

Abstract
In humans, DNA methylation at specific CpG sites can be used to estimate the ‘epigenetic clock’, a biomarker of aging and health. The mechanisms that regulate the aging epigenome and level of conservation are not entirely clear. We performed affinity-based enrichment with methyl-CpG binding domain protein followed by high-throughput sequencing (MBD-seq) to assay DNA methylation in mouse samples. Consistent with previous reports, aging is associated with increase in methylation at CpG islands that likely overlap regulatory regions of genes that have been implicated in cancers (e.g., C1ql3, Srd5a2 and Ptk7). The differentially methylated regions in mice have high sequence conservation in humans and the pattern of methylation is also largely conserved between the two species. Based on human ENCODE data, these sites are targeted by polycomb proteins, including EZH2. Chromatin immunoprecipitation confirmed that these regions interact with EZH2 in mice as well, and there may be reduction in EZH2 occupancy with age at C1ql3. This adds to the growing evidence that EZH2 is part of the protein machinery that shapes the aging epigenome. The conservation in both sequence and methylation patterns of the age-dependent CpGs indicate that the epigenetic clock is a fundamental feature of aging in mammals.
Keywords: Epigenetics, DNA methylation, MBD sequencing, polycomb repressive complex, EZH2
1. Introduction
The genome undergoes extensive epigenetic remodeling over the course of life. Starting from conception, epigenetic processes such as chromatin modification and DNA methylation play important roles in development and cell differentiation, and on the other end of the spectrum, cellular senescence and aging. In vertebrates, a common epigenetic modification is the addition of a methyl group to the 5th carbon of cytosine at CpG dinucleotides. Several human studies have confirmed that methylation levels at particular CpG sites are strongly correlated with age and also associated with health and fitness, and could potentially be an early predictor of lifespan (Hannum et al., 2013; Horvath, 2013; Marioni et al., 2015a; Marioni et al., 2015b; Teschendorff et al., 2010; Weidner et al., 2014). Furthermore, the age prediction from DNA methylation appears to be relatively robust to cell heterogeneity (Horvath, 2013).
This close association between the methylome and aging has led investigators to postulate an “epigenetic clock”, that cells somehow encode the passage of time in DNA methylation and these patterns collectively reflect the chronological age and possibly the biological age or aging rate of an individual (Hannum et al., 2013; Horvath, 2013; Mitteldorf, 2013). Based on the enrichment in polycomb target genes among the age-dependent CpGs, members of the polycomb repressive complex (PRC), particularly PRC2 (e.g., EZH2, SUZ2), have been implicated as potential regulators of the aging methylome (Beerman et al., 2013; Benayoun et al., 2015; Dozmorov, 2015; Sun et al., 2014). However, the mechanisms, and how these impact health and aging trajectories remain unclear.
Another question is, what is the level of conservation in age-dependent methylation at specific CpGs across mammalian species? Majority of the reports on the aging methylome in humans comes from cross-sectional studies in cohorts representing a wide age range. These studies have been largely reliant on high quality microarray platforms that provide an efficient means to assay DNA methylation from large cohorts. The most widely used arrays are the different versions of the Illumina Infinium HumanMethylation BeadChips, which provide reliable coverage of selected CpGs. Methods have been developed to conveniently calculate the epigenetic age (or alternatively, the biological age) from ~350 to as few as 3 probes on the HumanMethylation arrays (Hannum et al., 2013; Horvath, 2013; Weidner et al., 2014) (see https://labs.genetics.ucla.edu/horvath/htdocs/dnamage/weidener2014/ for a comparison of methods). There is however no equivalent method to calculate the epigenetic age in rodents and only few studies have directly compared age-dependent methylation at specific CpGs between humans and mice (Maegawa et al., 2010; Spiers et al., 2016). Generally, there appears to be some conservation, and given the relevance to health and disease, defining an epigenetic measure of age in mice that is comparable to that developed for humans will provide a powerful basis to study the mechanisms and effects of these methylation changes, and provide a means to test agents and interventions that could modify (i.e., delay or even reverse) the epigenetic clock.
Here we performed affinity-based enrichment of methylated CpGs using methyl-CpG binding domain (MBD) protein and high-throughput sequencing (MBD-seq) to profile DNA methylation in aging mouse liver. For the age-dependent differentially methylated regions (age-DMRs), we compared sequence conservation in humans and examined methylation patterns in existing human microarray datasets. We then followed this up with a chromatin immunoprecipitation (ChIP) analysis to examine if EZH2 (enhancer of zeste homologue 2), a member of PRC2 that has been shown to regulate DNA methylation (Vire et al., 2006), interacts with the conserved age-DMRs.
2. Materials and Methods
2.1. Animals and sample preparation
Tissue samples were provided by Dr. Robert W. Williams and were harvested from mice that were part of an aging colony at the University of Tennessee Health Science Center. All mice are housed 3–5 per cage in a temperature-controlled vivarium maintained at a 12 hour light/dark cycle. Mice are given free access to water and standard chow. All animal procedures were in accordance to the guidelines set by the UTHSC Animal Care and Use Committee.
For the DNA methylation study, liver was collected from 11 mice of two different strains: C57BL/6J (B6), and DBA/2J (D2). Mice were at ages ~4 months (mos; young), ~12 mos (mid), and ~24 mos (old). The colony was originally set up to study aging in females and most of the mice that contributed to this study were females. All mice were euthanized on the same day within a 3-hour timeframe. Mice were deeply anesthetized with avertin (250 to 500 mg/kg of a 20 mg/ml solution), then euthanized by cardiac puncture and exsanguination. Liver samples were snap-frozen and stored at −80 degree C until use. We no longer had tissue remaining from the samples used for DNA methylation assay, and for the ChIP study, we used biobanked liver tissue from three B6 female mice (ages at 6 mos, 12 mos, and 24 mos) and three D2 female mice (ages at 6 mos, 12 mos, and 24 mos). These mice were also part of the same aging colony and were euthanized according to the same protocol and tissue snap frozen and stored at −80 degree C.
DNA was purified using the Qiagen AllPrep kit (http://www.qiagen.com) on the QIAcube system. Nucleic acid quality was checked using a NanoDrop spectrophotometer (http://www.nanodrop.com).
2.2. Methylated DNA enrichment
We performed affinity-based enrichment using the MethylCap kit from Diagenode (https://www.diagenode.com). This relies on the methyl binding domain of methyl-CpG- binding protein 2 (MECP2) to capture DNA fragments containing methylated CpGs. DNA in 110 μl TE buffer was ultrasonicated on a Covaris S2 machine (http://covarisinc.com) to an average fragment size of 300–400 bp. The sonication settings were: duty cycle = 10%; intensity = 5; cycles/burst = 100; total of 12 minutes. DNA quality and fragment size were evaluated on the Agilent Bioanalyzer (http://www.agilent.com). Prior to processing with MethylCap, ~1μg of fragmented DNA was concentrated using the DNA Clean & Concentrator™-5 product from Zymo Research (http://www.zymoresearch.com). Enrichment was performed on these samples according to the standard MethylCap protocol and captured DNA eluted in a single step using the High Elution Buffer. Final purified DNA was eluted with 32 μl of TE buffer and 2 μl of this was used to check for DNA concentration on the Qubit fluorometer (https://www.thermofisher.com). The remaining 30 μl was used for library preparation and sequencing.
