

ABSTRACT
Infectious diseases caused by bacterial pathogens reduce the fitness of their associated host but are generally limited in duration. In order for the diseased host to regain any lost fitness upon recovery, a variety of molecular, cellular, and physiological processes must be employed. To better understand mechanisms underlying the recovery process, we have modeled an acute Pseudomonas aeruginosa infection in C. elegans using brief exposures to this pathogen and subsequent antibiotic treatment. To identify host genes altered during recovery from P. aeruginosa infection, we performed whole genome expression profiling. The analysis of this dataset indicated that the activity of the host immune system is down-regulated upon recovery and revealed shared and pathogen-specific host responses during recovery. We determined that the GATA transcription factor ELT-2 and the p38 MAP kinase PMK-1 are necessary for animals to successfully recover from an acute P. aeruginosa infection. In addition, we found that ELT-2 plays a more prominent and earlier role than PMK-1 during recovery. Our data sheds further light on the molecular mechanisms and transcriptional programs involved in recovery from an acute bacterial infection, which provides a better understanding of the entire infectious disease process.
Keywords: acute, C. elegans, disease, ELT-2, GATA, host, immunity, infection, infectious, innate, recovery, resolution, P. aeruginosa, p38 MAPK, pathogen, PMK-1
Introduction
Certain microbes, including bacteria, are capable of causing infectious disease — either acute or chronic — in their hosts. In humans, acute bacterial infections are controlled by a combination of immune responses, physiological alterations, and, if necessary, supplementary antibiotic treatment. The combined actions of these measures usually leads to the elimination of the disease-causing bacteria and the restoration of host health. The return to a healthy set point, or recovery, is crucial to avoid entering a state of reduced fitness that manifests in various pathogenic forms including recurrent infections, fibrosis, autoimmune disease, and chronic inflammatory disorders.
Recovery is a set of 3 loosely overlapping processes: resolution of inflammation, elimination of any residual harmful molecules, and repair of damaged tissue. Specific molecules that govern one or more of these processes have been revealed through various in vitro studies and, more recently, through a handful of in vivo studies.1-5 Signaling from a variety of molecules, including TGF-β, IL-10, arachidonic acid (AA)-derived lipid mediators, and resolution-associated molecular patterns (RAMPs), enhances host recovery through the induction of anti-inflammatory or pro-resolving pathways.6-10 Although many of these processes have been described and dissected, the full complement of resolution and repair pathways and the coordination of these pathways in the context of the host-microbe holobiont is not fully understood.
The nematode Caenorhabditis elegans has been utilized to study various aspects of the infectious disease process upon exposure to gram negative and gram positive bacteria.11-17 Many of these bacterial species, including Pseudomonas aeruginosa, Salmonella enterica, Serratia marcescens, Staphylococcus aureus, and Enterococcus faecalis, have a significant impact on human morbidity and mortality. Studies in C. elegans have identified the mechanistic details of conserved host pathways that protect the host from infection by these pathogens, from pathogen sensing to cellular surveillance to tolerance.18-22 Moreover, using the C. elegans-pathogen model, a variety of microbial virulence factors and processes, which could potentially be targeted to disrupt the infectious disease process, have been identified.22-24 Other recent studies have characterized the extent of host damage caused by these pathogens in C. elegans, further adding to our understanding of bacterial pathogenesis in this model system.25 Overall, these studies have enabled us to better understand both conserved and novel pathways that could be modulated in human patients to parry the effects of bacterial pathogens.
Two of the most well-characterized infectious disease processes in C. elegans include those caused by P. aeruginosa and S. enterica.11,14,26 The opportunistic pathogen P. aeruginosa can kill C. elegans in a “slow” colonization-dependent manner or “fast” colonization-independent manner. Both of these killing modalities depend on the secretion of various virulence factors — including phenazines, pyoverdins, elastases, exotoxin A, HCN, and NO – that contribute to pathogenesis in the host.23,27-29 In slow killing, specific P. aeruginosa-secreted virulence factors interfere with host proteins and pathways leading to the induction of a robust immune response in C. elegans in as little as 4 hours.30,31 In an uncontrolled infection, readily apparent damage at the tissue level is evident 8–24 hours after exposure25 and death of the animal comes shortly thereafter. S. enterica kills C. elegans in a colonization-dependent manner.26 Like P. aeruginosa, S. enterica also uses a wide variety of virulence factors — including LPS, SptP, KdpD, and RamA — to colonize and kill C. elegans.24,32-36 One major mechanism of pathogen-dependent killing is disseminated oxidative stress, a byproduct of host immune system activation,37 although other pathogenic mechanisms are surely involved. Studies of these and other infectious disease processes should enable the development of interventions that ameliorate the effects of bacterial pathogenesis in humans.
Recently, we extended the use of the C. elegans-pathogen model to study pathways that facilitate host recovery from an acute infection.4 By studying acute S. enterica infection of C. elegans, we demonstrated that, upon recovery, the innate immune response is downregulated and various cytoprotective pathways are up-regulated.4 We also identified a role for the GATA transcription factor ELT-2 in controlling the expression of genes that are markers of recovery from S. enterica infection.4 In this study, we modeled acute P. aeruginosa infection and recovery in C. elegans and characterized host whole genome expression profiles across multiple phases of the acute infection disease process, from exposure to early recovery to late recovery. We compared and contrasted transcriptional programs that respond to P. aeruginosa and S. enterica recovery to elucidate shared and pathogen-specific recovery pathways. We also determined that recovery from acute P. aeruginosa infection requires the action of ELT-2, as is the case with recovery from S. enterica. Finally, we showed that p38-MAPK PMK-1 is required for recovery from both P. aeruginosa and S. enterica infections. In contrast to ELT-2, PMK-1 seems to be involved at later stages of recovery. In summary, our results help to elucidate potentially conserved pathways that contribute to the re-establishment of host homeostasis, or recovery, after acute infection by bacterial pathogens.
Results
Modeling acute P. aeruginosa infection and recovery in C. elegans
We have previously utilized C. elegans as a model to investigate host transcriptional networks and pathways that are involved in recovery from acute S. enterica infection.4 Upon recovery from acute S. enterica infection, genes that are markers of innate immunity are down-regulated, while genes involved in xenobiotic detoxification, redox regulation, and cellular homeostasis are upregulated.4 Here, we examined host recovery after acute infection by P. aeruginosa to learn more about shared and pathogen-specific recovery pathways.
To study recovery from acute P. aeruginosa infection, we employed fer-1(b232ts) animals, which are fertilization defective at the restrictive temperature. The use of this strain, rather than the wild-type N2 strain, prevents losing track of the initially exposed animals among their progeny. To model acute P. aeruginosa infection and recovery, we exposed larval stage 4 (L4) or adult fer-1(b232ts) animals to lawns of P. aeruginosa for 12 or 18 hours. After the initial exposure, animals were rinsed with M9 containing the antibiotic Neomycin (Neo) and then transferred to plates seeded with P. aeruginosa or to plates containing the antibiotic Streptomycin (Sm) seeded with SmR Escherichia coli. We found that the survival of adult animals exposed to P. aeruginosa for 12 or 18 hours and then treated with Neo/Sm is significantly higher than that of animals continuously exposed to P. aeruginosa (Fig. 1, yellow vs red lines) over a 3 day survival analysis period. Nearly all adult animals exposed to P. aeruginosa for 12 hours and then treated with Neo/Sm were alive at Day 3 (Fig. 1, yellow open squares). For adult animals exposed to P. aeruginosa for 18 hours and then treated with Neo/Sm, a smaller but still large percentage (∼80%) of animals were alive at Day 3 (Fig. 1, yellow filled squares). In addition, the survival of adult animals exposed to P. aeruginosa for 12 hours and then treated with Neo/Sm was not statistically different (p < 0.5637) from the survival of adult animals feeding on their normal E. coli food source (Fig. 1, yellow open squares vs black closed circles). Exposing L4 animals to P. aeruginosa for 12 or 18 hours followed by treatment with Neo/Sm did not lead to a shortening of lifespans relative to animals feeding on E. coli over the 3 day survival analysis period (Fig. S1). This data confirms previous results11 and indicates that we are capable of modeling acute P. aeruginosa infection in C. elegans using a combination of antibiotic treatment and a shift in bacterial food source.
