

Abstract
The amplitude of transforming growth factor‐β (TGF‐β) signal is tightly regulated to ensure appropriate physiological responses. As part of negative feedback loop SMAD7, a direct transcriptional target of downstream TGF‐β signaling acts as a scaffold to recruit the E3 ligase SMURF2 to target the TGF‐β receptor complex for ubiquitin‐mediated degradation. Here, we identify the deubiquitinating enzyme USP26 as a novel integral component of this negative feedback loop. We demonstrate that TGF‐β rapidly enhances the expression of USP26 and reinforces SMAD7 stability by limiting the ubiquitin‐mediated turnover of SMAD7. Conversely, knockdown of USP26 rapidly degrades SMAD7 resulting in TGF‐β receptor stabilization and enhanced levels of p‐SMAD2. Clinically, loss of USP26 correlates with high TGF‐β activity and confers poor prognosis in glioblastoma. Our data identify USP26 as a novel negative regulator of the TGF‐β pathway and suggest that loss of USP26 expression may be an important factor in glioblastoma pathogenesis.
Keywords: Smad7, TGF‐β, USP26
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
TGF‐β is a pleiotropic cytokine that plays a key role in a number of cellular processes regulating both embryogenesis and tissue homoeostasis. Aberrant TGF‐β signaling has also been associated with various diseases including cancer. In normal epithelial cells, TGF‐β acts a tumor suppressor eliciting a potent cytostatic response inhibiting tumor growth. In contrast, during tumor progression, the TGF‐β tumor suppressor function is lost, and in certain advanced cancers, TGF‐β becomes an oncogenic factor enhancing cellular proliferation, invasion, and metastasis. As a result, inhibition of the TGF‐β pathway is currently considered as a potential therapeutic option in advanced cancers, and encouragingly, several anti‐TGF‐β agents in clinical trials have shown promising results 1, 2. Despite this, overall patient response remains unpredictable. High TGF‐β activity confers poor prognosis in patients, suggesting that the identification of biomarkers which lead to enhanced TGF‐β signaling may stratify patients as potential responders to TGF‐β‐targeted therapies 3.
TGF‐β signal is transmitted through the binding of the TGF‐β ligand to a heteromeric receptor complex containing two type I and two type II receptors. Phosphorylation of type I receptors (TβRI) by the type II receptor (TβRII) opens up a docking site on TβRI permitting the binding and phosphorylation of receptor SMADs (R‐SMADs) 4. Phosphorylation in the C‐terminal tail of R‐SMADs by TβRI creates an interaction interface which induces oligomerization with the co‐SMAD, SMAD4 and translocation to the nucleus where this complex regulates expression of hundreds of genes in a cell type‐ and context‐specific manner 4, 5. As part of a negative feedback loop, the inhibitor SMAD (I‐SMAD) protein SMAD7 is transcriptionally induced by TGF‐β and functions to downregulate TGF‐β signaling through multiple mechanisms including acting as a key node for ubiquitin‐mediated regulation of the TGF‐β pathway 6. Mechanistically, SMAD7 serves as scaffold to recruit the SMAD‐specific E3 ubiquitin protein ligase 2 (SMURF2) to the TGF‐β receptor complex to facilitate receptor polyubiquitination and complex degradation 7. Moreover, the binding of SMAD7 to the TβRI occurs at the same site as the R‐SMADs further limiting R‐SMAD activation 8, 9. Besides acting as a scaffold for SMURF2, SMAD7 performs a similar function for the E3 ligases neural precursor cell expressed developmentally downregulated protein 4 (NEDD4) and its homolog WW domain‐containing E3 ubiquitin protein ligase 1 (WWP1), which also target the type I TGF‐β receptors for ubiquitin‐mediated degradation 10, 11. SMAD7 can also act as scaffold for SMURF1/2 and NEDD4 targeting of R‐SMADs 7, 12. Furthermore, SMAD7 itself is also targeted for ubiquitylation‐ and proteasomal‐mediated degradation by the E3 ligases ARKADIA, Jun activation domain‐binding protein 1 (JAB1), and ITCH 13, 14, 15.