2.3. High throughput sequencing
Sequencing was done on the Life Technologies Ion Proton platform at the UTHSC Molecular Resource Center. The DNA fragments were used to prepare barcoded libraries employing the Ion Xpress™ Plus Fragment Library Kit and the Ion Xpress™ Barcode Kit from Life Technologies (http://www.thermofisher.com) according to protocol supplied by the manufacturer. After library preparation, the barcoded libraries were screened on an Agilent High Sensitivity DNA chip for size distribution. As an initial step, 1ul of each barcoded library was pooled and sequenced on an Ion Torrent PGM 314 chip. The read counts from the 314 chip were then used to prepare a final equalized pool. The final library pool was quantified by real-time polymerase chain reaction (PCR), used to prepare beads, and finally sequenced using the P1 chip on the Ion Torrent Proton sequencer. To avoid batch artifacts related to fragment library-processing steps and chip runs, we multiplexed the 11 samples and ran multiple chips to reach depth of ~30 million raw reads per sample. In total, seven P1 chips were used.
2.4. Read alignment and data quality control
FASTQ files were processed with FastQC tools (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to generate chip level reports with statistics on quality score distributions, complexity, length distributions, and PCR duplication level. This ensures that there are fewer technical problems that might be difficult to correct at a later stage. Low-quality ends of reads were trimmed with Trimmomatic v0.30 (Bolger et al., 2014) using a Phred score of 15 (Q15), a 10 bp sliding window and a minimal read length of 40 bp. Trimmed reads were aligned against the mouse mm10 reference genome (Ensemble GRCm38) using the TMAP (https://github.com/iontorrent/TMAP) aligner with default settings. TMAP is commonly distributed with the Ion Torrent technology and designed specifically to align Ion Torrent reads with high homopolymer length errors and is able to handle reads with varied lengths. Samtools was used to sort and index aligned (bam) files (Li et al., 2009). Reads with mapping quality of less than 10 were filtered from the bam file using ‘samtools –q 10’ command. For each sample, there were seven high quality bam files (each from the seven separate runs) and these were merged and indexed using samtools. The full MBD-seq data is available from the NCBI NIH Gene Expression Omnibus (GEO accession ID GSE95361 at http://www.ncbi.nlm.nih.gov/geo/). Raw sequence data is available from the NCBI NIH Sequence Repository Archive (SRA accession ID SRP100703 at https://www.ncbi.nlm.nih.gov/sra).
The generated Bam files were then analyzed using the MEDIPS R package and visualized on the Integrative Genomics Viewer (Chavez et al., 2010; Lienhard et al., 2014; Robinson et al., 2011; Rodriguez et al., 2012; Thorvaldsdottir et al., 2013). To assess if we have sufficient coverage for reliable and reproducible results, we first performed a saturation analysis (MEDIPS.saturation) and this showed sufficient sequencing depth. We then examined pair-wise correlation among the samples, and calculated CpG coverage and enrichment in the sequenced fragments (Data S1). Sequenced reads were then counted for every 500 bp non-overlapping window with normalization to the local CpG density (i.e., coupling factor CF). We used only the high quality uniquely aligned reads and excluded duplicate reads. The percent of uniquely aligned and non-duplicate reads is similar to what has been previously reported for MBD-seq (Aberg et al., 2015; De Meyer et al., 2013). This process was done according to the following MEDIPS parameters: uniq=1, extend=300, shift=0, ws=500. This divided the mouse genome into 5451088 total bins with coverage counts at each bin.
2.5. Statistics for differential methylation analysis
The read counts generated on the MEDIPS package were loaded to the EdgeR R package for differential methylation analysis (Robinson et al., 2010). Prior to statistical tests, we excluded the sex chromosomes. We also removed all bins with low coverage as these can provide no reliable statistics but will only add to the penalty for multiple testing. We filtered out regions with coupling factor (CF) below the genome-wide median of 3, and bins that had less than one count per million in 2 or more samples. This resulted in 466039 bins that have an average CpG count of 9.5 (minimum of 3 and maximum of 91) and mean count of 16.7 (minimum of 4 and maximum of 1167). We applied a generalized linear regression model on this set of 466039 bins to evaluate the effect of age. We classified the mice into three age categories: young, mid and old. A regression model was then applied to detect differential methylation across the three age groups with adjustment for strain differences (~strain + ageGroups) (full results and R codes are provided in Data S2). For this we applied the EdgeR function glmFit function followed by glmLRT to calculate the likelihood ration statistics.
2.6. Bioinformatics analysis of age-DMRs
We used RefSeq annotations (http://www.ncbi.nlm.nih.gov/refseq/) to assign gene names and gene features to each bin (i.e., 5′ UTR, exon1, intron1, exons, introns, 3′UTR). Only regions within annotated gene boundaries were considered. To examine enrichment in specific gene features among the age-DMR, we applied the hypergeometric test in R (phyper) and compared the percent represented in the age-DMR set relative to the genome-wide background set (the total 466039 bins on which statistical test was performed). Gene function enrichment analysis was done using DAVID 6.7 (https://david.ncifcrf.gov).
For the genome-wide significant age-DMRs, degree of sequence conservation between mice and humans was examined using the BLAT tool provided by the UCSC Genome Browser (http://genome.ucsc.edu/). For Srd5a2, Rfx4, and Ptk7, adjacent age-DMRs bins were combined to a 1000 bp window. The 500 bp or 1000 bp sequences in the mouse GRCm38/mm10 reference genome were compared to the human GRCh37/hg19 reference genome. We also utilized the UCSC Genome Table Browser interface and computed the average conservation scores for the age-DMRs using the phastCons method for placental mammals (Siepel et al., 2005). Conservation scores for these 500 bp age-DMRs was then compared to a set of ~13500 randomly selected 500 bp bins that are not age-DMRs. ChIP-seq annotation on DNA binding protein factors were then retrieved from the ENCODE Transcription Factor ChIP-seq (161 factors) track that is accessible through the UCSC Genome Browser. We also retrieved probes in the Illumina Infinium HumanMethylation450 BeadChips that map to the conserved regions. DNA methylation beta values were then extracted from four human datasets (GEO accession IDs GSE61258, GSE48325, GSE41169, GSE40279). These consist of two liver and two blood HumanMethylation450 datasets (Ahrens et al., 2013; Hannum et al., 2013; Horvath et al., 2014; Horvath et al., 2012). Using these resources, we performed Pearson correlations to evaluate if there is consistent and significant correlation between DNA methylation and human age at these conserved sites.
2.7. ChIP-qPCR protocol and analysis
Samples were sent to Active Motif (http://www.activemotif.com) for ChIP-qPCR. In brief, liver tissue was submerged in PBS + 1% formaldehyde, cut into small pieces and incubated at room temperature for 15 minutes. Fixation was stopped by the addition of 0.125 M glycine. The tissue pieces were then treated with a Tissue Tearer homogenizer and spun down and washed 2x in PBS. Chromatin was isolated by adding lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and DNA sheared to an average length of 300–500 bp with Active Motif’s EpiShear Probe Sonicator and the Cooled Sonication Platform. The input genomic DNA was prepared by treating aliquots of chromatin with RNase, proteinase K and heated for decrosslinking, followed by ethanol precipitation. Pellets were re-suspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantification of the total chromatin yield. An aliquot of mouse liver chromatin (40 ug) was precleared with protein A agarose beads (Invitrogen; https://www.thermofisher.com). Genomic DNA regions of interest were isolated using an antibody against EZH2 (http://www.activemotif.com, cat # 39901). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubating overnight at 65 degree C, and the ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation.
Nine primer pairs were designed to cover the 5′ region age-DMRs in C1ql3 (mm10 chr2:13009968-13011298), Srd5a2 (chr17:74047228-74047931), and Ptk7 (chr17:46628561-46629567), and the intronic age-DMR in Rfx4 (chr10:84758977-84760490). Primers were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) and ordered from IDT (https://www.idtdna.com). Depending on the size of the test site, each CGI was assayed by 1 (for Srd5a2), 2 (Ptk7), or 3 (C1ql3 and Rfx4) primer pairs (primers listed in Data S5). A positive and negative control sites were also included. The positive control was a site in Ccnd2 that is known to be bound by EZH2 in the mouse, and the negative control was a region on the genome with no annotated genes within 100–200 kb and known to give very low binding signal. Quantitative PCR (qPCR) reactions were carried out in triplicates for each primer pair using SYBR Green Supermix (http://www.bio-rad.com). The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using input DNA.
