the absence of BRAF-X8 and BRAF-X9 transcription.
For BRAF-003, the similar expression levels between exons 1–9 and exons 10–19 suggests it is transcribed at negligible levels.
BRAF-004 is reported as a truncated transcript variant in which the last exon is NE6, a 548 bp longer version of E3 (Additional file 1: Figure S6a, the NE6-specific region is named NE6p).
Since E3 and NE6 exons collect reads that belong to both full length transcripts and the BRAF-004 truncated transcript, we reasoned that the higher number of reads mapping to these two exons compared to the other exons (Figs. 1b and 2 left panels and Additional file 1: Figures S3 and S4) is consistent with BRAF-004 being transcribed. Sanger sequencing of the PCR band obtained from A375 cDNA using a forward primer located on E1/2 and a reverse primer located on NE6p confirms the transcription of NE6 (Additional file 1: Figure S6b). We also assessed the relative contribution of the BRAF-004 transcript variant to the pool of BRAF transcripts. As reported in Additional file 1: Figure S6c, we performed real-time PCR on melanoma cell lines using primers that span: the E2-E3 junction, as measure of the cumulative expression levels of full length BRAF transcripts and BRAF-004 (grey); the E3-E4 junction, as measure of the expression levels of full length BRAF transcripts (red); and the E3-NE6p junction, as measure of the expression levels of BRAF-004 (orange). We discovered that BRAF-004 accounts for up to 30% of the pool of BRAF transcripts.
Next, we assessed whether BRAF-004 is indeed truncated. The observation that NE6 and E4 cannot be amplified in the same PCR band (Additional file 1: Figure S6d) indicates BRAF-004 as truncated. This is further supported by the analysis of exon-spanning reads: as shown in Additional file 1: Figure S6e-m, NE6p-E4 spanning reads are undetectable. Due to its truncation, and thus inability to encode for a full-length protein, the BRAF-004 transcript variant is not analyzed further, even if it is transcribed.
The expression of the very long exon 19 (Additional file 1: Figures S3 and S4 right panels) is consistent with the transcription of the BRAF-X1 (XM_005250045.1) and BRAF-X2 (XM_005250046.1) variants. Compared to the reference sequence, in BRAF-X1 the shorter exon 18 (E18.2, 154 bp) does not contain the very last part of the CDS, the STOP codon and the 3’UTR. Such features are all located in the more downstream exon 19 and are therefore different [16, 17]. Compared to BRAF-X1, BRAF-X2 lacks exon 18.2 (exon 17 is spliced directly with exon 19) (Fig. 1c).
In June 2016, a new annotation (GRCh38.p8) of BRAF transcript variants was published in NCBI. The new BRAF-X3 (XM_005250045.2) is in fact indistinguishable from the old BRAF-X1. The new BRAF-X1 (XM_017012558.1) and BRAF-X2 (XM_017012559.1) are 120 nt longer than the corresponding old versions, due to the presence of an additional exon located between the 9th and the 10th (exon coordinates: 140785808–140785689). We remapped raw reads for all nine cancer types and discovered very few mapped reads on this novel exon. Therefore, we kept the annotation we were already using (GRCh38.p1).
In conclusion, this first round of analysis allowed us to establish that, in addition to the reference, two more transcript variants of BRAF exist, namely BRAF-X1 and BRAF-X2. To evaluate the relative contribution of the identified BRAF variants to the pool of BRAF transcripts, we again used exon-spanning reads, according to the strategy summarized in Fig. 1c. We counted the number of paired reads spanning on: exon 17 and exon 18.2, as a measure of the expression of BRAF-ref and BRAF-X1 (yellow); exon 18.2 and the BRAF-ref-specific part of exon 18.3 (from now on E18b), as a measure of the expression of BRAF-ref (grey); exon 18.2 and exon 19, as a measure of the expression of BRAF-X1 (blue); and exon 17 and exon 19, as a measure of the expression of BRAF-X2 (green).
For primary and metastatic melanoma samples, the results of this analysis are reported in Fig. 1d. We discovered that: 1, BRAF-X1 is expressed at higher levels than BRAF-ref (blue line vs grey line) and BRAF-X2 (blue line vs green line); and 2, BRAF-ref and BRAF-X2 are expressed at similar levels (grey line vs green line).
The RNA-seq data presented above are consistent with those we previously reported on melanoma cell lines. By performing real-time PCR detection of the reference 3’UTR and the non-canonical 3’UTR transcribed from exon 19, we obtained the ratio between them as ~1:3 [16]. To assess the relative abundance of BRAF-X1 and BRAF-X2 in the pool of exon 19-containing transcripts, we again performed real-time PCR on some of the same cell lines previously tested. However, this time we used not only the primers that detect BRAF-X1 and BRAF-X2 combined (BRAF-E19-1), but also those that detect the BRAF-X1 and BRAF-X2 isoforms distinctly (Additional file 1: Figure S7). As shown in Additional file 1: Figure S8a, we confirmed that irrespective of their mutational status, all the melanoma cell lines show that the expression of the exon 19-derived 3’UTR is higher than the expression of BRAF-ref (grey vs black). In addition, we found that the expression of the exon 19-derived 3’UTR is mostly accounted for by BRAF-X1 (black vs blue), while BRAF-X2 levels are similar to those of BRAF-ref (green vs grey).
From other tumors in which BRAF mutations are frequently observed, we obtained different results compared to melanoma: in colon cancer, the BRAF-ref and BRAF-X1 isoforms are expressed at similar levels (Fig. 2b); while in lung adenocarcinoma (Fig. 2d) and in thyroid cancer (Fig. 2f), BRAF-ref is in fact expressed at higher levels compared to the BRAF-X1 and BRAF-X2 isoforms. Among the other cancer types analyzed, we found that BRAF-ref is the most abundant isoform in breast cancer, head and neck cancer, lung SCC, and DLBCL, while BRAF-X1 is the most abundant isoform in AML (Additional file 1: Figure S9). Using the real-time primer pairs described above, we measured the relative expression levels of the BRAF-ref, BRAF-X1, and BRAF-X2 isoforms on cell lines derived from breast, cervix, colon, lung, and prostate cancer (Additional file 1: Figure S8b), as well as on leukemia/lymphoma cell lines and patient samples (Additional file 1: Figure S8c-d). Overall, we found that BRAF-X1 is the most expressed isoform. However, we did find cases, such as the T47D breast cancer cells and the CaCo2 colon cancer cells, in which BRAF-ref prevails compared to the X1 and X2 isoforms, in agreement with the RNA-seq data.
Finally, we assessed whether the difference in expression levels between BRAF-ref and BRAF-X1 is at least in part attributable to a differential stability of their RNA molecules. By treating A375 melanoma cells with actinomycin D and measuring the decay rate of the short reference 3’UTR compared to the long E19-derived 3’UTR, we discovered that the former undergoes a faster decay compared to the latter (Additional file 1: Figure S10), a finding that is consistent with the lower BRAF-ref vs BRAF-X1 levels observed in melanoma cells.
The expression levels of BRAF-X1/X2 are inversely correlated with those of BRAF-ref
We next assessed whether there are correlations among the expression levels of the different isoforms. First, by counting reads mapped on all BRAF transcripts, we found that total BRAF levels are similar across cancer patients belonging to the same group (Fig. 3, left panels and Additional file 1: Figure S11, left panels), though a few outliers were observed. We then sorted patients by the number of reads that span on E18.2-E18b (increasing expression level of the BRAF-ref variant). Finally, we calculated the E18.2-E19/E18.2-E18b ratio (the BRAF-X1/BRAF-ref ratio, in red), as well as the E17-E19/E18.2-E18b ratio (the BRAF-X2/BRAF-ref ratio, in blue) (Fig. 3, middle panels and Additional file 1: Figure S11, middle panels). The overall distribution of the data points suggest that the expression levels of BRAF-X1 and BRAF-X2 are highest where the expression level of BRAF-ref is lowest, and vice versa, which equals to say that the expression levels of BRAF-X1 and BRAF-X2 are inversely correlated with that of BRAF-ref. Furthermore, the distribution of the blue and red data points across the zero line (which in the log scale marks the BRAF-X1/BRAF-ref ratio = 1 and the BRAF-X2/BRAF-ref ratio = 1) indicates there are tumor types, such as melanoma, in which a substantial number of patients express more BRAF-X1 than BRAF-ref, and there are others, such as lung adenocarcinoma and thyroid cancer, in which a substantial number of patients express more BRAF-ref than the X1 isoform. These data confirm at the individual level those reported in Figs. 1d and 2b, d, f and Additional file 1: Figure S9.
Fig. 3.
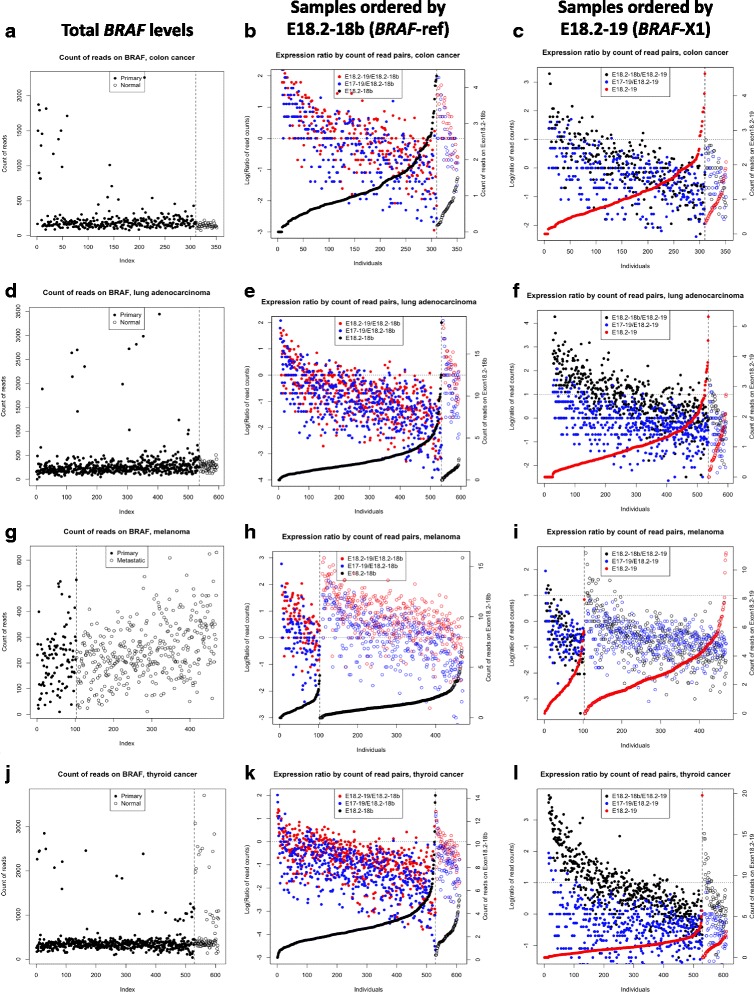
Correlation among the expression levels of the different BRAF isoforms in colon cancer, lung adenocarcinoma, melanoma and thyroid cancer. a, d, g, j Total number of BRAF reads across patients. b, e, h, k Expression ratios over the ref spanning reads. Samples were sorted by reads spanning E18.2-E18.b (BRAF-ref, in black). Red dots are E18.2-E19/E18.2-E18b ratios (which means the X1/ref ratio) and blue dots are E17-E19/E18.2-E18b ratios (which means the X2/ref ratio). The data points are log transformed and the dotted line marks the 0, which means X1/ref ratio = 1 and X2/ref ratio = 1. c, f, i, l Expression ratios over the X1 spanning reads. Samples were sorted by reads spanning E18.2-E19 (BRAF-X1, in red). Black dots are E18.2-E18b/E18.2-E19 ratios (which means the ref/X1 ratio) and blue dots are E17-E19/E18.2-E19 ratios (which means the X2/X1 ratio). The data points are log transformed and the dotted line marks the 0, which means ref/X1 ratio = 1 and X2/X1 ratio = 1. In the left and middle panels the samples are presented in the same order
We also sorted patients by the number of reads that span on E18.2-E19 (increasing expression of the BRAF-X1 variant, in red) and calculated the E18.2-E18b/E18.2-E19 ratio (the BRAF-ref/BRAF-X1 ratio, in black), as well as the E17-E19/E18.2-E19 ratio (the BRAF-X2/BRAF-X1 ratio, in blue) (Fig. 3, right panels and Additional file 1: Figure S11, right panels). The distribution of the black data points confirms that BRAF-X1 is prevalent where BRAF-ref is least expressed, and vice versa. Conversely, the distribution of the blue data points suggests that the expression of the X2 isoform, although always lower, follows the trend of that of the X1 isoform.