The addition of ubiquitin moieties either as monomers or as polyubiquitin chains generates varying molecular signals determining overall protein localization and fate. The most well studied of these ubiquitin chain topologies include lysine 48 (Lys48) and lysine 63 (Lys63) polyubiquitin linkages. Lys48 chains predominantly target proteins for proteasomal‐mediated degradation, while Lys63 chains regulate kinase activation, signal transduction, protein trafficking, and endocytosis 16.
Ubiquitylation is a reversible process whereby ubiquitin moieties can be removed from polypeptides by deubiquitinating enzymes (DUBs). There are approximately 80 DUBs in the human proteome, and a number of these have been implicated in human diseases, including cancer 17, 18. Recently, a number of DUBs have been identified that regulate the TGF‐β pathway including 3 DUBs (USP11, USP15, and UCH37) that directly deubiquitinate the TGF‐β receptor (TβR) through complex formation with SMAD7 19, 20, 21, 22, 23. However, no DUB has been identified which directly regulates SMAD7 levels. Here, we identify the DUB USP26 as a key modulator of TGF‐β activity by regulating the stability of SMAD7.
Results
Identification of USP26 as an activator of TGF‐β signaling
We have recently performed a shRNA deubiquitinating enzyme screen in the TGF‐β pathway and identified a number of DUBs as potent regulators of TGF‐β activity 21. This shRNA library consists of 100 pools of four non‐overlapping shRNAs targeting known or putative DUBs. However, a proportion of DUBs were excluded from our final analyses due to inconsistencies in transfection efficiency. We reevaluated these remaining DUB pools and analyzed their effect on the activity of the TGF‐β‐responsive luciferase reporter (CAGA‐Luc) following the addition of TGF‐β (Fig EV1A and B). To our surprise the DUB pool, targeting the DUB USP26 greatly enhanced CAGA‐luciferase activity (Fig 1A). We isolated the two hairpin vectors from the USP26 DUB pool, which had previously been demonstrated to most effectively inhibit USP26 expression and tested their ability to knockdown USP26 24. Both shRNA vectors, denoted here as shRNA 1 and shRNA 2, efficiently inhibited the ectopic expression of a GFP fusion of USP26 in HEK293T cells and decreased endogenous USP26 mRNA levels as determined by quantitative reverse transcriptase PCR (qRT–PCR) (Fig 1B and C). Unfortunately, we were unable to identify any antibody that could detect endogenous USP26 by immunoblot. Next, we tested whether both verified knockdown vectors targeting USP26 could enhance CAGA‐Luc activity. As seen in Fig 1D, both USP26kd1 and USP26kd2 efficiently augmented luciferase levels in the presence and absence of TGF‐β compared to relevant controls. To further substantiate the role of USP26 in the TGF‐β pathway, we analyzed the expression of the TGF‐β target genes in cell lines depleted for USP26. Similar to the effects observed with the CAGA‐Luc reporter, knockdown of USP26 significantly enhanced the expression of TGF‐β target genes in both the absence and presence of TGF‐β ligand (Figs 1E and EV1C). To address whether the enhanced TGF‐β activity observed by USP26 inhibition was due to enzymatic activity USP26, we generated a catalytically inactive mutant USP26, USP26 C/S 25. As observed with USP26 knockdown, ectopic expression of the DUB dead mutant recapitulated the augmentation of SMAD7 induction by TGF‐β whereas wild‐type USP26 significantly diminished the induction of SMAD7 in the presence of TGF‐β (Fig 1F). Intriguingly, following the addition of TGF‐β, USP26 mRNA expression levels were significantly enhanced with USP26 mRNA levels showing an eightfold increase after 3 h of TGF‐β stimulation (Fig 1G). Together, these results demonstrate that USP26 expression is regulated by TGF‐β and acts as a critical negative regulator of TGF‐β signaling.