The normalized values for the technical triplicates were averaged (Data S6). For CpG regions that were interrogated by more than one primer pair, we pooled the normalized data and applied a linear regression analysis to evaluate binding signal as a function of two age categories (group 1: 6 mos and 12 mos; group 2: 24 mos) with strain as covariate.
3. Results
3.1. MBD-seq data quality
DNA methylation assay was done in mouse liver tissue at three age groups: young at 4 mos, mid at 10–12 mos, and old at 24 mos. Sample details are provided in Table 1. Starting with ~1 μg of fragmented input DNA (average fragment size ranging from 300–400 bp), we captured an average of 18 ± 8 ηg of methylated DNA. Sequencing resulted in 10.5–15.8 million uniquely aligned reads per sample. Quality metrics including pair-wise correlation between samples, read coverage, and CpG enrichment profiles are provided in Data S1. The average correlation between each pair of samples is high at 0.85. The MBD protein has higher affinity for methylated CpGs and the captured samples were enriched for CpGs (average CpG enrichment score in sequenced fragments relative to reference genome = 2.24, SD ± 0.18). We calculated the number of read fragments that cover each “CG” pattern in the mouse mm10 reference genome. On average, 55% of CGs was covered by at least 1 read, and of these, 15% was covered by more than 5 reads (Data S1).
Table 1.
Sample information, MBD-based DNA yield, and alignment counts
| Sample | Strain | Age (months) | Sex | Captured DNA (ng)1 | Uniquely aligned reads (UAR) | Non-duplicate UAR |
|---|---|---|---|---|---|---|
| Liver1 | DBA/2J | 4 | F | 9.51 | 13,369,898 | 10,659,297 |
| Liver2 | DBA/2J | 4 | F | 9.84 | 11,018,283 | 8,860,034 |
| Liver3 | DBA/2J | 11 | F | 20.49 | 13,394,600 | 10,468,283 |
| Liver4 | DBA/2J | 11 | F | 15.54 | 13,533,994 | 10,837,155 |
| Liver5 | DBA/2J | 12 | M | 35.7 | 10,463,616 | 8,495,368 |
| Liver6 | DBA/2J | 24 | F | 12.93 | 15,770,977 | 12,691,701 |
| Liver7 | C57BL/6J | 4 | F | 16.38 | 11,649,128 | 9,109,375 |
| Liver8 | C57BL/6J | 4 | F | 26.67 | 15,831,564 | 12,564,343 |
| Liver9 | C57BL/6J | 10 | M | 19.29 | 12,674,509 | 10,058,270 |
| Liver10 | C57BL/6J | 24 | F | 19.05 | 14,227,699 | 11,147,770 |
| Liver11 | C57BL/6J | 24 | F | 13.23 | 13,279,919 | 10,571,366 |
Amount of captured DNA from ~1 μg total fragmented input DNA
3.2. Differentially methylated regions
For quantification, we counted read fragments at every 500 bp non-overlapping bins. As expected, read coverage was directly proportional to the CF, which is the number of CpGs in a given 500 bp window (Down et al., 2008). Regions with CF below the genome-wide median of 3 had correspondingly low coverage with only 0 to 3 reads (Data S1). We excluded these bins with low CpGs density and little to no coverage. Statistical test was then applied to the remaining 466039 bins that had an average CF of 9.5, and covered by an average of 17 reads per window.
As samples were obtained from two different mouse strains (see Table 1), we used a multiple regression model to examine effect of age group with adjustment for strain effect. At a stringent Bonferroni threshold of 5%, 9 sites located within 6 different genes have significant age-dependent differential methylation (Table 2). The top 8 most significant age-DMRs showed increase in methylation with age (age-hypermethylation). For C1ql3 (Fig. 1A), Srd5a2 (Fig. 1B), Rfx4, and Ptk7, the age-hypermethylation occurs at CpG islands (CGIs) overlapping the 5′ features of genes and likely to encompass transcription regulatory regions. The intronic region in Zfand3 showed decrease in methylation with age (age-hypomethylation). Strain did not have a significant effect on methylation at these top age-DMRs (Data S3). For strain specific effect, at a Bonferroni threshold of 5%, we find a total of 4104 sites with differential methylation between B6 and D2. We refer to these as strain-DMRs. We must note that since the alignment was done to the B6 mouse reference genome, such purported differences in methylation may be partly due to underlying sequence variation between strains. As expected, we find a much larger proportion of strain-DMRs that have higher methylation in B6 relative to D2 (3541 out of the 4104). These sites may be either due to true differences in methylation level, or due to sequence variation that results in more efficient alignment for the B6 reads. The full statistics for the complete set of methylation bins are provided in Data S2. We however focus on the age-DMRs for further analysis.
Table 2.
Genome-wide significant age-dependent differentially methylated regions at Bonferroni corrected p-value < 0.05
| Coordinate (mm10) | Gene | Gene location1 | CF2 | logFC (young vs mid)3 | logFC (young vs old)4 | p-value | Bonf p-value |
|---|---|---|---|---|---|---|---|
| chr2:13010001-13010500 | C1ql3 | intron1, exon1; CGI | 35 | 1.84 | 3.38 | 4.4E-13 | 2.0E-07 |
| chr17:74047501-74048000 | Srd5a2 | intron1, exon1, 5′UTR; CGI | 31 | 4.31 | 5.89 | 5.9E-13 | 2.7E-07 |
| chr10:84760001-84760500 | Rfx4 | intron1; CGI | 33 | 2.27 | 3.22 | 1.2E-09 | 5.5E-04 |
| chr17:74047001-74047500 | Srd5a2 | intron1; CGI | 18 | 2.30 | 3.31 | 1.8E-09 | 8.3E-04 |
| chr17:46629001-46629500 | Ptk7 | intron1, exon1, 5′UTR; CGI | 59 | 3.67 | 4.17 | 5.5E-09 | 0.003 |
| chr17:46628501-46629000 | Ptk7 | intron1; CGI | 30 | 1.79 | 2.17 | 6.0E-09 | 0.003 |
| chr10:84759501-84760000 | Rfx4 | intron1 | 11 | 2.87 | 4.20 | 7.8E-08 | 0.036 |
| chr6:28832501-28833000 | Snd1 | intron16; CGI | 68 | 2.51 | 1.85 | 1.0E-07 | 0.048 |
| chr17:30116501-30117000 | Zfand3 | intron2 | 11 | −1.49 | −1.24 | 1.1E-07 | 0.049 |
CGI is CpG island
Coupling factor or the number of CpGs in the 500 bp bin
Log2 fold change between young and middle aged mice (positive is increase in methylation with age, negative is decrease in with age)
Log2 fold change between young and old aged mice (positive is increase in methylation with age, negative is decrease in with age)
Fig. 1.

Methylation patterns in (A) C1ql3 (full gene shown) and (B) Srd5a2 (only 5′ region shown). The 5′ regions of both genes harbor CpG islands (CGI; shown as green block) that undergo progressive increase in methylation with age (Bonferroni p < 0.05). Number of mice per age group are young = 4, mid = 4, old = 3. Combined read coverage is depicted for young (yellow), mid (green), and old mice (maroon). Horizontal bars below the coverage map show the aligned read fragments. These CGIs are conserved in humans and are targeted by CpG probes in the Illumina Infinium HumanMethylation450K BeadChips. Most show significant positive correlation with age in human liver tissue and correlation between age and example probes for C1QL3 (Pearson R = 0.66, p-value < 0.0001) (C) and SRD5A2 C1QL3 (Pearson R = 0.44, p-value < 0.0001) (D) are shown.