Figure 1.
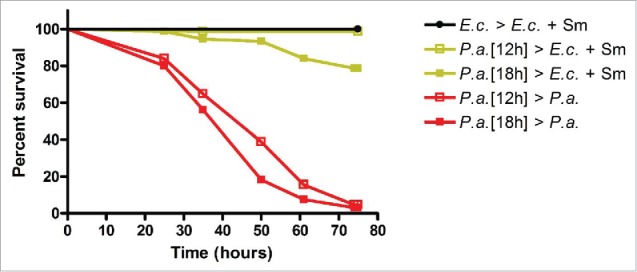
Modeling recovery from acute P. aeruginosa infection in C. elegans. fer-1(b232ts) adult animals were exposed to E. coli or P. aeruginosa for 12 or 18 hours, treated with Neomycin, and then transferred to E. coli plus Streptomycin or P. aeruginosa plates and scored for survival. Scoring began 12 or 18 hours post initial exposure to E. coli or P. aeruginosa. n = 1.
For subsequent acute infection and recovery experiments, we decided to use adult fer-1(b232ts) animals exposed to P. aeruginosa-GFP for 12 or fewer (see below) hours. We used adult animals to avoid the effects of C. elegans larval development on recovery. For the length of exposure, 12 hours was the longest exposure time tested that did not alter survival over the 3 day survival analysis period (Fig. 1). Importantly, adult animals exposed to P. aeruginosa for 12 hours and then treated with Neo/Sm were not visibly colonized by P. aeruginosa-GFP at any point during the 3 day survival analysis period (data not shown). Exposure times shorter than 12 hours in duration should not alter host survival nor lead to P. aeruginosa intestinal colonization. In summary, we have established an acute P. aeruginosa infection and recovery system that allows us to study host processes leading to the restoration of homeostasis.
Profiling gene expression changes in C. elegans upon P. aeruginosa acute infection and recovery
To investigate pathways that mediate C. elegans recovery from P. aeruginosa infection, we utilized Agilent microarrays to identify changes in gene expression over the entire time course of acute infection and recovery. We utilized hypochlorite treatment of gravid fer-1(b232ts) adults to developmentally synchronize a suitable number of animals for the gene expression profiling experiments. A 4 hour exposure to P. aeruginosa is sufficient for virulence factor-induced host cell alterations and for C. elegans to generate a robust immune response.25,30,38 Thus, for the gene expression profiling experiments, we utilized a 4 hour exposure of adult animals to P. aeruginosa. We collected cohorts of fer-1(b232ts) animals grown on E. coli for 72 hours (72 h), immediately following a 4 hour exposure to P. aeruginosa (76 h), and at 3 times during the recovery period (R1, R2, and R3) (Fig. 2A). As a control, we collected animals that were not exposed to P. aeruginosa but were exposed to the antibiotics Neo/Sm.
Figure 2.
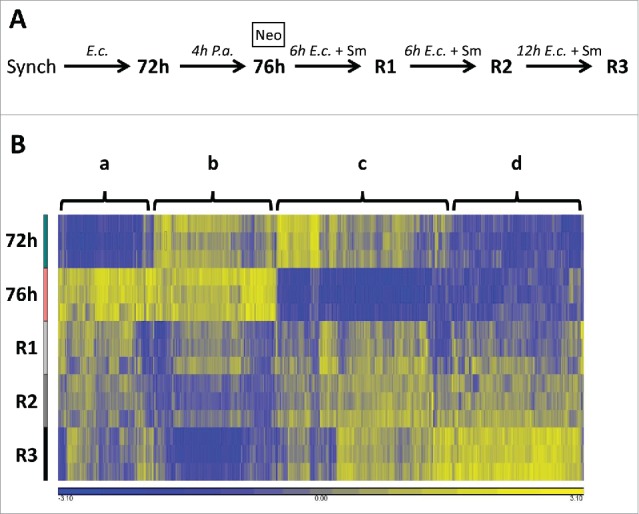
Gene expression profiling reveals dynamic transcriptional responses over the entire course of acute infection and recovery. (A) Flowchart of animal cohorts collected for the microarray analysis. (B) Heat map depicting all 1,323 genes whose expression is downregulated or upregulated in one or more comparisons over the entire course of acute infection and recovery. Columns correspond to the 1,323 genes and are clustered. Rows correspond to the 5 time points collected and are arranged chronologically. Four broad clusters of genes, a-d, are indicated. Each column is normalized by shifting to a mean of zero and scaling to an SD of 1. Yellow hues indicate higher relative expression and blue hues indicate lower relative expression.
Log2-transformed data from the microarray is given in Table S1. A total of 84 genes are regulated by exposure to Neo/Sm antibiotics alone (Table S2); these genes were excluded from subsequent analyses. Overall, the expression profiles of 1,323 genes, or 6.35% of the genome, were altered more than 2-fold (p < 0.05 with FDR) in one or more relevant comparisons over the entire course of acute infection and recovery. These comparisons are infected versus uninfected (76 h vs 72 h) and recovered vs. infected animals (R1 vs 76 h, R2 vs 76 h, and R3 vs 76 h) (Table S3). A hierarchical clustering analysis of transcript abundance over time for these 1,323 genes is depicted in Figure 2B. The clustering analysis indicates that there are 4 major groupings of genes that respond to acute infection and recovery. One group of genes is strongly up-regulated after 4 hours of exposure to P. aeruginosa and then generally downregulated close to pre-exposure levels by R3 (Fig. 2B, cluster a). One group of genes is weakly upregulated after 4 hours of exposure to P. aeruginosa and then strongly down-regulated by R3 (Fig. 2B, cluster b). A third group of genes is strongly downregulated after 4 hours of exposure to P. aeruginosa and then generally up-regulated close to pre-exposure levels by R3 (Fig. 2B, cluster c). The final group of genes do not change upon exposure to P. aeruginosa but are upregulated during the recovery process (Fig. 2B, cluster d). The identity of each gene in clusters a-d is given in Table S4. In conclusion, we have identified more than 1,300 C. elegans genes that respond to acute P. aeruginosa infection and recovery.
A subset of the genes in our acute infection and recovery gene set are down- or up-regulated upon P. aeruginosa exposure, independent of their behavior during recovery. A total of 542 genes – 268 down-regulated and 276 upregulated – were altered upon P. aeruginosa exposure relative to animals feeding on E. coli (p < 0.05 with FDR) (Table S3). Comparison of our P. aeruginosa-regulated gene set to a P. aeruginosa-regulated gene set described in a previous study30 reveals a high degree of overlap between the 2 data sets (Fig. S2, Table S5). The representation factor, a measure of the correlation between 2 populations, is significantly higher than 1, indicating a high degree of overlap (Fig. S2). Thus, as previously noted,15 C. elegans has a highly stereotypic transcriptional response to P. aeruginosa.
We next assessed the transcriptional activity of these 542 pathogen-regulated genes during the recovery period. We found that 60% (156/268) of the genes downregulated upon pathogen exposure, were conversely up-regulated 24 hours post-treatment, at the R3 time point. Similarly, we found that 36% (98/276) of genes upregulated upon pathogen exposure, were down-regulated post-treatment. We therefore conclude that the rapid host transcriptional response that occurs after a 4 hour exposure to P. aeruginosa is quickly dampened upon recovery.