Next, we looked at melanoma samples to check whether the levels of BRAF-ref, BRAF-X1, and BRAF-X2 and/or their ratios are associated with clinical variables. As shown in Additional file 1: Figures S12-13, this does not seem to be the case, at least when the age, gender, and stage at diagnosis are considered.
The 3’UTR of BRAF-X1 and BRAF-X2 is up to 7 kb long
We then investigated whether in melanoma cells the 3’UTR of BRAF-X1 and BRAF-X2, which is transcribed from exon 19, is indeed as long as 7 kb (Additional file 1: Figure S1). RNA-seq data suggested us that this is the case, since reads distribution is homogeneous across the entire length of E19 (Fig. 4a). Nevertheless, we verified this piece of evidence by performing PCR on A375 (homozygous for BRAFV600E), 501Mel (heterozygous for BRAFV600E), and MeWo (wtBRAF) melanoma cell lines, using multiple primer pairs that are located across the entire 3’UTR length (Fig. 4b). As shown in Fig. 4c, we detected a PCR product with each of the four primer pairs used. Furthermore, using the chained PCR approach reported in Fig. 4d we established that the amplified fragments are linked together, hence they belong to the same RNA molecule.
Fig. 4.
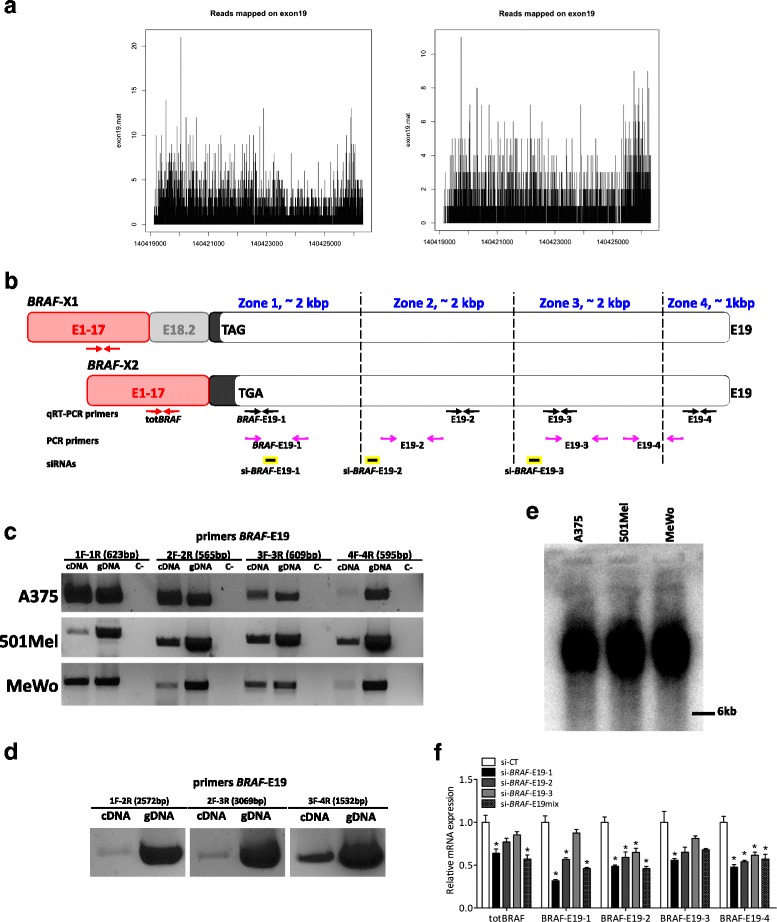
Length of BRAF-X1 and BRAF-X2 3’UTR in melanoma. a The analysis of the reads mapping to exon 19 across its entire length indicates that the 3’UTR of BRAF-X1 and BRAF-X2 is as long as 7 kb. A representative example of a primary (left) and a metastatic (right) melanoma case is reported. b Cartoon summarizing the position of the primers and the siRNAs used to determine the length of the 3’UTR of BRAF-X1 and BRAF-X2 in melanoma cell lines. The 4 primer pairs used for real-time PCR amplification of BRAF-X1 plus X2 (BRAF-E19-1/2/3/4 qRT-PCR F/R) are represented as black arrows. The 4 primer pairs used for PCR amplification of BRAF-X1 plus X2 (BRAF-E19-1/2/3/4 F/R) are represented as open pink arrows. BRAF-E19-1 qRT-PCR F and BRAF-E19-1 F have the same sequence. The siRNAs used to knock-down BRAF-X1 plus X2 (si-BRAF-E19-1/2/3) are represented as yellow and black rectangles. The primers used for real-time PCR amplification of all BRAF isoforms (totBRAF qRT-PCR F/R) are represented as red arrows. c PCR performed on A375, 501Mel and MeWo melanoma cells using BRAF-E19-1/2/3/4 primer pairs. Genomic DNA (gDNA) is used as positive control. d Chained PCR performed on A375 melanoma cells using “hybrid” primer pairs: BRAF-E19-1 F with BRAF-E19-2 R, BRAF-E19-2 F with BRAF-E19-3 R, and BRAF-E19-3 F with BRAF-E19-4 R. Genomic DNA (gDNA) is used as positive control. e Northern blot of total RNA extracted from A375, 501Mel, and MeWo cells and hybridized with a labeled probe for BRAF CDS. Considering that the size of BRAF CDS is about 2.3 kb, the band detected in all cell lines is consistent with a 7 kb long 3’UTR. f Expression levels of BRAF CDS (detected using the totBRAF qRT-PCR primers) and of different regions of the 3’UTR transcribed from E19 (detected using the BRAF-E19-1/2/3/4 qRT-PCR primer pairs) after the transfection of the indicated siRNAs. The pictures are taken from 1 out of 3 independent experiments performed, all with comparable outcome. The graphs represent the mean ± SEM of 3 independent experiments. *p < 0.05
To prove that complete BRAF transcripts containing the CDS and the 7 kb long E19-derived 3’UTR do exist, we carried out a PCR reaction using 3 forward primers that are located within BRAF CDS and a reverse primer located in the E19 3’UTR. As shown in Additional file 1: Figure S14, we obtained a doublet of PCR bands, which are consistent with the amplification of the BRAF-X1 and BRAF-X2 isoforms. The transcription of BRAF CDS plus the ~7 kb long E19-derived 3’UTR was further confirmed by Northern Blot. Using a 2.3 kb long probe that recognizes BRAF CDS, we were able to detect a net band of ~10 kb (Fig. 4e). Finally, by transfecting A375 cells with siRNAs that recognize three different regions along the E19 3’UTR, we invariably found a downregulation of BRAF CDS (Fig. 4b and f). The comparison among the three BRAF-E19 siRNAs allowed us to establish that the most efficient is si-BRAF-E19-1, since it achieves a stronger BRAF downregulation than the other 2 siRNAs, and stronger even than the mix of the 3. Therefore we used si-BRAF-E19-1 only for the following studies. In Additional file 1: Figure S15, we report that the transfection of si-BRAF-E19-1 in 501Mel and MeWo cells produces similar results to those obtained in A375 cells.
BRAF transcript variants together account for BRAF functions
To establish the relative contribution of the BRAF-ref, BRAF-X1, and BRAF-X2 isoforms to BRAF functions, we compared the effects obtained in A375 melanoma cells upon the knock-down of: the reference variant using si-refBRAF (Fig. 5a, grey), the BRAF-X1 plus BRAF-X2 variants using si-BRAF-E19-1 (black), and all BRAF variants using the combo si-refBRAF plus si-BRAF-E19-1 or si-totBRAF (red). As reported in Fig. 5b, we confirmed that si-refBRAF and si-BRAF-E19-1 are able to cause the specific knock-down of the isoform(s) they are designed to target. However, we observed that the effects of si-refBRAF on total BRAF levels are negligible. The effects caused by si-BRAF-E19-1 are much stronger and comparable to those caused by si-totBRAF (black bar vs red bar), while the combination with si-refBRAF does not produce a further downregulation (black bar vs checked bar). These results are consistent with our findings about the higher expression levels of BRAF-X1 vs BRAF-ref mRNA, and were also confirmed in other melanoma cell lines such as 501Mel and MeWo cells (Additional file 1: Figure S16).
Fig. 5.
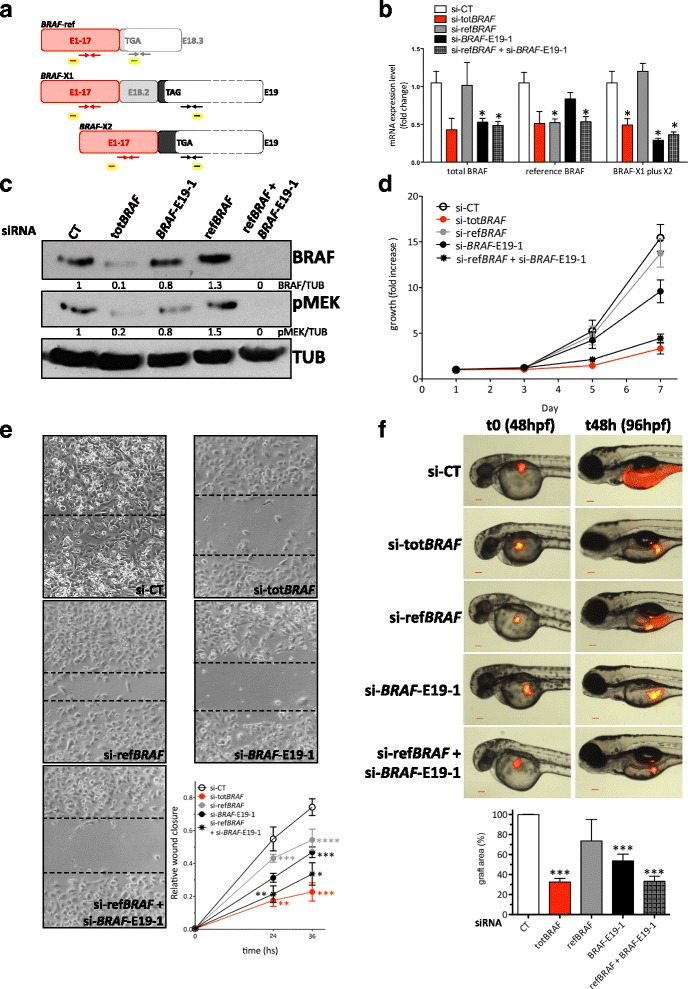
BRAF isoforms together account for BRAF functions in melanoma cells. a Cartoon summarizing the position of the primers and the siRNAs used to determine the contribution of reference, X1 and X2 isoforms to BRAF activities in melanoma cell lines. The primers used for real-time PCR amplification of all BRAF isoforms (totBRAF qRT-PCR F/R), BRAF-ref (refBRAF qRT-PCR F/R), and BRAF-X1 plus X2 (BRAF-E19-1 qRT-PCR F/R) are represented as red, grey and black arrows, respectively. The siRNAs used for the knock-down of the different BRAF isoforms are schematically represented as rectangles: yellow and red, for the knock-down of all BRAF isoforms (si-totBRAF); yellow and grey, for the knock-down of BRAF-ref (si-refBRAF); and yellow and black for the knock-down of BRAF-X1 plus X2 (si-BRAF-E19-1). b Real-time PCR detection of total BRAF, BRAF-ref, and BRAF-X1 plus X2 24 h after the transfection of the indicated siRNAs in A375 cells. c Western blot of BRAF and of its substrate pMEK 48 h after the transfection of the indicated siRNAs in A375 cells. d Growth curve of A375 cells after the transfection of the indicated siRNAs. e Wound healing assay performed using A375 cells transfected with the ndicated siRNAs. The pictures were taken 24 and 36 h after the removal of the silicone inserts. f Xenograft in zebrafish embryos of A375 cells stably expressing mCherry and transfected with the indicated siRNAs. The pictures are taken from 1 out of 3 independent experiments performed, all with comparable outcome. Hpf: hours post fertilization. Scale bar: 100 um. The graphs represent the mean ± SEM of 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
At the protein level we observed a consistent scenario, with BRAF levels going sensibly down only upon si-BRAF-E19-1 and not upon si-refBRAF transfection. However, we observed that si-BRAF-E19-1 is not sufficient to recapitulate the effects obtained using si-totBRAF, and the combination with si-refBRAF is required (Fig. 5c).