3.3. Characteristics of age-DMRs
To survey the overall trend, we lowered the stringency to a nominal p-value ≤0.0005 (FDR of 0.60) and 402 regions passed this lenient statistical threshold. To minimize type 2 error, we considered only the 330 regions that showed consistent direction of change (i.e., either hyper- or hypomethylation with age) in comparisons between young vs. mid, and young vs. old age groups (Data S3). While the strongest effect of age (i.e., those significant above Bonferroni threshold in Table 2) involve increase in methylation, 63% (209 sites) of the age-DMRs selected at nominal p-value showed decreased methylation, and only 37% (121 sites) showed increased methylation with age. On average, there is higher CpG density in hypermethylated regions compared to hypomethylated regions (mean and SD of 13.12 ± 0.76 vs. 11 ± 0.58; p-value = 0.028), and the fold-increase in methylation from young to old is also positively correlated with CpG density (Pearson correlation between CF vs. log-fold change in methylation at the 330 sites is 0.26, p-value < 0.0001; Data S3).
About 68% of the age-DMRs (226 out of 330) are within annotated genes (Fig. 2). In particular, intragenic regions are overrepresented among age-hypermethylated DMRs (enrichment p-value = 0.0007 based on hypergeometric test). For a general overview of gene features among the age-DMRs, we used a simple classification based on RefSeq annotations and intragenic regions were classified as 5′ UTR, exon1, intron1, exon (all exons other than the exon1), intron (all introns other than intron1), or 3′ UTR. The percent of bins that overlap these gene features in the background set (the total 466039 bins), the age-hypermethylated, and the age-hypomethylated regions are shown in Fig. 2. Compared to the background, the set of age-hypermethylated DMRs is significantly enriched in bins that overlap exon1 and intron1 (enrichment p-value = 1.5 × 10−5 and 0.02, respectively) and contain annotated CGIs and have higher average CpG density (Fig. 2). The age-hypomethylated set is enriched in regions overlapping 5′ UTRs (enrichment p-value = 0.0003), intron1 (p-value =0.03), and 3′ UTRs (p-value = 0.01).
Fig. 2.

Genomic features and intergenic regions among sequenced CpGs. RefSeq gene annotations were classified as 5′ UTR, exon1, intron1, all introns except intron1, all exons except exon1, and 3′ UTR. Average CpG density and standard deviation (within parenthesis) are shown below percentages. CpG bins that increase in methylation with age (age-hypermethylated DMRs) have relatively higher proportion of exon1 and intron1 and higher CpG density compared to the background set of 466039 bins. Age-hypomethylated DMRs is enriched in 5′ UTRs and intron1, and 3′ UTRs.
These genes belong to a variety of gene ontology categories (Data S4). For the smaller set of genes with hypermethylated CpGs, we find no significant enrichment by gene function other than 12 genes that are in the GO “cytoskeleton” category (Benjamini corrected enrichment p = 0.07). For the set of genes with hypomethylated CpGs, there are 53 phosphoprotein genes (Benjamini corrected enrichment p = 0.01), including Apoe and Apoa1. There is also slight enrichment in HDAC pathway genes (Benjamini corrected enrichment p = 0.03) and genes involved in vasculogenesis (Benjamini corrected enrichment p = 0.04).
3.4. Conservation in sequence and methylation pattern of age-DMRs in humans
For the genome-wide significant age-DMRs in Table 2, we examined (1) the level of sequence conservation, and (2) whether methylation patterns at the conserved sites show consistent effect of aging in humans. With the exception of the intronic site in Zfand3, all other sites have between 92% to 100% conservation in base sequence in humans and overlap conserved CGIs (Table 3). We used existing human methylation data from two liver (Ahrens et al., 2013; Horvath et al., 2014) and two blood datasets (Hannum et al., 2013; Horvath et al., 2012) to examine correlation between methylation at these CpGs and human age. Since the age-hypomethylated region in Zfand3 is not conserve, all the conserved age-DMRs in Table 2 are age-hypermethylated regions. To evaluate if negative correlations are also consistent between the two species, we selected the next two conserved sites that showed age-hypomethylation. These are both bins overlapping 3′UTR and last exon regions of Apoa1 (uncorrected p-value = 6.8E-07 and Bonferroni p-value = 0.3) and Serpina1b (uncorrected p-value = 6.9E-07 and Bonferroni p-value = 0.3).
Table 3.
Mouse-human comparison of age-DMRs
| Gene | Mouse mm10 | Human hg191 | Conservation (mm10-hg19)1 | CGI?2 | #450K probes3 | Protein Factors (ENCODE data) 4 |
|---|---|---|---|---|---|---|
| C1QL3 | chr2:13010001-13010500 | chr10:16,562,204-16,562,717 | 100.0% bases, 100.0% span | 5′UTR, Exon1 CGI | 8 | EZH2, RBBP5, CTBP2, CTCF |
| SRD5A2 | chr17:74047001-74048000 | chr2:31,805,006-31,806,119 | 95.8% bases, 100.0% span | 5′UTR, Exon1 CGI | 8 | EZH2, TBP |
| PTK7 | chr17:46628501-46629500 | chr6:43044006-43045132 | 96.0% bases, 100.0% span | 5′UTR, Exon1 CGI | 5 | CHD2, CTBP2, E2F1, EGR1, ELF1, HDAC1, HDAC2, HDAC6, HMGN3, JUN, JUND, MXI1, PHF8, POLR2A, RAD21, RBBP5, REST, RFX5, SIN3A, SIN3AK20, SP1, TAF1, TBP, UBTF, YY1, ZBTB7A, ZNF143 |
| ZFAND3 | chr17:30116501-30117000 | na | Not conserved | na | na | na |
| RFX4 | chr10:84759501-84760500 | chr12:106980246-106981266 | 92.9% bases, 100.0% span | Intron1 CGI | 9 | EZH2, SUZ12, RBBP5 |
| SND1 | chr6:28832501-28833000 | chr7:127671585-127672065 | 95.6% bases, 100.0% span | Intron CGI | 9 | RBBP5, CHD1, RUNX3, SUZ12, POLR2A, E2F1, CTCF, RAD21, SIN3AK20, FOS, GATA2, TAF1, MAFK |
| SERPINA1B | chr12:103728001-103728500 | chr14:94844558-94845035 | 92.4% bases, 100.0% span | Last exon, 3′UTR | 1 | POLR2A |
| APOA1 | chr2:13010001-13010500 | chr11:116706431-116706931 | 98.4% bases, 100.0% span | Last exon, 3′UTR CGI | 4 | POLR2A, JUND, RBBP5, PHF8, PML, TAF1, HMGN3, KAP1, REST, ZNF263, CCNT2, TCF7L2, MAX, CTBP2, E2F6, MYC, MXI1, ELF1, FOXM1, SP1, NRF1, PAX5, TCF3, E2F4, TRIM28, BACH1, GABPA, MAZ, ZBTB7A, FOXP2, BHLHE40, HDAC6, YY1, SRF, IRF4, CTCF, TCF12, GATA3, ZNF143, RUNX3, RCOR1, SMC3, RAD21, CTCFL, CBX3, NR3C1, ESR1 |
Sequence conservation between mouse reference genome (mm10) and human reference genome (hg19)
Gene features and whether site overlaps CpG island (CGI) or not
Number of probes in the Illumina Infinium HumanMethylation450 BeadChips that map to the conserved region
Protein factors that bind to the age-DMRs; data from transcription factor ChIP-seq for 161 factors generated by ENCODE Consortium for human cell lines
A total of 44 probes in the Illumina Infinium HumanMethylation450 platform map to these conserved CpGs. We note that the CpG probe cg19502744, which targets the SRD5A2 CGI, has been previously reported as an age-DMR in human blood by Horvath et al. (2012) and by Teschendorff et al. (2010). The probe cg21663580, which targets the PTK7 CGI, has also been associated with age by Horvath et al. (2012). This indicates a conserved effect of age that is not just specific to liver but also seen in blood. We performed correlation analysis between methylation beta values detected by these probes and age in the four human datasets. The block of CpG probes in the 5′ regions of C1QL3 (Fig. 1C), SRD5A2 (Fig. 1D) and RFX4 have particularly strong positive correlation with age in liver and this effect is also replicated in blood (Fig. 3; Data S5 provides correlation results for all 44 probes). For the probe in the SERPINA1B 3′UTR, there is significant negative correlation with age in the liver data from Ahrens et al. (2013). However, the negative association with age at the 3′UTR region of Apoa1 is not replicated in any of the human data sets. For these large effect age-DMRs, our results show that age dependent methylation patterns are largely conserved between mice and humans, and while more pronounced in liver, the positive correlation can also be detected in non-liver tissue, suggesting a level of cross-tissue effect of aging.
