

Abstract
The shrinking antibiotic development pipeline together with the global increase in antibiotic resistant infections requires that new molecules with antimicrobial activity are developed. Traditional empirical screening approaches of natural and non-natural compounds have identified the majority of antibiotics that are currently available, however this approach has produced relatively few new antibiotics over the last few decades. The vast amount of bacterial genome sequence information that has become available since the sequencing of the first bacterial genome more than 20 years ago holds potential for contributing to the discovery of novel antimicrobial compounds. Comparative genomic approaches can identify genes that are highly conserved within and between bacterial species, and thus may represent genes that participate in key bacterial processes. Whole genome mutagenesis studies can also identify genes necessary for bacterial growth and survival under different environmental conditions, making them attractive targets for the development of novel inhibitory compounds. In addition, transcriptomic and proteomic approaches can be used to characterize RNA and protein levels on a cellular scale, providing information on bacterial physiology that can be applied to antibiotic target identification. Finally, bacterial genomes can be mined to identify biosynthetic pathways that produce many intrinsic antimicrobial compounds and peptides. In this review, we provide an overview of past and current efforts aimed at using bacterial genomic data in the discovery and development of novel antibiotics.
Keywords: antibiotic resistance, antibiotic discovery, genomics, essential genes, bacteriocins
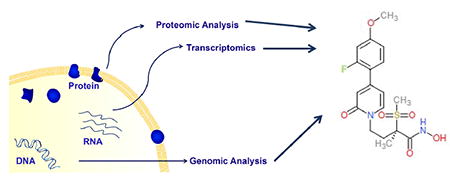
1. Introduction
Widespread antibiotic resistance poses an important threat to modern medicine. Infections caused by antibiotic resistant strains from multiple bacterial species continue to increase in incidence, and in some cases there are only a few antibiotics with appropriate antimicrobial activity that continue to be effective. Recent reports describing the identification and spread of antibiotic resistance genes that reduce susceptibility to “last resort” antibiotics such carbapenems and colistin add to concerns that infections that are untreatable with currently available antibiotics may become a reality [1,2]. The United States Centers for Disease Control and Prevention published a “Threat Report” in which it was estimated that more than 2 million individuals acquire antibiotic resistant infections every year in the U.S. alone, resulting in approximately 23,000 deaths [3]. The future global impact of antimicrobial resistance has potential to be devastating, as a government commissioned study in the United Kingdom estimated that by the year 2050, 10 million deaths per year could result from antimicrobial resistance in certain bacterial, viral and parasitic infections if current trends continue [4]. The economic implications of antimicrobial resistance also warrant concern given estimates from the same study indicating that by 2050 the unabated increase in the incidence of resistant infectious and their associated mortality could result in a reduction in the global gross domestic product of between 2% and 3.5% (approximately $100 trillion between 2014 and 2050).
In light of these estimates, it may seem obvious that efforts to develop new antibiotics with novel mechanisms of action should be a priority. However, in spite of the fact that the need for new antibiotics has been outlined by a number of experts [5,6,7], there has been a dearth of novel molecules approved for clinical use over the last three decades [8,9]. A major factor contributing to the dwindling antibiotic pipeline is the decreasing number of companies within the pharmaceutical sector that continue to pursue the development of novel antimicrobials [10]. While decreased profitability compared to other clinical indications that require longer term therapy is commonly cited as a major contributing factor to a reduction in active antibiotic development programs, there are other factors that likely also play a role in this decline. The antibiotics that have been developed to date using traditional discovery methods that employ high throughput screening of natural compounds may be those molecules that are most readily identified using these methodologies [11]. For example, the screening of natural products synthesized by actinomycetes and different fungal species has been successful in identifying novel antimicrobial compounds. However, the molecules that were previously identified may represent the antimicrobial compounds that are most commonly produced by these microorganisms, and were thus more easily detected using these traditional approaches. Although natural product screening can continue to be optimized to identify less abundant molecules, the yield of these techniques in terms of the number of new compounds that are identified may not return to levels seen during the decades between the 1930’s and 1970’s, which saw the introduction of multiple new antibiotics with novel mechanisms of action. For these reasons, the chemical modification of previously identified molecules with known targets is increasingly being employed for antibiotic development [12,13]. An obvious limitation of this approach is that only a finite number of modifications can be made to existing compounds. Perhaps more worrisome, however, is the fact that this approach may not result in the development of new compounds with novel mechanisms of action, an issue of critical importance given that resistance to one compound within an antibiotic class often produces cross resistance to other antibiotics within the same class.
The increasing number of bacterial genome sequences, together with techniques that permit global transcriptional and proteomic profiling of bacterial cells under different conditions, may provide information that can be applied to the development of novel antimicrobials. In this review, we provide an overview of how bacterial genomes have been employed in the search for new antibiotics since the first complete bacterial genome sequence was available in 1995 [14], and provide examples from the recent literature that demonstrate current efforts related to antimicrobial development that are being undertaken in order to exploit the massive amount of information that the genomic era has provided.
In this context, a number of recent initiatives aim to facilitate the development of novel antimicrobials and their introduction into the clinic. In 2014 an Executive Order signed by President Obama entitled Combating Antibiotic-Resistant Bacteria aimed to take a comprehensive approach toward preventing the emergence of antimicrobial resistance and developing next generation antibiotics (www.whitehouse.gov/the-press-office/2014/09/18/executive-order-combating-antibiotic-resistant-bacteria). This initiative provides 1.2 billion USD to fund a coordinated effort involving multiple government agencies in the fight against antibiotic resistance. Additionally, the Innovative Medicines Initiative’s New Drugs for Bad Bugs initiative, which is funded by the European Commission, aims to facilitate the development and evaluation of novel antimicrobials by creating public private partnerships that take advantage of capacities within the public sector to advance products in the pharmaceutical industry’s antimicrobial pipeline [15]. In 2016, the CARB-X initiative was launched in order to accelerate development of new antimicrobials in the preclinical stages of development with the goal of advancing them into clinical testing. Over the first five years, the CARB-X initiative aims to promote the advancement of at least 20 new compounds into human trials [16].
2. Target based antibiotic discovery
Traditionally, the identification and development of compounds with antibacterial activity has relied upon the empiric testing of both natural and non-natural compounds with exponentially growing bacterial cells. This approach, together with the development of analogues of compounds showing antibiotic activity through chemical modification, has been the mainstay of the pharmaceutical industry and has produced the majority of antibiotics approved for clinical use. It is worth noting that the screening of compound libraries and the modification of existing antimicrobials continue to be used extensively within the pharmaceutical and biotechnology sectors for the identification of compounds with antibacterial activity [11,13]. Over the last two decades, the availability of genome sequencing techniques and transcriptomic and proteomic approaches that permit the global characterization of bacterial components have raised the possibility that antibiotic discovery can be performed by first identifying high value bacterial targets, and then developing compounds that inhibit these targets. Once bacterial targets with the desired characteristics are identified (discussed below), these targets can be incorporated into in vitro biochemical assays that permit high throughput screening of chemical libraries for the identification of inhibitory compounds (Figure 1). The antibacterial activity of hits identified during high throughput screening can then be characterized using standard assays with bacterial cells. Leads that demonstrate adequate antimicrobial activity can then be further developed in preclinical and clinical studies similarly to molecules identified using traditional discovery methods.
Figure 1. Target based antibiotic discovery.
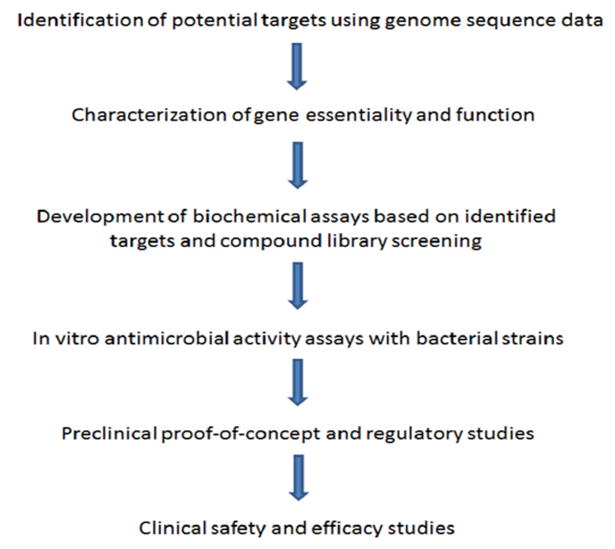
The schematic shows a general model of target based antibiotic discovery from initial target identification through the use of genomic data through clinical testing.
The major advantage of target based antibiotic discovery is that, at least in theory, it facilitates the identification of compounds that target bacterial components with desirable characteristics. This approach thus prompts the question, “What characteristics should ideal targets for the development of new antibiotics have?” A number of criteria that novel antibiotic targets should meet have been proposed and used as the basis for studies aiming to identify novel bacterial targets [13,17]. A key characteristic of novel targets is that they are present in the majority of pathogenic strains within a species, and that they are highly conserved at the sequence level between different strains. Fortunately, the increasing use of molecular typing methods and the availability of dozens to thousands of bacterial genomes for many pathogenic species facilitate these studies [18]. It is also desirable to exclude potential targets that have human counterparts in order to avoid potential toxicity. While toxicity studies must clearly be performed on any new compound intended for human use, comparative analyses of potential bacterial targets and human genome sequences can identify bacterial components that share homology, and may thus produce undesired effects on the host. An additional critical characteristic of an antibiotic is that its target is expressed and active during human infection, thus ensuring that inhibitory compounds have antimicrobial activity under physiologic conditions. As described in detail later in this review, advances in transcriptomic and proteomic techniques have greatly facilitated the global characterization of bacterial gene expression under different environmental conditions. A final consideration for the identification of novel targets for the development of antibiotics is the essentiality of the bacterial component being targeted. It seems logical to assume that bacterial components necessary for growth and survival would serve as ideal targets for the identification of inhibitory compounds that demonstrate antibacterial activity. As described below, the identification of so-called “essential genes” in bacterial genomes has been an area of intensive study.