Next, the consequences of the siRNA-mediated knock-down of the different variants were investigated at the functional level. To this end, we took A375 cells transfected with si-refBRAF, si-BRAF-E19-1, si-totBRAF, and si-refBRAF + si-BRAF-E19-1 and performed three different cellular assays: two in vitro (growth curve (Fig. 5d), wound healing assay (Fig. 5e)), and one in vivo (xenograft in zebrafish embryos (Fig. 5f)). In agreement with the degree of BRAF protein downregulation observed by western blot, we discovered that the impairment in growth and motility caused by si-refBRAF is very mild, while si-BRAF-E19-1 has much more pronounced effects. However, it is only the combination of si-refBRAF and si-BRAF-E19-1 that can fully recapitulate the effects of si-totBRAF. To explain the discrepancy between the results we obtained at the RNA and at the protein levels, we hypothesize that BRAF protein isoforms positively regulate each other, so that when they are concomitantly knocked-down the effects on total BRAF protein levels become more dramatic.
Based on these results, we conclude that BRAF activity in melanoma cells is fully accounted for by BRAF-ref and by the E19-containing BRAF isoforms, primarily BRAF-X1.
The relative contribution of BRAF-ref, BRAF-X1, and BRAF-X2 transcript variants to total BRAF levels is maintained in the context of acquired resistance to BRAF and MEK inhibitors
We next studied whether the composition of the cocktail of BRAF transcript variants changes in different conditions. Specifically, we analyzed BRAF gene amplification [19] and BRAF splicing variants [20] in the context of acquired resistance to BRAF and MEKi in melanoma cells.
451Lu-MR cells derive from the parental 451Lu melanoma line and show acquired resistance to both MEK and BRAF inhibitors, due to the focal amplification of BRAF gene [21]. As a consequence, total BRAF levels are increased (Fig. 6a, left). However, the composition of BRAF transcript variants remains quite similar (Fig. 6a, right).
Fig. 6.
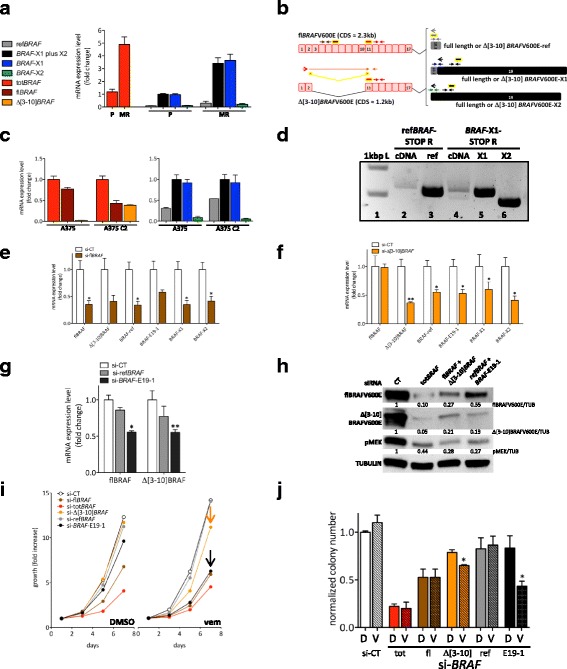
BRAF transcript variants in the context of acquired resistance to BRAF and MEK inhibitors. a Real-time PCR detection of total BRAF (red), BRAF-ref (grey), BRAF-X1 plus X2 (black), BRAF-X1 (blue), and BRAF-X2 (green) in 451Lu parental cells (P) and in 451Lu-MR resistant cells (MR). The latter show acquired resistance to BRAF and MEK inhibitors due to the focal amplification of the BRAF gene. b Cartoon summarizing the position of the primers and the siRNAs used to determine the presence and the level of the Δ[3–10] variant of BRAF. For details, please refer to Additional file 1: Figure S17. c Real-time PCR detection of total BRAF (red), full length BRAF (brown), Δ[3–10]BRAF (orange, left panel), BRAF-ref (grey), BRAF-X1 plus X2 (black), BRAF-X1 (blue), and BRAF-X2 (green, right panel) in A375 parental cells and in A375 C2 cells. The latter show acquired resistance to vemurafenib due to the presence of Δ[3–10]BRAFV600E splicing variant. d PCR amplification of the reference, X1, and X2 Δ[3–10]BRAF splicing variants from the cDNA of A375 C2 cells. Lane 1: 1 kbp ladder. Lane 2: Δ[3–10]BRAF-ref amplification was obtained using BRAF-E1/2 F primer and refBRAF-STOP R primer (open red and grey arrows in b). Lane 3: Δ[3–10]BRAF-ref CDS was amplified from pMSCVHygro-Δ[3–10]BRAFV600E-ref plasmid and used as positive control. Lane 4: the amplification of Δ[3–10]BRAF-X1 (upper band) and Δ[3–10]BRAF-X2 (lower band) was obtained using BRAF-E1/2 F primer and BRAF-X1-STOP R primer (open red and black arrows in b). Lane 5: Δ[3–10]BRAF-X1 CDS was amplified from pMSCVHygro-Δ[3–10]BRAFV600E-X1 plasmid and used as positive control. Lane 6: Δ[3–10]BRAF-X2 CDS was amplified from pMSCVHygro-Δ[3–10]BRAFV600E-X2 plasmid and used as positive control. e-f Real-time PCR detection of full length BRAF, Δ[3–10]BRAF, BRAF-ref, BRAF-X1 plus X2, BRAF-X1, and BRAF-X2 24 h after the transfection of si-flBRAF (e) and si-Δ[3–10]BRAF (f) in A375 C2 cells. g Real-time PCR detection of full length and Δ[3–10] BRAF 24 h after the transfection of si-refBRAF and si-BRAF-E19-1 in A375 C2 cells. h Western blot of full length and Δ[3–10] BRAFV600E, as well as of pMEK 48 h after the transfection of the indicated siRNAs or siRNA mixes in A375 C2 cells. i Growth curve of A375 C2 cells after the transfection of the indicated siRNAs. Throughout the experiment, the cells were kept in DMSO (left panel) or in 2 uM vemurafenib (right panel). The arrows highlight the increased sensitivity displayed by A375 C2 cells to si-Δ[3–10]BRAF (orange) and si-BRAF-E19-1 (black), when grown in vemurafenib. The graph represents the mean only of 3 independent experiments. (j) Colony formation assay of A375 C2 cells after the transfection of the indicated siRNAs. Throughout the experiment, the cells were kept in DMSO (clean bars) or in 2 uM vemurafenib (dashed bars). The pictures are taken from 1 out of 3 independent experiments performed, all with comparable outcome. The graphs represent the mean ± SEM (or mean ± SD in a and c) of 3 independent experiments. *p < 0.05, **p < 0.01
A375 C2 cells derive from the parental A375 melanoma line and show acquired resistance to the BRAF inhibitor vemurafenib, due to the presence of a BRAF splicing variant that lacks exons 3 to 10 (Δ[3–10]BRAFV600E) [5]. Contrary to the parental A375 cells, where total BRAF levels are almost completely accounted for by full length BRAF, in A375 C2 cells the full length and the Δ[3–10] variants are expressed at similar levels (Fig. 6b, Additional file 1: Figure S17 and Fig. 6c, left). Nevertheless, the composition of BRAF transcripts remains the same: BRAF-X1 is more expressed than BRAF-ref and BRAF-X2, which in turn are expressed at similar levels (Fig. 6c, right). The analysis of additional vemurafenib-resistant clones and clonal populations obtained from A375 and 501Mel parental cells and carrying BRAF splicing variants produced comparable results (Additional file 1: Figure S18). These findings suggest that BRAF splicing variants are themselves a mixture of BRAF-ref, BRAF-X1, and BRAF-X2 isoforms. Indeed, when we used a BRAF-ref-specific reverse primer (refBRAF-STOP R) to perform PCR amplification of A375 C2 cDNA, we obtained a band whose size is compatible with the reference Δ[3–10] splicing variant. However, when we used a BRAF-X1/X2-specific reverse primer (BRAF-X1-STOP R), we obtained a doublet of bands whose sizes are compatible with the X1 and X2 Δ[3–10] splicing variants (Fig. 6d). PCR results were confirmed using an siRNA that targets exon 6, causing the specific knock-down of full length BRAF (si-flBRAF, brown in Fig. 6b) and an siRNA that spans the E2-E11 junction and therefore specifically targets the Δ[3–10] variant (si-Δ[3–10]BRAF, orange in Fig. 6b). As shown in Fig. 6e-f, we found that not only si-flBRAF, but also si-Δ[3–10]BRAF can cause a decrease in the levels of all three BRAF isoforms (BRAF-ref, BRAF-X1, and BRAF-X2). Conversely, si-refBRAF, and to a bigger extent si-BRAF-E19-1, can cause a decrease not only in full length BRAF levels but also in the levels of the Δ[3–10] splicing variant (Fig. 6g). In addition, we found that si-Δ[3–10]BRAF causes a decrease in the levels of the E19-derived 3’UTR across its entire length (Additional file 1: Figure S19a) and that, conversely, Δ[3–10]BRAF levels decrease upon the transfection not only of si-BRAF-E19-1 but also of more “downstream” siRNAs (Additional file 1: Figure S19b). We hypothesize that, analogously to full length BRAF, the Δ[3–10] BRAF-X1 and BRAF-X2 splicing variants have a 3’UTR that is up to 7 kb long.
Real-time results were further confirmed at the protein level: the strong decrease in both full length and Δ[3–10] BRAF protein isoforms observed with si-totBRAF was also obtained by mixing si-flBRAF with si-Δ[3–10]BRAF, as well as by mixing si-refBRAF with si-BRAF-E19-1 (Fig. 6h).
We next aimed at assessing whether the BRAF-X1 is the most prevalent Δ[3–10] variant, as it happens for full length BRAF. Since it is known that resistant cells rely on BRAF splicing variants in order to counteract vemurafenib activity, we reasoned that if the X1 splicing variant is expressed at higher levels than the reference one, then si-BRAF-E19-1 should cause a much stronger increase in the sensitivity of A375 C2 cells to vemurafenib compared to si-refBRAF. As shown in Fig. 6i-j, the experimental results confirmed our hypothesis. When grown in vemurafenib, A375 C2 cells are very sensitive to the knock-down of Δ[3–10]BRAF and BRAF-X1, as indicated by the decreased proliferation (i) and ability to form colonies (j) that they display upon the transfection with si-Δ[3–10]BRAF (orange) and si-BRAF-E19-1 (black).
BRAF-X1 is translated into protein
As mentioned above, BRAF-X1 mRNA derives from the splicing of exon 18.2 with exon 19, which contains the very last part of the CDS, the STOP codon and the 3’UTR (Fig. 1b). Not only does the BRAF-X1 mRNA differ from the BRAF-ref mRNA in its 3’UTR sequence, the BRAF-X1 protein (XP_005250102.1, GYGEFAAFK) also differs in its C-terminal sequence from the BRAF-ref protein (NP_004324.2, GYGAFPVH) (Fig. 7a and Additional file 1: Figure S20a, b). We also observed that BRAF-X1 appears to be more conserved across species than BRAF-ref (Additional file 1: Figures S21-23).
Fig. 7.

Identification and characterization of BRAF protein isoforms. a Schematic representation of the 3’ terminal region of reference, X1, and X2 BRAF mRNAs, as well as of the corresponding C-terminal regions of reference, X1, and X2 BRAF proteins. b Immunoprecipitation of BRAF protein in A375 cells. Endogenous BRAF was immunoprecipitated using a specific antibody that recognizes the N-terminal domain (IP-BRAF). As negative control, no antibody was used (No Ab). The basal level of BRAF in the cell lysate is shown in Input. c Identification by mass spectrometry of the C-terminal peptides of BRAF-ref and BRAF-X1. Immunoprecipitated BRAF was subjected to LC-MS analysis. The presence of both isoforms is revealed by the detection of isoform-specific peptides (in green). d Best transitions (BRAF-ref: 352 and 904; BRAF-X1: 1046 and 1117) of the two BRAF protein isoforms by mass spectrometry (MRM based method). e-f Upon the transient transfection of PIG-BRAFV600E-ref, X1, and X2 plasmids in HEK293T cells, western blot indicates that only reference and X1 BRAFV600E are efficiently translated and able to phosphorylate MEK, while X2 is not (e). This occurs in spite of the fact that according to real-time PCR for total BRAF levels, all 3 mRNAs are transcribed at similar levels (f). g-i Upon the stable infection of pMSCVHygro-Δ[3–10]BRAFV600E-ref, X1, and X2 plasmids in A375 cells, real-time PCR for total BRAF indicates that all 3 mRNAs are transcribed at similar levels (g), but western blot indicates that reference and X1 Δ[3–10]BRAFV600E are efficiently translated and able to phosphorylate MEK even in the presence of vemurafenib, while X2 is not (h). Consistently, only Δ[3–10]BRAFV600E-ref and -X1 are able to decrease the sensitivity of A375 cells to vemurafenib (i). The pictures are taken from 1 out of 3 independent experiments performed, all with comparable outcome. The graphs represent the mean ± SEM of 3 independent experiments
The difference between the BRAF-ref and BRAF-X1 proteins on one side and the BRAF-X2 protein (XP_005250103.1) on the other side is even more pronounced. The skipping of exon 18 and the direct splicing of exon 17 with exon 19 causes a distortion of the RNA sequence (Additional file 1: Figures S24-25) and a shift of the frame. Therefore, it results in the translation of a completely different C-terminal domain (Fig. 7a and Additional file 1: Figure S20c).