Fig. 3.

Correlation between age and DNA methylation at conserved age-dependent differentially methylated regions (age-DMR) in humans. The top age-DMRs in mice have high sequence conservation in humans and probes in the Illumina Infinium HumanMethylation450K BeadChips that target these sites were extracted from two liver and two blood datasets. The plots show Pearson correlations between human age (x-axis) and methylation beta-values (y-axis) for some of the probes. Datasets and GEO accession numbers are Ahrens et al. GSE48325 (first column of scatter plots), Horvath et al. GSE61258 (second column), Horvath et al. GSE41169 (third column), and Hannum et al. GSE40279 (forth column). Gene names are shown on the right.
We next considered the average conservation scores for the larger set of 330 age-DMRs and examined if the degree of conservation relates to level of age-dependent methylation (average conservation scores in Data S3). Based on comparison between these age-DMRs and randomly selected methylation bins that are not age-DMRs, we see no significant difference in average conservation. However, among the 330 age-DMRs, there is significant association between conservation score and level of age-dependent methylation. Specifically, we find that associated p-value of age effect (−log10P) and fold-increase in methylation are higher in more conserved regions (Pearson R = 0.35, p-value = 0.0001 for −log10 of age effect p-value, and Pearson R = 0.28, p-value = 0.0025 for fold-change from young to old; Fig. 4). We see no such correlation between levels of conservation and methylation at age-hypomethylated sites.
Fig. 4.
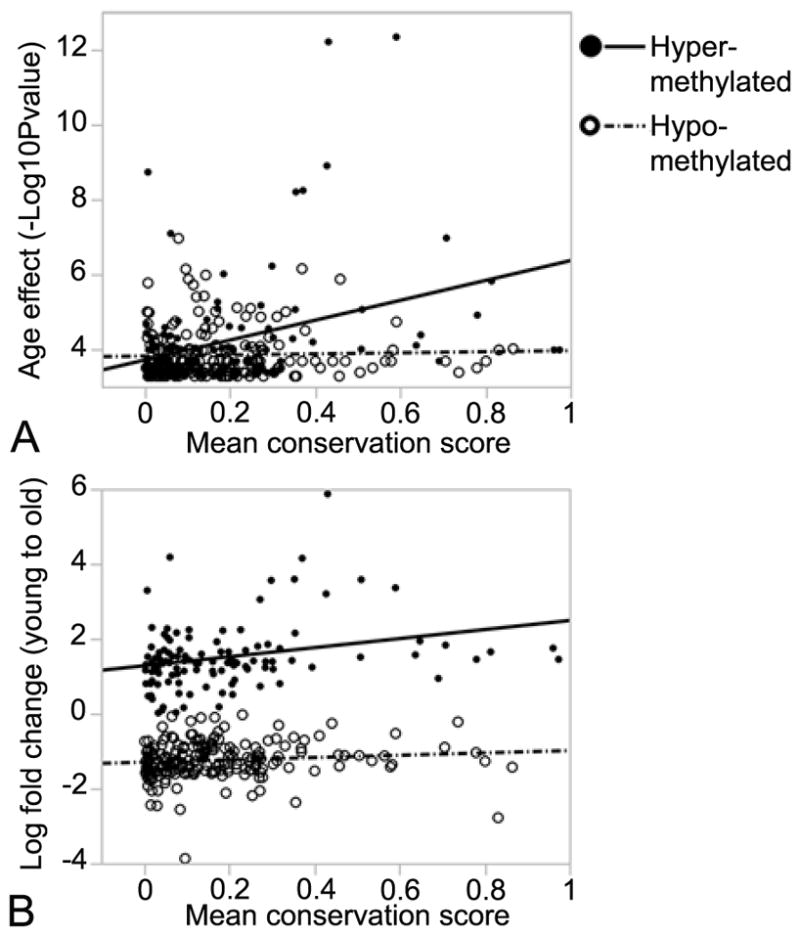
Correlation between conservation score and level of age-dependent methylation in mouse liver. For the 330 age-dependent differentially methylated regions (age-DMRs), mean conservation scores based on phastCons method was calculated for each of the 500 bp bins. For age-DMRs that increase in methylation with age (age-hypermethylated), there is significant positive correlation with (A) −log10P of age effect (Pearson R = 0.35, p-value = 0.0001), and (B) log2 fold-change in methylation (Pearson R = 0.28, p-value = 0.0025). For age-DMRs that decrease in methylation with age (age-hypomethylated), there is no significant correlation with level of conservation.
3.5. Binding of EZH2 to age-DMRs
To identify potential regulators of the aging methylome, we examined protein factors that bind to these conserved sites using ChIP-seq data generated by the ENCODE Consortium (Consortium, 2012). This data provides DNA peak intensities for 161 DNA binding factors summarized from 91 human cell lines. For the top age-DMRs that are highly conserved in humans, we catalogued different factors that interact with these sites (Table 3). In agreement with previous reports (Beerman et al., 2013; Dozmorov, 2015; Taiwo et al., 2013), we find that EZH2 and SUZ12 (both members of the PCR2 complex) and RBBP5 associate with the age-hypermethylated CGIs. For the conserved age-hypomethylated regions in SERPINA1B and APOA1, POLR2A is the common factor that binds to both sites.
We selected EZH2 for ChIP analysis to evaluate (1) if EZH2 associates with these age-DMRs in mice, and (2) if there is age effect in binding signal. We selected the conserved age-hypermethylated CGIs in C1ql3, Srd5a2, Ptk7, and Rfx4 for targeted ChIP analysis. We used liver samples from three female B6 mice (ages at ~6 mos, 12 mos, and 24 mos) and three female D2 mice (ages at ~6 mos, 12 mos, and 24 mos). On average, EZH2 binding was detected at over 2- to 5-fold above background in all four test sites (Data S6). The exception was the liver sample from the oldest D2 (~24 mos old), which showed low EZH2 binding signal that was comparable to background at all test sites but showed strong signal at the positive control site.
This is a small samples size with only two samples per age group. For the target sites in C1ql3 and Srd5a2, average EZH2 binding was the lowest at age 24 mos compared to the other age groups (Fig. 5). We grouped the samples into two categories (6 mos and 12 mos as age group 1, and 24 mos as age group 2) and performed a regression analysis to examine the effect of age with strain as a covariate (Table 4). At a lenient statistical threshold of nominal p-value < 0.05, we find significantly lower binding at age 24 mos in the C1ql3 and Srd5a2 CGIs. The effect of age was not significant for Rfx4 and Ptk7. Strain also showed an effect on EZH2 binding levels at Srd5a2, Ptk7, and Rfx4 with on average lower binding in D2 compared to B6, particularly for Rfx4. Our results show that EZH2 associates with these conserved age-hypermethylated CGIs. While the sample size is small and the p-values nominal, there is modest evidence that EZH2 binding state may decrease with age in the CGIs located in the 5′ regions of C1ql3 and Srd5a2.
