

Abstract
The assembly of long reads from Pacific Biosciences and Oxford Nanopore Technologies typically requires resource-intensive error-correction and consensus-generation steps to obtain high-quality assemblies. We show that the error-correction step can be omitted and that high-quality consensus sequences can be generated efficiently with a SIMD-accelerated, partial-order alignment–based, stand-alone consensus module called Racon. Based on tests with PacBio and Oxford Nanopore data sets, we show that Racon coupled with miniasm enables consensus genomes with similar or better quality than state-of-the-art methods while being an order of magnitude faster.
With the advent of long-read sequencing technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), the ability to produce genome assemblies with high contiguity has received a significant fillip. However, to cope with the relatively high error rates (>5%) of these technologies, assembly pipelines have typically relied on resource-intensive error-correction (of reads) and consensus-generation (from the assembly) steps (Chin et al. 2013; Loman et al. 2015). More recent methods such as FALCON (Chin et al. 2016) and Canu (Koren et al. 2017) have refined this approach and have significantly improved runtimes but are still computationally demanding for large genomes (Sović et al. 2016a). Recently, Li (2016) showed that long erroneous reads can be assembled without the need for a time-consuming error-correction step. The resulting assembler, miniasm, is an order of magnitude faster than other long-read assemblers but produces sequences that can have more than 10 times as many errors as other methods (Sović et al. 2016a). As fast and accurate long-read assemblers can enable a range of applications, from more routine assembly of mammalian and plant genomes to structural variation detection, improved metagenomic classification, and even online, “read until” assembly (Loose et al. 2016), a fast and accurate consensus module is a critical need. This was also noted by Li (2016), highlighting that fast assembly was only feasible if a consensus module matching the speed of minimap and miniasm was developed as well.
Here we address this need by providing a very fast consensus module called Racon (for Rapid Consensus), which when paired with a fast assembler such as miniasm, enables the efficient construction of genome sequences with high accuracy even without an error-correction step. Racon provides a stand-alone, platform-independent consensus module for long and erroneous reads and can also be used as a fast and accurate read correction tool.
Results
Racon is designed as a user-friendly stand-alone consensus module that is not explicitly tied to any de novo assembly method or sequencing technology. It reads multiple input formats (GFA, FASTA, FASTQ, SAM, MHAP, and PAF), allowing simple interoperability and modular design of new pipelines. Even though other stand-alone consensus modules, such as Quiver (Chin et al. 2013) and Nanopolish (Loman et al. 2015) exist, they require sequencer-specific input and are intended to be applied after the consensus phase of assembly to further polish the sequence. Racon is run with sequencer-independent input (only uses base quality values), is robust enough to work with uncorrected read data, and is designed to rapidly generate high-quality consensus sequences. These sequences can be further polished with Quiver or Nanopolish or by applying Racon for more iterations.
Racon takes as input a set of raw backbone sequences, a set of reads, and a set of mappings between reads and backbone sequences. Mappings can be generated using any mapper/overlapper that supports the MHAP, PAF, or SAM output formats, such as minimap (Li 2016), MHAP (Berlin et al. 2015), or GraphMap (Sović et al. 2016b). In our tests, we used minimap as the mapper as it was the fastest and provided reasonable results. Racon uses the mapping information to construct a partial-order alignment (POA) graph, using a single instruction multiple data (SIMD) implementation to accelerate the process (SPOA). More details on Racon and SPOA can be found in the Methods section.
For the purpose of evaluation, we paired Racon with minimap (as an overlapper) and miniasm to form a fast and accurate de novo assembly pipeline (referred to here as miniasm+Racon for short). Our complete pipeline is thus composed of four steps: (1) minimap for overlap detection, (2) miniasm layout for generating raw contigs, (3) minimap for mapping of raw reads to raw contigs, and (4) Racon for generating high-quality consensus sequences. Steps three and four can be run multiple times to achieve further iterations of the consensus. We then compared the miniasm+Racon pipeline to other state-of-the-art de novo assembly tools for third-generation sequencing data (i.e., FALCON and Canu). Note that FALCON and Canu have previously been benchmarked with other assembly methods such as PBcR and a pipeline from Loman et al. (2015) and shown to produce high-quality assemblies with improved running times (Sović et al. 2016a). Assembly pipelines were evaluated in terms of consensus sequence quality (Table 1), runtime and memory usage (Table 2; Fig. 1), and scalability with respect to genome size (Fig. 2) on several PacBio and Oxford Nanopore data sets (see Methods).
Table 1.