Although target based approaches permit the identification of bacterial components with characteristics that are desirable for the development of novel antibiotics, they are not without their disadvantages. Perhaps the primary limitation associated with target based approaches is the potential for compounds that demonstrate inhibitory activity against their cognate target in biochemical assays to demonstrate poor antimicrobial activity when used with bacterial cells. While biochemical assays with purified targets can provide very precise data regarding the inhibitory capacity of compounds, they do not provide information regarding the action of a compound on its target in the physiological context of a bacterial cell. A number of possibilities could limit the activity of hits identified using in vitro assays in experiments that directly assess antimicrobial activity on bacterial cells. One possibility is limited access of the inhibitory compound to its target due to low permeability of the bacterial membrane, or the action of efflux pumps that remove the compound from the interior of the cell. A second possibility is functional redundancy in which the function of the inhibited target can be carried out by other bacterial components. An additional limitation associated with the use of biochemical assays that are employed in target based approaches is that certain bacterial processes, some of which have been shown to be essential for viability, are difficult to reconstitute with in vitro assays. This may be especially relevant for some of the more complex bacterial processes, such as translation and membrane biosynthesis, targets of previously developed antibiotic classes. A final limitation associated with target based approaches is that, in contrast to empirical approaches, a large amount of information (e.g. genome sequences from different strains/species) is typically required in order to initiate studies aiming to identify high value targets.
3. Comparative Genomics
With the sequencing of the first bacterial genome in the mid-1990s [14] came the hope that genomic information could be interrogated to identify novel targets that would facilitate antibiotic discovery. In the following two decades, advances in sequencing technologies enabled the rapid sequencing (a few hours) of microbial genomes at a cost that continues to decrease, and can now be achieved for a few hundred dollars per genome [19,20]. Together with the development of bioinformatic applications that facilitate assembly and annotation [21,22,23], these advances have permitted the sequencing and analysis of thousands of bacterial genomes. This is evidenced by the fact that in a span of only five years between 2009 and 2014 the number of prokaryotic genome sequence submissions to NCBI increased from less than 1,000 to more than 14,000 [18].
As described above, antimicrobial compounds should ideally target highly conserved bacterial components that are expressed in the majority of pathogenic strains within a species. Comparative genomic approaches can facilitate the rapid identification of targets that meet these criteria and provide information regarding the putative functions of identified targets by identifying sequence homology to proteins with known functions. This purely bioinformatic approach may be only the first step in elucidating genes of interest, as subsequent experimentation aimed at characterizing the role of the identified target in pathogenesis, the extent to which they are expressed during infection, and their essentiality can provide additional information necessary for identifying high value targets. The availability of numerous genome sequences for a given bacterial species permits the determination of the species’ core genome [24,25], those genes that are present in all, or the majority, of sequenced strains. Based on the idea that the presence of these genes in the majority of strains indicates that they are likely to participate in key bacterial processes, the elucidation of core genomes can provide an initial step for antibiotic target identification. Recently, core genomes have been proposed for multiple bacterial species associated with multidrug resistance. Two studies with Escherichia coli have identified approximately 3000 genes as those comprising the core genome of the roughly 5000 genes that are typically present in a single E. coli strain [18,26]. The potential that core genome analysis has in excluding genes that may not represent ideal targets for antibiotic development is demonstrated by that fact that in one of these studies, the E. coli pan-genome, the collection of genes that are present in at least one of the strains used for analysis, consisted of approximately 90,000 genes [18]. Interestingly, roughly one third of the genes making up the pangenome were present in only one of the more than 2,000 genomes employed in the study. A separate study employing 17 reference strains of Pseudomonas aeruginosa included 5,233 genes in the core genome [27]. These genes represented approximately 88% of the number of genes in the strains used in the study, and consisted of genes encoding enzymes involved in basic metabolic processes, virulence factors and antibiotic resistance determinants. It is of note that of the 2304 genes encoding metabolic functions in the strains included in the study, 1840 were identified as part of the core genome. Although these studies exemplify the ability of core genome analysis to identify genes that are widespread within a bacterial species, the number of genes identified using core genome analysis alone (typically a few thousand) may not sufficiently reduce the number of identified genes to a number that makes further characterization of all identified genes feasible. For this reason, additional studies may provide information that can be used to focus subsequent development on high value targets that meet desired criteria. These additional studies can be purely bioinformatic in nature, for example determining the predicted function of core genome genes by comparison with homologues in other species, or can be based on experimental results, for example studies that identify genes that play a critical role in infection. It is also worth noting that, in addition to intraspecies analyses, such as core genome identification, cross- species comparative genomic approaches can also be employed to identify genes conserved across bacterial species based on the idea that these targets may be involved in critical bacterial functions [13,28]. For both intraspecies and interspecies analyses, the quality of the results depends upon the epidemiologic diversity of the strains and the number of sequenced genomes included in the analysis.
It is important to point out that, in spite of the initial enthusiasm that was generated in the late 90s and early 2000s regarding the potential impact of comparative genomic approaches on antibiotic development, these methods have led to the identification of few lead compounds and have yet to result in a clinically approved antibiotic. An excellent summary of the efforts undertaken by GlaxoSmithKline (GSK) between 1995 and 2001 gives a detailed account of the use of comparative genomics in identifying targets for the development of broad spectrum antimicrobials [13]. Over this seven year period, researchers at GSK mined the genomes of Haemophilus influenza, Moraxella catarrhalis, Streptococcus pneumonia, Staphylococcus aureus and Enterococcus fecalis to identify highly conserved genes. Subsequent mutagenesis experiments were used to characterize gene essentiality in order to narrow the number targets that would developed for high throughput screening. In total, more than 300 genes were identified as potential targets, resulting in 67 high throughput screens. These screens led to the identification of sixteen hits that gave a positive result in vitro, five of which were developed as lead compounds. However, none of these compounds advanced into clinical testing. As a consequence of these results, GSK subsequently reorganized its antibiotic discovery program to employ more traditional methods based on using chemical synthesis to develop novel agents that inhibit known targets using novel mechanisms.
Potential reasons for the low yield of promising candidates obtained using approaches based on the use of genomics for the identification of novel targets are (i) an insufficient number of genomes used in the identification of potential targets, (ii) a lack of sufficient complexity in the compound libraries that are employed in biochemical screening assays for hit identification, and (iii) many bacterial processes cannot be effectively adapted to in vitro screening methodologies [11,13,17]. These considerations, together with reports describing the lack of viable candidates that have emerged from target based screens, have led some to suggest that these approaches may not be as robust as the traditional screening of natural products for the identification of new compounds [11]. It is also worth considering the extent to which genome based target identification will be able to identify novel targets in light of analyses suggesting that there are few bacterial metabolic targets that remain unknown [29,30]. In this context, it is probably justifiable to consider that genome based target identification has not lived up to the original expectations that were widely expressed upon entering into the genomics era [31,32,33]. In spite of these initial disappointments, however, the use of genomic information for target identification continues to shed light on multiple aspects of bacterial physiology and has potential to contribute to continued efforts aimed at the identification and characterization of targets for antibiotic development using the method described below.
The small number of targets that have been identified using comparative genomics has led to the suggestion that additional screening methodologies may increase the yield of viable hits [34]. Over the last two decades, multiple molecular modelling platforms have been developed that permit the in silico screening of potential inhibitors based on the proposed 3-dimensional structures of identified targets [35,36,37]. These structural biology-based approaches can identify potential inhibitors with predicted high-affinity binding to the active site or allosteric sites of bacterial targets. “Hits” identified by in silico screening can then be tested using in vitro biochemical assays to directly assess their ability to inhibit targets. This approach has the advantage of being rapid and comparatively inexpensive compared to high throughput biochemical approaches, however the results are dependent upon the availability of high quality structural information for both targets and potential inhibitors. Hit identification and lead optimization using in silico-based modelling approaches have already contributed to the identification of multiple antiviral compounds [38,39,40]. An additional structure-based screening approach that combines the purified target with potential inhibitors and employs NMR in order to determine if binding occurs between molecules has been developed [41]. This approach was recently used to characterize inhibitors of β-hydroxydecanoyl-acyl carrier protein dehydratase from Pseudomonas aeruginosa [42]. These structure-based approaches, together with the biochemical characterization of potential targets together with high throughput screening of potential inhibitory molecules may facilitate the identification of novel lead compounds.
The availability of thousands of complete genome sequences from hundreds of bacterial species has provided evidence that a large number of biosynthetic pathways are cryptic, meaning that they are not highly expressed under commonly-used culture conditions. The identification of these cryptic gene clusters in antibiotic-producing soil bacteria (e.g. Streptomyces species) raise the possibility that these pathways may code for enzymes involved in the synthesis of molecules with antimicrobial activity [11]. A number of approaches have been developed to identify these cryptic genes and characterize the molecules that they produce. Sequence-based approaches have been developed that permit the identification of these cryptic gene clusters and their promotors and then predict characteristics of their biosynthetic products based on conserved enzyme motifs [43,44]. Culturing microorganisms that harbor these gene clusters under different conditions can then be employed in order to facilitate the expression of the cryptic genes, and the identification of their biosynthetic products [43,44]. A second approach is to introduce the cryptic gene clusters into culturable microorganisms to facilitate their expression. This approach was demonstrated to produce high yields of the antibiotic daptomycin in Streptomyces lividans after introduction of the 128 Kb biosynthetic gene cluster [45]. The development of methodologies that permit the identification of antimicrobial compounds from unculturable microorganisms also holds potential for identifying novel target compounds. A recent example is the iChip technology that was recently used to identify the new antibiotic teixobactin [46]. This technology permits the isolation of single bacteria from environmental sources, such as soil samples, and allows metabolic products (including antimicrobial compounds) to diffuse through a permeable barrier for detection. This approach thus permits the identification of molecules that are produced by microorganisms that are not culturable under standard conditions.
4. Essential Genes as Targets for Antibiotic Discovery
Genes that are required for bacterial growth and survival are attractive targets for the development of novel antibiotics since the development of compounds that inhibit the functions encoded by these genes are, at least theoretically, more likely to demonstrate antimicrobial activity than compounds targeting genes that encode non-essential functions. As described above, comparative genomic approaches employ genome sequence data together with bioinformatic approaches to identify genes that are likely to encode essential functions based on their conservation and distribution within and across different bacterial species. However, these sequence based analyses do not directly test the essentiality of genes. Multiple techniques have been developed that empirically characterize the essentiality of bacterial genes.