Fig. 5.
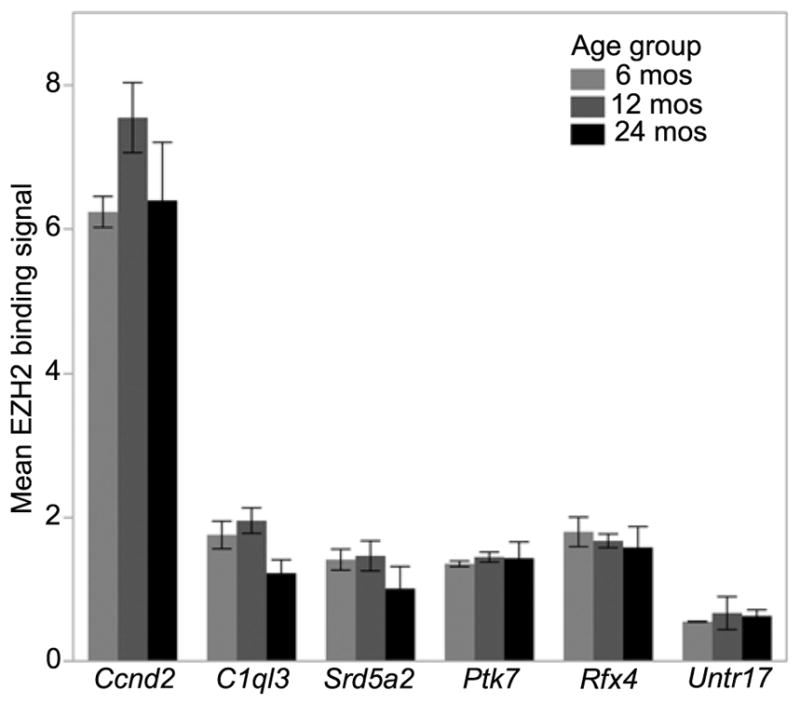
Binding signal for EZH2. Average binding signal (± standard errors) are shown for the three age groups at the four differentially methylated CpG Islands in C1ql3, Srd5a2, Ptk7, and Rfx4. The region in Ccnd2 provides the positive control and Untr17 provides the negative control. Two mice were used for each age group.
Table 4.
EZH2 binding as a function of age
| Gene | Age effect | Strain effect | ||||
|---|---|---|---|---|---|---|
| Estimate | t-ratio | p-value | Estimate | t-ratio | p-value | |
| C1ql3 | −0.63 | −2.74 | 0.015 | 0.05 | 0.48 | 0.64 |
| Srd5a2 | −0.43 | −5.07 | 0.015 | 0.22 | 5.53 | 0.012 |
| Ptk7 | 0.03 | 0.24 | 0.82 | 0.15 | 2.29 | 0.048 |
| Rfx4 | −0.15 | −0.91 | 0.38 | 0.36 | 4.55 | 0.0004 |
Regression model was EZH2 binding as a function of age + strain; negative t-ratio for age effect indicates decreased binding at age 24 mos. Positive t-ration for strain effect indicates higher binding in C57BL/6J compared to DBA/2J
4. Discussion
In summary, we find robust effect of aging on DNA methylation in differentiated mouse somatic tissue. Despite the relatively small sample size (3–4 animals per age group), we detect age-DMRs at stringent genome-wide significance threshold. These top age-DMRs have high sequence conservation in humans. Furthermore, the pattern in DNA methylation with age is largely conserved between the two species, and we find that age-dependent increase in methylation is more pronounced in methylation bins with conserved sequences. Our results further add to the growing evidence that member of PRC2 play a role in shaping the aging methylome. Specifically, we show that CGIs that undergo age-hypermethylation associate with EZH2, and there may be age-dependent decline in EZH2 binding at two of the most significant age-DMRs in C1ql3 and Srd5a2.
4.1. MBD-seq of the aging methylome highlight cancer-related genes
For this work, we used liver samples, an organ that is reported to be particularly sensitive to the effects of aging and health (Horvath et al., 2014). The liver also presents a relatively homogeneous cellular composition, at least compared to more complex tissues such as blood or brain. Among the genome-wide significant age-DMRs, only one showed age-dependent decrease, and the remaining were hypermethylation at specific CGIs that were in 5′ features of genes (i.e., transcription start sites, 5′UTRs, and first exons and introns). However, majority of the sites selected at a nominally significant threshold involved decrease in methylation with age. Our observations are consistent with previous reports that aging is associated with (1) an overall decrease in methylation, particularly at low CpG density regions, and (2) a contrasting site-specific increase at promoter CGIs (Maegawa et al., 2010; McClay et al., 2014; Teschendorff et al., 2010). The aging methylome therefore appears to represent a reversal of what occurs during development, and resembles methylation features associated with tumor development (Illingworth et al., 2010; Teschendorff et al., 2010). Interestingly, among the 6 genes with the most significant age-DMRs (Table 2), C1QL3, SRD5A2, PTK7 and SND1 are linked to the development and progression of different cancers. SRD5A2 (steroid 5 alpha-reductase 2) is an androgen metabolic gene linked to prostrate and breast cancer and shown to undergo age-dependent hypermethylation and downregulation of expression (Ge et al., 2015; Hein et al., 2012). PTK7 (protein tyrosine kinase 7) plays a role in the Wnt signaling pathway and linked to several different cancers and reported to undergo hypermethylation in hepatic cancer cells (Dunn and Tolwinski, 2016; Hishida et al., 2015). SND1 (staphylococcal nuclease and tudor domain containing 1), a transcriptional co-activator, is implicated in different cancers and methylation status of this gene is considered as a potential biomarker of colorectal cancer (Naumov et al., 2013; Navarro-Imaz et al., 2016). At least one recent study has also implicated hypermethylation of C1QL3 (complement C1q like 3) as a biomarker of metastatic breast cancer (Legendre et al., 2015). These specific examples suggest that DNA methylation may be the molecular mediator that links development with aging and the increased susceptibility to tumorigenesis with age.
4.2. Technical considerations
The aging samples we used came from two mouse strains: B6 and D2. Our main focus was on the effect of age, and we applied a regression model with age as a categorical variable blocked by strain. Both strains are also represented in all three age groups and we are confident that for the strong age-DMRs we identified are true effects of age and not confounded by strain differences. While we report the strain effect, these should be considered with the caveat that underlying sequence differences between the two strains can result in differential efficiency in alignment and spurious quantitative differences in methylation levels. Our study sample also included two males in the middle-age category (see Table 1). To mitigate sex effect, we excluded the sex chromosomes and based our analysis on contrasts between young (all females) vs. mid (2 males, 2 females), and young (all females) vs. old (all females) groups. We considered only the 330 age-DMRs that showed consistent direction of effect in both the middle and old aged groups relative to the young.
We should note that we used a broad window to count read fragments (500 bp non-overlapping bins) and the genomic feature annotations are somewhat imprecise since a bin is likely to overlap multiple adjacent gene features (for instance, 5′ features such as the UTR and first exon and intron). Additionally, we considered only one representative RefSeq transcript and did not account for alternative isoform variants. We used a broad bin since the fragment sizes of the input DNA were on average 300–400 bp. While MBD-seq lacks the resolution provided by other methods such as bisulfite sequencing, it is a cost- and computationally-efficient approach to measure CpG methylation on a genome-wide scale (Aberg et al., 2012; Bock et al., 2010; De Meyer et al., 2013; McClay et al., 2014). Our goal here was to provide a broad overview of the aging mouse methylome and since methylation of CpGs within 500 bp is generally correlated (Li et al., 2010; Lovkvist et al., 2016; Zhang et al., 2015), this approach successfully captures the overall pattern within a circumscribed region.