To validate the results of the microarray, we performed quantitative real-time PCR (qRT-PCR) on a subset of the 1,323 genes altered during recovery. This subset includes 5 downregulated genes (Fig. 3A) and 10 up-regulated genes (Fig. 3B) that spanned early to late responders and a variety of time course expression profiles (Fig. 3, white bars). We performed qRT-PCR on RNA harvested from C. elegans that were subjected to the same conditions as in the microarray experiments. Changes in gene expression as assessed by qRT-PCR were comparable to those observed by microarray profiling after 4 hours of exposure to P. aeruginosa (76 h vs 72 h), at recovery time point 1 (R1 vs 76 h), and at recovery time point 3 (R3 vs 76 h) (Fig. 3, colored vs white bars). Statistical analysis indicated that the gene expression changes determined by qRT-PCR and microarray profiling were not statistically different in 39 out of 45 measurements (p > 0.05) (Fig. 3). Thus, the microarray data accurately reflects the majority of gene expression differences over the acute P. aeruginosa infection and recovery period.
Figure 3.
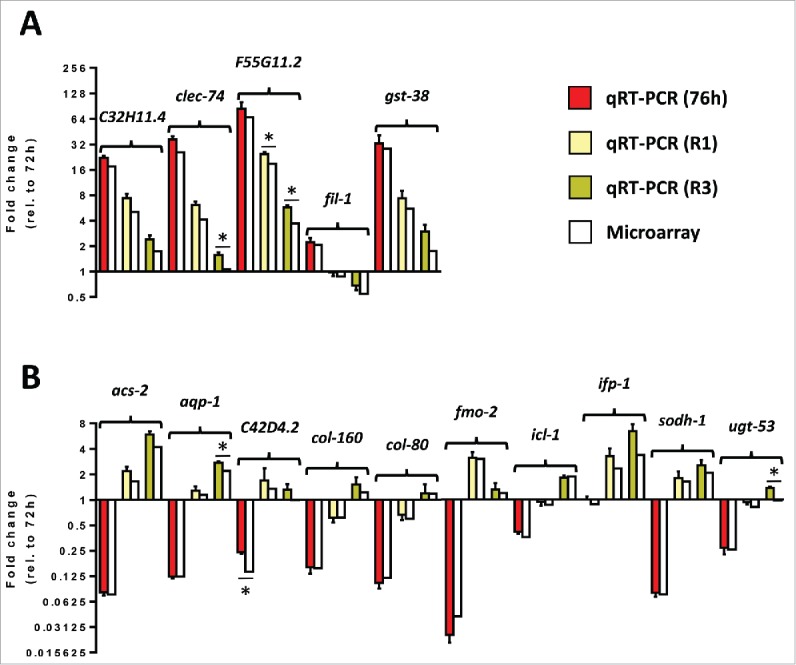
qRT-PCR confirms microarray gene expression data for a select subset of regulated genes. (A) Transcript levels of 5 selected down-regulated genes from adult animals exposed to P. aeruginosa for 4 hours, adult animals exposed to P. aeruginosa for 4 hours and then treated with Neomycin/Streptomycin for 6 hours, or adult animals exposed to P. aeruginosa for 4 hours and then treated with Neomycin/Streptomycin for 24 hours relative to L1 animals grown on E. coli for 72 hours. (B) Transcript levels of 10 selected up-regulated genes from adult animals exposed to P. aeruginosa for 4 hours, adult animals exposed to P. aeruginosa for 4 hours and then treated with Neomycin/Streptomycin for 6 hours, or adult animals exposed to P. aeruginosa for 4 hours and then treated with Neomycin/Streptomycin for 24 hours relative to L1 animals grown on E. coli for 72 hours. Red, yellow, and mustard correspond to fold changes determined using qRT-PCR at 76 h, R1, and R3, respectively. n = 3. SEM is shown. White bars correspond to fold changes determined using microarrays. Statistical significance is indicated (p < 0.05: *).
Shared and pathogen-specific host pathways regulate recovery from acute P. aeruginosa and S. enterica infections
One of the goals of our study was to identify shared and pathogen-specific host responses during recovery from acute bacterial infection. To do this, we compared sets of genes regulated during P. aeruginosa recovery (Table S3) to sets of genes regulated during S. enterica recovery.4 In our previous analysis using recovery from acute S. enterica infection, we analyzed gene expression profiles 24 hours post-treatment to determine genes regulated during recovery.4 Thus, we began by focusing on P. aeruginosa recovery genes that have significantly altered expression 24 hours post-treatment (R3 vs 76 h). This time point also showed the greatest number of genes with altered transcription among all P. aeruginosa recovery gene sets (Table S3). The overlap of the P. aeruginosa and S. enterica recovery gene sets is shown in Figure 4A. The representation factors are 6.2 and 6.0 for down-regulated and upregulated gene sets, respectively (Fig. 4A). These values are less than that for the overlap between 2 studies of the transcriptional response of C. elegans exposed to P. aeruginosa for 4 hours (Fig. S2) but more than 1 indicating both common and distinct transcriptional responses of C. elegans to P. aeruginosa and S. enterica recovery. Shared and pathogen-specific recovery genes are given in Table S6. A similar comparative analysis at 6 hours post-treatment (R1 vs 76 h), also revealed shared and distinct host responses during recovery (Fig. S3A). Taken together, our data indicate that a small subset of genes are important for recovery from both of these 2 bacterial pathogens while a larger subset of genes are pathogen-specific.
Figure 4.
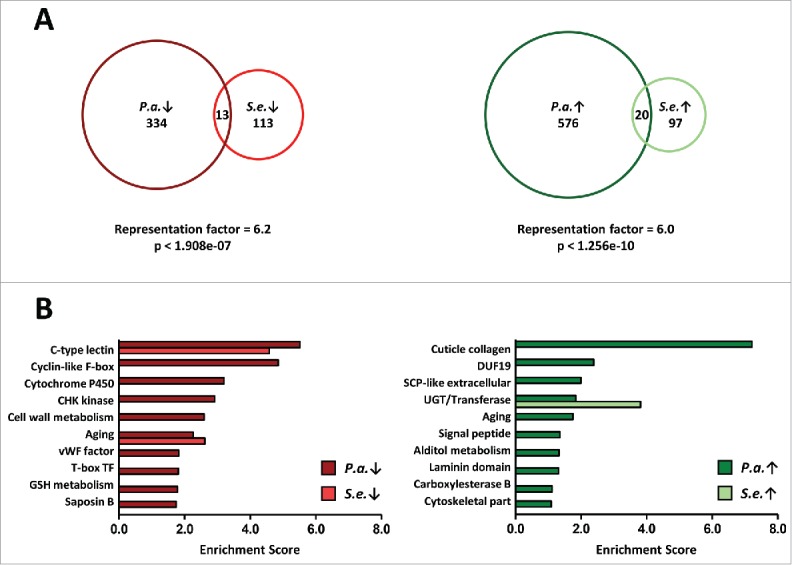
Shared and pathogen-specific responses during recovery from acute bacterial infection. (A) Proportional Venn diagrams showing the overlap of P. aeruginosa R3 and S. enterica downregulated recovery genes, left, and overlap of P. aeruginosa R3 and S. enterica upregulated recovery genes, right. (B) Gene ontology analysis of P. aeruginosa R3 and S. enterica recovery genes using the DAVID Bioinformatics Database. Enrichment scores of the down-regulated clusters are shown in the left panel. Enrichment scores of the up-regulated clusters are shown in the right panel. The top 10 P. aeruginosa recovery clusters are shown. Only the top 10 S. enterica recovery clusters that overlap with one of the top 10 P. aeruginosa recovery clusters are shown.