Traditionally, in order to determine if a gene of interest was essential for bacterial growth or survival, bacterial mutants lacking the gene could be engineered using well-known techniques for genome manipulation. An inability to obtain mutants that grow in the absence of the target gene is suggestive of essentiality. A complementary approach places the gene of interest under the control of an inducible promoter that, if the target gene is critical for growth/survival, results in no or impaired growth in the absence of induction. These targeted approaches have been widely used, and are well suited for characterizing the essentiality of a relatively small number of target genes that have been identified using complementary methods, such as comparative genomics. Methods that aim to globally identify essential genes, as opposed to characterizing the essentiality of a single gene, have also been developed. One such approach is to generate mutant libraries using transposon mutagenesis by transforming bacterial cells with a transposon that inserts itself into the bacterial chromosome. Due to the fact that transposon insertion throughout the bacterial genome is random, if a large enough number of mutants are characterized, genes that are essential can be identified as they will not give rise to viable mutants. An example of this type of study performed by Jacobs et al. generated more than 30,000 mutants of Pseudomonas aeruginosa by “saturating” the genome with transposon mutations such that each gene would be expected to be interrupted in multiple individual transposon mutants [47]. The analysis of viable mutants demonstrated that approximately 12% of all predicted open reading frames were not represented in the collection of mutants, suggesting that these genes had a high probability of being essential for survival. These data, in additional to further analysis, led the authors to estimate that between 300 and 400 P. aeruginosa genes are essential for growth on rich laboratory media. A separate study employing a different strain of P. aeruginosa generated an ordered, non-redundant collection of all interruptible (non-essential) genes by transposon mutagenesis [48]. Comparison of the data sets generated by both studies permitted further prediction of essential genes across different strains. In a later study, the essential genes identified with these collections of transposon mutants were used to identify conserved P. aeruginosa genes that are located within the cell membrane or secreted from the bacterium [49]. Deletion mutants and strains that conditionally express target genes were used to identify LptH, which participates in lipopolysaccharide transport, and LolA, which plays a role in lipoprotein transport, as genes that are essential for growth in vitro. One of the gene products, LptH was also shown to be necessary for producing infection in experimental models of P. aeruginosa infection. LolA, on the other hand, only moderately affected the ability of P. aeruginosa to produce infection. It is important to keep in mind that whole genome mutagenesis approaches that aim to identify essential genes based on the absence of viable transposon mutants only provide statistical evidence that a gene is essential based on the likelihood that a sequence of a given length will be disrupted by random mutagenesis. For this reason, determining that these genes are truly essential must typically be confirmed using the targeted gene mutation methods described above.
A key aspect that must be taken into consideration when characterizing the essentiality of a gene or performing studies aimed at the identification of essential genes is the environmental context of the bacteria. Due to the fact that bacteria have evolved to survive and grow under multiple different conditions, genes that are essential in one environment may not be essential in a different environment. It may therefore be misleading in some cases to classify genes as essential or non-essential since, for some genes, essentiality may be determined by the environmental context. While it is likely that a subset of genes that encode critical functions required for bacterial survival under all conditions are truly essential, some genes may encode functions that are essential for growth/survival under certain conditions. It may therefore be more appropriate to refer to the latter as conditionally essential genes. This aspect may be of special importance with respect to the identification of essential genes for the development of antibiotic targets since the laboratory conditions that are often employed to identify essential genes may exclude genes that are essential during host infection, or include genes that are necessary for growth under laboratory conditions but not essential for growth in the host. Multiple techniques have been developed for identifying conditionally essential genes, and they have been used extensively to identify genes that play a role in different aspects of host infection. One approach that has been broadly applied is to create mutant libraries that contain strains that grow under laboratory conditions either through random transposon mutagenesis or through the directed interruption of predicted bacterial open reading frames. These mutants can then be individually screened for growth under conditions that mimic the environment encountered by bacterial cells during colonization and infection of the host. A salient example is a study describing the use of a collection of Acinetobacter baumannii transposon mutants to identify genes that are essential for survival/growth on media consisting of human ascites as an approach to elucidating genes necessary for establishing infection [50]. Thirty-four of the genes that demonstrated impaired growth on human ascites were evaluated in a rat model of A. baumannii infection, resulting in the identification of 18 genes essential for growth in vivo. The essential genes that were identified included genes involved in nucleic acid synthesis, protein transport, metabolic genes, two component systems and structural proteins. Importantly, the genes identified as essential in vivo overlapped poorly with genes that were previously identified as essential in other Gram negative species under laboratory conditions, and none of them are targeted by clinically used antibiotics or antimicrobial molecules that are currently being developed. In a later study, the same authors validated one of the essential genes identified in this study, the gene encoding the response regulator BfmR, as a potential target for the development of novel antibiotics [51].
Recently, next generation sequencing technology has resulted in the development of methods that can identify conditionally essential genes without the necessity of isolating and phenotypically testing individual bacterial mutants. Techniques such as transposon sequencing (Tn-seq), insertion sequencing (INseq) and high throughput insertion tracking by deep sequencing (HITS) can characterize mutants in complex mixtures containing thousands to tens of thousands of individual mutants [52,53], thus providing a powerful platform for the identification of conditionally essential genes. These methods all employ random insertion libraries that are typically generated using rich laboratory media. The massive sequencing of the bacterial DNA adjacent to the transposon/insertion of the mixture of mutants, and the mapping of these reads to the bacterial genome permits the identification and quantification of the abundance of mutants corresponding to each gene in the mutant library (Figure 2). Exposing this mixture of mutants to certain environmental conditions, such as infection of experimental animals, followed by the identification and quantification of the mutants that have survived under these conditions allows for the identification of conditionally essential genes by detecting those genes that significantly decrease in abundance after exposure to a given condition. The underlying basis for these approaches is that mutants that encode functions necessary for growth or survival under a given condition will be less abundant after exposure to this condition, as these strains will be less able to thrive compared to those mutants containing interrupted genes that encode non-essential functions. A key advantage of these approaches compared to the phenotypic characterization of individual transposon mutants described above is that they are well suited to identifying genes that are essential for host infection since a mixture containing thousands of mutants is used to infect a single animal [53]. A number of recent studies have employed these methods to identify genes that play a role in different aspects of bacterial infection. Dembek et al. used transposon mutagenesis to create a mutant library containing more than 70,000 unique mutants in an epidemic strain of Clostridium difficile [54]. The authors then used transposon-directed insertion site sequencing to identify approximately 400 genes that were essential for growth of C. difficile in vitro and 798 genes that participate in C. difficile sporulation. In a separate study, more than 25,000 random insertion mutants were generated in the Klebsiella pneumniae reference strain KPPR1 and genes that participate in establish lung infection in a mouse model were identified by comparing the relative abundance of mutants in the initial inoculum and in the lung after infection [55]. This study identified more than 300 genes that resulted in at least a two-fold reduction in abundance in lung tissue when compared to the inoculum, and 69 genes with at least a 10-fold decrease in abundance. Directed mutagenesis was used to create mutants in six of the identified genes, and their necessity for full bacterial fitness during experimental infection was confirmed by comparison with the wild type parent strain. The genes that contributed to fitness included genes encoding enzymes that participate in amino acid synthesis, the transcription factor RfaH, and a copper efflux pump CopA. In addition, a recent study employing approximately 300,000 transposon mutants in P. aeruginosa used INSeq to identify genes involved in infection [56]. Interestingly, this study demonstrated that 90% of the genes that were nonessential for growth under laboratory conditions contributed to bacterial survival during infection. These results underscore the importance of the conditions used to identify and classify essential genes, as for some genes essentiality depends on the environmental context.
Figure 2. Identification of conditionally essential genes.
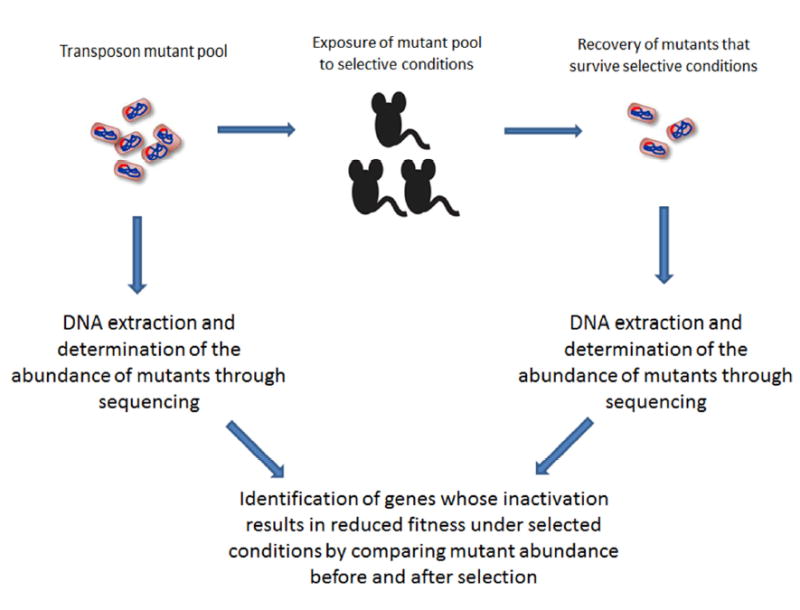
The figure shows a general schematic based on techniques such as HITS, Tn-seq and INseq that employ pools of bacterial transposon mutants to identify genes that contribute to bacterial growth and survival under defined experimental conditions. The pool of transposon mutants is exposed to a selective condition (such as infection in an experimental animal model) and the mutants that survive are recovered. DNA is then extracted from both the original mutant pool and the pool of mutants surviving selective conditions and subjected to next generation sequencing in order to determine the abundance of mutants in each gene present in the pools. The abundance of mutants is compared between pools, and genes that contribute to growth and survival are identified based on the idea that mutants in these genes will demonstrate decreased abundance after selection.
It is important to point out that many of the studies aiming to identify essential and conditionally essential genes were not carried out with the specific intention of elucidating targets for the development of antibiotics, but with the goal of characterizing bacterial physiology. Identifying genes that are essential under various conditions that mimic host infection may provide additional genes that could serve as high value targets for the antibiotic development. Notably, these approaches may also identify non-traditional targets, such as bacterial virulence factors, which have recently been proposed as targets for the development of antimicrobials with novel mechanisms of action [57,58].