To identify BRAF protein isoforms that are translated in A375 melanoma cells, we immunoprecipitated endogenous BRAF using an antibody that recognizes the N-terminal domain (Fig. 7b). We then ran the immunoprecipitated BRAF on SDS-PAGE and subjected the corresponding band to trypsin digestion followed by mass spectrometry analysis, using an LTQ-Orbitrap XL and a 5600 TripleTOF mass spectrometers. Several peptides belonging to “human BRAF” were identified (Additional file 1: Figure S26), among them the BRAF-ref and the BRAF-X1 C-terminal peptides (Fig. 7c). Therefore we can conclude that BRAF-X1 is translated as a protein variant of BRAF. On the contrary, the BRAF-X2 protein variant was not detected. When other melanoma cell lines were tested, such as WM793B, similar results were obtained (Additional file 1: Figure S27).
We also estimated the ratio between the BRAF-ref and the BRAF-X1 proteins. To this end, we synthesized two peptides with the sequence of the C-terminal peptides identified by mass-spectrometry (see Fig. 7c, green). We then determined their spectra (Fig. 7d, upper) and confirmed that they are the same as those of the experimental peptides obtained from the immunoprecipitated BRAF (Fig. 7d, lower). The best MRM transitions proposed by SWATH analysis are 708/352 and 708/904 for the reference C-terminal peptide (TPIQAGGYGAFPVH) and 779/1046 and 779/1117 for the X1 C-terminal peptide (TPIQAGGYGEFAAFK). Since the transitions are retrieved using the synthetic peptides in equimolar concentration (10 uM, Fig. 7d, upper), we can infer that a most intense ionization of the X1 isoform occurs (response factor ref/X1 = 0.75). Furthermore, since a similar response factor (ref/X1 = 0.83) is observed in Fig. 7d lower, we can conclude that in the experimental sample the ref and X1 BRAF protein variants are expressed at similar concentration.
We next investigated whether the comparable BRAF-ref and BRAF-X1 levels, which we detected in spite of the fact that BRAF-X1 mRNA levels are markedly higher, are attributable to the fact that the translation of BRAF-ref mRNA is more efficient [22]. In order to answer to this question, we took advantage of the analysis that Obenauf and colleagues [23] performed on A375 cells and that is available at GEO (GSE64741). Specifically, we counted the paired reads that span E18.2-E18b, E18.2-E19 and E17-E19 as a measure of BRAF-ref, −X1, and -X2 mRNA levels, respectively, and we used them to compare total RNA samples (CT) and RNA samples obtained by TRAP (translating ribosome affinity purification). This technique enables for the enrichment of actively translated RNA molecules that are complexed with polysomes (PS). We found that BRAF-ref mRNA translation is in fact the most efficient (Additional file 1: Figure S28) and we propose that this might compensate for its lower stability (Additional file 1: Figure S10). It might also contribute to explain why the siRNA-mediated knock-down of BRAF-ref mRNA has at the protein level less pronounced effects than the knock-down of BRAF-X1 plus X2 mRNAs (Fig. 5c).
The C-terminal domain of BRAF-X2 favors its degradation through the proteasome pathway
We next decided to investigate the absence of BRAF-X2 protein, as observed by mass spectrometry. By transiently transfecting BRAFV600E-ref, BRAFV600E-X1, and BRAFV600E-X2 coding sequences in HEK293T cells using the PIG-NotI plasmid, we discovered that the first two are expressed into proteins and can phosphorylate MEK (Fig. 7e, upper). However, we found that the BRAFV600E-X2 protein is expressed much less efficiently despite having similar mRNA levels to the other two variants (Fig. 7f). We also tested the Δ[3–10] splicing variants and obtained comparable results. The stable infection of pMSCVHygro-Δ[3–10]BRAFV600E-ref and -X1 in A375 cells leads to an increase in the corresponding RNA (Fig. 7g) and protein (Fig. 7h) levels. This results in a sustained activation of the MAPK pathway (MEK gets phosphorylated even in the presence of vemurafenib, Fig. 7h) and in a decreased sensitivity to the drug (Fig. 7i). On the contrary, Δ[3–10]BRAFV600E-X2 is efficiently expressed only at the RNA but not at the protein level and fails to protect A375 cells from vemurafenib activity (Fig. 7g-i). Altogether, these results suggest there is an impairment of BRAF-X2 expression that occurs at the post-transcriptional level.
To assess whether such impairment is dependent on the X2-specific domain, we devised two complementary strategies. First, we cloned the C-terminal CR3 region of BRAFV600E-ref, BRAFV600E-X1, and BRAFV600E-X2 in frame with EGFP coding sequence, taking advantage of the pEGFP-C1 plasmid (Fig. 8a). The pEGFP-CR3-ref, −X1, and -X2 plasmids were then transiently transfected in A375 cells. We obtained that, despite the fact that all transcripts are expressed at similar levels (Fig. 8b), the presence of the CR3-X2 domain causes a decrease in EGFP protein levels, as detected by western blot (Fig. 8c), FACS analysis (Fig. 8d), and confocal microscopy analysis (Fig. 8e). Then, we cloned the BRAFV600E-ΔCterm sequence, which is the BRAFV600E-X2 coding sequence minus the isoform-specific region that derives from the translation of exon 19 (Fig. 8f), in the PIG-NotI plasmid and used it to transfect HEK293T cells. As shown in Fig. 8g-h, we obtained a rescued expression of BRAFV600E compared to the full length BRAFV600E-X2. Therefore, we conclude that the X2-specific C-terminal domain negatively affects BRAFV600E-X2 protein levels.
Fig. 8.
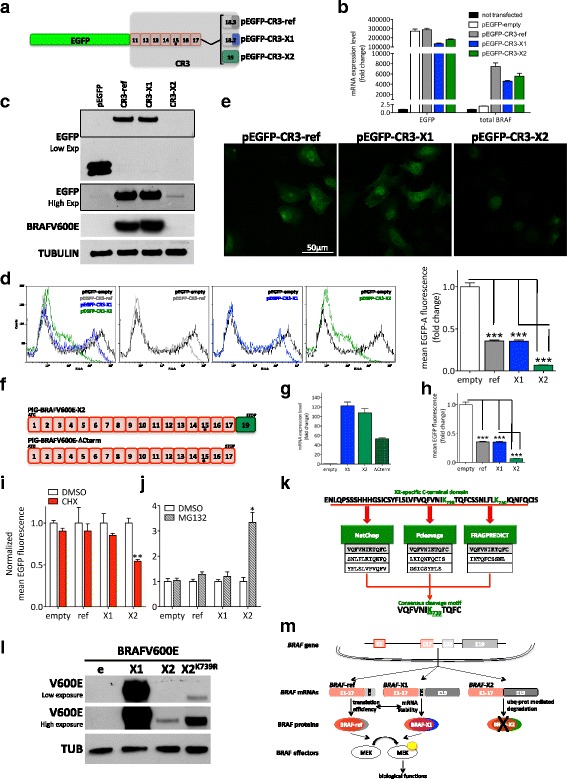
The X2 isoform displays a faster decay due to increased proteosomal-mediated degradation. a Schematic representation of the chimerical protein derived from the fusion of EGFP coding sequence with the CR3 domain of BRAFV600E-ref, X1, and X2 within the pEGFP-C1 plasmid. The asterisk indicates the presence of the V600E mutation. b-e Upon the transient transfection of pEGFP-C1 empty (pEGFP-empty), pEGFP-CR3-ref, pEGFP-CR3-X1, and pEGFP-CR3-X2 plasmids in A375 cells, real-time PCR performed with primers for EGFP and for total BRAF indicates that the chimerical mRNAs are all transcribed at similar levels (b), but western blot (c), flow cytometry (d) and confocal microscopy analysis (e) indicate that, when fused with CR3-X2, EGFP protein is expressed at lower levels. The dotted box shows a higher exposure of the anti-EGFP antibody. f-h When PIG-BRAFV600E-ΔCterm plasmid, which lacks the nucleotides encoding for the X2-specific C-terminal domain (f), is transiently transfected in HEK293T cells, not only BRAF mRNA (g), but also BRAF protein is detectable (h). e: empty PIG-NotI; X1: PIG-BRAFV600E-X1 (used as positive control); X2: PIG-BRAFV600E-X2; ΔCterm: PIG-BRAFV600E-ΔCterm. i-j Upon the transient transfection of pEGFP-empty, pEGFP-CR3-ref, pEGFP-CR3-X1, and pEGFP-CR3-X2 plasmids, A375 cells were treated with 100 ug/ml cicloheximide (CHX) (i) or 20 uM MG132 (j) for 8 h. The CHX treatment indicates that the decay rate of CR3-X2 is faster than that of CR3-ref and CR3-X1, while the MG132 treatment suggests that this is due to higher degradation rate through the ubiquitin-proteasome pathway. k The prediction of potential proteasomal cleavage sites using 3 different algorithms retrieves the indicated X2-specific consensus peptide. (l) The mutagenesis of Lys739 into a proteasome-insensitive Arg rescues the expression of the X2 isoform of BRAF protein. e: empty PIG-NotI; X1: PIG-BRAFV600E-X1 (used as positive control); X2: PIG-BRAFV600E-X2; X2K739R: PIG-BRAFV600E-X2 in which Lys(K)739 has been substituted with Arg(R) (AAA to AGA triplet change). m Cartoon that summarizes the main findings of this article (details in the text). The pictures are taken from 1 out of 3 independent experiments performed, all with comparable outcome. The graphs represent the mean ± SEM of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
To assess if the X2-specific C-terminal domain negatively affects BRAFV600E-X2 protein levels by increasing protein degradation, we transiently transfected A375 cells with the pEGFP-CR3-ref, −X1, and -X2 plasmids and treated them with 100 ug/ml cycloheximide for 8 h. In so doing, we discovered that the decay rate of the X2 isoform is faster than that of the other two isoforms (Fig. 8i). Furthermore, we discovered that the treatment of transfected A375 cells with 20 uM MG132 rescues the expression of the chimerical EGFP-CR3-X2 protein (Fig. 8j and Additional file 1: Figure S29). Therefore, we conclude that the X2 C-terminal domain causes BRAFV600E protein degradation through the ubiquitin-proteasome pathway.
Next, we aimed at predicting proteasomal cleavage sites located within the X2-specific C-terminal domain (aa710–758, green in Additional file 1: Figure S20c). To this end, we interrogated three different in silico algorithms: NetChop, Pcleavage, and FRAGPREDICT. The consensus analysis of the output of these three algorithms allowed us to identify the peptide VQFVNIK 739TQFC as a very high-scoring cleavage-determining amino acid motif (Fig. 8k and Methods). Of note, when the reference-specific (aa 710–766) and the X1-specific (aa 710–767) C-terminal domains were analyzed using the same parameters, no notable proteasomal cleavage site was retrieved. This confirms the C-terminal region of BRAF-X2 is the most sensitive to proteasome-mediated degradation. To experimentally prove that the predicted cleavage-determining aminoacid motif contributes to increase the degradation of the X2 protein isoform through the ubiquitin-proteasome pathway, we mutagenized the Lysine at position 739 into a ubiquitin-insensitive Arginine and obtained the expected rescue in BRAFV600E-X2 protein expression (Fig. 8l).
Conclusions
Our work sheds new light on BRAF, a crucially important gene in human cancer, by unveiling the repertoire of its mRNA and protein variants.
Using RNA-seq data obtained from more than 4800 patients and 9 cancer types, we demonstrate that BRAF mRNA is not a single transcript, but rather a pool of 3 transcripts (reference, X1, and X2), which differ in the length and sequence of their 3’UTR. Specifically, we show that BRAF-ref 3’UTR is as short as 76 nt, while the E19-derived 3’UTR that is shared by BRAF-X1 and BRAF-X2 extends for ~7 kb.