Assembly and consensus results across six data sets of varying genome length and sequencing data type
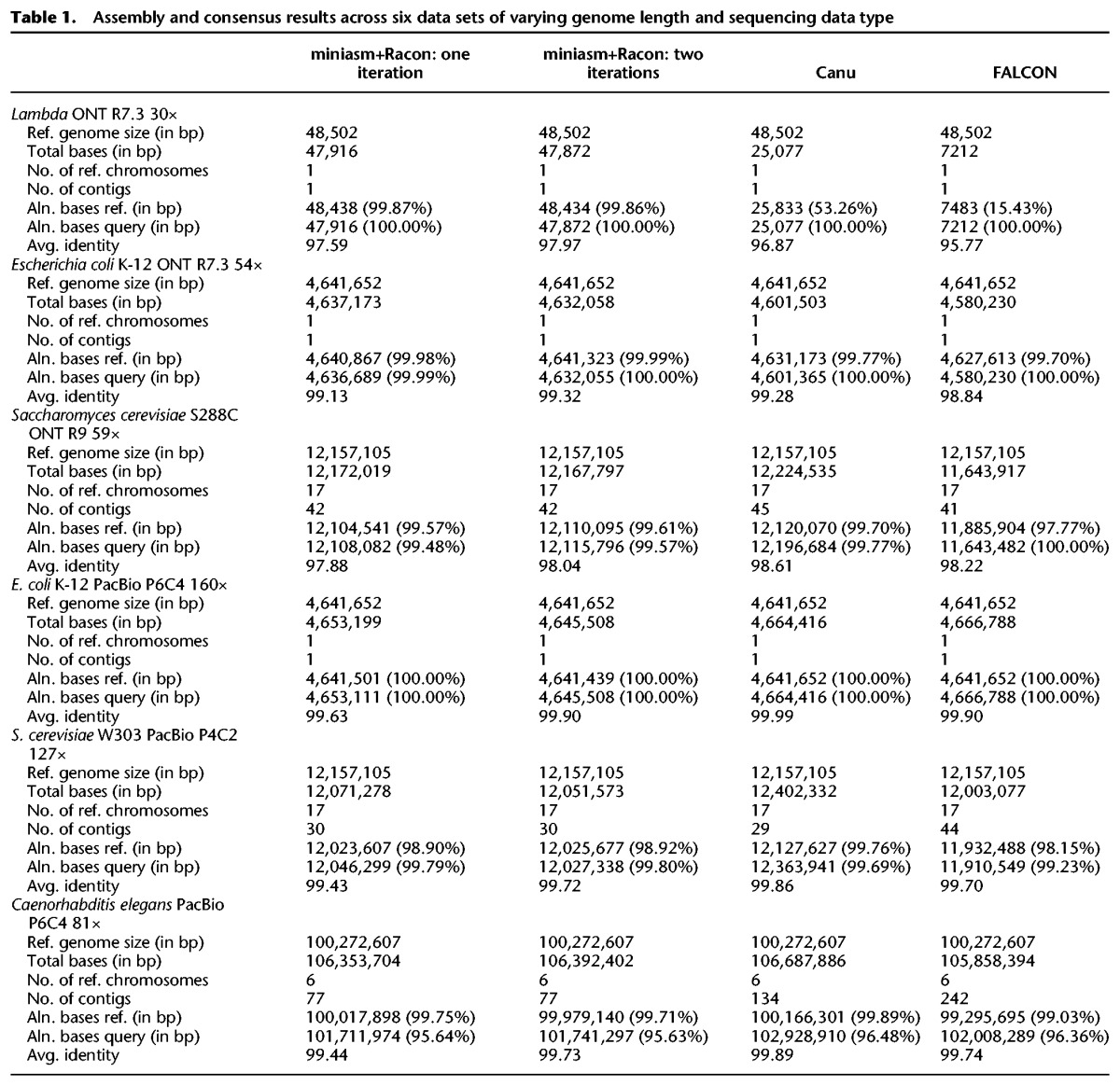
Table 2.
Time/memory consumption for all data sets
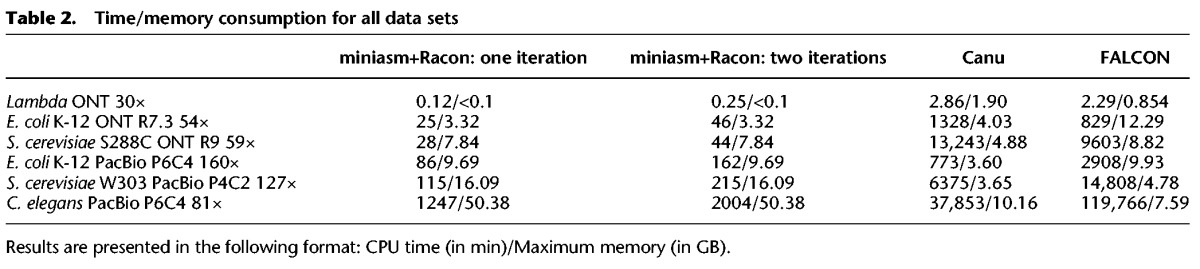
Figure 1.

Racon's speedup compared with FALCON and Canu.
Figure 2.