4.3. Mouse-human comparison
A chief motivation of this study was to compare sequence conservation and consistency in DNA methylation between mice and human. Considerable work has been done on aging and DNA methylation in humans and it is well established that there is extensive modification over the course of life. There is also evidence that the biological age estimated by the “epigenetic clock” is associated with health and fitness, and purportedly, human lifespan (Marioni et al., 2015a; Marioni et al., 2015b). In one recent report, Spiers and colleagues (2016) performed targeted analysis of DNA methylation at four loci in the mouse genome that are homologous to age-associated CpGs in humans. The analysis was done in tissues from blood, lung, hippocampus and cerebellum. They found conservation in age-dependent methylation particularly in blood, which is generally the tissue used in human studies. Another study found partial conservation in age-dependent methylation in intestinal tissue between humans and mice (Maegawa et al., 2010).
Based on average conservation scores for the 330 age-DMRs, we find that regions that are more conserved tend to show higher effect of age-dependent hypermethylation. These age-hypermethylated sites are also enriched for annotated gene features and have higher CpG densities (i.e., the CF values provided in Data D2 and S3). Our results are consistent with previous report that in higher vertebrates, conserved functional sequences have, on average, higher density of CpG dinucleotides compared to the rest of the genome (Jiang et al., 2014). Taken together, these observations suggest that age-dependent increase in methylation occurs at specific CpG rich and conserved regions of the genome, and may be a conserved feature of the aging epigenome in mammals. We however do not find a correlation between age-hypomethylation and degree of conservation.
We performed a more detailed sequence comparison between mice and humans for the few sites that were age-DMRs in mouse liver at a genome-wide p-value threshold. For these, we also examined the pattern of age-dependent methylation in human tissue at corresponding regions using data from Illumina Infinium HumanMethylation450. It is noteworthy that at least two probes that we identified (cg19502744 in SRD5A2 CGI, and cg21663580 in PTK7) have been previously reported in the human literature as age-DMRs, indicating the robust and replicable effect of age. Our results show that strong age-dependent methylation occurs in conserved regions, and the pattern of methylation is also generally consistent between mice and humans. While the replication is better in the same tissue (i.e., liver), we detect weaker but consistent effects in human blood. This conveys some level of tissue-independent cross-species effect. The stronger concurrence in the same tissue may explain why none of the four genes analyzed by Spiers et al. (2016) and the 3 blood specific age calculator used by Weidner et al. (2014) are in our list of significant age-DMRs as these are mostly from blood-based studies. Additionally, none of the 44 CpGs are among the list of 353 tissue-independent CpGs used for age calculation by Horvath’s method.
4.4. EZH2 and PRC2 members
The mechanistic link between aging, cellular senescence and DNA methylation has not been elucidated (Lowe et al., 2016). Given the consistency and reliability in age prediction, these methylation patterns are unlikely to be entirely due to stochastic noise during aging. Instead, conserved regulatory protein networks are likely to play a key role. Evidence from a number of different studies is converging on the polycomb proteins, particularly PRC2, as a potential regulator of the aging methylome. This comes mainly from bioinformatics analysis and observations that many of the age-DMRs in both stem cells and differentiated tissues overlap binding sites for PRC2 members such as EZH2 and SUZ2 (Beerman et al., 2013; Dozmorov, 2015; Maegawa et al., 2010; Sun et al., 2014; Taiwo et al., 2013). While the exact mechanism is not known, the emerging view is that occupancy by PRC2 may maintain a hypomethylated state during development, and a decline in PRC2 occupancy during aging may facilitate the hypermethylation of these sites (Beerman et al., 2013; Jung and Pfeifer, 2015). Alternatively, it may be the occupancy by PRC2 that directly recruits DNA methylation enzymes and results in hypermethylation (Cedar and Bergman, 2012; Vire et al., 2006).
The present study takes this a step further by performing assay for EZH2-DNA interaction. Based on the ChIP analysis of the conserved age-hypermethylated sites, we confirm that these regions associate with EZH2 in mouse liver. We also find a slight decrease in EZH2 occupancy at the CGIs in C1ql3 and Srd5a2 in the aged samples. This supports the view that there is antagonistic interaction between PRC members and DNA methylation. However, these are not conclusive as sample sizes are small and further work must be done to confirm this.
4.5. Conclusion
In conclusion, the work presented here demonstrates that age-dependent DNA methylation occurs in regions that are conserved and show consistent patterns between humans and mice. Many of the associated genes are implicated in tumor development and progression, and methylation at these sites is altered in cancer cells. We also provide further evidence that EZH2 is an important candidate regulator of the aging methylome. Further study is required to evaluate the regulatory protein networks that interact with EZH2 and other polycomb members, and potential impact this will have of health, aging and lifespan.
Supplementary Material
Quality metrics for MBD-seq data (.xlsx)
Full set of methylation bin coordinates, test statistics, and gene feature and CpG island annotations (.csv)
Top 330 age-DMRs and R codes for differential methylation analysis (.xlsx)
Enriched functional categories among age-DMRs (.xlsx)
Data S5. Comparison of age-DMRs in humans (.xlsx)
Data S6. ChIP-qPCR for EZH2 (.xlsx)
Highlights.
We performed cross-species comparison of DNA methylation and aging in mice and humans.
DNA methylation was assayed in aging mouse liver using affinity-based enrichment and high-throughput sequencing.
Age-dependent methylation occur at highly conserved CpG islands of genes such as C1ql3, Srd5a2 and Ptk7, which are implicated in cancers.
Comparison with microarray-based data from humans shows consistent age-dependent methylation patterns.
ChIP assay confirms that member of the polycomb repressive complex 2, EZH2, binds to these conserved age-dependent sites.
Acknowledgments
This work was supported by funds from UTHSC Faculty Award UTCOM-2013KM. We are very grateful to Robert W. Williams for providing advice and access to the aging colony that is funded by NIA NIH grant R01AG043930. We gratefully acknowledge Megan K. Mulligan for her help, and her comments on the manuscript. Finally, we are thankful to Trevor Houseal for his technical assistance, and to Jesse Ingles for providing help and expert advice in the laboratory and for showing how to innovatively use a 1 ml pipette tip to keep the rotisserie tube rotator from falling apart during the MBD capture.