Figure EV1. USP26 enhances SMAD2 phosphorylation and TGF‐β‐mediated transcription.
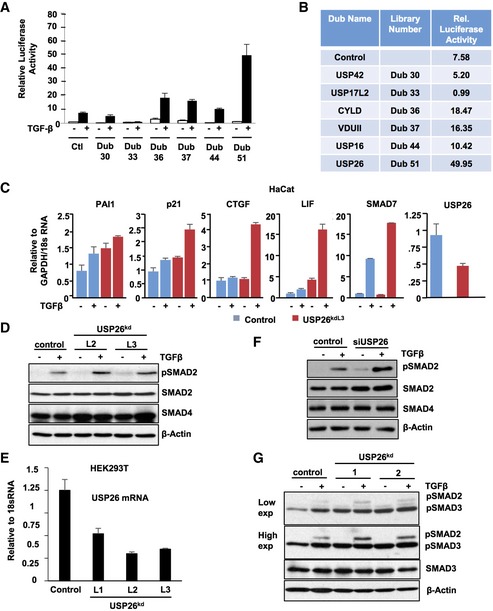
- Graph representing relative luciferase values obtained from DUB screen. 293T cells were transfected with CAGA‐Luc and indicated DUB pools. Forty‐eight hours later, cells were treated with TGF‐β for 16 h and a luciferase assay was performed. Data are mean ± SD of triplicate samples.
- Table indicates relative luciferase value of each gene tested in (A).
- HaCat cells stably transduced with a hairpin targeting USP26 or vector control were stimulated with TGF‐β for 3 h. PAI1, CDKN1A (p21), CTGF, LIF, SMAD7, and USP26 mRNA levels relative to GAPDH or 18S are shown as evaluated by quantitative real‐time PCR. Data are mean ± SD of triplicate samples.
- 293T cells were stably infected with two independent hairpins (L2 and L3) targeting USP26 and treated with TGF‐β overnight. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were stably infected with three independent hairpins (L1, L2, and L3) targeting USP26. USP26 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are mean ± SD of triplicate samples.
- 293T cells transfected with siRNA targeting USP26 or control vector were treated with TGF‐β overnight. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells expressing knockdown vectors targeting USP26 or control vector were treated with TGF‐β overnight. Whole‐cell extracts were probed with the indicated antibodies.
Source data are available online for this figure.
Figure 1. Identification of USP26 as a regulator of TGF‐β signaling.
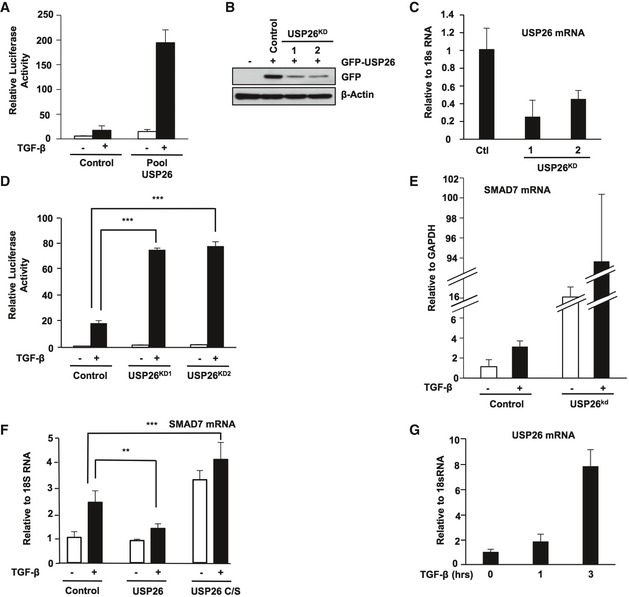
- 293T cells were co‐transfected with a CAGA‐luciferase reporter and the USP26 shRNA pool from the DUB knockdown library. Cells were stimulated with TGF‐β overnight, and luciferase activity was measured. Data are mean ± SD of triplicate samples from a representative experiment performed three times.