To provide further insight into the biological functions of the genes that are transcriptionally altered during recovery from P. aeruginosa, we performed gene ontology (GO) analyses using the DAVID Bioinformatics Resource (DAVID, http://david.abcc.ncifcrf.gov/). The 10 gene ontology clusters with the highest enrichment score for down- and up-regulated P. aeruginosa recovery genes at 24 hours post-treatment (R3 vs 76 h) are shown in Fig. 4B (dark red, dark green). For the downregulated gene set, the highest scoring cluster is C-type lectins, a class of genes that is involved in C. elegans pathogen defense.18 Other high scoring down-regulated clusters include genes with F-box, cytochrome P450, CHK kinase, and cell wall metabolism ontologies. For the upregulated gene set, the highest scoring ontology cluster is collagen, a class of genes important for extracellular matrix integrity, tissue repair, and longevity responses.39,40 Other up-regulated clusters include genes with DUF-19, SCP-like extracellular domains, and UDP-glucuronosyltransferase (UGT) ontologies. A GO analysis of genes regulated 6 hours post-treatment (R1 vs 76 h) yields similar lists of high-scoring clusters (Fig. S3B). Our analysis indicates that, upon recovery from P. aeruginosa infection, C. elegans down-regulates genes that are important for controlling pathogen abundance and up-regulates a variety of other genes that are potentially involved in inflammation resolution, damage control, or tissue repair.
A comparison between GO clusters that are altered during P. aeruginosa recovery to GO clusters that are altered during S. enterica recovery,4 confirms the idea that there are both shared and pathogen-specific recovery pathways (Fig. 4B, dark red vs red, dark green vs green). The highest scoring cluster in both P. aeruginosa and S. enterica downregulated gene sets is C-type lectins, indicating that C. elegans dampen the inflammatory response upon recovery (Fig. 4B, left panel). Comparison of the other top 10 GO clusters for the down-regulated gene set reveals distinct functions of genes that are downregulated upon recovery from the 2 bacterial pathogens (Fig. 4B, left panel). This would be expected as both P. aeruginosa and S. enterica provoke distinct C. elegans immune responses, outside of C-type lectin induction.15,30,31,41,42 For the upregulated gene sets, the highest scoring clusters are collagens for P. aeruginosa and UGTs for S. enterica (Fig. 4B, right panel). Collagen genes are not up-regulated during recovery from S. enterica recovery. However, UGTs, Phase II detoxification enzymes, are a key component of recovery from both acute infections. As with the down-regulated comparison, the GO data reveals a distinct set of functions for upregulated genes, outside of the UGT genes, during recovery from P. aeruginosa and S. enterica. We conclude that C. elegans uses both shared and distinct pathways during recovery from acute bacterial infection.
Recovery from acute P. aeruginosa infection requires early ELT-2 activity and late PMK-1 activity
To further explore pathways that mediate the expression of genes involved in recovery from P. aeruginosa infection, we compared the set of genes regulated during recovery from P. aeruginosa infection to previously identified gene sets that are known to be controlled by various signaling pathways, transcription factors, or treatments (Table S7). We found a significant overlap between P. aeruginosa recovery genes and gene sets regulated by ELT-2/GATA, PMK-1/p38 MAPK, DAF-16/FOXO, or SKN-1/Nrf (Table S7). Gene sets for HIF-1, hypoxia, and SMA-6/TGF-β did not have a significant overlap with P. aeruginosa recovery genes. Our analysis suggests that ELT-2, PMK-1, DAF-16, and SKN-1 may be involved in orchestrating gene expression programs that mediate C. elegans recovery from acute P. aeruginosa infection.
The GATA transcription factor ELT-2 is required for full recovery from acute S. enterica infection4 and may also be involved in recovery from acute P. aeruginosa infection (Table S7). Thus, we chose to examine the survival of fer-1(b232ts) elt-2(RNAi) animals during recovery and upon continuous exposure to P. aeruginosa or E. coli. Because ELT-2 is essential for C. elegans larval development43 we performed RNAi on L4 animals. This approach has been used successfully to inhibit elt-2 expression for at least 6 days.31,44 As with acute S. enterica infection, RNAi of elt-2 completely prevented the recovery of animals acutely exposed to P. aeruginosa for 12 hours over the 3 day analysis period (Fig. 5A, yellow lines, Fig. S4). RNAi of elt-2 also reduced the survival of animals continuously exposed to P. aeruginosa (Fig. 5A, red lines, Fig. S4), which is in line with ELT-2s role in the generation of an early transcriptional response to P. aeruginosa infection.31,45 RNAi of elt-2 did not alter the survival of animals growing on live E. coli over the 3 day analysis period, confirming that elt-2 RNAi at the L4 stage does not simply induce a general debilitation of the animals (Fig. 5A, black lines, Fig. S4).4,31,45 We conclude that, similar to the requirement of ELT-2 for S. enterica recovery, the function of ELT-2 is necessary for C. elegans to recover from an acute P. aeruginosa infection.
Figure 5.
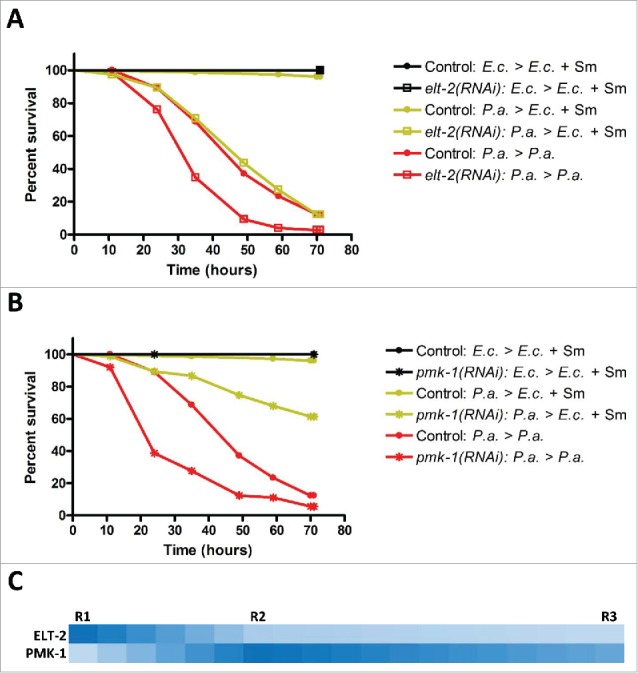
Sequential requirement for ELT-2 and PMK-1 activity during recovery from acute P. aeruginosa infection. (A) Control fer-1(b232ts) or fer-1(b232ts) elt-2(RNAi) adult animals were exposed to E. coli or P. aeruginosa for 12 hours and then transferred to E. coli plus Streptomycin or P. aeruginosa plates and scored for survival. (B) Control fer-1(b232ts) or fer-1(b232ts) pmk-1(RNAi) adult animals were exposed to E. coli or P. aeruginosa for 12 hours and then transferred to E. coli plus Streptomycin or P. aeruginosa plates and scored for survival. Scoring began 12 hours post initial exposure to E. coli or P. aeruginosa. Representative graph of n = 3 independent experiments. See also Figure S4. (C) Image depicting the relative intensity of the overlap between ELT-2-regulated and PMK-1-regulated gene sets and gene sets regulated during recovery from acute P. aeruginosa infection. A darker shade of blue is representative of a higher representation factor while a lighter shade of blue is representative of a lower representation factor.
ELT-2 is known to regulate the transcription of genes in the intestine via the TGATAA (extended GATA) cis-regulatory motif.43,46 We used an in silico approach to examine the prevalence of TGATAA sites in our acute infection and recovery gene sets – P. aeruginosa exposure, 76 h, and 3 sequential recovery time points, R1, R2, and R3 (Table S8). For genes regulated upon exposure to P. aeruginosa, 64% have at least 1 TGATAA site in the 1.5 kb region upstream of their transcriptional start site. The same percentage, 64%, of R1 genes contain at least 1 TGATAA site in the upstream region. In subsequent time points, the percentage of genes with at least 1 TGATAA declines to 60%. This data also correlates to an overall decrease in the number of TGATAA sites per gene over the course of the acute infection and recovery time course (Table S8). A randomly selected set of genes from the C. elegans genome exhibited a lower enrichment of TGATAA sites by either percentage of genes or number of sites per gene (Table S8). This in silico analysis highlights the fact that ELT-2 plays a role at multiple phases of the acute infectious disease process, but is more important for early gene expression programs.