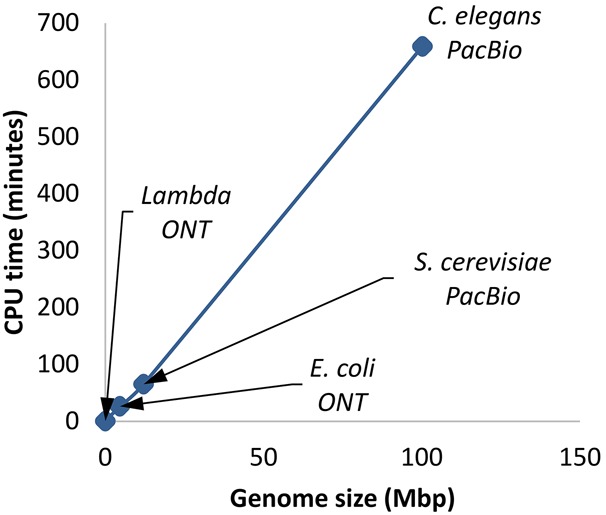
Scalability of Racon as a function of genome size. Read coverage was subsampled to be 81× (limited by the Caenorhabditis elegans data set), and the figure shows results for one iteration of Racon.
As can be seen from Table 1, all assembly pipelines were able to produce assemblies with high coverage of the reference genome and in a few contigs. Canu, FALCON, and the miniasm+Racon pipeline also constructed sequences with comparable sequence identity to the reference genome, with the iterative use of Racon serving as a polishing step for obtaining higher sequence identity. In addition, the miniasm+Racon pipeline was found to be significantly faster for all data sets, with a 5× to 301× speedup compared with Canu and 9× to 218× speedup compared with FALCON (Fig. 1).
Racon's speedup was more pronounced for larger genomes and is likely explained by the observation that it scales linearly with genome size (for fixed coverage) (Fig. 2).
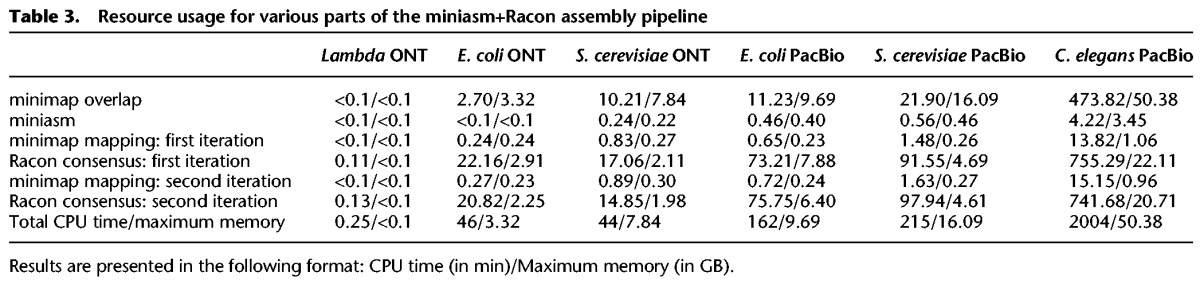
The runtime of the miniasm+Racon pipeline was dominated by the time for the consensus-generation step in Racon, highlighting that this step is still the most compute intensive one for small genomes (Table 3). However, the results in Table 3 suggest that for larger genomes the mapping computation stage can catch up in terms of resource usage. Furthermore, if a polishing stage is used, this would typically be more resource intensive.
Table 3.
Resource usage for various parts of the miniasm+Racon assembly pipeline
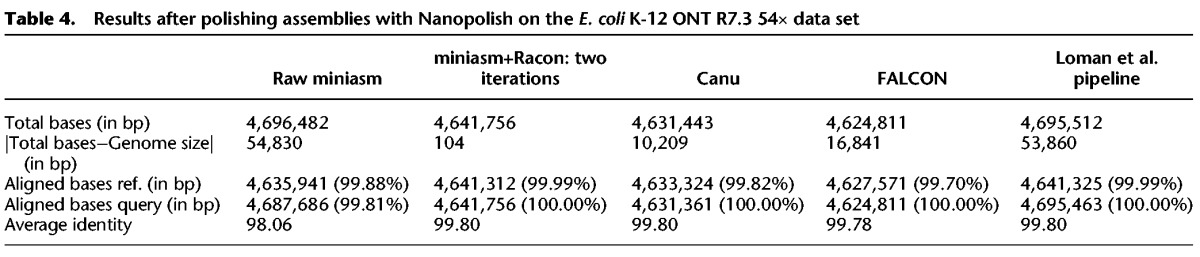
Comparison of the results of the various assembly pipelines after a polishing stage confirmed that the use of Racon provided better results than just the miniasm assembly (average identity of 99.80% vs. 98.06%) and that the miniasm+Racon assembly matched the best reported sequence quality for this data set (from the Loman et al. pipeline) (Sović et al. 2016a), while providing a better match to the actual size of the reference genome (4,641,652 bp) (Table 4). We additionally observed that Nanopolish executed more than 6× faster on miniasm+Racon contigs than on raw miniasm assemblies (248.28 CPUh vs. 1561.80 CPUh), and the miniasm+Racon+Nanopolish approach achieved the same sequence quality as the original Loman et al. pipeline, while being much faster.