We also show that the total levels of BRAF mRNA are kept constant, while the levels of BRAF-ref on one side and those of BRAF-X1 plus X2 on the other side are inversely related.
In addition, we show that the ratio among the three transcripts is different in different cancer types. In melanoma in particular, the BRAF-X1 isoform is more expressed than the reference and the X2 isoforms, which in turn are expressed at similar levels. Remarkably, the presence and relative expression levels of the different BRAF isoforms are maintained when acquired resistance to vemurafenib occurs due to BRAF gene amplification [19] or BRAF splicing variants that lack the RAS-binding domain [20].
We demonstrate that BRAF-X1 mRNA, both the full length and the BRAFi-resistant Δ[3–10] splicing variant, is translated into a fully functional protein that differs from the reference one in the last few amino acids at the C-terminal domain. Together with BRAF-ref, BRAF-X1 protein accounts for BRAF functional activities.
We also show that the levels of the BRAF-ref and BRAF-X1 proteins are similar, despite the fact that BRAF-X1 mRNA is expressed at higher levels than BRAF-ref mRNA. We provide evidence that this might be due to a balance between mRNA stability (higher for the X1 isoform) and translation efficiency (higher for the reference isoform).
In contrast with our X1 variant experiments, we failed to detect endogenous BRAF-X2 proteins. We elucidate the underlying molecular mechanism: Lys739 in the X2-specific C-terminal domain is specifically recognized by the ubiquitin-proteasome pathway and thus causes a net impairment in protein stability.
Taken together, the results presented in this study are novel and highly relevant to basic cancer biology. They unveil that each step of BRAF expression (including transcription, splicing, mRNA stability, translation efficiency, and protein stability, Fig. 8m) is subjected to a very tight and sophisticated regulation that warrants further investigations. For example, we hypothesize that the 3’UTR that characterizes BRAF-X1 and -X2 is bound by microRNAs and RNA-binding proteins that are reasonably different from those that bind to the reference 3’UTR [24], and is therefore subjected to a different post-transcriptional regulation. We also hypothesize that, through microRNA binding, the extremely long E19-derived 3’UTR is actively involved in ceRNA networks and therefore confers coding-independent activities to BRAF mRNA [25]. In human cancer, amplification events can coexist with BRAFV600E mutation (www.cbioportal.org). Furthermore, the selective amplification of the BRAFV600E mutant allele has been reported as one of the mechanisms responsible for acquired resistance to BRAFi and/or MEKi [26–28]. Finally, the direct relationship that we observed between BRAF-X1 and BRAF-X2 levels is consistent with a reciprocal sponging effect. Therefore, we speculate that the unrestrained activity of the mutant BRAF protein can be further boosted by an increase in the levels of BRAF mRNA itself and, consequently, by an increase in the levels/activity of its oncogenic ceRNA partners.
Our results are important from a translational point of view as well.
The presence of different combinations of BRAF isoforms might contribute to explaining the highly heterogeneous degree of responses to BRAFi commonly observed among patients carrying different cancer types, and even among patients carrying the same cancer type [29].
Furthermore, the currently available BRAF-targeting drugs should be tested to determine whether they are equally effective against all BRAF protein variants. If this is not the case, isoform-specific drugs should be developed and used as cocktails.
Finally, we speculate that the combination of synthetic drugs that target BRAF proteins and RNA-based drugs that target BRAF mRNAs might result in increased efficacy, since this would concomitantly impair both the coding-dependent and the coding-independent activities of this gene.
Methods
Primers
All qRT-PCR and PCR primers were purchased from Eurofins Genomics. Sequences are reported in Additional file 2: Tables S3 and S4, respectively and are mapped in Additional file 3.
siRNAs were purchased from Shanghai GenePharma and their sequences are reported in Additional file 2: Table S5.
Plasmids
PIG-NotI
The PIG-NotI retroviral plasmid was obtained from pMSCV-PIG plasmid (PIG, kind gift from Prof. Pandolfi, BIDMC-HMS), by adding a NotI site between the BglII and the XhoI site, so that the expanded multicloning site results composed of BglII, NotI, XhoI, and HpaI sites. PIG-NotI was used as backbone for the cloning of the plasmids listed below and as empty vector control for transient transfection/stable infection experiments.
PIG-BRAFV600E-ref, −X1, and -X2
BRAFV600E-ref CDS was amplified by PCR from pcDNA3.1-BRAFV600E-FLAG (kind gift from Dr. Pao, Vanderbilt University Medical Center) and cloned into the HindIII and SacI sites of pYES2 plasmid (kind gift from Dr. Galli, CNR-IFC), which was used as a cloning intermediate. The primers used for PCR amplification were the following:
BRAF HindIII Kozak Fw 5′-CATAAGCTTGCCACC ATGGCGGCGCTGAGCGGTGG-3′ (ATG underlined; Kozak sequence in bold);
BRAF SacI Rv 5′-GCAGAGCTCTCAGTGGACAGGAAACGCACCATA-3′ (STOP codon underlined).
The FLAG tag was not PCR amplified together with the CDS.
The pYES2-BRAFV600E-ref plasmid was then used as backbone for the generation of pYES2-BRAFV600E-X1 and pYES2-BRAFV600E-X2 plasmids: the X1- and X2-specific sequences were PCR amplified from A375 cDNA, using the common forward primer BglII CDS Fw 5′-GCAAGATCTCAGTAAGGTACGGAGT-3′ and the X1-specific reverse primer (BRAF X1 SacI Rv 5′-CATGAGCTCCTACTTGAAGGCTGCAAATTCTC-3′, STOP codon underlined) or the X2-specific reverse primer (BRAF X2 SacI Rv 5′-CATGAGCTCTCAGCTTATGCATTGGAAATTTTG-3′, STOP codon underlined). Subsequently, the reference specific sequence was substituted with the X1- and X2-specific sequences in the pYES2-BRAFV600E-ref plasmid, using BglII and SacI restriction sites.
At this point, BRAFV600E-ref, −X1, and -X2 CDS were PCR amplified from pYES2-BRAFV600E-ref, −X1, and -X2 plasmid using the common BRAF NotI Kozak Fw primer (5′-CATGCGGCCGCCACC ATGGCGGCGCTGAGCG-3′, ATG underlined, Kozak in bold) and isoform specific reverse primers:
BRAF SalI Rv 5′-GCAGTCGACTCAGTGGACAGGAAACGCACCATATC-3′
BRAF X1 SalI Rv 5′-GCAGTCGACCTACTTGAAGGCTGCAAATTCTC-3′
BRAF X2 SalI Rv 5′-GCAGTCGACTCAGCTTATGCATTGGAAATTTTG-3′
(STOP codon underlined).
Finally, the CDS were cloned in PIG-NotI using the NotI and XhoI restriction sites.
PIG-BRAFV600E-ΔCterm
The BRAFV600E CDS lacking the X2-specific triplets was PCR amplified from PIG-BRAFV600E-X2 plasmid, using BRAF NotI Kozak Fw primer and the BRAF X2deltaCterm SalI Rv primer (5′-CATGTCGACTCATTGGGGAAAGAGTGGTCTCTCAT-3′, STOP codon underlined). It was then cloned in PIG-NotI using the NotI and XhoI restriction sites.
PIG-BRAFV600E-X2K739R
The BRAFV600E-X2 CDS that is mutagenized at the 739 aa position was PCR amplified from pYES2-BRAFV600E-X2K739R plasmid (see “Mutagenesis” below), using the BRAF NotI Kozak Fw primer and the BRAF X2 SalI Rv primer, then cloned in PIG-NotI using the NotI and XhoI restriction sites.
pMSCVHygro-NotI
The pMSCVHygro retroviral plasmid was obtained from pMSCVHygro plasmid (kind gift from Prof. Pandolfi, BIDMC-HMS), by adding a NotI site between the BglII and the XhoI site, so that the expanded multicloning site results composed of BglII, NotI, XhoI, and HpaI sites. pMSCVHygro-NotI was used as backbone for the cloning of the plasmids listed below and as empty vector control for transient transfection/stable infection experiments.
pMSCVHygro-Δ[3–10]BRAFV600E-ref, −X1 and -X2
These plasmids were obtained following the same strategy already described for PIG-BRAFV600E-ref, −X1, and -X2. However, the starting point was the Δ[3–10]BRAFV600E-ref cDNA that was amplified by PCR from A375 C2 cDNA using the BRAF HindIII Kozak Fw and the BRAF SacI Rv primers mentioned above.
pEGFP-CR3-ref, −X1 and -X2
These plasmids were obtained by cloning the CR3 domain of reference, X1, and X2 BRAFV600E (from exon 11 to the exon containing the STOP codon) in frame with EGFP in the pEGFP-C1 plasmid (Clontech). The CR3 domains were obtained by PCR amplification from PIG-BRAFV600E-ref, −X1, and -X2 plasmids, using a common forward primer BRAFCR3 HindIII Fw (5′-CTCAAGCTTTGAAAACACTTGGTAGACGGGAC-3′) and isoform-specifc reverse primers (BRAF SalI Rv, BRAF X1 SalI Rv, BRAF X2 SalI Rv, see above for the sequences).
Mutagenesis
The BRAFV600E-X2K739R CDS was obtained using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). 100 ng of pYES2-BRAFV600E-X2 plasmid were used as a template, along with the primers for mutagenesis listed in Additional file 2: Table S4, following the manufacturer’s instructions.
Cell Culturing
Cells were grown at 37 °C in a humidified atmosphere with 5% CO2 and they were routinely tested for mycoplasma contamination. The cell lines derived from solid tumors were cultured and fingerprinted as reported in ref [16].
The resistant clones and clonal populations were obtained by prolonged exposure of A375 and 501Mel parental cells to 2 uM vemurafenib as reported in [5].
HEK293T cells were cultured in DMEM low glucose (Euroclone) supplemented with 10% fetal bovine serum (FBS, Euroclone), 1% glutamine (Sigma-Aldrich) and 1% Penicillin/Streptomycin (Euroclone).
RNA extraction and retrotranscription
RNA was extracted using QIAzol (Qiagen), following the manufacturer’s instructions. Before retrotranscription, 1ug of total RNA was subjected to DNAse treatment using DNaseI amplification grade (Invitrogen), following the manufacturer’s protocol. 0.25ug of DNAse-treated RNA were retrotranscribed on a S1000 Thermal Cycler (Bio-Rad) using iScript cDNA Synthesis Kit (Bio-Rad).
PCR
To determine the length of the E19-derived 3’UTR, the PCR reactions were performed using the PCR Master Mix (Thermo Fisher Scientific) in a S1000 Thermal Cycler. 1 ul of cDNA or 10 ng of gDNA were used as templates. The primers are listed in Additional file 2: Table S4 (BRAF-E19-1/2/3/4 F/R). The reaction condition were: 98 °C 10 s, (98 °C 1 s, 57 °C 5 s, 72 °C 55 s)× 35 cycles, 72 °C 1 min. The “chained PCR” were performed using Phusion Flash High Fidelity Master mix (Thermo Fisher Scientific) with the following reaction conditions: 98 °C 10 s, (98 °C 1 s, 55 °C 5 s, 72 °C 45 s)× 35 cycles, 72 °C 7 min).
To determine the expression of the NE6 exon, 1 ul of cDNA obtained from A375 cells was used as template in PCR reactions, using Phusion Flash High Fidelity Master mix (Thermo Fisher Scientific). The primers used were: BRAF-E1/2 F, BRAF-E3 qRT-PCR F, BRAF-NE6p qRT-PCR F, BRAF-E4a qRT-PCR R, BRAF-E4b R, and BRAF-NE6p qRT-PCR R (Additional file 2: Tables S3 and S4). The PCR conditions were: 98 °C 1 s, (98 °C 1 s, 57 °C 5 s, 72 °C 15 s)× 35 cycles, 72 °C 1 min.
To determine the expression of Δ[3–10] splicing variant in its reference, X1, and X2 isoforms, 1ul of cDNA obtained from A375 C2 cells was used as template, while 10 ng of pMSCVHygro-Δ[3–10]BRAFV600E-ref, X1, and X2 plasmids were used as positive controls. The reaction condition using Phusion Flash High Fidelity Master mix and primers BRAF-E1/2 F, refBRAF-STOP R, BRAF-X1-STOP R (Additional file 2: Table S4) were: 98 °C 10 s, (98 °C 1 s, 57 °C 5 s, 72 °C 35 s)× 35 cycles, 72 °C 1 min.