We next examined the effects of RNAi inhibition of the stress response kinase and innate immunity regulator PMK-1 during recovery. Using the same L4 RNAi treatment protocol employed in the elt-2 RNAi experiments above, we found that RNAi of pmk-1 significantly impaired but did not completely prevent the recovery of fer-1(b232ts) animals acutely exposed to P. aeruginosa for 12 hours (Fig. 5B, yellow lines, Fig. S4). RNAi of pmk-1 significantly reduced the survival of animals continuously exposed to P. aeruginosa but did not affect survival of animals on E. coli, as previously reported (Fig. 5B, red and black lines, Fig. S4).47 We found a similar effect on recovery using pmk-1 mutant animals (Fig. S5A). We conclude that host PMK-1 activity is required to both generate an immune response against P. aeruginosa and, also, to orchestrate recovery from an acute infection by this pathogen.
We also observed a significant enrichment of DAF-16- and SKN-1-regulated targets in the set of genes altered during recovery from P. aeruginosa infection (Table S7). Consequently, we explored whether the activity of these 2 transcription factors, in addition to PMK-1 and ELT-2, is required for recovery from acute P. aeruginosa infection. Neither RNAi of daf-16 nor that of skn-1 prevented the full recovery of fer-1(b232ts) animals from acute P. aeruginosa infection (Fig. S6, yellow lines). Importantly, the daf-16 RNAi and skn-1 RNAi protocols used for the survival analysis completely eliminated visible GFP fluorescence in animals expressing DAF-16::GFP and SKN-1::GFP translational reporters, respectively, indicating that the RNAi treatments were effective in reducing levels of DAF-16 and SKN-1 (data not shown). The daf-16 RNAi and skn-1 RNAi results also demonstrate the specificity of elt-2 RNAi and pmk-1 RNAi treatments in preventing recovery from P. aeruginosa infection. We conclude that, in contrast to ELT-2 and PMK-1, neither DAF-16 nor SKN-1 activity are required for recovery from acute P. aeruginosa infection.
The role of PMK-1 in recovery from P. aeruginosa infection was unexpected. We previously found that pmk-1 RNAi at the L4 stage does not affect recovery from acute S. enterica infection.4 Thus, we chose to reexamine the role of PMK-1 in recovery from acute S. enterica infection using pmk-1 mutant animals. Mutant pmk-1 animals were unable to recover from acute S. enterica infection (Fig. S5B), showing a stronger recovery defect relative to pmk-1(RNAi) animals.4 To examine whether genes required for immunity are generally required for recovery from acute S. enterica infection, we tested animals with a loss of function mutation in fshr-1. We used fshr-1 mutants because they are more sensitive than wild-type animals to killing by both Gram-negative and Gram-positive bacteria.48 We found that while fshr-1 mutants are susceptible to S. enterica infection they are capable of fully recovering from infection by this pathogen (Fig. S5B-C). In summary, PMK-1 is involved in recovery from both P. aeruginosa and S. enterica, though its role in P. aeruginosa recovery is more prominent.
Further analysis of the role of ELT-2 and PMK-1 in recovery from P. aeruginosa reveals additional details about the sequence of molecular events that may take place during recovery. The effects of elt-2 RNAi on recovery are evident at the earliest time points indicating that ELT-2 is required early in the recovery process (Fig. 5A, yellow lines, Fig. S4). In contrast, the effects of pmk-1 RNAi on recovery are evident at later time points (Fig. 5B, yellow lines, Fig. S4). As shown in Figure 5, 50% of elt-2 RNAi animals failed to recover at 48 hours while more than 50% of pmk-1 RNAi animals remain alive at 72 hours. Our in silico analysis of overlaps between recovery and published gene sets (Table S7) supports this sequential requirement for ELT-2 followed by PMK-1 during recovery (Fig. 5C). We conclude that although both ELT-2 and PMK-1 are both required for recovery from acute P. aeruginosa infection, ELT-2 plays a more prominent and earlier role than PMK-1.
Discussion
We have utilized C. elegans to investigate host pathways that contribute to recovery from acute P. aeruginosa infection. Gene expression profiling of animals at multiple time points during the recovery phase shows that a broad class of genes, some expected and others novel, are altered during this phase. Further analysis of P. aeruginosa recovery genes using an in silico approach enabled the identification of 2 intracellular factors that regulate gene expression during recovery. We show that these factors, the GATA transcription factor ELT-2 and the p38 MAP kinase PMK-1, are required for recovery from acute P. aeruginosa infection, though ELT-2 plays a more prominent and early role than PMK-1.
In our study, we employed a slow killing protocol to investigate P. aeruginosa pathogenesis, as opposed to fast or liquid killing protocols.29 Slow killing involves accumulation of P. aeruginosa in the intestine of C. elegans and requires several virulence factors, including the secreted Exotoxin A molecule.23,49-51 Importantly, these virulence factors are utilized by P. aeruginosa to cause disease in other organisms, including in mammals.23 In fast and liquid killing, P. aeruginosa has different modes of killing that involve molecules of the pyocanin-phenazine class or siderophores, respectively. These molecules contribute to the rapid killing of C. elegans in part by eliciting oxidative stress or a hypoxic response, respectively.28,52 In this study, there is no evidence of a transcriptional signature indicating oxidative stress or hypoxia upon exposure or during recovery (Table S7), confirming the differences between slow, fast, and liquid killing. Further studies are necessary to determine whether C. elegans could recover from other modes of P. aeruginosa pathogenesis and what host pathways might be involved in those instances.
We previously investigated pathways that mediate C. elegans recovery from acute S. enterica infection.4 We have taken advantage of our previously published results by comparing and contrasting P. aeruginosa and S. enterica recovery processes to learn about shared and pathogen-specific recovery pathways. The recovery phase for both infections is characterized by the down-regulation of C-type lectins and the up-regulation of Phase II detoxification genes (Fig. 4B). Outside of these 2 shared signatures, most genes regulated during recovery from P. aeruginosa and S. enterica are pathogen-specific. This is not surprising as both the immune response and the type of damage that P. aeruginosa and S. enterica pathogens produce during an infection is quite different. As mentioned above, P. aeruginosa modulates host biology through translation inhibition, oxidative stress, hypoxia, mitochondrial toxicity, and through other known and unknown mechanisms.23,27,28,50-53 S. enterica, on the other hand, has been shown to induce organism-wide oxidative stress in C. elegans.37 Our data indicates a complex cellular and physiological response of C. elegans during recovery from either of these 2 pathogens.
In the context of P. aeruginosa infection, multiple cuticular collagen genes are up-regulated during recovery (Fig. 4B). As P. aeruginosa slow killing is known to involve pathogen accumulation in the lumen of the intestine and subsequent damage to intestinal epithelial cells,11,25 it is interesting to note the upregulation of collagens in the cuticle, or skin, of C. elegans. It is likely that during the slow killing process, P. aeruginosa also induces damage to the cuticle, either directly or indirectly. It is also possible that many of these collagens are components of both the cuticle and the intestinal extracellular matrix. Further studies on these collagen genes would help to determine the exact mechanisms underlying slow killing pathogenicity and the repair processes important for recovery. No matter the location, rebuilding the extracellular matrix and restoration of tissue integrity is likely to be an important for recovery. Recently, collagen remodeling by SKN-1 has been shown to be important for longevity.40 Although we have ruled out the action of SKN-1 in recovery from P. aeruginosa acute infection, perhaps infection resolution signals feed into the collagen remodeling pathway downstream of SKN-1 and facilitate tissue repair.