Recently identified antibiotics with novel mechanisms of action support the idea that essential bacterial functions may be high value targets for the development of new antimicrobials. One example is the isolation and characterization of teixobactin (Figure 3A), an antibiotic that has shown activity against Gram positive bacteria and mycobacteria without producing resistance [46]. Teixobactin inhibitis peptidoglycan biosynthesis by binding to both lipid II (a peptidoglycan precursor) and lipid III (a teichoic acid precursor) and inhibits their incorporation into the bacterial cell wall. Interestingly, teixobactin was isolated using iChip technology, a method that permits the identification of antibiotics produced by unculturable microorganisms [46]. A second example is the identification and characterization of inhibitors of the LpxC enzyme (Figure 3B), a zinc-dependent deacetylase that catalyzes the first committed step in lipid A biosynthesis in Gram negative bacteria [59,60]. LpxC inhibitors have shown potent in vitro activity against the multidrug resistant Gram negative species Klebsiella pneumoniae, Escherichia coli and Pseudomonas aeruginosa [61,62], and have been demonstrated to potentiate the activity of currently-used antibiotics on Acinetobacter baumannii [63]. Teixobactin and LpxC inhibitors both target essential bacterial functions, peptidoglycan synthesis and lipopolysaccharide biosynthesis, respectively, to achieve antimicrobial activity.
Figure 3. Chemical structures of recently identified antibiotics with novel mechanisms of action.
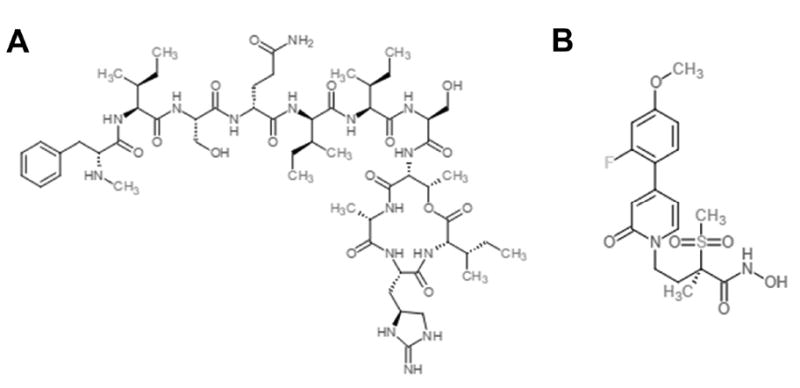
Chemical structures of teixobactin (A) and the LpxC inhibitor PF-5081090 (B).
5. Transcriptomics and Proteomics in Antibiotic Target Discovery
The availability of complete genome sequences has facilitated the development of methods that can quantify expression levels of all bacterial genes. While these transcriptomic techniques have not been specifically applied to antibiotic target identification to the same extent of comparative genomics, they may provide information useful for elucidating high value targets. As mentioned above, for an antibiotic to be effective, it most likely must target a bacterial component that is active during infection. Transcriptomic analyses can identify genes that are highly expressed during conditions that mimic infection, providing evidence that they contribute to bacterial processes that are important for establishing and/or maintaining infection. Transcriptomic studies may also be useful for characterizing the bacterial response to different antibiotics, and thus may facilitate that identification of pathways that can be exploited for the development of compounds that potentiate the effects of existing antibiotics [64,65].
Microarrays have been widely used for characterizing bacterial gene expression under different conditions and in response to multiple environmental stimuli since their development in the mid-1990s [66,67,68]. More recently, techniques such as RNA-seq, which employs next generation sequencing technology in order to sequence individual RNAs in a sample, have been employed in global transcriptional profiling studies [69]. For RNA-seq, total RNA is extracted from samples, and after removal of high abundance ribosomal RNAs, cDNAs are generated and sequenced, typically producing millions of individual reads. Once sequenced, these RNAs can then be mapped to predicted open reading frames in a reference sequence, allowing the quantification and identification of transcripts. Unlike microarrays, RNA-seq does not employ predetermined probes based on predicted open reading frames. For this reason, RNA-seq has the potential to detect and quantify transcripts that are not predicted based on sequence analysis. In addition, RNA-seq can provide information regarding individual transcripts that microarrays are unable to provide, such as transcriptional start sites [69].
Multiple recent studies have used RNA-seq to characterize the transcriptional regulation of bacterial species associated with antibiotic resistance under conditions that mimic human infection. Scaria et al. used RNA-seq to perform global transcriptional profiling of virulent C. difficile strains under physiologically relevant conditions related to nutrient concentration and osmotic stress [70]. Significant transcriptional changes occurred in many core genes related to multiple different bacterial processes including carbohydrate and nucleic acid biosynthesis, and amino acid metabolism. In a separate study, RNA-seq was employed to compare gene expression in 151 isolates of P. aeruginosa, and characterize transcriptional regulation in response to 14 different environmental stimuli with a P. aeruginosa reference strain [71]. The study identified multiple gene families that demonstrated significant changes in different environmental conditions, some of which are relevant to those encountered by bacteria during human infection, and concluded that environmental changes have a greater effect on gene expression levels than the genetic background of the stain. The global regulator LasR was also identified as a key regulator of P. aeruginosa gene expression in response to environmental cues, and thus may represent a novel target for antibiotic development. RNA-seq was recently employed to identify S. aureus genes whose expression was upregulated during infection [72]. An experimental model of S. aureus osteomyelitis was employed in order to characterize RNA levels during infection versus growth in laboratory media. A total of 180 genes were found to be significantly overexpressed during infection, and included genes involved in diverse processes such as iron metabolism, the bacterial stress response, and carbohydrate metabolism. These recent studies represent only a handful of those that have characterized global transcriptional profiles in bacterial species associated with multidrug resistance. However, they provide clear examples of how this technology can provide information useful for the elucidation of potentially novel antibiotic targets.
The use of genome sequences together with mass spectrometry-based methods can identify, and in some cases quantify, peptides within a complex sample. As with global transcriptional profiling, these proteomic approaches can provide information regarding the presence and level of expression of potential antibiotic targets in infection-like conditions. For example, the technique isobaric tags for relative and absolute quantification (iTRAQ®) involves differentially labelling peptides from separate proteolytically cleaved samples, which are then identified and quantified using mass spectrometry [73]. The conjugation of isobaric tags to the peptides from different samples permits their quantification. A recent study employed iTRAQ® to identify P. aeruginosa surface proteins that are overexpressed during infection [74]. Protein levels in outer membrane extracts were quantified in a reference strain and three isolates obtained from patients with cystic fibrosis after growth in either M9 minimal media or in a defined media that mimics the composition of the lung extracellular lining fluid. Protein quantification showed significant differences in multiple membrane proteins between the reference strain and the clinical isolates, most notably in multiple proteins associated with antibiotic resistance, such as MexY, MexB and MexC. A separate study used iTRAQ® to compare protein levels in C. difficile grown in vitro and in a pig ileal-ligated loop model [75]. Of the 705 proteins for which quantitative data was obtained, 109 were differentially expressed between experimental conditions. Analysis of clusters of orthologous group demonstrated that the differentially expressed proteins participated in multiple key physiological functions such as energy production, cell division, amino acid and carbohydrate transport and cell wall biogenesis. Taken together, these studies demonstrate the potential of quantitative proteomic techniques in the identification of bacterial gene products that participate in important bacterial processes during infection.
In addition to transcriptomic and proteomic methods, there are multiple emerging techniques that can provide global information regarding different bacterial process. Examples include metabolomics, phosphoproteomics and lipidomics, and their development and application have been described elsewhere [76,77,78]. While these methods have not been widely used for antibiotic target elucidation, they may facilitate the identification of bacterial components that participate in key aspects of bacterial physiology that have not yet been explored as potential targets for antibiotic development.
6. Identification and use of novel ribosomally encoded peptide antibiotics
In addition to novel antibiotic targets, advances in metagenomics and rapid DNA sequencing have made it possible to mine bacterial genomes for peptide antibiotics. As more genomes are sequenced, the possibility of discovering peptides with novel mechanisms of action increases. By capitalizing upon the competition that occurs between bacterial species, bacteriocins (genetically encoded bacterial antimicrobial peptides) can be identified as potential antibiotic lead compounds. Bacteriocins can have a narrow or broad spectrum of activity. This activity can be further tuned as appropriate for specific molecular targets, using gene-based protein engineering and rational design with library screening methods. This makes bacteriocins an underutilized and extremely rich source of potential novel antibiotic scaffolds.
6.1 Bacteriocin Classification
Bacteriocins are small, ribosomally synthesized, antimicrobial peptides produced by bacteria. They fall into two main classes: modified (class I) and unmodified (class II) (Figure 4). Class I peptides, also known as ribosomally synthesized and post-translationally modified peptides (RiPPS), undergo extensive post-translational modification prior to export (PTM) [79,80]. The precursor peptide, transporters, modifying enzymes, and immunity proteins involved in bacteriocin production are often encoded in the same biosynthetic operon. All RiPPS precursor peptides contain a leader sequence and a core peptide domain. The leader sequence is thought to bind specifically to modification enzymes that install PTMs on the cognate peptide. Upon modification, the leader sequence is cleaved from the modified core peptide and the mature bacteriocin is then exported from the bacterial cell. Class I bacteriocins are subdivided according to the nature of their PTMs. The lanthipeptides, or lantibiotics, such as nisin, contain unusual amino acids such as lanthionine or methyllanthionine. The cyclized peptides, such as enterocin AS-48, contain a N- to C-terminus peptide bond. This creates a circular peptide that consists of α-helical segments. The sactibiotics, such as thuricin CD, contain a sulfur-to-α-carbon bond. The thiopeptides, such as lactocillin, contain peptide macrocycles containing thiazole rings. Linear azol(in)e-containing peptides (LAPs) or the thiazole/oxazole modified microcins (TOMMs) are peptides that contain thiazole and (methyl)oxazole heterocycles. Prominent LAPs are microcin B17 (MccB17), a bacterial DNA gyrase inhibitor from E. coli, and streptolysin S (SLS), a cytolysin, from Group A Streptococcus. Glycosins are type I bacteriocins that specifically contain glycosylated residues. Finally, the lasso peptides are type I bacteriocins characterized by an amide bond between the first amino acid and an acidic residue at position +7 to +9 in the peptide chain. This creates a “loop” through which the rest of the C-terminal portion of the peptide is threaded [79,80,81].