Abbreviations
- MBD-seq
methyl-CpG binding domain protein-enriched sequencing
- PRC
polycomb repressive complex
- age-DMR
age-dependent differentially methylated regions
- mos
months
- CF
coupling factor
- CGI
CpG island
- ChIP-qPCR
chromatin immunoprecipitation quantitative polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberg KA, McClay JL, Nerella S, Xie LY, Clark SL, Hudson AD, Bukszar J, Adkins D, Hultman CM, Sullivan PF, Magnusson PK, van den Oord EJ Consortium SS. MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case--control samples. Epigenomics. 2012;4:605–621. doi: 10.2217/epi.12.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg KA, Xie L, Chan RF, Zhao M, Pandey AK, Kumar G, Clark SL, van den Oord EJ. Evaluation of Methyl-Binding Domain Based Enrichment Approaches Revisited. PloS one. 2015;10:e0132205. doi: 10.1371/journal.pone.0132205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, Erhart W, Egberts J, Sipos B, Schreiber S, Hasler R, Stickel F, Becker T, Krawczak M, Rocken C, Siebert R, Schafmayer C, Hampe J. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell metabolism. 2013;18:296–302. doi: 10.1016/j.cmet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, Rossi DJ. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell stem cell. 2013;12:413–425. doi: 10.1016/j.stem.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nature reviews Molecular cell biology. 2015;16:593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nature biotechnology. 2010;28:1106–1114. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Programming of DNA methylation patterns. Annual review of biochemistry. 2012;81:97–117. doi: 10.1146/annurev-biochem-052610-091920. [DOI] [PubMed] [Google Scholar]
- Chavez L, Jozefczuk J, Grimm C, Dietrich J, Timmermann B, Lehrach H, Herwig R, Adjaye J. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome research. 2010;20:1441–1450. doi: 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer T, Mampaey E, Vlemmix M, Denil S, Trooskens G, Renard JP, De Keulenaer S, Dehan P, Menschaert G, Van Criekinge W. Quality evaluation of methyl binding domain based kits for enrichment DNA-methylation sequencing. PloS one. 2013;8:e59068. doi: 10.1371/journal.pone.0059068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Backdahl L, Herberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJ, Durbin R, Tavare S, Beck S. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nature biotechnology. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov MG. Polycomb repressive complex 2 epigenomic signature defines age-associated hypermethylation and gene expression changes. Epigenetics: official journal of the DNA Methylation Society. 2015;10:484–495. doi: 10.1080/15592294.2015.1040619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn NR, Tolwinski NS. Ptk7 and Mcc, Unfancied Components in Non-Canonical Wnt Signaling and Cancer. Cancers. 2016;8 doi: 10.3390/cancers8070068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge R, Wang Z, Bechis SK, Otsetov AG, Hua S, Wu S, Wu CL, Tabatabaei S, Olumi AF. DNA methyl transferase 1 reduces expression of SRD5A2 in the aging adult prostate. The American journal of pathology. 2015;185:870–882. doi: 10.1016/j.ajpath.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein R, Abbas S, Seibold P, Salazar R, Flesch-Janys D, Chang-Claude J. Polymorphism Thr160Thr in SRD5A1, involved in the progesterone metabolism, modifies postmenopausal breast cancer risk associated with menopausal hormone therapy. Breast cancer research and treatment. 2012;131:653–661. doi: 10.1007/s10549-011-1772-z. [DOI] [PubMed] [Google Scholar]
- Hishida M, Inokawa Y, Takano N, Nishikawa Y, Iwata N, Kanda M, Tanaka C, Kobayashi D, Yamada S, Nakayama G, Fujii T, Sugimoto H, Koike M, Fujiwara M, Kodera Y, Nomoto S. Protein tyrosine kinase 7: a hepatocellular carcinoma-related gene detected by triple-combination array. The Journal of surgical research. 2015;195:444–453. doi: 10.1016/j.jss.2014.12.045. [DOI] [PubMed] [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome biology. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, Becker T, Rocken C, Schafmayer C, Hampe J. Obesity accelerates epigenetic aging of human liver. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:15538–15543. doi: 10.1073/pnas.1412759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue. Genome biology. 2012;13:R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS genetics. 2010;6:e1001134. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Wang L, Chen J, Wang L, Leach L, Luo Z. Conserved and divergent patterns of DNA methylation in higher vertebrates. Genome biology and evolution. 2014;6:2998–3014. doi: 10.1093/gbe/evu238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Pfeifer GP. Aging and DNA methylation. BMC biology. 2015;13:7. doi: 10.1186/s12915-015-0118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre C, Gooden GC, Johnson K, Martinez RA, Liang WS, Salhia B. Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clinical epigenetics. 2015;7:100. doi: 10.1186/s13148-015-0135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, Zheng H, Yu J, Wu H, Sun J, Zhang H, Chen Q, Luo R, Chen M, He Y, Jin X, Zhang Q, Yu C, Zhou G, Sun J, Huang Y, Zheng H, Cao H, Zhou X, Guo S, Hu X, Li X, Kristiansen K, Bolund L, Xu J, Wang W, Yang H, Wang J, Li R, Beck S, Wang J, Zhang X. The DNA methylome of human peripheral blood mononuclear cells. PLoS biology. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014;30:284–286. doi: 10.1093/bioinformatics/btt650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovkvist C, Dodd IB, Sneppen K, Haerter JO. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic acids research. 2016;44:5123–5132. doi: 10.1093/nar/gkw124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D, Horvath S, Raj K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget. 2016;7:8524–8531. doi: 10.18632/oncotarget.7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome research. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, Martin NG, Montgomery GW, Feinberg AP, Fallin M, Multhaup ML, Jaffe AE, Joehanes R, Schwartz J, Just AC, Lunetta KL, Murabito JM, Starr JM, Horvath S, Baccarelli AA, Levy D, Visscher PM, Wray NR, Deary IJ. DNA methylation age of blood predicts all-cause mortality in later life. Genome biology. 2015a;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, Corley J, Taylor A, Murphy L, Starr JM, Horvath S, Visscher PM, Wray NR, Deary IJ. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. International journal of epidemiology. 2015b;44:1388–1396. doi: 10.1093/ije/dyu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClay JL, Aberg KA, Clark SL, Nerella S, Kumar G, Xie LY, Hudson AD, Harada A, Hultman CM, Magnusson PK, Sullivan PF, Van Den Oord EJ. A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Human molecular genetics. 2014;23:1175–1185. doi: 10.1093/hmg/ddt511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitteldorf JJ. How does the body know how old it is? Introducing the epigenetic clock hypothesis. Biochemistry Biokhimiia. 2013;78:1048–1053. doi: 10.1134/S0006297913090113. [DOI] [PubMed] [Google Scholar]
- Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, Alekseev MV, Shelygin YA, Govorun VM. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics: official journal of the DNA Methylation Society. 2013;8:921–934. doi: 10.4161/epi.25577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Imaz H, Rueda Y, Fresnedo O. SND1 overexpression deregulates cholesterol homeostasis in hepatocellular carcinoma. Biochimica et biophysica acta. 2016;1861:988–996. doi: 10.1016/j.bbalip.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez BA, Frankhouser D, Murphy M, Trimarchi M, Tam HH, Curfman J, Huang R, Chan MW, Lai HC, Parikh D, Ball B, Schwind S, Blum W, Marcucci G, Yan P, Bundschuh R. Methods for high-throughput MethylCap-Seq data analysis. BMC genomics. 2012;13(Suppl 6):S14. doi: 10.1186/1471-2164-13-S6-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome research. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiers H, Hannon E, Wells S, Williams B, Fernandes C, Mill J. Age-associated changes in DNA methylation across multiple tissues in an inbred mouse model. Mechanisms of ageing and development. 2016;154:20–23. doi: 10.1016/j.mad.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, Gu H, Bock C, Meissner A, Gottgens B, Darlington GJ, Li W, Goodell MA. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell stem cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo O, Wilson GA, Emmett W, Morris T, Bonnet D, Schuster E, Adejumo T, Beck S, Pearce DJ. DNA methylation analysis of murine hematopoietic side population cells during aging. Epigenetics: official journal of the DNA Methylation Society. 2013;8:1114–1122. doi: 10.4161/epi.26017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome research. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jockel KH, Erbel R, Muhleisen TW, Zenke M, Brummendorf TH, Wagner W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome biology. 2014;15:R24. doi: 10.1186/gb-2014-15-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Spector TD, Deloukas P, Bell JT, Engelhardt BE. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome biology. 2015;16:14. doi: 10.1186/s13059-015-0581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quality metrics for MBD-seq data (.xlsx)
Full set of methylation bin coordinates, test statistics, and gene feature and CpG island annotations (.csv)
Top 330 age-DMRs and R codes for differential methylation analysis (.xlsx)
Enriched functional categories among age-DMRs (.xlsx)
Data S5. Comparison of age-DMRs in humans (.xlsx)
Data S6. ChIP-qPCR for EZH2 (.xlsx)