The GATA transcription factor ELT-2 contributes to recovery from acute pathogen infection (Fig. 5A).4 ELT-2-mediated transcription also specifies the formation of the C. elegans intestine43 and it participates in the generation of a robust immune response upon exposure of C. elegans to various pathogens.31,45,54 For ELT-2 to specify these diverse transcriptional outputs, it must act on the genome with other nuclear factors. We previously observed an up-regulation of many DAF-16- and SKN-1-regulated genes, in addition to ELT-2-regulated genes, during recovery from acute S. enterica infection.4 This suggested that ELT-2 may modulate transcription during recovery in parallel to or in sequence with these transcription factors. We find that these factors do not play a role in recovery from acute P. aeruginosa infection (Fig. S6), although they may still be involved in recovery from S. enterica infection. Other as yet unknown factors must therefore be involved in specifying ELT-2 transcriptional activity during recovery from acute pathogen infection.
Our study shows that C. elegans initiates shared and distinct transcriptional responses that mediate recovery from different pathogens. By virtue of their molecular nature, many of the genes we have identified are likely to act in a local or paracrine manner to facilitate recovery. Other recovery genes, including a litany of genes encoding secreted proteins or proteins that are capable of synthesising a long-range response, may influence recovery in a systemic or endocrine manner. Future studies should seek to ascertain the mechanism by which these genes influence recovery. Combining these studies with the data presented here will help to shed light on the complex nature by which a host returns to homeostasis following an acute infection.
Materials and methods
Strains and reagents
C. elegans strains HH142 [fer-1(b232ts)], N2 [wild-type], KU25 [pmk-1(km25)], RB911 [fshr-1(ok778)], LD1 [SKN-1::GFP], and TJ356 [DAF-16::GFP] were provided by the Caenorhabditis Genetics Center. C. elegans strains were maintained at 15°C on NGM-OP50 plates without antibiotics. The following bacterial strains were used for maintenance or experiments: E. coli strain OP50-1 (OP50) [SmR],4 E. coli strain HT115 (HT115) [TetR],4 E. coli strain HT115 pL4440-RNAi (HT115-RNAi) [AmpR, TetR],4 P. aeruginosa strain PA14 (PA14) [RifR],11 P. aeruginosa strain PA14 expressing GFP (PA14-GFP) [KanR RifR],11 S. enterica Typhimurium strain 1344 (S. enterica) [SmR].4 Bacteria were grown overnight for 13-15 hours in 3 ml LB broth at 37°C except for HT115-RNAi bacteria (see below). Ampicillin (Merck, #171254-25GM), Carbenicillin (Cellgro, #46-100-RG), Neomycin (Sigma, #N1876-25G), Streptomycin (Sigma, S6501-100G), and isopropyl-β-D-1-thiogalactopyranoside (Anatrace, I1003) stocks were kept frozen at −20°C until needed.
P. aeruginosa survival assays
Plate preparation: HT115, OP50, or PA14-GFP were grown overnight for 13-15 hours in 3 ml LB broth at 37°C. A total of 200 µl of the resulting HT115 culture was seeded on 6.0 cm modified (0.35% peptone) NGM plates without antibiotics and allowed to grow for 1-2 d at 25°C. A total of 50 µl (exposure) or 30 µl (scoring) of the resulting PA14-GFP culture was seeded on 3.5 cm modified (0.35% peptone) NGM plates without antibiotics and allowed to grow overnight at 37°C. A total of 30 µl of the resulting OP50 culture was seeded on 3.5 cm modified (0.35% peptone) NGM plates with 50 µg/ml Streptomycin and allowed to grow for 1-2 d at 25°C.
Survival assay: Gravid fer-1(b232ts) animals were allowed to lay eggs for 4 hours at 25°C on 6.0 cm HT115 plates. Gravid animals were removed and the eggs/plates were incubated for 36 or 72 hours at 25°C. Synchronized L4 or adult animals were transferred to full lawn 3.5 cm OP50 or PA14-GFP plates and exposed to these bacteria for 12 or 18 hours at 25°C. After the designated amount of time, animals were washed off OP50 or PA14-GFP plates using M9, rinsed with several changes of M9, treated with 200 µg/ml Neomycin in M9 for 20 minutes, and subsequently transferred to 3.5 cm OP50 or PA14-GFP plates. The animals/plates were incubated at 25°C. Animals were scored every day and were considered dead when they failed to respond to touch. Animals were transferred to fresh plates every other day for the entire length of the experiment. Survival was plotted using Kaplan-Meier survival curves and analyzed by the log-rank test using GraphPad Prism (GraphPad Software, Inc.). Survival curves resulting in P values of < 0.05 were considered significantly different. A total of 25-75 (uninfected = 25, continuously infected = 75, recovered = 75) animals per condition were used.
RNAi-coupled P. aeruginosa survival assays
Plate preparation: HT115, OP50, and PA14-GFP plates were prepared as described above. HT115-RNAi strains were grown for 9 hours in 5 ml LB broth containing 50 µg/ml Ampicillin at 37°C. A total of 250 µl of the resulting culture was seeded on 6.0 cm NGM plates containing 50 µg/ml Carbenicillin and 3 mM isopropyl-β-D-1-thiogalactopyranoside. HT115-RNAi bacteria were allowed to grow for 2 d at 25°C to produce a thick lawn and to generate dsRNA. The daf-16 (sjj_R13H8.1), elt-2 (mv_AAC36130), pmk-1 (sjj_B0218.3), and skn-1 (sjj_T19E7.2) RNAi vectors were verified by DNA sequencing.
Survival assay: Gravid fer-1(b232ts) animals were allowed to lay eggs for 4 hours at 25°C on 6.0 cm HT115 plates. Gravid animals were removed and the eggs/plates were incubated for 36 hours at 25°C. Synchronized L4 animals were transferred to 6.0 cm RNAi or vector control plates and incubated for an additional 36 hours at 25°C. Adult RNAi or vector control animals were transferred to full lawn 3.5 cm OP50 or PA14-GFP plates and exposed to these bacteria for 12 hours at 25°C. Animals were washed off OP50 or PA14-GFP plates using M9, rinsed with several changes of M9, treated with 200 µg/ml Neomycin in M9 for 20 minutes, and subsequently transferred to 3.5 cm OP50 or PA14-GFP plates. The animals/plates were incubated at 25°C. Animals were scored every day and were considered dead when they failed to respond to touch. Animals were transferred to fresh plates every other day for the entire length of the experiment. Survival was plotted using Kaplan-Meier survival curves and analyzed by the log-rank test using GraphPad Prism (GraphPad Software, Inc.). Survival curves resulting in P values of < 0.05 were considered significantly different. A total of 30-75 (uninfected = 30, continuously infected = 75, recovered = 75) animals per condition were used.
S. enterica survival assays
Assays were performed as previously described.4
RNA isolation for microarray analysis
Plate preparation: OP50 or PA14-GFP were grown overnight for 13-15 hours in 3 ml LB broth at 37°C. A total of 500 µl of the resulting OP50 or PA14-GFP cultures was seeded on 10.0 cm modified (0.35% peptone) NGM plates with or without 50 µg/ml Streptomycin and allowed to grow for 1-2 d at 25°C to produce a thick lawn.
RNA isolation: fer-1(b232ts) animals were synchronized by treating gravid adults with sodium hydroxide and bleach. Approximately 2,000 synchronized L1 animals were transferred to full lawn 10.0 cm OP50 plates and incubated for 72 hours at 25°C. At 72 hours, adult animals were washed off OP50 plates using M9, rinsed with several changes of M9, and transferred to full lawn 10.0 cm PA14-GFP plates. At 76 hours, animals were washed off PA14-GFP plates using M9, rinsed with several changes of M9, treated with 200 µg/ml Neomycin in M9 for 20 minutes, and subsequently transferred to 10.0 cm OP50 or PA14-GFP plates. The animals/plates were incubated at 25°C. At designated harvest time points (72, 76, 82, 88, and 100 hours), animals were washed off plates using M9, rinsed with several changes of M9, and flash-frozen in QIAzol (Qiagen, #79306). Adult animals treated with Neomycin and Streptomycin for 6 hours served as a control for antibiotic exposure. Total RNA was extracted using the RNeasy Plus Universal Kit (Qiagen, #73404).