Table 4.
Results after polishing assemblies with Nanopolish on the E. coli K-12 ONT R7.3 54× data set
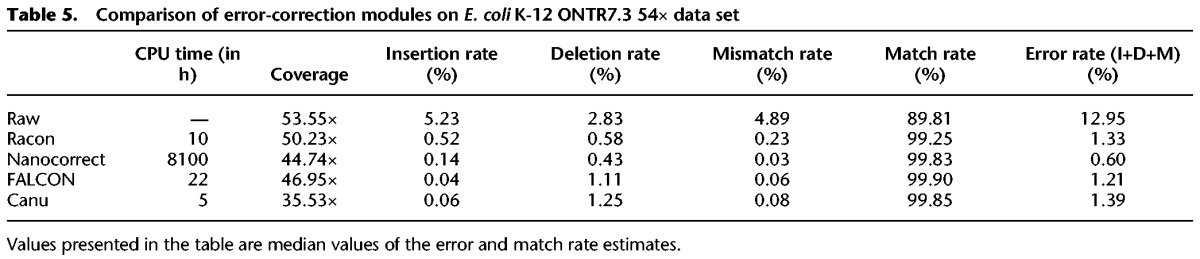
We also evaluated Racon's use as an error-correction module. The three main conceptual differences between error-correction and consensus modes in Racon are that (1) instead of mapping reads to a reference, overlapping of reads needs to be performed (e.g., using minimap with overlap parameters instead of defaults); (2) multiple overlaps are not filtered like mappings (when mapping, we only want to find one best mapping position); and (3) the parallelization is not conducted on a per-window level, but on a per-read level to reduce the system time; i.e., each read consumes one thread. We noted that Racon–corrected reads had error rates comparable to FALCON and Canu but provided better coverage of the genome (Table 5). Overall, Nanocorrect (Loman et al. 2015) had the best results in terms of error rate, but it had lower reference coverage and was more than two orders of magnitude slower than Racon.
Table 5.
Comparison of error-correction modules on E. coli K-12 ONTR7.3 54× data set
Finally, the default mode of Racon determines the consensus by splicing results of nonoverlapping windows together. We additionally attempted to assess the influence of using overlapping windows, where we extended each window by 10% on both of its ends. Neighboring windows were then aligned and clipped to form the final consensus sequence. By using this approach, we re-evaluated the consensus quality (Supplemental Table S1), the speed (Supplemental Table S2), and the quality of error correction (Supplemental Table S3) to the default Racon mode on all data sets. When using the window overlapping mode, the consensus quality increased (up to 0.06%) in all but one case. The most significant increase was obtained on the E. coli PacBio data set, where average identity rose from 99.90% to 99.94% This improvement came at the expense of higher running time (≈10%–15%) and an additional heuristic (window overlap length), which is why we enabled this mode as a nondefault option within the Racon module.
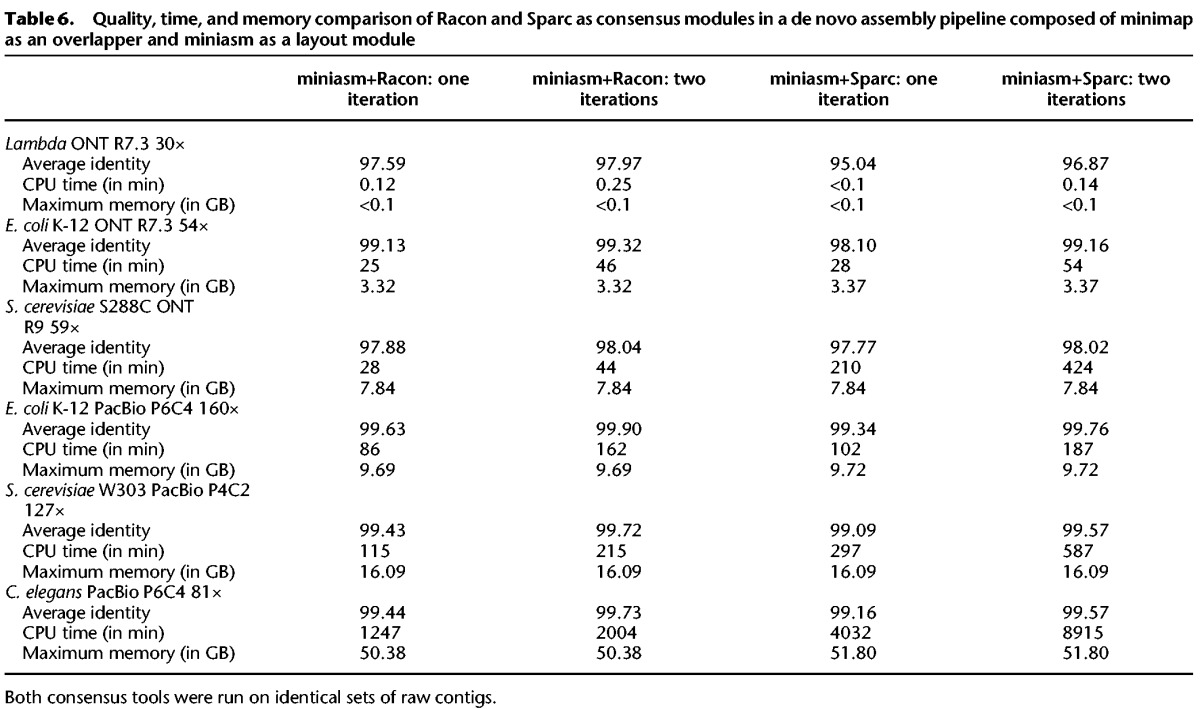
Recently, another stand-alone consensus module called Sparc (Ye and Ma 2016) emerged. We evaluated Sparc in a similar manner to Racon, with the only difference being in the mapping step (step 3 in the miniasm+Racon pipline) where Sparc uses BLASR (Chaisson and Tesler 2012). We named this pipeline miniasm+Sparc accordingly. The results of the comparison are shown in Table 6, which contains consensus sequence quality, runtime, and memory usage. It can be seen that the consensus quality of the miniasm+Sparc pipeline is consistently lower than that of the miniasm+Racon pipeline. In addition, miniasm+Sparc is up to 10× slower on larger genomes. Also, we noticed that Sparc crashed on some contigs due to unknown reasons.