Figure 4. Biosynthetic gene clusters of model bacteriocins.
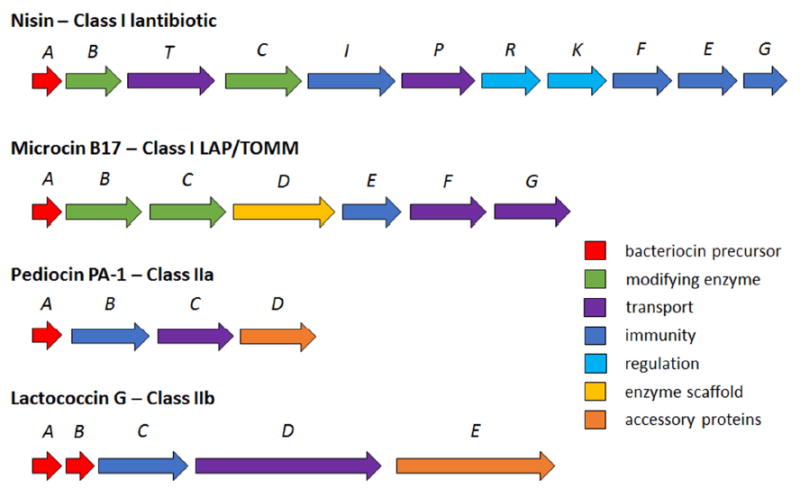
The figure illustrates the arrangement of bacteriocin biosynthetic gene clusters (BGC). These BGCs contain genes for the bacteriocin precursor peptides, modifying enzymes, immunity proteins, transporters, regulatory proteins, and accessory proteins. The BGCs of class I modified bacteriocins, such as nisin and microcin B17, contain multiples enzymes for the installment of PTMs. The BGCs of the class II unmodified bacteriocins, such as pediocin PA-1 and lactococcin G, lack these modifying enzymes.
Class II bacteriocins are unmodified peptides that are located in gene clusters encoding an ATP-binding cassette (ABC) transporter, immunity proteins, accessory proteins, and bacteriocin structural genes. The bacteriocin precursor gene encodes a peptide consisting of a leader sequence and core peptide domain. This leader sequence is cleaved from the core peptide by the action of the peptidase domain of the ABC transporter complex during its export from the cell. The class II unmodified peptides are subdivided into four main categories. The class IIa, or pediocin-like bacteriocins, are characterized by a YGNGV motif. Class IIb peptides comprise bacteriocins whose activity relies on the presence of two unmodified peptides. The class IIc peptides consist of bacteriocins that lack a leader domain. Finally, the class IId bateriocins are composed of the non-pediocin like, single peptide bacteriocins [79,81].
6.2 Genome Mining for Bacterocins
Given the current diversity of bacteriocin compounds, genes that specifically encode bacteriocin peptide products can be identified via BLAST searches against a known bacteriocin database. Online tools, such as BACTIBASE and BAGEL, mine target genomes for bacteriocin precursor genes [82,83,84]. Other tools such as antiSMASH expand their search parameters to identify not only bacteriocin-like precursor genes but other non-ribosomal classes of antibiotics and secondary metabolites [82,85] (Figure 5). Recently, these tools were combined to mine the genomes of anaerobic bacteria for RiPPs. This method detected novel RiPPs precursors including unique lanthipeptides, sactipeptides, thiopeptides, LAPs, and lasso peptides [86]. Unfortunately, these mining approaches are limited in that novel bacteriocins that do not have significant similarity to those in the database cannot be discovered. In addition, the small size of these bacteriocins (30-70aa) makes ORF prediction difficult. Genome mining approaches using the associated modifying enzymes located in the bacteriocin biosynthetic operon have been developed recently to expand the repertoire of discovered bacteriocins using such approaches. Using BLAST algorithms and Hidden Markov Model (HMM) profiles, known modifying enzymes can be used to search for homologues in other organisms. Since many of these enzymes are functionally redundant and largely conserved throughout organisms, they can be used to improve the discovery of novel bacteriocin candidates that are difficult to identify based solely on bacteriocin precursor genes. For example, using the conserved sequences of bacteriocin modifying enzymes for the TOMM family of bacteriocins, SLS and MccB17, researchers were able to identify potential novel RiPPs among a variety of prokaryotes [87].
Figure 5. Genome mining strategies for bacteriocin discovery.
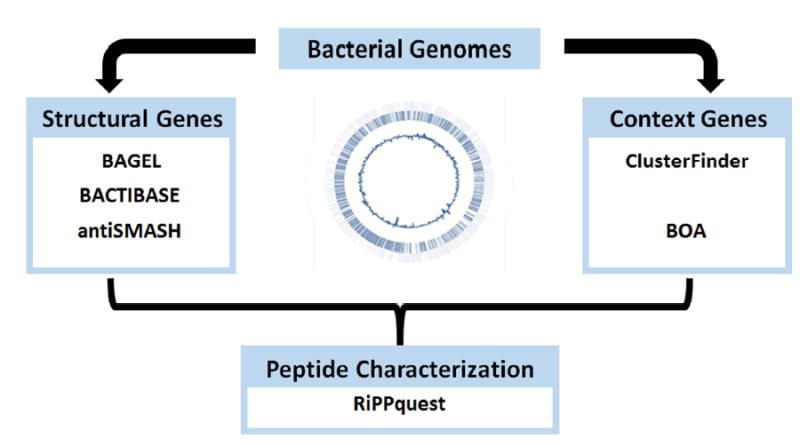
The figure depicts the approaches available for genome based discovery of novel bacteriocins. This includes mining for “structural genes” using programs such as BAGEL, BACTIBASE, and antiSMASH or by mining for “context genes” using programs such as ClusterFinder and BOA. These mining efforts can be followed by RiPPquest database curation for tandem MS detection of the peptide product.
A recent genomics-based bacteriocin mining program, the bacteriocin operon and gene block associator (BOA), utilizes this approach to mine for novel bacteriocin candidates. Genes that are contained a bacteriocin biosynthetic operon that process the maturation, export, or other processing of the bacteriocin candidate are identified as bacteriocin “context genes.” These “context genes” are thereby used to perform homology-based genome searches for bacteriocin operons. Using a curated set of gene blocks representative of lantibiotics, thiopeptides, TOMMs, lasso-peptides, and circular bacteriocins, BOA analysis identified 95% of previous BAGEL-annotated bacteriocins, but also identified an additional 1003 RiPPs in genomes not identified by BAGEL or other bacteriocin mining approaches. Many of these bacteriocins were identified in bacteria across diverse ecological niches including the human respiratory tract, soil, and plants [88]. Therefore, mining the genomes of bacteria living in competitive environments, such as the human microbiome, is likely to serve as a rich reservoir for the identification of novel bacteriocin compounds through genome searching algorithms. Similar studies using context-based gene searching methods have expanded the discovery of bacteriocin families, including the TOMM/LAP family of bacteriocins. This includes a new subfamily, the faecalisins, whose modifying enzymes share some similarity to MccB17 [89]. The faecalisin operon also contains hypothetical proteins which could further modify the precursor peptide precursor and install PTMs heretofore not observed. An additional new class of bacteriocins, the thermoacidophisins, share enzyme similarities to the bacteriocins helibactin and plantazolicin. The thermoacidophisin gene clusters also contain many hypothetical proteins which could confer additional modifications to the mature peptide [89]. In summary, genome mining approaches utilizing conserved ‘context’ genes have been established as a powerful tool to greatly expand the family of novel bacteriocin biosynthetic gene clusters.
6.3 Connecting putative bacteriocin genes to their peptide product
Genome mining methods to identify novel bacteriocins offer a quick “prediction step” when looking for novel peptide natural products. However, the “connection step” in which the peptide, or chemotype, is associated with the gene cluster, or genotype often is time-extensive and requires greater experimental rigor. This process requires natural isolation of the peptide from bacterial cultures followed by mass spectrometry analysis to identify the peptides and subsequently the gene associated with the production of the specific bacteriocin peptide. This approach can be highly limiting in that common mass spectrometry methods and proteomic databases (Sequest and Mascot) have difficulty identifying highly modified, poorly fragmented, or nonlinear peptides [90]. Recently, a new tandem mass spectrometry database called RiPPquest was developed to detect novel RiPPs from mass spectrometry data [91]. In this database, genome mining is used to limit the search to areas of putative RiPP biosynthetic gene clusters from the target bacteria. Next, all MS/MS spectra of possible mature peptides are computed based off of possible biosynthetic transformations. Finally, peptide spectrum matches (PSMs) are scored to identify all possible chemotype to genotype matches between the LC-MS/MS data and the RiPP database. This procedure was first applied to the identification of novel lanthipeptides from Streptomyces viridochromogenes, a known producer of the potent antibiotic avilamysin A [92]. The researchers were able to identify the novel lanthipeptide, informatipeptin, using the RiPPquest approach [91]. Although this approach streamlines the connection between mature peptide and biosynthetic gene cluster, it may not be able to detect novel modifications that have not been described or included in their database generation step.