Microarray analysis
Amplification, hybridization, and scanning were performed as previously described.4 Scanned data was log2 transformed and quantile normalized using Partek Genomics Suite (Partek Inc.). P value and fold-change calculations were performed using Partek Genomics Suite (Partek Inc.). A P value of < 0.05 using the FDR linear step-up procedure55 was considered significant. Hierarchical clustering (Euclidean, average linkage) was performed on the 1,323 genes that were altered during the acute infection and recovery time course using Partek Genomics Suite (Partek Inc.). For each of the 5 acute infection and recovery time points, 3 biological replicates were assessed. For the antibiotic exposure control time points, 82 hOP and 82 hOPC, 2 or 3 biological replicates were assessed. The microarray data was deposited in the NCBI Gene Expression Omnibus database: GSE81592.
RNA isolation for qRT-PCR
Total RNA was extracted from animals that were subjected to the same conditions as those described in the RNA isolation for microarray analysis section above.
Quantitative real-time PCR (qRT-PCR)
Quantitative real-time PCR assays were performed as previously described.4 To compare qPCR and microarray data, a one sample, 2-tailed t-test was performed.
Bioinformatics
All gene lists, including those culled from the literature, were passed through WormBase Converter19 using the WS220 genome release as the output (references are noted in Table S7). A total of 20,834 WS220 genes are represented by 1 or more probes in the Agilent C. elegans V2 array (Agilent Technologies). Gene ontology analysis was performed using the DAVID Bioinformatics Database (david.abcc.ncifcrf.gov/).56 Overlap of 2 gene sets was performed using Partek Genomics Suite (Partek Inc.). Statistical significance of the overlap between 2 gene sets was calculated using the following on-line program: nemates.org/MA/progs/overlap_stats.html. The representation factor (RF) represents the number of overlapping genes divided by the expected number of overlapping genes drawn from 2 independent groups. A background gene list of 20,834 was used for the calculation. P values were calculated using the exact hypergeometric probability. 1.5 kb cis-regulatory sequences were identified using WormMart (wormbase.org). Detailed bioinformatics protocols are available upon request.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
C. elegans strains were provided by the Caenorhabditis Genetics Center (University of Minnesota), which is funded by the NIH-ORIP. We thank Dr. Danielle Ferraz Mello for technical support and helpful discussions and the Duke GCB Microarray Facility for technical advice.
Funding
B.H. is funded by NIH Grant GM103122. A.A. is funded by grants from NIGMS GM070977 and NIAID AI117911.
References
- [1].Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol 2013; 13:59-66; PMID:23197111; http://dx.doi.org/ 10.1038/nri3362 [DOI] [PubMed] [Google Scholar]
- [2].Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014; 40:315-27; PMID:24656045; http://dx.doi.org/ 10.1016/j.immuni.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014; 510:92-101. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24899309; PMID:24899309; http://dx.doi.org/ 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Head B, Aballay A. Recovery from an acute infection in C. elegans requires the GATA transcription factor ELT-2. PLoS Genet 2014; 10:e1004609; PMID:25340560; http://dx.doi.org/ 10.1371/journal.pgen.1004609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Torres BY, Oliveira JHM, Thomas Tate A, Rath P, Cumnock K, Schneider DS. Tracking Resilience to Infections by Mapping Disease Space. PLoS Biol 2016; 14:e1002436; PMID:27088359; http://dx.doi.org/ 10.1371/journal.pbio.1002436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wahl SM. TGF-beta in the evolution and resolution of inflammatory and immune processes. Introduction. Microbes Infect 1999; 1:1247-9; PMID:10611751; http://dx.doi.org/ 10.1016/S1286-4579(99)00261-0 [DOI] [PubMed] [Google Scholar]
- [7].Filippi CM, von Herrath MG. IL-10 and the resolution of infections. J Pathol 2008; 214:224-30; PMID:18161757; http://dx.doi.org/ 10.1002/path.2272 [DOI] [PubMed] [Google Scholar]
- [8].Shields AM, Panayi GS, Corrigall VM. Resolution-associated molecular patterns (RAMP): RAMParts defending immunological homeostasis? Clin Exp Immunol 2011; 165:292-300; PMID:21671907; http://dx.doi.org/ 10.1111/j.1365-2249.2011.04433.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Laidlaw BJ, Cui W, Amezquita RA, Gray SM, Guan T, Lu Y, Kobayashi Y, Flavell RA, Kleinstein SH, Craft J, et al.. Production of IL-10 by CD4(+) regulatory T cells during the resolution of infection promotes the maturation of memory CD8(+) T cells. Nat Immunol 2015; 16:871-9; PMID:26147684; http://dx.doi.org/ 10.1038/ni.3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol 2015; 7:a016311; http://dx.doi.org/ 10.1101/cshperspect.a016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tan MW, Mahajan-Miklos S, Ausubel FM. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 1999; 96:715-20. PMID:9892699; http://dx.doi.org/ 10.1073/pnas.96.2.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shivers RP, Youngman MJ, Kim DH. Transcriptional responses to pathogens in Caenorhabditis elegans. Curr Opin Microbiol 2008; 11:251-6. PMID:18567532; http://dx.doi.org/ 10.1016/j.mib.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Partridge FA, Gravato-Nobre MJ, Hodgkin J. Signal transduction pathways that function in both development and innate immunity. Dev Dyn 2010; 239:1330-6; PMID:20131356 [DOI] [PubMed] [Google Scholar]
- [14].Marsh EK, May RC. Caenorhabditis elegans, a model organism for investigating immunity. Appl Environ Microbiol 2012; 78:2075-81; PMID:22286994; http://dx.doi.org/ 10.1128/AEM.07486-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Simonsen KT, Gallego SF, Faergeman NJ, Kallipolitis BH. Strength in numbers: “Omics” studies of C. elegans innate immunity. Virulence 2012; 3:477-84; PMID:23076279; http://dx.doi.org/ 10.4161/viru.21906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pukkila-Worley R, Ausubel FM. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr Opin Immunol 2012; 24:3-9; PMID:22236697; http://dx.doi.org/ 10.1016/j.coi.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ewbank JJ, Pujol N. Local and long-range activation of innate immunity by infection and damage in C. elegans. Curr Opin Immunol 2016; 38:1-7; PMID:26517153; http://dx.doi.org/ 10.1016/j.coi.2015.09.005 [DOI] [PubMed] [Google Scholar]
- [18].Wong D, Bazopoulou D, Pujol N, Tavernarakis N, Ewbank JJ. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol 2007; 8:R194. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17875205; PMID:17875205; http://dx.doi.org/ 10.1186/gb-2007-8-9-r194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Engelmann I, Griffon A, Tichit L, Montanana-Sanchis F, Wang G, Reinke V, Waterston RH, Hillier LW, Ewbank JJ. A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS One 2011; 6:e19055. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21602919; PMID:21602919; http://dx.doi.org/ 10.1371/journal.pone.0019055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 2012; 149:452-66. PMID:22500807; http://dx.doi.org/ 10.1016/j.cell.2012.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aballay A. Role of the nervous system in the control of proteostasis during innate immune activation: insights from C. elegans. PLoS Pathog 2013; 9:e1003433; PMID:23950707; http://dx.doi.org/ 10.1371/journal.ppat.1003433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cohen LB, Troemel ER. Microbial pathogenesis and host defense in the nematode C. elegans. Curr Opin Microbiol 2015; 23:94-101. PMID:25461579; http://dx.doi.org/ 10.1016/j.mib.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 1999; 96:47-56. PMID:9989496; http://dx.doi.org/ 10.1016/S0092-8674(00)80958-7 [DOI] [PubMed] [Google Scholar]