Table 6.
Quality, time, and memory comparison of Racon and Sparc as consensus modules in a de novo assembly pipeline composed of minimap as an overlapper and miniasm as a layout module
Discussion
The principal contribution of this work is to take the concept of fast, error-correction-free, long-read assembly, as embodied by the recently developed program miniasm, to its logical end. Miniasm is remarkably efficient and effective in taking erroneous long reads and producing contig sequences that are structurally accurate (Sović et al. 2016a). However, assemblies from miniasm do not match up in terms of sequence quality compared with the best assemblies that can be produced with existing assemblers. This serves as a significant barrier for adopting this “light-weight” approach to assembly, despite its attractiveness for greater adoption of de novo assembly methods in genomics. In this work, we show that the sequence quality of a correction-free assembler can indeed be efficiently boosted to a quality comparable to other resource-intensive, state-of-the-art assemblers. This makes the tradeoff offered much more attractive, and the concept of a correction-free assembler more practically useful. Racon is able to start from uncorrected contigs and raw reads and still generate accurate sequences efficiently because it exploits the development of a SIMD version of the robust POA framework. This makes the approach scalable to large genomes and general enough to work with data from very different sequencing technologies. With the increasing interest in the development of better third-generation assembly pipelines, we believe that Racon can serve as useful plug-in consensus module that enables software reuse and modular design.
We would also like to note that while miniasm is a very fast tool that produces correct genome structures, its current implementation is not optimized for long repetitive genomes (Li 2016). Miniasm loads all overlaps into RAM, which might cause crashes due to insufficient memory resources. Although the focus of this study is on the miniasm+Racon pipeline, Racon is designed as a general purpose consesus module that can be coupled with other assembly pipelines as a consensus or polishing step.
Methods
Racon is based on the Partial Order Alignment (POA) graph approach (Lee et al. 2002; Lee 2003), and we report the development of a SIMD version that significantly accelerates this analysis. An overview of Racon's steps is given in Figure 3. The entire process is also shown in detail in Algorithm 1.
Figure 3.
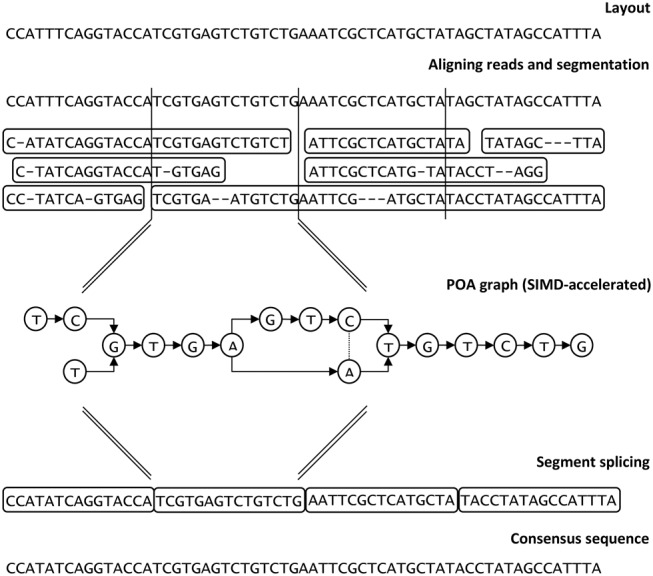
Overview of the Racon consensus process.