6.4 Connecting peptide to function
Although genomic mining and recent proteomic approaches have been highly successful in expanding the library of bacteriocin compounds, these approaches are limited in that predictions about the functions of these bacteriocin candidates cannot be deduced. Many bacteriocins have been shown to have antimicrobial, antiviral and antitumor properties. Others have been shown to function as bacterial signaling molecules such as the competence signaling peptide (Csp) of Streptococcus intermedius or to serve as host virulence factors, such as SLS and Listeriolysin S from Group A Streptococcus and Listeria monocytogenes, respectively [80,82,93]. Although the functions of novel RiPPs may be inferred by gene gazing via sequence similarity to known bacteriocins and modifying enzymes, similarly modified bacteriocin candidates have been shown to have divergent functions when tested in vivo [80,94]. In silico approaches to identify bacteriocins will need to be supported with in vitro characterization of peptide function. A recent study followed bacteriocin discovery from in silico detection to in vitro characterization. Using the ClusterFinder algorithm to mine the genomes of human microbiomes, it was revealed that biosynthetic gene clusters encoding RiPPs were widely distributed across the human microbiome [95]. Lantibiotics and TOMMs were identified across microbiome members despite having only been described in rare commensals and pathogens. Interestingly, thiopeptides were also present throughout the human microbiotia as well as microbiota members of the porcine gut. Following the in silico analysis, the authors used a genome-guided approach to isolate a novel thiopeptide antibiotic, lactocillin, from the vaginal commensal, Lactobacillus gasseri. This thiopeptide was previously identified using their in silico mining methods. Latocillin showed potent activity against the pathogens Staphylococcus aureus, Enterococcus faecalis, and Gardnerella vaginalis [96]. The study emphasizes that notion that the microbiomes of humans and other animals are likely to serve as a rich source of novel bacteriocins. Bacteriocin candidates that have specific antimicrobial functions are likely to be abundant in the human microbiome, as these bacteria must produce substances to compete in these environments. Just as the vaginally derived lactocillin inhibits the growth of the vaginal pathogen G. vaginalis but not vaginal commensals, the microbiome may be a rich source of bacteriocins effective against pathogens but not microbiota members.
Lactocillin’s selective spectrum of antibacterial activity is one of the many advantages that bacteriocins have over tradition antibiotics. Bacteriocins can exist as narrow or broad spectrum antimicrobial peptides. Narrow spectrum peptides can be used to target specific infections without harming the microbiota, while broad spectrum peptides can be used when the cause of the infection is unknown [81,97]. For example, the sactibiotic thuricin CD displays potent specific activity against antibiotic resistant Clostridium difficile without adversely affecting members of the gut microbiome [98]. Nisin, a lantibiotic, broadly inhibits the growth of many gram positive food borne pathogens leading to its current use a naturally derived food preservative [80,81,97]. Since many bacteriocins are naturally produced by lactic acid bacteria present in fermented foods and microbiome communities, they have a precedent for low toxicity towards mammalian cells. Nisin has been shown to be non-toxic to intestinal epithelial cells [99]. In addition, class II unmodified bacteriocins, show low to no toxicity against mammalian cells in culture [81,100,101]. Many bacteriocins show robust bacteriocidal activity in vitro; to be considered for antibiotic development they must also show bacteriocidal activity in vivo. Nisin-F, a nisin variant, and the unmodified class II bacteriocin, ST4SA, have been incorporated into bone cements to prevent the colonization of Staphylococcus aureus [102,103]. Thuricin CD has also been shown to be bioavailable upon rectal administration in mice [98]. Thiopeptide bacteriocins are currently a focus of antibiotic development. For example, a thiopeptide from Planobispora rosea, GE2270 A, has been selected as a lead compound for antibiotic development. Upon chemical modification, the GE2270 derivative, LFF571, showed significant oral bioavailability making it useful for the treatment of C. difficile. The compound also showed effective antibiotic properties in a hamster model of the disease [104]. This compound, currently in clinical trails through Novartis Pharmaceuticals, exhibits only mild gastrointestinal side effects [81,105].
The modification of GE2270 A to LFF571 to improve activity demonstrates the potential for bacteriocins to be subject to further chemical modifications or additional bioengineering strategies to increase diversity [81,104,106]. These bioengineering strategies can include chemical synthesis of the peptide, genetic manipulation of bacteriocin precursors and enzymes, or in vitro biosynthetic approaches [81]. The model lantibiotic, nisin, has been the subject of many of these bioengineering strategies. In a recent study using site saturation mutagenesis, nisin derivatives were created by randomizing 19 residues to all other naturally occurring amino acids. One derivative, nisin I4V, showed increased bacteriocidal activity against Staphylococcus pseudintermedius, a causative agent of antibiotic resistant infection in dogs. This novel nisin derivative also inhibits the formation of S. pseudintermedius biofilms [107]. Pairing these bioengineering strategies with probiotic approaches could also be used as an alternative to antibiotics through delivery of prolific bacteriocin producing organisms to the site of infection. This concept has already been applied to treatment of dental caries. A bioengineered strain of non-pathogenic Streptococcus mutans with decreased lactic acid production and increased production of the lantibiotic mutacin 1140 is currently used as a replacement therapy by excluding pathogenic S. mutans in the mouth [108,109].
Despite these advantages, there is still the possibility of the development of resistance. Nisin and other lantibiotics prevent the incorporation of lipid II during cell wall synthesis, but resistance to nisin has been noted in Streptococcus agalactiae via the production of the nisin resistance protein, NSR, which cleaves the lanthione rings at the C-terminus [110]. Resistance to bacteriocins with proteinaceous cellular targets, such as MccB17, which targets DNA gyrase, or lactococcin G, which targets the cell wall synthesis enzyme UppP, can arise through mutations in their targets [111,112]. Although bacteriocins are a rich and novel source of antibiotics, they are likely to have similar issues as traditional antibiotics with regard to the development of resistance, and mechanisms to investigate resistance should be taken into account as these novel compounds are used.
7. Conclusions
The sequencing of the first bacterial genome more than twenty years ago led to the expectation that genomic data would transform antibiotic discovery by elucidating high value bacterial targets. Initial efforts based primarily on comparative genomic approaches have not lived up to these initial expectations, as few leads have been identified and new molecules discovered using these approaches have entered the market. Despite these initial disappointments, bacterial genome sequences still have potential to contribute significantly to antibiotic development. The development of novel technologies, many based on next generation sequencing methods, can be complementary to the information obtained using comparative genomics for the identification of targets for antibiotic development. Genome sequence information now permits the identification of genes that are essential for bacterial growth and survival under different environmental conditions, and thus may be attractive targets for the development of new antimicrobial compounds. Additionally, transcriptomic and proteomic methods, as well as emerging techniques such as metabolomics and lipidomics, are beginning to shed light on bacterial processes that may elucidate potential targets. Bioinformatic tools have also been developed that are facilitating the identification of pathways in bacterial genomes that encode functions for the biosynthesis of antibacterial compounds and peptides, such as bacteriocins. These intrinsically encoded antimicrobial molecules may be of special interest given that they have evolved to produce both potent and selective antibacterial activity.
In summary, the current antibiotic resistance crisis requires that multiple strategies are undertaken for the development of new antibiotics. Although approaches based on genome sequencing data have yet to yield significant results, their potential for contributing to antibiotic discovery may only be beginning to be realized.
Acknowledgments
This work was funded by the Ministerio de Economía y Competitividad, Instituto de Salud Carlos III - co-financed by European’s Development Regional Fund “A way to achieve Europe” ERDF, Spanish Network for the Research in Infectious Diseases (REIPI RD06/0008/0000). MJM is supported by the Subprograma Miguel Servet from the Ministerio de Economía y Competitividad of Spain (CP11/00314). SWL is supported by an NIH Innovator Grant (1DP2OD008468-01) and Monahan Family Professorship in Rare and Neglected Diseases at the University of Notre Dame. FRF is supported by an NSF GRFP National Graduate Fellowship and GEM Graduate Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16:161–168. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 2.Walsh TR, Weeks J, Livermore DM, Toleman MA. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis. 2011;11:355–362. doi: 10.1016/S1473-3099(11)70059-7. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Threat Report 2013: Antibiotic/Antimicrobial Resistance. Centers for Disease Control and Prevention; 2013. [Google Scholar]
- 4.Review on Antimicrobial Resistance. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. 2014 http://amrreview.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf.