- [24].Tenor JL, McCormick BA, Ausubel FM, Aballay A. Caenorhabditis elegans-based screen identifies Salmonella virulence factors required for conserved host-pathogen interactions. Curr Biol 2004; 14:1018-24. http://dx.doi.org/ 10.1016/j.cub.2004.05.050 [DOI] [PubMed] [Google Scholar]
- [25].Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog 2010; 6:1-24. http://dx.doi.org/ 10.1371/journal.ppat.1000982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aballay A, Yorgey P, Ausubel FM. Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr Biol 2000; 10:1539-42. PMID:11114525; http://dx.doi.org/ 10.1016/S0960-9822(00)00830-7 [DOI] [PubMed] [Google Scholar]
- [27].Gallagher LA, Manoil C. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 2001; 183:6207-14; PMID:11591663; http://dx.doi.org/ 10.1128/JB.183.21.6207-6214.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cezairliyan B, Vinayavekhin N, Grenfell-Lee D, Yuen GJ, Saghatelian A, Ausubel FM. Identification of Pseudomonas aeruginosa Phenazines that Kill Caenorhabditis elegans. PLoS Pathog 2013; 9:e1003101. PMID:23300454; http://dx.doi.org/ 10.1371/journal.ppat.1003101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kirienko N V, Cezairliyan BO, Ausubel FM, Powell JR. Pseudomonas aeruginosa PA14 pathogenesis in Caenorhabditis elegans. Methods Mol Biol 2014; 1149:653-69; PMID:24818940; http://dx.doi.org/ 10.1007/978-1-4939-0473-0_50 [DOI] [PubMed] [Google Scholar]
- [30].Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2006; 2:e183. PMID:17096597; http://dx.doi.org/ 10.1371/journal.pgen.0020183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan MW. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci U S A 2006; 103:14086-91. PMID:16968778; http://dx.doi.org/ 10.1073/pnas.0603424103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aballay A, Drenkard E, Hilbun LR, Ausubel FM. Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr Biol 2003; 13:47-52. PMID:12526744; http://dx.doi.org/18221392 10.1016/S0960-9822(02)01396-9 [DOI] [PubMed] [Google Scholar]
- [33].Alegado RA, Campbell MC, Chen WC, Slutz SS, Tan MW. Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell Microbiol 2003; 5:435-44. http://dx.doi.org/ 10.1046/j.1462-5822.2003.00287.x [DOI] [PubMed] [Google Scholar]
- [34].Alegado RA, Tan MW. Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell Microbiol 2008; 10:1259-73; Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18221392; PMID:18221392 [DOI] [PubMed] [Google Scholar]
- [35].Bailey AM, Ivens A, Kingsley R, Cottell JL, Wain J, Piddock LJ V. RamA, a member of the AraC/XylS family, influences both virulence and efflux in Salmonella enterica serovar Typhimurium. J Bacteriol 2010; 192:1607-16; PMID:20081028; http://dx.doi.org/ 10.1128/JB.01517-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Alegado RA, Chin CY, Monack DM, Tan MW. The two-component sensor kinase KdpD is required for Salmonella typhimurium colonization of Caenorhabditis elegans and survival in macrophages. Cell Microbiol 2011; 13:1618-37; PMID:21790938; http://dx.doi.org/ 10.1111/j.1462-5822.2011.01645.x [DOI] [PubMed] [Google Scholar]
- [37].Sem X, Rhen M. Pathogenicity of Salmonella enterica in Caenorhabditis elegans relies on disseminated oxidative stress in the infected host. PLoS One 2012; 7:e45417. PMID:23028994; http://dx.doi.org/ 10.1371/journal.pone.0045417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sun J, Singh V, Kajino-Sakamoto R, Aballay A. Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science (80- ) 2011; 332:729-32. PMID:21474712; http://dx.doi.org/ 10.1126/science.1203411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kramer JM. Basement membranes. WormBook 2005; 1-15; PMID:18050423; http://dx.doi.org/ 10.1895/wormbook.1.16.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ewald CY, Landis JN, Abate JP, Murphy CT, Blackwell TK. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature 2014; PMID: 25517099; http://dx.doi.org/11226309 10.1038/nature14021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Aballay A, Ausubel FM. Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc Natl Acad Sci U S A 2001; 98:2735-9. PMID:11226309; http://dx.doi.org/ 10.1073/pnas.041613098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Haskins KA, Russell JF, Gaddis N, Dressman HK, Aballay A. Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev Cell 2008; 15:87-97. PMID:18606143; http://dx.doi.org/ 10.1016/j.devcel.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fukushige T, Hawkins MG, McGhee JD. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol 1998; 198:286-302. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9659934; PMID:9659934 [PubMed] [Google Scholar]
- [44].Lee SH, Wong RR, Chin CY, Lim TY, Eng SA, Kong C, Ijap NA, Lau MS, Lim MP, Gan YH, et al.. Burkholderia pseudomallei suppresses Caenorhabditis elegans immunity by specific degradation of a GATA transcription factor. Proc Natl Acad Sci U S A 2013; 110:15067-72; PMID:23980181; http://dx.doi.org/ 10.1073/pnas.1311725110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kerry S, TeKippe M, Gaddis NC, Aballay A. GATA transcription factor required for immunity to bacterial and fungal pathogens. PLoS One 2006; 1:e77. PMID:17183709; http://dx.doi.org/ 10.1371/journal.pone.0000077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McGhee JD, Fukushige T, Krause MW, Minnema SE, Goszczynski B, Gaudet J, Kohara Y, Bossinger O, Zhao Y, Khattra J, et al.. ELT-2 is the predominant transcription factor controlling differentiation and function of the C. elegans intestine, from embryo to adult. Dev Biol 2009; 327:551-65. PMID:19111532; http://dx.doi.org/ 10.1016/j.ydbio.2008.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, et al.. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science (80- ) 2002; 297:623-6. Available from: http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&dopt=r&uid=12142542; PMID:12142542 [DOI] [PubMed] [Google Scholar]
- [48].Powell JR, Kim DH, Ausubel FM. The G protein-coupled receptor FSHR-1 is required for the Caenorhabditis elegans innate immune response. Proc Natl Acad Sci U S A 2009; 106:2782-7. PMID:19196974; http://dx.doi.org/ 10.1073/pnas.0813048106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci U S A 1999; 96:2408-13. PMID:10051655; http://dx.doi.org/22520465 10.1073/pnas.96.5.2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 2012; 11:375-86; PMID:22520465; http://dx.doi.org/ 10.1016/j.chom.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].McEwan DL, Kirienko N V. Ausubel FM. Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 2012; 11:364-74; PMID:22520464; http://dx.doi.org/ 10.1016/j.chom.2012.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kirienko NV, Kirienko DR, Larkins-Ford J, Wahlby C, Ruvkun G, Ausubel FM, Wählby C, Ruvkun G, Ausubel FM. Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe 2013; 13:406-16. PMID:23601103; http://dx.doi.org/ 10.1016/j.chom.2013.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kirienko N V, Ausubel FM, Ruvkun G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 2015; 112:1821-6. PMID:25624506; http://dx.doi.org/ 10.1073/pnas.1424954112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Block DHS, Twumasi-Boateng K, Kang HS, Carlisle JA, Hanganu A, Lai TY-J, Shapira M. The Developmental Intestinal Regulator ELT-2 Controls p38-Dependent Immune Responses in Adult C. elegans. PLoS Genet 2015; 11:e1005265; PMID:26016853; http://dx.doi.org/ 10.1371/journal.pgen.1005265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Benjamini Y, Hochberg Y. Controlling the False Discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995; 57:289-300. Available from: http://www.jstor.org/stable/2346101 [Google Scholar]
- [56].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57. PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.