Algorithm 1.

The Racon algorithm for consensus generation.
To perform consensus calling (or error-correction), Racon depends on an input set of query-to-target mappings (query is the set of reads, while a target is either a set of contigs in the consensus context or a set of reads in the error-correction context) as well as quality values of the input reads. Racon then loads the mappings and performs simple filtering (Algorithm 1, lines 1–3; Algorithm 2): (1) at most one mapping per read is kept in consensus context (in error-correction context this particular filtering is disabled), and (2) mappings that have a high error rate (i.e. |1 − min(dq,dt)/max(dq,dt)| ≥ e, where dq and dt are the lengths of the mapping in the query and the target, respectively, and e is a user-specified error-rate threshold) are removed. For each mapping that survived the filtering process, a fast edit-distance–based alignment is performed (Algorithm 1, lines 4–10; Myers 1999). We used the Edlib implementation of the Myers algorithm (Šošić and Šikić 2016). This alignment is needed only to split the reads into chunks that fall into particular nonoverlapping windows on the backbone sequence. Chunks of reads with an average quality lower than a predefined threshold are removed from the corresponding window. With this quality filter, we are able to use only high-quality parts of reads in the final consensus. Each window is then processed independently in a separate thread by constructing a POA graph using SIMD acceleration and calling the consensus of the window. Quality values are used again during POA graph construction, where each edge is weighted by the sum of qualities of its source and destination nodes (bases; logarithmic domain). The final consensus sequence is then constructed by splicing the individual window consensuses together (per contig or read to be corrected).
Algorithm 2.

Functions for filtering mappings/overlaps in Racon.
POA and SIMD vectorization
POA performs multiple sequence alignment (MSA) through a directed acyclic graph (DAG), where nodes are individual bases of input sequences, and weighted, directed edges represent whether two bases are neighboring in any of the sequences. Weights of the edges represent the multiplicity (coverage) of each transition. Alternatively, weights can be set according to the base qualities of sequenced data. The alignment is carried out directly through dynamic programming (DP) between a new sequence and a prebuilt graph. While the regular DP for pairwise alignment has time complexity of O(3nm), where n and m are the lengths of the sequences being aligned, the sequence to graph alignment has a complexity of O((2np + 1)n|V|), where np is the average number of predecessors in the graph and |V| is the number of nodes in the graph (Lee et al. 2002).
Consensus sequences are obtained from a built POA graph by performing a topological sort and processing the nodes from left to right. For each node v, the highest-weighted in-edge e of weight ew is chosen, and a score is assigned to v such that scores[v] = ew + scores[w], where w is the source node of the edge e (Lee 2003). The node w is marked as a predecessor of v, and a final consensus is generated by performing a traceback from the highest-scoring node r. In case r is an internal node (r has out edges), Lee (2003) proposed the idea of branch completion, where all scores for all nodes except scores[r] would be set to a negative value, and the traversal would continue from r as before, with the only exception that nodes with negative scores could not be added as predecessors to any other node.
One important advantage of POA is its speed, with its linear time complexity in the number of sequences (Lee et al. 2002). However, implementations of POA in current error-correction modules, such as Nanocorrect, are prohibitively slow for larger data sets. In order to increase the speed of POA while retaining its robustness, we explored a SIMD version of the algorithm (SPOA).
SPOA (Fig. 4; Algorithm 3) is inspired by the Rognes and Seeberg Smith-Waterman intra-set parallelization approach (Rognes and Seeberg 2000). It places the SIMD vectors parallel to the query sequence (the read), while placing a graph on the other dimension of the DP matrix (Fig. 4). In our implementation, the matrices used for tracking the maximum local-alignment scores ending in gaps are stored entirely in memory (Algorithm 3, lines 8 and 10). These matrices are needed to access scores of predecessors of particular nodes during alignment. Unlike a regular Gotoh alignment (Gotoh 1982), for each row in the POA DP matrix, all its predecessors (via in-edges of the corresponding node in graph) need to be processed as well (Algorithm 3, line 17). All columns are then processed using SIMD operations in a query-parallel manner, and the values of Gotoh's vertical matrix (Algorithm 3, line 20) and a partial update to Gotoh's main scoring matrix (Algorithm 3, line 24) are calculated. SIMD operations in Algorithm 3 process eight cells of the DP matrix at a time (16-bit registers). A temporary variable is used to keep the last cell of the previous vector for every predecessor (Algorithm 3, lines 21–23), which is needed to compare the upper-left diagonal of the current cell to the cell one row up. Processing the matrix horizontally is not performed using SIMD operations due to data dependencies (each cell depends on the result of the cell to the left of it) and is instead processed linearly (Algorithm 3, lines 25–33). SPOA uses shifting and masking to calculate every particular value of a SIMD vector individually (Algorithm 3, lines 29–31). After the alignment is completed, the traceback is performed (Algorithm 3, line 39) and integrated into the existing POA graph (Algorithm 3, line 40).