Real-time PCR
For real-time PCR (qRT-PCR) reactions, 1 ul of diluited cDNA (1:5), the SsoAdvanced Universal Supermix (Bio-Rad) and appropriate primers (Additional file 2: Table S3) were used in a CFX96 Real-Time System (Bio-Rad). The reaction conditions were the following: 98 °C 30 s, (98 °C 3 s, 58 °C 20 s, 72 °C 10 s)× 40 cycles. In order to confirm the specificity of the reaction, a melting curve was performed after each PCR (from 65 °C to 95 °C with an increase of temperature of 0.5 °C/s). All reactions were performed in duplicate, and the average of the two Ct values was used to calculate the expression of the different transcripts by the “2-ΔΔCt” method, using the geometrical square mean of three housekeeping genes as a reference.
siRNA transfection
2×105 A375 or 2,5×105 MeWo and 501 Mel cells/6well plate were seeded. The day after, 6ul of 20 uM siRNA stock solution (see Additional file 2: Table S5 for siRNA sequences) per well were added to 250 ul of OptiMEM I® (Invitrogen), while 10 ul of 1 mg/ml LIPOFECTAMINE 2000™ (Invitrogen) were added to additional 250 ul of OptiMEM I®. These two solutions were then combined together and the siRNA-LIPOFECTAMINE 2000™ complexes were allowed to form for 20 min at room temperature. In the meantime, the medium from each well was aspirated and replaced with 1.5 ml of fresh OptiMEM I®. The OptiMEM I®/siRNA/LIPOFECTAMINE 2000™ mixture was then added to the wells, let stand for 6 h and replaced with complete medium for 24 h (for qRT-PCR analyses) or 48 h (for western blot analyses). In alternative, at the end of the 6 h, the cells were trypsinized and used for cellular assays.
Plasmid transfection
The transfection of plasmids in HEK293T cells was performed using PEI. 5×105 cells/6well plate were seeded. In the afternoon of the day after, 167 ul of DMEM low glucose were mixed with 4ug of plasmidic DNA and 7.5 ul of 0.02 mM PEI (Sigma-Aldrich). The mixture was then left at room temperature for 20 min. Meanwhile, the cell medium was replaced with 833 ul of fresh DMEM low glucose containing 2%FBS. The DNA/PEI/DMEM mixture was then added to the wells and the next morning was replaced with fresh complete DMEM low glucose medium. Cell pellets were collected after 24 additional hours.
The transfection of plasmids in A375 cells was performed using LIPOFECTAMINE 2000™ (Invitrogen). 5×104 A375 cells/24well plate were seeded and 24 h later they were transfected with 0.8 ug of plasmidic DNA and 2 ul of 1 mg/ml LIPOFECTAMINE 2000™, following the manufacturer’s protocol.
Stable infection
The stable infections of retroviral plasmids were carried out as described in [30].
Proliferation assay
6 h after the transfection mix was added, A375 or A375 C2 siRNA-transfected cells were seeded at 3000 cells/well in 12well plates (3 wells per condition per time point). Each time point (1, 3, 5, and 7 days post transfection) was fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. All time points were then stained together using 0.5 ml/well of crystal violet solution (0.1% crystal violet, 20% methanol) for 15 min at room temperature on a rocking plate. After the excess crystal violet solution was removed, the plates were washed with tap water and dried. Then, they were de-stained using a 10% acetic acid solution. Finally, the absorbance of each well was read in duplicate at 590 nm. Each sample was normalized on its first time point (day 1) and the results of three independent experiments were averaged for statistical analyses.
Clonogenicity assay
6 h after the transfection mix was added, 2×102 siRNA-transfected A375 C2 cells were seeded in 6 cm plates in triplicate and treated with vehicle (DMSO) or 2 uM vemurafenib. After 8 days, cells were fixed and stained with a 0.1% crystal violet, 4% formaldehyde solution. The number of colonies of the si-CT transfected and DMSO-treated cells was used as normalizer and the results of three independent experiments were averaged for statistical analyses.
Migration assay
6 h after the transfection mix was added, 1.5×104 siRNA-transfected A375 cells were seeded on a plate using silicone inserts (Culture-Insert Family, IBIDI). 24 h later (t0), when the confluency was ~70–80%, the inserts were removed and the quality of the covered surface was evaluated. Then, the cell-free gaps were monitored at different time points after insert removal, using 10× and 20× objective lens. Images were captured using Leica DM IL LED microscope. The measure of cell-free gaps was taken with Image J software (http://rsb.info.nih.gov). The migratory rate was determined as a relative percentage of gap closure compared to the t0 area. For each condition, ‘wounds’ from three independent experiments were measured.
Western blot analysis
Cells were harvested and protein were extracted with 5 mM Tris pH8, 1% TritonX 100, 0.25% Sodium Deoxycholate, 10% proteinase inhibitor, 2% PMSF, 0.5% Ortovanadate. The samples were then heated at 95 °C for 5 min, separated on 10% SDS-polyacrylamide gels (Mini-PROTEAN Precast gel, Bio-Rad) and electrotransferred to polyvinylidene difluoride (PVDF) membranes using Trans-Blot Turbo system (Bio-Rad).
Membranes were blocked at room temperature for 1 h using 3% milk in TBST for the detection of BRAFV600E, BRAF, α-TUBULIN and EGFP or 3% BSA in TBST for the detection of pMEK. They were then incubated overnight at 4 °C with the following primary antibodies:
anti-BRAF (F-7, #5284, Santa Cruz Biotechnology; mouse monoclonal antibody, dilution 1:1000 in 5% milk in TBST);
anti-human BRAFV600E (VE1, #E19290, Spring Bioscience; mouse monoclonal antibody, dilution 1:400 in 1% milk in TBST);
anti-pMEK (#9154, Cell Signaling; rabbit monoclonal antibody, dilution 1:1000 in 3% BSA in TBST);
anti-α-TUBULIN (#T9026, Sigma-Aldrich; mouse monoclonal antibody, dilution 1:20000 in 5% milk in TBST);
anti-EGFP (#A111-22, Molecular Probes, rabbit polyclonal antibody, dilution 1:2000 in 5% milk in TBST).
According to the manufacturers’ indications, all the primary antibodies used have been tested for their ability to recognize the relevant human proteins.
The detection of primary antibodies was performed using alkaline phosphatase-conjugated secondary antibodies and enhanced chemiluminescence reagents (GE Healthcare, Life Sciences, Milano, Italy).
FACS analysis
The day after transfection, A375 cells were treated with 20 uM MG132 (Sigma-Aldrich) or 100 ug/ml cycloheximide (CHX, Sigma-Aldrich) or both in combination for 8 h. At the end of the treatment, cells were collected and washed once with PBS. The mean fluorescence of EGFP was then measured by flow cytometry using Accuri C6 (BD Biosciences).
Confocal microscopy analysis
5 × 104 A375 cells were seeded on circular 12 mm coverslips and transfected with pEGFP-C1 or pEGFP-CR3-ref/X1/X2 plasmids as reported in “Plasmid transfection”. The day after, the cells were washed twice with PBS and fixed with 4% formaldehyde for 10 min at room temperature. The coverslips with fixed cells were then mounted on microscope glass using Vectashield® and EGFP was visualized by confocal microscopy (inverted confocal laser scanning microscopy Leica TCS SP8 microscope).
Actinomycin D assay
2.5 × 105 cells were seeded in 6well plates. The day after, the cells seeded in 1 well were collected and used as t0, while the others were treated with 10 ug/ml actinomycin D (Sigma-Aldrich) for 2/4/8/12/24 h. Total RNA was then extracted and qRT-PCR performed as described above.
In vitro BRAF transcription
The in vitro transcription of the [a32P]UTP-labeled antisense BRAF probe was performed using T7 RNA polymerase on a linearized plasmid containing BRAF CDS (2.3 kbp). The MEGAshortscript™ T7 Transcription kit (Thermo Fisher Scientific) was used.
Northern blot
A total of 30 ug of each RNA sample was resolved in a 1% formaldehyde-agarose gel, transferred to a GeneScreen Plus membrane (Perkin-Elmer, Waltham, MA), hybridized with a 32P-labeled antisense RNA probe for BRAF CDS at 45° on ULTRAhyb® Ultrasensitive Hybridization Buffer (Thermo Fisher Scientific), washed and, finally, detected and quantified with a Cyclone Phosphoimager (Perkin-Elmer).
Immunoprecipitation
A375 cells were lysed on ice for 1 h by incubating them in Pierce Lysis buffer (25 mM Tris, 0.15 M NaCl, 1 mM EDTA, 1% NP40, 5% glycerol, pH 7.4) supplemented with protease inhibitors. They were then subjected to sonication 3 times for 30 s and centrifuged at 16000 g for 30 min at 4 °C in order to remove cellular debris. Protein concentration was determined by bicinchoninic assay (#23223 and #23224, Pierce). Subsequently, 1 mg of proteins in a final volume of 500 ul were mixed with 10 ug of BRAF antibody (F-7, #5284, Santa Cruz Biotechnology, see above) and 25 ul of Protein G magnetic beads (Pierce) overnight at 4 °C. The immunoprecipitate was washed once with water and boiled in loading buffer in order to release the BRAF-antibody complexes from the beads. Western blot was performed using again F7 BRAF antibody (dilution 1:500 in 5% milk in TBST) and overnight incubation.
In-gel tryptic digestion and LC-mass spectrometry
Immunoprecipitates were separated by SDS-PAGE using 10% polyacrylamide gel. Gel was subject to coomassie staining and the bands corresponding to BRAF size were excised. For each gel lane, a single band was excised, dehydrated with acetonitrile, and dried under vacuum. Gel was incubated with 10 mM dithiothreitol (Fluka) in 25 mM NH4HCO3 for 45 min at 56 °C. After removing the reducing solution, alkylation was performed with 55 mM iodoacetamide (Sigma-Aldrich) dissolved in 25 mM NH4HCO3 for 30 min with constant shaking, at room temperature and in the dark. In-gel protein digestion was performed overnight by treating the samples with 5 ng/ul sequencing-grade modified trypsin (Promega) in 25 mM NH4HCO3 at 37 °C. Peptides were extracted from the gel with 50% acetonitrile/0.1% formic acid (v/v) and analyzed with a LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled with a nano-HPLC Ultimate 3000 (Dionex-Thermo Fisher Scientific). Peptides were loaded onto a homemade pico-frit column packed with C18 material (Aeris Peptide 3.6 um XB-C18; Phenomenex) and separated using a linear gradient of acetonitrile/0.1% formic acid from 3 to 40% in 20 min at a flow rate of 250 nl/min. Spray voltage was set to 1.2 kV with an ion source capillary temperature of 200 °C. The instrument was operating in data-dependent mode with a top-ten acquisition method (a full scan at 60,000 resolution on the Orbitrap followed by MS/MS fragmentation in the linear trap of the ten most intense ions).
Analysis of LC-mass spectrometry data
The acquired raw data files were processed using Proteome Discoverer 1.4 software (Thermo Fisher Scientific) coupled with a Mascot server (version 2.2.4; Matrix Science). Protein identification was performed against the Uniprot Human protein database (release April 2015; 90411 sequences) modified in-house with the addition of three FASTA sequences from NCBI database: Serine/threonine-protein kinase BRAF (ref. NP_004324.2); Predicted: Serine/threonine-protein kinase BRAF isoform X1 (ref. XP_005250102.1); predicted: Serine/threonine-protein kinase BRAF isoform X2 (ref. XP_005250103.1).
Enzyme specificity was set to trypsin, and a maximum of one missed cleavage was allowed. The precursor and fragment mass tolerances were set to 10 ppm and 0.6 Da, respectively. Carbamidomethylation of Cys was set as a static modification, whereas oxidation of Met was set as variable modification. Percolator (Wright et al., 2012) was used to assess peptide and protein identification confidence: only proteins identified with at least 2 unique peptides with high confidence (q ≤ 0.01) were considered as positive hits. Proteins were grouped into protein families according to the principle of maximum parsimony.
Protein preparation for subsequent LC-MS/MS SWATH
The proteins present in the gel slices from immunoprecipitates were reduced with dithiothreitol 5 mM at 80 °C for 30 min and alkylated using iodoacetamide 10 mM at 37 °C for 20 min (both in 50 mM NH4HCO3). Digestion was performed by incubating the proteins with 10ug/ul trypsin (#11418033001, Roche) in 25 mM NH4HCO3 at 37 °C overnight. The peptide solution was recovered by sonication and centrifugation at 16000 g and then loaded on a C18 column. Once eluted from the column, the peptides were diluted in a 2% Acetonitrile, 0.1% Formic acid buffer and filtered with a 0.22 um filter.