- 5.Kostyanev T, Bonten MJ, O’Brien S, Steel H, Ross S, Francois B, et al. The Innovative Medicines Initiative’s New Drugs for Bad Bugs programme: European public-private partnerships for the development of new strategies to tackle antibiotic resistance. J Antimicrob Chemother. 2016;71:290–295. doi: 10.1093/jac/dkv339. [DOI] [PubMed] [Google Scholar]
- 6.Roca I, Akova M, Baquero F, Carlet J, Cavaleri M, Coenen S, et al. The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect. 2015;6:22–29. doi: 10.1016/j.nmni.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thabit AK, Crandon JL, Nicolau DP. Antimicrobial resistance: impact on clinical and economic outcomes and the need for new antimicrobials. Expert Opin Pharmacother. 2015;16:159–177. doi: 10.1517/14656566.2015.993381. [DOI] [PubMed] [Google Scholar]
- 8.Barbachyn MR, Ford CW. Oxazolidinone structure-activity relationships leading to linezolid. Angew Chem Int Ed Engl. 2003;42:2010–2023. doi: 10.1002/anie.200200528. [DOI] [PubMed] [Google Scholar]
- 9.Kern WV. Daptomycin: first in a new class of antibiotics for complicated skin and soft-tissue infections. Int J Clin Pract. 2006;60:370–378. doi: 10.1111/j.1368-5031.2005.00885.x. [DOI] [PubMed] [Google Scholar]
- 10.Cooper MA, Shlaes D. Fix the antibiotics pipeline. Nature. 2011;472:32. doi: 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- 11.Baltz RH. Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol. 2008;8:557–563. doi: 10.1016/j.coph.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Bush K, Pucci MJ. New antimicrobial agents on the horizon. Biochem Pharmacol. 2011;82:1528–1539. doi: 10.1016/j.bcp.2011.07.077. [DOI] [PubMed] [Google Scholar]
- 13.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 14.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 15.Kostyanev T, Bonten MJ, O’Brien S, Goossens H. Innovative Medicines Initiative and antibiotic resistance. Lancet Infect Dis. 2015;15:1373–1375. doi: 10.1016/S1473-3099(15)00407-7. [DOI] [PubMed] [Google Scholar]
- 16.Outterson K, Rex JH, Jinks T, Jackson P, Hallinan J, Karp S, et al. Accelerating global innovation to address antibacterial resistance: introducing CARB-X. Nat Rev Drug Discov. 2016;15:589–590. doi: 10.1038/nrd.2016.155. [DOI] [PubMed] [Google Scholar]
- 17.Wecke T, Mascher T. Antibiotic research in the age of omics: from expression profiles to interspecies communication. J Antimicrob Chemother. 2011;66:2689–2704. doi: 10.1093/jac/dkr373. [DOI] [PubMed] [Google Scholar]
- 18.Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, Ahn TH, et al. Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics. 2015;15:141–161. doi: 10.1007/s10142-015-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dark MJ. Whole-genome sequencing in bacteriology: state of the art. Infect Drug Resist. 2013;6:115–123. doi: 10.2147/IDR.S35710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sintchenko V, Roper MP. Pathogen genome bioinformatics. Methods Mol Biol. 2014;1168:173–193. doi: 10.1007/978-1-4939-0847-9_10. [DOI] [PubMed] [Google Scholar]
- 21.Kalkatawi M, Alam I, Bajic VB. BEACON: automated tool for Bacterial GEnome Annotation ComparisON. BMC Genomics. 2015;16:616. doi: 10.1186/s12864-015-1826-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar K, Desai V, Cheng L, Khitrov M, Grover D, Satya RV, et al. AGeS: a software system for microbial genome sequence annotation. PLoS One. 2011;6:e17469. doi: 10.1371/journal.pone.0017469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 24.Huang K, Brady A, Mahurkar A, White O, Gevers D, Huttenhower C, et al. MetaRef: a pan-genomic database for comparative and community microbial genomics. Nucleic Acids Res. 2014;42:D617–624. doi: 10.1093/nar/gkt1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Tonder AJ, Mistry S, Bray JE, Hill DM, Cody AJ, Farmer CL, et al. Defining the estimated core genome of bacterial populations using a Bayesian decision model. PLoS Comput Biol. 2014;10:e1003788. doi: 10.1371/journal.pcbi.1003788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaas RS, Friis C, Ussery DW, Aarestrup FM. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics. 2012;13:577. doi: 10.1186/1471-2164-13-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valot B, Guyeux C, Rolland JY, Mazouzi K, Bertrand X, Hocquet D. What It Takes to Be a Pseudomonas aeruginosa? The Core Genome of the Opportunistic Pathogen Updated. PLoS One. 2015;10:e0126468. doi: 10.1371/journal.pone.0126468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan PF, Macarron R, Payne DJ, Zalacain M, Holmes DJ. Novel antibacterials: a genomics approach to drug discovery. Curr Drug Targets Infect Disord. 2002;2:291–308. doi: 10.2174/1568005023342227. [DOI] [PubMed] [Google Scholar]
- 29.Becker D, Selbach M, Rollenhagen C, Ballmaier M, Meyer TF, Mann M, et al. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature. 2006;440:303–307. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- 30.Schmid MB. Do targets limit antibiotic discovery? Nat Biotechnol. 2006;24:419–420. doi: 10.1038/nbt0406-419. [DOI] [PubMed] [Google Scholar]
- 31.Buysse JM. The role of genomics in antibacterial target discovery. Curr Med Chem. 2001;8:1713–1726. doi: 10.2174/0929867013371699. [DOI] [PubMed] [Google Scholar]
- 32.Loferer H. Mining bacterial genomes for antimicrobial targets. Mol Med Today. 2000;6:470–474. doi: 10.1016/s1357-4310(00)01815-3. [DOI] [PubMed] [Google Scholar]
- 33.Rosamond J, Allsop A. Harnessing the power of the genome in the search for new antibiotics. Science. 2000;287:1973–1976. doi: 10.1126/science.287.5460.1973. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Meng Q, Bai L, Zhou H. In silico discovery of aminoacyl-tRNA synthetase inhibitors. Int J Mol Sci. 2014;15:1358–1373. doi: 10.3390/ijms15011358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim KH, Kim ND, Seong BL. Pharmacophore-based virtual screening: a review of recent applications. Expert Opin Drug Discov. 2010;5:205–222. doi: 10.1517/17460441003592072. [DOI] [PubMed] [Google Scholar]
- 36.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, et al. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- 39.Reich SH, Melnick M, Davies JF, 2nd, Appelt K, Lewis KK, Fuhry MA, et al. Protein structure-based design of potent orally bioavailable, nonpeptide inhibitors of human immunodeficiency virus protease. Proc Natl Acad Sci U S A. 1995;92:3298–3302. doi: 10.1073/pnas.92.8.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 41.Koehnke A, Friedrich RE. Review: Antibiotic discovery in the age of structural biology - a comprehensive overview with special reference to development of drugs for the treatment of Pseudomonas aeruginosa infection. In Vivo. 2015;29:161–167. [PubMed] [Google Scholar]
- 42.Moynie L, Leckie SM, McMahon SA, Duthie FG, Koehnke A, Taylor JW, et al. Structural insights into the mechanism and inhibition of the beta-hydroxydecanoyl-acyl carrier protein dehydratase from Pseudomonas aeruginosa. J Mol Biol. 2013;425:365–377. doi: 10.1016/j.jmb.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 43.McAlpine JB, Bachmann BO, Piraee M, Tremblay S, Alarco AM, Zazopoulos E, et al. Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J Nat Prod. 2005;68:493–496. doi: 10.1021/np0401664. [DOI] [PubMed] [Google Scholar]
- 44.Zazopoulos E, Huang K, Staffa A, Liu W, Bachmann BO, Nonaka K, et al. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat Biotechnol. 2003;21:187–190. doi: 10.1038/nbt784. [DOI] [PubMed] [Google Scholar]
- 45.Penn J, Li X, Whiting A, Latif M, Gibson T, Silva CJ, et al. Heterologous production of daptomycin in Streptomyces lividans. J Ind Microbiol Biotechnol. 2006;33:121–128. doi: 10.1007/s10295-005-0033-8. [DOI] [PubMed] [Google Scholar]
- 46.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandez-Pinar R, Lo Sciuto A, Rossi A, Ranucci S, Bragonzi A, Imperi F. In vitro and in vivo screening for novel essential cell-envelope proteins in Pseudomonas aeruginosa. Sci Rep. 2015;5:17593. doi: 10.1038/srep17593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Umland TC, Schultz LW, MacDonald U, Beanan JM, Olson R, Russo TA. In vivo-validated essential genes identified in Acinetobacter baumannii by using human ascites overlap poorly with essential genes detected on laboratory media. MBio. 2012;3 doi: 10.1128/mBio.00113-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russo TA, Manohar A, Beanan JM, Olson R, MacDonald U, Graham J, et al. The Response Regulator BfmR Is a Potential Drug Target for Acinetobacter baumannii. mSphere. 2016;1 doi: 10.1128/mSphere.00082-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol. 2013;11:435–442. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Opijnen T, Lazinski DW, Camilli A. Genome-Wide Fitness and Genetic Interactions Determined by Tn-seq, a High-Throughput Massively Parallel Sequencing Method for Microorganisms. Curr Protoc Microbiol. 2015;36(1E 3):1–24. doi: 10.1002/9780471729259.mc01e03s36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dembek M, Barquist L, Boinett CJ, Cain AK, Mayho M, Lawley TD, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio. 2015;6:e02383. doi: 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bachman MA, Breen P, Deornellas V, Mu Q, Zhao L, Wu W, et al. Genome-Wide Identification of Klebsiella pneumoniae Fitness Genes during Lung Infection. MBio. 2015;6:e00775. doi: 10.1128/mBio.00775-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skurnik D, Roux D, Aschard H, Cattoir V, Yoder-Himes D, Lory S, et al. A comprehensive analysis of in vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog. 2013;9:e1003582. doi: 10.1371/journal.ppat.1003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lopez-Rojas R, Smani Y, Pachon J. Treating multidrug-resistant Acinetobacter baumannii infection by blocking its virulence factors. Expert Rev Anti Infect Ther. 2013;11:231–233. doi: 10.1586/eri.13.11. [DOI] [PubMed] [Google Scholar]
- 58.Maura D, Ballok AE, Rahme LG. Considerations and caveats in anti-virulence drug development. Curr Opin Microbiol. 2016;33:41–46. doi: 10.1016/j.mib.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barb AW, Zhou P. Mechanism and inhibition of LpxC: an essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr Pharm Biotechnol. 2008;9:9–15. doi: 10.2174/138920108783497668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tomaras AP, McPherson CJ, Kuhn M, Carifa A, Mullins L, George D et al. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid A biosynthesis in Gram-negative pathogens. MBio. 2014;5:e01551–01514. doi: 10.1128/mBio.01551-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown MF, Reilly U, Abramite JA, Arcari JT, Oliver R, Barham RA, et al. Potent inhibitors of LpxC for the treatment of Gram-negative infections. J Med Chem. 2012;55:914–923. doi: 10.1021/jm2014748. [DOI] [PubMed] [Google Scholar]
- 62.Montgomery JI, Brown MF, Reilly U, Price LM, Abramite JA, Arcari J, et al. Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections. J Med Chem. 2012;55:1662–1670. doi: 10.1021/jm2014875. [DOI] [PubMed] [Google Scholar]