Figure 4.
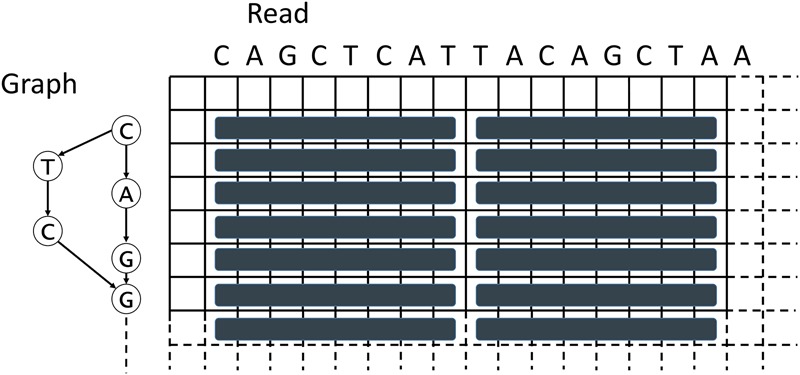
Depiction of the SIMD vectorization approach used in SPOA.
Algorithm 3.

Pseudocode for the SPOA algorithm. The displayed function aligns a sequence to a preconstructed POA graph using SIMD intrinsics. Capitalized variables are SIMD vectors. Alignment mode is Needleman-Wunsch.
SIMD intrinsics decrease the time complexity for alignment from O((2np + 1)n|V|) to roughly O((2np/k + 1)n|V|), where k is the number of variables that fit in a SIMD vector. SPOA supports Intel SSE version 4.1 and higher, which embed 128-bit registers. Both short (16 bits) and long (32 bits) integer precisions are supported (therefore k equals eight and four variables, respectively). Eight-bit precision is insufficient for the intended application of SPOA and is therefore not used. Alongside global alignment displayed in Algorithm 3, SPOA supports local and semi-global alignment modes, in which SIMD vectorization is implemented as well.
Implementation and reproducibility
Racon and SPOA are both implemented in C++. All tests were run using Ubuntu-based systems with two six-core Intel Xeon E5645 CPUs at 2.40 GHz with hyperthreading, using 12 threads where possible. The versions of various methods used in the comparisons reported here are shown in Table 7. On PacBio data sets, Canu was run using the “-pacbio-raw” option, FALCON using the config file presented in Supplemental Table S4, and Sparc using “k 1 c 2 g 1 t 0.2”. On ONT data sets, Canu was run using the “-nanopore-raw” option, FALCON using the config file presented in Supplemental Table S5, and Sparc using “k 2 g 2 t 0.2”. To provide initial alignments for Sparc, BLASR was run using “-bestn 1 -m 5 -minMatch 19”. Racon was run with the default parameters on all data sets except S. cerevisiae S288c ONT R9, where we increased the base quality value threshold to 20 using the parameter “--bq 20” (the default value is 10). To provide the initial raw assemblies for both Racon and Sparc, minimap (as an overlapper) and miniasm were used. Minimap was run using the “-Sw5 -L100 -m0” options on all data sets (PacBio and ONT); miniasm, with default parameters. For error-correction purposes, Canu was run using “-correct -nanopore-raw”, FALCON with an additional config entry “target = pre-assembly,” and Racon with the “--erc” option.
Table 7.
Versions of various tools used in this paper

Data sets
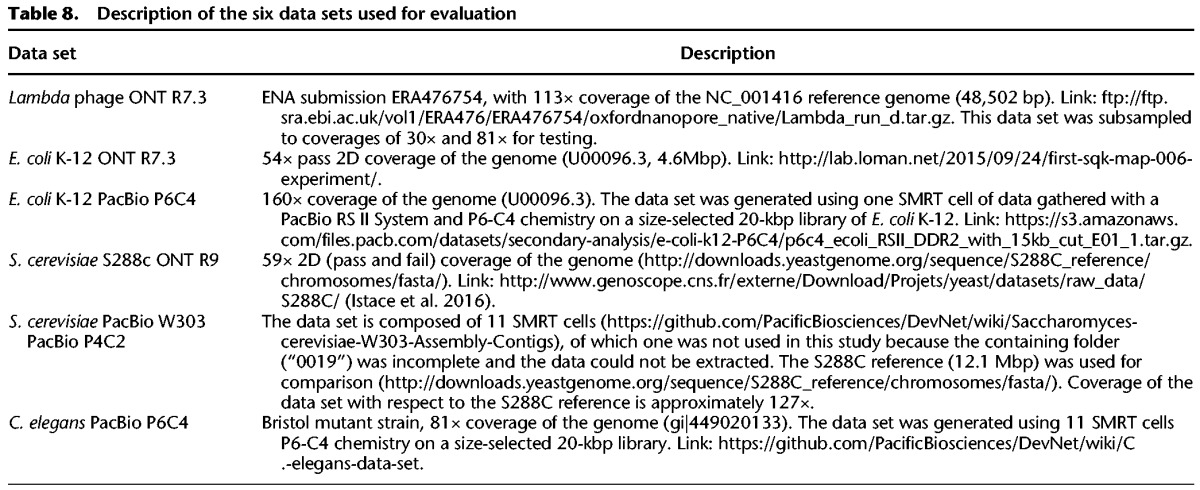
Six publicly available PacBio and Oxford Nanopore data sets were used for evaluation. These are listed in Table 8.