Analysis of LC-MS/MS SWATH data
Peptides were subject to chromatographic separation using a nano-HPLC system (Eksigent, ABSciex). The samples were pre-concentrated in a pre-column cartridge by the loading pump (PepMap-100 C18 um, 100 Å, 0.1 × 20 mm, Thermo Fisher Scientific) and then separated in a C18 PepMap-100 column (3 um, 75 um × 250 mm, Thermo Fisher Scientific) at 300 nl/min flow rate. Each run was done with eluent A (ultrapure water, 0.1% Formic acid) with 60 min linear gradient from 5 to 40% of eluent B (Acetonitrile, 0.1% Formic acid) followed by a purge step of 10 min and a re-equilibration step of 20 min. Eluted peptides were directly processed by 5600 TripleTOF™ mass spectrometer (ABSciex) equipped with a Duo Spray™ ion source (ABSciex). Information dependent acquisition (IDA) analysis was performed acquiring survey scans in 250 ms and collecting 25 products ion scans if a threshold of 125 counts per second was exceeded. For each scan four time bins were summed at a pulser frequency value of 11 kHz through monitoring of the 40 GHz multichannel TDC detector with 4-anode/channel detection. Dynamic exclusion was set for 1/2 of peak width (~8 s) and the precursor was refreshed off the exclusion list. We performed three runs for each sample. MS/MS data were processed with ProteinPilot software (ABSciex), using the Paragon and Pro Group Algorithms against the database containing all the human protein sequences from NCBI Reference Sequences. The false discovery rate (FDR) was analyzed by the integrated tools in ProteinPilot software with a set confidence level of 95%. Data were also acquired using the new Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH™) method for shotgun data independent MRM quantification. We performed three runs for each sample. The SWATH MS spectral library was generated by ProteinPilot Software. PeakView4.5 Software (ABSciex) with MS/MS(ALL) with SWATH™ Acquisition MicroApp 2.0 and MarkerViewTM (ABSciex) was used for label free statistical comparative analysis. Peptides from top score proteins were selected for retention time alignment with a processing settings of 7 peptides per protein, 7 transitions per peptide, 95% peptide confidence, 5% FDR, XIC (Extracted-Ion Chromatogram) extraction window of 10 min, width 50 ppm and 0.1 Da. Global normalization of profiles based on total protein content was applied.
Peptide synthesis and purification
The two peptides TPIQAGGYGAFPVH (C-terminal of BRAF-ref) and TPIQAGGYGEFAAFK (C-terminal of BRAF-X1) were prepared by solid-phase synthesis using Fmoc chemistry on an automatic Liberty Blue Peptide Synthesizer with an integrated microwave system (CEM). Standard couplings suggested by the producer were used thorough the synthesis. The resin was Rink-amide, loading 0.46 mmol/g (Bachem). The deprotected peptide was precipitated with cold diethyl ether after removing in vacuo most of the cleavage cocktail. In turn, the cleavage cocktail was composed of: TFA/Tri(isopropylsilane)/H2O/Ethanedithiol/thioanisole 93/2/2/2/1; cleavage was performed according to manufacturer conditions (30 min at 30 °C under microwave irradiation). HPLC analyses and purifications were performed on a Dionex Ultimate 3000 HPLC system or on a Shimadzu Nexera HPLC system. Crude peptides were purified by RP-HPLC on a Jupiter Proteo 90 Å column (4 uM, 250 × 10 mm; Phenomenex) using water:TFA 100:0.01 v/v (eluent A) and acetonitrile:water:TFA 95:5:0.01 v/v (eluent B) as mobile phase. The identity of the purified product was confirmed by electrospray mass spectroscopy, using an API3200QTRAP Hybrid Triple Quadrupole/Linear Ion Trap (ABSciex).
Mass spectrometry characterization of synthetic peptides
Synthetic peptides were analysed by LC-MSMS using a microHPLC (Eksigent LC-300) interfaced with 5500 QTRAP (ABSciex). 5 ul of a 10 uM solution of each peptide was injected in a Jupiter 4u Proteo 90A chromatographic column (250 × 0.3 mm, Phenomenex) at a flow rate of 5 ul/min. Runs were performed with eluent A (ultrapure water, 0.1% Formic acid) under 10 min linear gradient from 5 to 95% of eluent B (Acetonitrile, 0.1% Formic acid) followed by 5 min of re-equilibration step. Peptides eluted from chromatography were directly processed using 5500 QTRAP™ mass spectrometer (ABSciex) equipped with an ESI ion source (ABSciex). Precursor Ions (MS spectra) and MSMS fragmentations (Product ions) were obtained. For C-terminal peptide of BRAF-ref, the m/z principal ion was m/z 708.00 (M + 2H+) and the Q3 selected ions were 904.44, 352.92, and 440.76, while for C terminal peptide of BRAF-X1 Q1 the m/z principal ion was m/z 779.04 (M + 2H+) and Q3 selected ions were 1046.49, 1117.53, and 974.52.
The best MRM transitions proposed by SWATH analysis were 708/352 and 708/904 for the C-terminal peptide of BRAF-ref and 779/1046 and 779/1117 for the C-terminal peptide from BRAF-X1. These ions were chosen for peptide quantitation analysis and for determining peptide area ratio at known concentration (10 uM solutions).
Xenograft in zebrafish embryos
24 h after siRNA transfection, mcherry-A375 cells were prepared by mixing 106 cells with 2ul of MATRIGEL (Cultrex Basement Membrane Extract, PathClear) and injected into zebrafish embryos of the wt AB line (RRID:ZIRC_ZL1, kind gift from Prof. Andreazzoli, Dipartimento di Biologia, Università di Pisa, Italy). The AB fishes were raised in the zebrafish facility at CNR-IFC and inbred. The protocol followed for the injection and the subsequent analyses was the one described in ref [31]. Briefly, 48 hpf embryos were anesthetizes with 0.04 mg tricaine (#A-5040, Sigma-Aldrich) and grafted in the perivitelline space with 500 cells in 1 nl. The fluorescence imaging was carried out 2 days after the injection using the Nikon Eclips E600 microscope. The embryos were carefully set down on glass slides embedded with 1% low melting agarose. The acquisitions were conducted with CoolSnap-CF camera using NIS-Elements software version 2.0. ImageJ software (http://rsb.info.nih.gov) was used to analyze the tumor area. The rcmdr package of R software was used for statistical analysis (http://www.rcommander.com/). 15–25 embryos were injected per experimental condition and the experiment was repeated 3 times.
Statistical analyses
Data were analyzed with unpaired and two-tailed t test (GraphPad Prism, GraphPad Software Inc.). Values of p < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, *** < 0.001, ****p < 0.0001).
Analysis of BRAF transcript variants
Data download and read mapping
RNA-seq paired-end raw reads of nine cancers were downloaded from the TCGA database. Raw reads were then mapped on human genome version GRCh37.75, using tophat2 on Pegasus cluster in the Center for Computational Science at University of Miami.
Count of reads on exons
The reads mapping on each exon of BRAF in each individual were extracted from alignment files using an in-house program. Only completely mapped reads were kept for the following analysis.
Count of exon-spanning reads
The exon-spanning reads were identified as reads of the same pair that are mapped one on an exon and the other on another exon, with the distance in between falling in a reasonable range as inferred by all read pairs.
Comparison among exons and transcripts
The comparison among exons and transcripts regarding read counts was conducted using R. The reads on each exon were normalized dividing them by the total number of reads mapped on all exons of each individual and then multiplying them by a 107 factor, which brings the normalized values into a suitable range for comparison. The normalized values falling outside 95% quantile were discarded as outliers.
Analysis of the secondary structure of BRAF-X1 and BRAF-X2 mRNAs
The prediction of the most stable secondary structure of BRAF-X1 mRNA (XM_005250045.1, 9464 nt starting from the ATG) and BRAF-X2 mRNA (XM_005250046.1, 9310 nt starting from the ATG) was performed using UNAfold program [32], version 3.8. Among all the possible ones, the secondary structure showing the lowest energy of pairing (dG) was taken for each of the 2 molecules. These structures were then compared using a dot plot. The conversion was made taking advantage of the Python interactive visualization library Bokeh, version 0.11.1 (http://bokeh.pydata.org/en/latest/).
Prediction of potential proteasomal cleavage sites
Three different algorithms have been used. NetChop 3.1 stand-alone software package [33] is a neuronal network method trained with a database consisting of 1260 publicly available C-terminal cleavage site of proteasome ligands (3.0 C-ter option). To select the most probable cleavage-determining amino acid motifs, we performed our in silico analyses using the very high 0.9 threshold. Pcleavage is a Support Vector Machine (SVM) method for the prediction of 20S proteasome cleavage sites based on 172 fragments generated by 184 distinct cleavage sites [34]. Also in this case, a very high stringency threshold (0.2) was used in our analysis. Finally, FRAGPREDICT combines the two strategies: it predicts potential proteasomal cleavage sites that are used as input for the selection of major proteolytic fragments, applying the kinetic model of the 20S proteasome [35, 36]. Also in this case a stringent threshold value was used (0.9). The final output was obtained through a consensus analysis, by considering the fragments selected by all three algorithms.
Additional files
Figure S1. Schematic representation of the exons belonging to the different variants of human BRAF reported in NCBI and Ensembl. Figure S2. Scan of exon 18 of BRAF in 8 cancer types. Figure S3. Count of the reads mapping to all BRAF exons, E19 included. Figure S4. Count of reads mapping to all BRAF exons (w/o E19 on the left and w E19 on the right). Figure S5. Count of E13-E16 exon-spanning reads. Figure S6. BRAF-004 transcript variant is expressed but it is truncated. Figure S7. Position of the primers/siRNAs used to detect/downregulate BRAF-ref, BRAF-X1, and BRAF-X2. Figure S8. Real-time PCR detection of BRAF-ref, BRAF-X1 plus X2, BRAF-X1, and BRAF-X2. Figure S9. Box plot of the reads that span E17-E18.2, E18.2-E18b, E18.2-E19, and E17-E19. Figure S10. Stability of reference and X1/X2 BRAF mRNA. Figure S11. Correlation among the expression levels of the different BRAF isoforms in breast cancer, head and neck cancer, lung SCC, AML, and DLBCL. Figure S12. Lack of association of reference, X1, and X2 BRAF levels with age (left panels), gender (middle panels), and stage at diagnosis in primary and metastatic melanoma patients. Figure S13. Lack of association of age (left panels), gender (middle panels), and stage at diagnosis (right panels) with the ratio between BRAF-X1 and BRAF-ref levels (upper panels, red) and with the ratio between BRAF-X2 and BRAF-ref levels (lower panels, blue) in primary and metastatic melanoma patients. Figure S14. Tools for the detection of BRAF CDS plus the E19-derived 3’UTR. Figure S15. Length of BRAF-X1 and BRAF-X2 3’UTR in melanoma. Figure S16. siRNA-mediated downregulation of BRAF isoforms in melanoma cells. Figure S17. Cartoon summarizing the position of the primers and the siRNAs used to determine the levels and the identity of the Δ[3–10] splicing variant of BRAF. Figure S18. Real-time PCR detection of BRAF-ref (grey) and BRAF-X1 plus X2 (black) in vemurafenib-resistant clones and clonal populations. Figure S19. The 3’UTR of X1 and X2 Δ[3–10]BRAF splicing variant is up to 7 kb long. Figure S20. Sequence of reference, X1, and X2 BRAF proteins. Figure S21. Alignment between the sequence of human BRAF-ref (NP_004324.2, left) or BRAF-X1 (XP_005250102.1, right) and mouse Braf-ref (NP_647455.3). Figure S22. Alignment between the sequence of human BRAF-ref (NP_004324.2, left) or BRAF-X1 (XP_005250102.1, right) and rat Braf-X1 (XP_001070228.2). Figure S23. Alignment between the sequence of human BRAF-ref (NP_004324.2, left) or BRAF-X1 (XP_005250102.1, right) and pig Braf-X1 (XP_005654324.1). Figure S24. Dot plot of the secondary structure of BRAF-X1 (blue) and BRAF-X2 (green) mRNA sequences. Figure S25. Enlargement of box1 as reported in Figure S24. Figure S26. BRAF peptides identified by an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, a) and by a 5600 TripleTOF mass spectrometer (ABSciex, b) in A375 melanoma cells. Figure S27. BRAF peptides identified by LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) in WM793B melanoma cells. Figure S28. Translation efficiency of reference, X1 and X2 BRAF mRNAs in A375 cells. Figure S29. BRAF-X2 protein displays a faster decay due to increased proteosomal-mediated degradation. (PDF 14500 kb)
Table S1. Coordinates of the exons of human BRAF analyzed in this study. Table S2. List of the human tumor samples used in this study. Table S3. Real-time PCR primers. Table S4. PCR primers. Table S5. siRNA sequences. (PDF 172 kb)
Primers and siRNAs location along BRAF-ref, BRAF-X1, and BRAF-X2 mRNAs. (PDF 67 kb)
Acknowledgements
The authors thank: Dr. K. Dawson for manuscript editing; Dr. R. D’Aurizio for insightful discussions; Dr. M. Rizzo for providing the RNA of leukemia/lymphoma cell lines; Dr. A. Cecchettini and Dr. C. Ippolito for their assistance with confocal microscopy; F. Rossari for his help with cloning of expression vectors; Cassa di Risparmio di Padova e Rovigo (Cariparo) holding for funding the acquisition of the LTQ-Orbitrap XL mass spectrometer; the advance computing group of UMCCS for providing HPC computational resources and expertise.