- 63.Garcia-Quintanilla M, Caro-Vega JM, Pulido MR, Moreno-Martinez, Pachon J, McConnell MJ. Inhibition of LpxC Increases Antibiotic Susceptibility in Acinetobacter baumannii. Antimicrob Agents Chemother. 2016;60:5076–5079. doi: 10.1128/AAC.00407-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howden BP, Beaume M, Harrison PF, Hernandez D, Schrenzel J, Seemann T, et al. Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob Agents Chemother. 2013;57:3864–3874. doi: 10.1128/AAC.00263-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hua X, Chen Q, Li X, Yu Y. Global transcriptional response of Acinetobacter baumannii to a subinhibitory concentration of tigecycline. Int J Antimicrob Agents. 2014;44:337–344. doi: 10.1016/j.ijantimicag.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 66.Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genomics. 2011;12:126. doi: 10.1186/1471-2164-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hanses F, Roux C, Dunman PM, Salzberger B, Lee JC. Staphylococcus aureus gene expression in a rat model of infective endocarditis. Genome Med. 2014;6:93. doi: 10.1186/s13073-014-0093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.LaBauve AE, Wargo MJ. Detection of host-derived sphingosine by Pseudomonas aeruginosa is important for survival in the murine lung. PLoS Pathog. 2014;10:e1003889. doi: 10.1371/journal.ppat.1003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scaria J, Mao C, Chen JW, McDonough SP, Sobral B, Chang YF. Differential stress transcriptome landscape of historic and recently emerged hypervirulent strains of Clostridium difficile strains determined using RNA-seq. PLoS One. 2013;8:e78489. doi: 10.1371/journal.pone.0078489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dotsch A, Schniederjans M, Khaledi A, Hornischer K, Schulz S, Bielecka A, et al. The Pseudomonas aeruginosa Transcriptional Landscape Is Shaped by Environmental Heterogeneity and Genetic Variation. MBio. 2015;6:e00749. doi: 10.1128/mBio.00749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szafranska AK, Oxley AP, Chaves-Moreno D, Horst SA, Rosslenbroich S, Peters G, et al. High-resolution transcriptomic analysis of the adaptive response of Staphylococcus aureus during acute and chronic phases of osteomyelitis. MBio. 2014;5 doi: 10.1128/mBio.01775-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Evans C, Noirel J, Ow SY, Salim M, Pereira-Medrano AG, Couto N, et al. An insight into iTRAQ: where do we stand now? Anal Bioanal Chem. 2012;404:1011–1027. doi: 10.1007/s00216-012-5918-6. [DOI] [PubMed] [Google Scholar]
- 74.Kamath KS, Pascovici D, Penesyan A, Goel A, Venkatakrishnan V, Paulsen IT, et al. Pseudomonas aeruginosa Cell Membrane Protein Expression from Phenotypically Diverse Cystic Fibrosis Isolates Demonstrates Host-Specific Adaptations. J Proteome Res. 2016;15:2152–2163. doi: 10.1021/acs.jproteome.6b00058. [DOI] [PubMed] [Google Scholar]
- 75.Janvilisri T, Scaria J, Teng CH, McDonough SP, Gleed RD, Fubini SL, et al. Temporal differential proteomes of Clostridium difficile in the pig ileal-ligated loop model. PLoS One. 2012;7:e45608. doi: 10.1371/journal.pone.0045608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gowda GA, Djukovic D. Overview of mass spectrometry-based metabolomics: opportunities and challenges. Methods Mol Biol. 2014;1198:3–12. doi: 10.1007/978-1-4939-1258-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hewelt-Belka W, Nakonieczna J, Belka M, Baczek T, Namiesnik J, Kot-Wasik A. Comprehensive methodology for Staphylococcus aureus lipidomics by liquid chromatography and quadrupole time-of-flight mass spectrometry. J Chromatogr A. 2014;1362:62–74. doi: 10.1016/j.chroma.2014.08.020. [DOI] [PubMed] [Google Scholar]
- 78.Pulido MR, Garcia-Quintanilla M, Gil-Marques ML, McConnell MJ. Identifying targets for antibiotic development using omics technologies. Drug Discov Today. 2016;21:465–472. doi: 10.1016/j.drudis.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 79.Alvarez-Sieiro P, Montalban-Lopez M, Mu D, Kuipers OP. Bacteriocins of lactic acid bacteria: extending the family. Appl Microbiol Biotechnol. 2016;100:2939–2951. doi: 10.1007/s00253-016-7343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cotter PD, Ross RP, Hill C. Bacteriocins - a viable alternative to antibiotics? Nat Rev Microbiol. 2013;11:95–105. doi: 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- 82.Flaherty RA, Freed SD, Lee SW. The wide world of ribosomally encoded bacterial peptides. PLoS Pathog. 2014;10:e1004221. doi: 10.1371/journal.ppat.1004221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hammami R, Zouhir A, Ben Hamida J, Fliss I. BACTIBASE: a new web-accessible database for bacteriocin characterization. BMC Microbiol. 2007;7:89. doi: 10.1186/1471-2180-7-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Heel AJ, de Jong A, Montalban-Lopez M, Kok J, Kuipers OP. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013;41:W448–453. doi: 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Letzel AC, Pidot SJ, Hertweck C. Genome mining for ribosomally synthesized and post-translationally modified peptides (RiPPs) in anaerobic bacteria. BMC Genomics. 2014;15:983. doi: 10.1186/1471-2164-15-983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, et al. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci U S A. 2008;105:5879–5884. doi: 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morton JT, Freed SD, Lee SW, Friedberg I. A large scale prediction of bacteriocin gene blocks suggests a wide functional spectrum for bacteriocins. BMC Bioinformatics. 2015;16:381. doi: 10.1186/s12859-015-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cox CL, Doroghazi JR, Mitchell DA. The genomic landscape of ribosomal peptides containing thiazole and oxazole heterocycles. BMC Genomics. 2015;16:778. doi: 10.1186/s12864-015-2008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duncan MW, Aebersold R, Caprioli RM. The pros and cons of peptide-centric proteomics. Nat Biotechnol. 2010;28:659–664. doi: 10.1038/nbt0710-659. [DOI] [PubMed] [Google Scholar]
- 91.Mohimani H, Kersten RD, Liu WT, Wang M, Purvine SO, Wu S, et al. Automated genome mining of ribosomal peptide natural products. ACS Chem Biol. 2014;9:1545–1551. doi: 10.1021/cb500199h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gruning BA, Erxleben A, Hahnlein A, Gunther S. Draft Genome Sequence of Streptomyces viridochromogenes Strain Tu57, Producer of Avilamycin. Genome Announc. 2013;1 doi: 10.1128/genomeA.00384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Drider D, Bendali F, Naghmouchi K, Chikindas ML. Bacteriocins: Not Only Antibacterial Agents. Probiotics Antimicrob Proteins. 2016 doi: 10.1007/s12602-016-9223-0. [DOI] [PubMed] [Google Scholar]
- 94.Bleich R, Watrous JD, Dorrestein PC, Bowers AA, Shank EA. Thiopeptide antibiotics stimulate biofilm formation in Bacillus subtilis. Proc Natl Acad Sci U S A. 2015;112:3086–3091. doi: 10.1073/pnas.1414272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014;158:412–421. doi: 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, et al. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158:1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Piper C, Cotter PD, Ross RP, Hill C. Discovery of medically significant lantibiotics. Curr Drug Discov Technol. 2009;6:1–18. doi: 10.2174/157016309787581075. [DOI] [PubMed] [Google Scholar]
- 98.Rea MC, Alemayehu D, Casey PG, O’Connor PM, Lawlor PG, Walsh M, et al. Hill, Bioavailability of the anti-clostridial bacteriocin thuricin CD in gastrointestinal tract. Microbiology. 2014;160:439–445. doi: 10.1099/mic.0.068767-0. [DOI] [PubMed] [Google Scholar]
- 99.Maher S, McClean S. Investigation of the cytotoxicity of eukaryotic and prokaryotic antimicrobial peptides in intestinal epithelial cells in vitro. Biochem Pharmacol. 2006;71:1289–1298. doi: 10.1016/j.bcp.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 100.Jasniewski J, Cailliez-Grimal C, Chevalot I, Milliere JB, Revol-Junelles AM. Interactions between two carnobacteriocins Cbn BM1 and Cbn B2 from Carnobacterium maltaromaticum CP5 on target bacteria and Caco-2 cells. Food Chem Toxicol. 2009;47:893–897. doi: 10.1016/j.fct.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 101.Murinda SE, Rashid KA, Roberts RF. In vitro assessment of the cytotoxicity of nisin, pediocin, and selected colicins on simian virus 40-transfected human colon and Vero monkey kidney cells with trypan blue staining viability assays. J Food Prot. 2003;66:847–853. doi: 10.4315/0362-028x-66.5.847. [DOI] [PubMed] [Google Scholar]
- 102.van Staden AD, Brand AM, Dicks LM. Nisin F-loaded brushite bone cement prevented the growth of Staphylococcus aureus in vivo. J Appl Microbiol. 2012;112:831–840. doi: 10.1111/j.1365-2672.2012.05241.x. [DOI] [PubMed] [Google Scholar]
- 103.van Staden AD, Heunis TD, Dicks LM. Release of Enterococcus mundtii Bacteriocin ST4SA from Self-Setting Brushite Bone Cement. Probiotics Antimicrob Proteins. 2011;3:119–124. doi: 10.1007/s12602-011-9074-7. [DOI] [PubMed] [Google Scholar]
- 104.LaMarche MJ, Leeds JA, Amaral A, Brewer JT, Bushell SM, Deng G, et al. Discovery of LFF571: an investigational agent for Clostridium difficile infection. J Med Chem. 2012;55:2376–2387. doi: 10.1021/jm201685h. [DOI] [PubMed] [Google Scholar]
- 105.Slayton ET, Hay AS, Babcock CK, Long TE. New antibiotics in clinical trials for Clostridium difficile. Expert Rev Anti Infect Ther. 2016:1–12. doi: 10.1080/14787210.2016.1211931. [DOI] [PubMed] [Google Scholar]
- 106.Field D, Cotter PD, Hill C, Ross RP. Bioengineering Lantibiotics for Therapeutic Success. Front Microbiol. 2015;6:1363. doi: 10.3389/fmicb.2015.01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Field D, Gaudin N, Lyons F, O’Connor PM, Cotter PD, Hill C, et al. A bioengineered nisin derivative to control biofilms of Staphylococcus pseudintermedius. PLoS One. 2015;10:e0119684. doi: 10.1371/journal.pone.0119684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hillman JD. Genetically modified Streptococcus mutans for the prevention of dental caries. Antonie Van Leeuwenhoek. 2002;82:361–366. [PubMed] [Google Scholar]
- 109.Hillman JD, Mo J, McDonell E, Cvitkovitch D, Hillman CH. Modification of an effector strain for replacement therapy of dental caries to enable clinical safety trials. J Appl Microbiol. 2007;102:1209–1219. doi: 10.1111/j.1365-2672.2007.03316.x. [DOI] [PubMed] [Google Scholar]
- 110.Khosa S, Lagedroste M, Smits SH. Protein Defense Systems against the Lantibiotic Nisin: Function of the Immunity Protein NisI and the Resistance Protein NSR. Front Microbiol. 2016;7:504. doi: 10.3389/fmicb.2016.00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kjos M, Oppegard C, Diep DB, Nes IF, Veening JW, Nissen-Meyer J, et al. Sensitivity to the two-peptide bacteriocin lactococcin G is dependent on UppP, an enzyme involved in cell-wall synthesis. Mol Microbiol. 2014;92:1177–1187. doi: 10.1111/mmi.12632. [DOI] [PubMed] [Google Scholar]
- 112.Vizan JL, Hernandez-Chico C, del Castillo I, Moreno F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J. 1991;10:467–476. doi: 10.1002/j.1460-2075.1991.tb07969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]