Table 8.
Description of the six data sets used for evaluation
Evaluation methods
The quality of called consensus sequences was evaluated primarily using Dnadiff (Delcher et al. 2003). The parameters we took into consideration for comparison include total number of bases in the query, aligned bases on the reference, aligned bases on the query, and average identity. In addition, we measured the time and memory required to perform the entire assembly process by each pipeline.
The quality of error-corrected reads was evaluated by aligning them to the reference genome using GraphMap (Sović et al. 2016b) with the settings “-a anchorgotoh” and counting the match, mismatch, insertion, and deletion operations in the resulting alignments.
Software availability
Racon and SPOA are available open source under the MIT license at https://github.com/isovic/racon and https://github.com/rvaser/spoa. In addition, the version of source codes used in this study can be found in the Supplemental Material.
Competing interest statement
M.S. has received a complimentary place at an Oxford Nanopore Technologies organized symposium. N.N. has received travel expenses and accommodation from Oxford Nanopore to speak at two organized symposia.
Supplementary Material
Acknowledgments
This work has been supported in part by Croatian Science Foundation under the project UIP-11-2013-7353. I.S. is supported in part by the Croatian Academy of Sciences and Arts under the project “Methods for alignment and assembly of DNA sequences using nanopore sequencing data.” N.N. is supported by funding from A*STAR, Singapore.
Author contributions: I.S., M.S., and R.V. devised the original concept for Racon. I.S. developed and implemented several prototypes for Racon. R.V. suggested and implemented the SIMD POA version. I.S. implemented a version of Racon that uses SPOA for consensus generation and error correction. I.S. and R.V. tested Racon and SPOA. M.S. and N.N. provided helpful discussions and guidance for the project. M.S. helped define applications and devised tests for the paper; I.S. ran the tests and collected and formatted the results. I.S. and R.V. wrote the paper with input from all authors. M.S. and R.V. designed the figures for the paper. N.N. proof-read and corrected parts of the paper. All authors read the paper and approved the final version. M.S. coordinated the project and provided computational resources.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214270.116.
References
- Berlin K, Koren S, Chin C-S, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. 2016. Phased diploid genome assembly with single molecule real-time sequencing. Nat Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Salzberg SL, Phillippy AM. 2003. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics Chapter 10: Unit 10.3. [DOI] [PubMed] [Google Scholar]
- Gotoh O. 1982. An improved algorithm for matching biological sequences. J Mol Biol 162: 705–8. [DOI] [PubMed] [Google Scholar]
- Istace B, Friedrich A, d'Agata L, Faye S, Payen E, Beluche O, Caradec C, Davidas S, Cruaud C, Liti G, et al. 2016. de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. bioRxiv 10.1101/066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (this issue). 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. 2003. Generating consensus sequences from partial order multiple sequence alignment graphs. Bioinformatics 19: 999–1008. [DOI] [PubMed] [Google Scholar]
- Lee C, Grasso C, Sharlow MF. 2002. Multiple sequence alignment using partial order graphs. Bioinformatics 18: 452–464. [DOI] [PubMed] [Google Scholar]
- Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32: 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. [DOI] [PubMed] [Google Scholar]
- Loose M, Malla S, Stout M. 2016. Real-time selective sequencing using nanopore technology. Nat Methods 13: 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers G. 1999. A fast bit-vector algorithm for approximate string matching based on dynamic programming. J ACM 46: 395–415. [Google Scholar]
- Rognes T, Seeberg E. 2000. Six-fold speed-up of Smith-Waterman sequence database searches using parallel processing on common microprocessors. Bioinformatics 16: 699–706. [DOI] [PubMed] [Google Scholar]
- Šošić M, Šikić M. 2016. Edlib: A C/C++ library for fast, exact sequence alignment using edit distance. bioRxiv 10.1101/070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sović I, Križanović K, Skala K, Šikić M. 2016a. Evaluation of hybrid and non-hybrid methods for de novo assembly of nanopore reads. Bioinformatics 32: 2582–2589. [DOI] [PubMed] [Google Scholar]
- Sović I, Šikić M, Wilm A, Fenlon SN, Chen S, Nagarajan N. 2016b. Fast and sensitive mapping of error-prone nanopore sequencing reads with GraphMap. Nat Commun 7: 11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Ma Z. 2016. Sparc: a sparsity-based consensus algorithm for long erroneous sequencing reads. PeerJ 4: e2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.