Funding
This work was supported by: start up funding awarded by Istituto Toscano Tumori and the GR-2011-02348535 grant awarded by the Italian Ministry of Health to LP; the OTTED grant awarded by State of Florida to NT and ZJ.
CF was supported by a grant from Collegio Ghislieri, Pavia.
Availability of data and materials
Some of the results presented here were built upon data generated by The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/). The dataset published in ref [24] and available at GEO under the accession number GSE64741 was also used. All the analyses performed are available from the corresponding authors on reasonable request.
Authors’ contributions
AM, ZJ, NT, and LP conceived the project, designed the experiments and analyzed the data. AM, MV, EG, LC, SS, SL, CF, IEV, AT, ME, EL, GS, and SR performed the experiments. ZJ, GC, and AM performed the bioinformatic analyses. PS provided reagents. EL, EC, JV, GA, GS, SR, LPi, NT, and LP supervised the research. LP wrote the paper with the help of AM, ZJ, and NT. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Blood samples from leukemia patients were collected under the supervision of PS at the University of Perugia, within a study conducted according to Declaration of Helsinki Principles. The study was approved by Comitato Universitario di Bioetica-Università degli Studi di Perugia (#2015-001) and all patients provided written consent at enrollment.
The in vivo experiments on zebrafish embryos were performed in compliance with authorization #1222/2015-PR granted by the Italian Ministry of Health to LPi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Nicholas Tsinoremas, Phone: +1 305 243 4962, Email: ntsinoremas@med.miami.edu.
Laura Poliseno, Phone: +39 050 315 2780, Email: laura.poliseno@gmail.com.
References
- 1.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–98. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 2.Flockhart RJ, Webster DE, Qu K, Mascarenhas N, Kovalski J, Kretz M, Khavari PA. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res. 2012;22:1006–14. doi: 10.1101/gr.140061.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang CC, Croft A, Tseng HY, Guo ST, Jin L, Hersey P, Zhang XD. Repression of microRNA-768-3p by MEK/ERK signalling contributes to enhanced mRNA translation in human melanoma. Oncogene. 2014;33:2577–88. doi: 10.1038/onc.2013.237. [DOI] [PubMed] [Google Scholar]
- 4.Forloni M, Dogra SK, Dong Y, Conte D, Jr, Ou J, Zhu LJ, Deng A, Mahalingam M, Green MR, Wajapeyee N. miR-146a promotes the initiation and progression of melanoma by activating Notch signaling. Elife. 2014;3:e01460. doi: 10.7554/eLife.01460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitiello M, Tuccoli A, D’Aurizio R, Sarti S, Giannecchini L, Lubrano S, Marranci A, Evangelista M, Peppicelli S, Ippolito C, Barravecchia I, Guzzolino E, Montagnani V, Gowen M, Mercoledi E, Mercatanti A, Comelli L, Gurrieri S, Wu LW, Ope O, Flaherty K, Boland GM, Hammond MR, Kwong L, Chiariello M, Stecca B, Zhang G, Salvetti A, Angeloni D, Pitto L, Calorini L, Chiorino G, Pellegrini M, Herlyn M, Osman I and Poliseno L. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget. 2017. doi: 10.18632/oncotarget.15915. [DOI] [PMC free article] [PubMed]
- 6.Cantwell-Dorris ER, O’Leary JJ, Sheils OM. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther. 2011;10:385–94. doi: 10.1158/1535-7163.MCT-10-0799. [DOI] [PubMed] [Google Scholar]
- 7.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, Constantino-Casas F, Eser S, Hieber M, Seidler B, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell. 2013;24:15–29. doi: 10.1016/j.ccr.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McFadden DG, Vernon A, Santiago PM, Martinez-McFaline R, Bhutkar A, Crowley DM, McMahon M, Sadow PM, Jacks T. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A. 2014;111:E1600–9. doi: 10.1073/pnas.1404357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–6. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 12.Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, Whittaker S, Saturno G, Viros A, Pedersen M, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27:85–96. doi: 10.1016/j.ccell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–67. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gautschi O, Milia J, Cabarrou B, Bluthgen MV, Besse B, Smit EF, Wolf J, Peters S, Fruh M, Koeberle D, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol. 2015;10:1451–7. doi: 10.1097/JTO.0000000000000625. [DOI] [PubMed] [Google Scholar]
- 15.Tiacci E, Park JH, De Carolis L, Chung SS, Broccoli A, Scott S, Zaja F, Devlin S, Pulsoni A, Chung YR, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373:1733–47. doi: 10.1056/NEJMoa1506583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marranci A, Tuccoli A, Vitiello M, Mercoledi E, Sarti S, Lubrano S, Evangelista M, Fogli A, Valdes C, Russo F, Monte MD, A Caligo M, Pellegrini M, Capobianco E, Tsinoremas N, Poliseno L. Identification of BRAF 3’UTR Isoforms in Melanoma. J Invest Dermatol. 2015;135(6):1694–7. [DOI] [PubMed]
- 17.Kemper K, Krijgsman O, Kong X, Cornelissen-Steijger P, Shahrabi A, Weeber F, van der Velden DL, Bleijerveld OB, Kuilman T, Kluin RJ, et al. BRAF(V600E) kinase domain duplication identified in therapy-refractory melanoma patient-derived xenografts. Cell Rep. 2016;16:263–77. doi: 10.1016/j.celrep.2016.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–9. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–50. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 23.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–72. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fattore L, Mancini R, Acunzo M, Romano G, Lagana A, Pisanu ME, Malpicci D, Madonna G, Mallardo D, Capone M, et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc Natl Acad Sci U S A. 2016;113:E5005–13. doi: 10.1073/pnas.1607753113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77-78:41–50. [DOI] [PubMed]
- 26.Solit DB, Rosen N. Towards a unified model of RAF inhibitor resistance. Cancer Discov. 2014;4:27–30. doi: 10.1158/2159-8290.CD-13-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27:240–56. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- 29.Fiskus W, Mitsiades N. B-Raf inhibition in the clinic: present and future. Annu Rev Med. 2016;67:29–43. doi: 10.1146/annurev-med-090514-030732. [DOI] [PubMed] [Google Scholar]
- 30.Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, Sportoletti P, Varmeh S, Egia A, Fedele G, et al. Identification of the miR-106b ~ 25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010;3:ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicoli S, Presta M. The zebrafish/tumor xenograft angiogenesis assay. Nat Protoc. 2007;2:2918–23. doi: 10.1038/nprot.2007.412. [DOI] [PubMed] [Google Scholar]
- 32.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 33.Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 34.Bhasin M, Raghava GP. Pcleavage: an SVM based method for prediction of constitutive proteasome and immunoproteasome cleavage sites in antigenic sequences. Nucleic Acids Res. 2005;33:W202–7. doi: 10.1093/nar/gki587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holzhutter HG, Frommel C, Kloetzel PM. A theoretical approach towards the identification of cleavage-determining amino acid motifs of the 20 S proteasome. J Mol Biol. 1999;286:1251–65. doi: 10.1006/jmbi.1998.2530. [DOI] [PubMed] [Google Scholar]
- 36.Holzhutter HG, Kloetzel PM. A kinetic model of vertebrate 20S proteasome accounting for the generation of major proteolytic fragments from oligomeric peptide substrates. Biophys J. 2000;79:1196–205. doi: 10.1016/S0006-3495(00)76374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic representation of the exons belonging to the different variants of human BRAF reported in NCBI and Ensembl. Figure S2. Scan of exon 18 of BRAF in 8 cancer types. Figure S3. Count of the reads mapping to all BRAF exons, E19 included. Figure S4. Count of reads mapping to all BRAF exons (w/o E19 on the left and w E19 on the right). Figure S5. Count of E13-E16 exon-spanning reads. Figure S6. BRAF-004 transcript variant is expressed but it is truncated. Figure S7. Position of the primers/siRNAs used to detect/downregulate BRAF-ref, BRAF-X1, and BRAF-X2. Figure S8. Real-time PCR detection of BRAF-ref, BRAF-X1 plus X2, BRAF-X1, and BRAF-X2. Figure S9. Box plot of the reads that span E17-E18.2, E18.2-E18b, E18.2-E19, and E17-E19. Figure S10. Stability of reference and X1/X2 BRAF mRNA. Figure S11. Correlation among the expression levels of the different BRAF isoforms in breast cancer, head and neck cancer, lung SCC, AML, and DLBCL. Figure S12. Lack of association of reference, X1, and X2 BRAF levels with age (left panels), gender (middle panels), and stage at diagnosis in primary and metastatic melanoma patients. Figure S13. Lack of association of age (left panels), gender (middle panels), and stage at diagnosis (right panels) with the ratio between BRAF-X1 and BRAF-ref levels (upper panels, red) and with the ratio between BRAF-X2 and BRAF-ref levels (lower panels, blue) in primary and metastatic melanoma patients. Figure S14. Tools for the detection of BRAF CDS plus the E19-derived 3’UTR. Figure S15. Length of BRAF-X1 and BRAF-X2 3’UTR in melanoma. Figure S16. siRNA-mediated downregulation of BRAF isoforms in melanoma cells. Figure S17. Cartoon summarizing the position of the primers and the siRNAs used to determine the levels and the identity of the Δ[3–10] splicing variant of BRAF. Figure S18. Real-time PCR detection of BRAF-ref (grey) and BRAF-X1 plus X2 (black) in vemurafenib-resistant clones and clonal populations. Figure S19. The 3’UTR of X1 and X2 Δ[3–10]BRAF splicing variant is up to 7 kb long. Figure S20. Sequence of reference, X1, and X2 BRAF proteins. Figure S21. Alignment between the sequence of human BRAF-ref (NP_004324.2, left) or BRAF-X1 (XP_005250102.1, right) and mouse Braf-ref (NP_647455.3). Figure S22. Alignment between the sequence of human BRAF-ref (NP_004324.2, left) or BRAF-X1 (XP_005250102.1, right) and rat Braf-X1 (XP_001070228.2). Figure S23. Alignment between the sequence of human BRAF-ref (NP_004324.2, left) or BRAF-X1 (XP_005250102.1, right) and pig Braf-X1 (XP_005654324.1). Figure S24. Dot plot of the secondary structure of BRAF-X1 (blue) and BRAF-X2 (green) mRNA sequences. Figure S25. Enlargement of box1 as reported in Figure S24. Figure S26. BRAF peptides identified by an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, a) and by a 5600 TripleTOF mass spectrometer (ABSciex, b) in A375 melanoma cells. Figure S27. BRAF peptides identified by LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) in WM793B melanoma cells. Figure S28. Translation efficiency of reference, X1 and X2 BRAF mRNAs in A375 cells. Figure S29. BRAF-X2 protein displays a faster decay due to increased proteosomal-mediated degradation. (PDF 14500 kb)
Table S1. Coordinates of the exons of human BRAF analyzed in this study. Table S2. List of the human tumor samples used in this study. Table S3. Real-time PCR primers. Table S4. PCR primers. Table S5. siRNA sequences. (PDF 172 kb)
Primers and siRNAs location along BRAF-ref, BRAF-X1, and BRAF-X2 mRNAs. (PDF 67 kb)
Data Availability Statement
Some of the results presented here were built upon data generated by The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/). The dataset published in ref [24] and available at GEO under the accession number GSE64741 was also used. All the analyses performed are available from the corresponding authors on reasonable request.