- 293T cells were co‐transfected with GFP‐tagged USP26 and two knockdown vectors (USP26KD) as indicated (1, 2) or a control vector. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected with knockdown vectors shUSP26KD1 and shUSP26KD2 or control vector. USP26 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are shown as the mean ± SD of triplicate samples from a representative experiment performed three times.
- 293T cells were co‐transfected with a CAGA‐luciferase reporter and two knockdown vectors targeting USP26 or control vector. Cells were stimulated with TGF‐β overnight and luciferase activity was measured. Data are shown as the mean ± SD of triplicate samples from a representative experiment performed three times. ***P < 0.001 using Student's t‐test.
- 293T cells were transfected with a hairpin targeting USP26 or vector control and stimulated with TGF‐β for 3 h. SMAD7 mRNA levels relative to GAPDH are shown as evaluated by quantitative real‐time PCR. Data are shown as the mean ± SD of triplicate samples from a representative experiment performed three times.
- 293T cells were transfected as indicated and stimulated with TGF‐β for 3 h. SMAD7 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are shown as the mean ± SD of triplicate samples from a representative experiment performed three times. **P < 0.01 and ***P < 0.001 using Student's t‐test.
- 293T cells were stimulated with TGF‐β for 1 and 3 h. USP26 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are shown as the mean ± SD of triplicate samples from a representative experiment performed three times.
Source data are available online for this figure.
USP26 deubiquitinates SMAD7
To elucidate the mechanisms behind the enhanced TGF‐β response in USP26 knockdown cells, we compared the intercellular responses of HEK293T or HEK293T USP26‐depleted cells following TGF‐β treatment. Loss of USP26 expression intensified SMAD2 phosphorylation while having no effect on total SMAD2, or SMAD4 levels (Fig 2A). Similar results were observed in HEK293T cells either stably expressing shRNA vectors or transfected with short interfering RNA (siRNA) targeting USP26 (Fig EV1D–F). Overexpression of the catalytically inactive USP26 C/S mutant also enhanced the levels of p‐SMAD2 whereas ectopic expression of USP26 slightly diminished the levels of phosphorylated SMAD2 (Fig 2B). Curiously, USP26 knockdown cells displayed higher levels of pSMAD3 in the absence of TGF‐β compared to relevant controls but did not show a further increase in the presence of TGF‐β. (Fig EV1G).
Figure 2. USP26 regulates SMAD2 activity and binds to SMAD7.
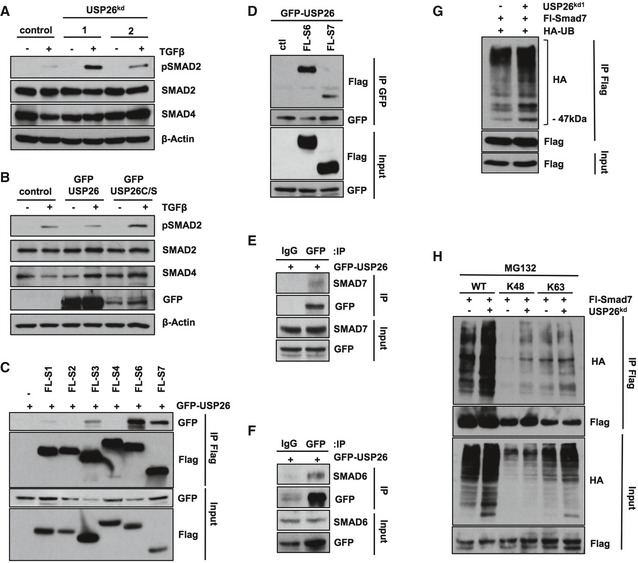
- 293T cells expressing knockdown vectors targeting USP26 or control vector were treated with TGF‐β overnight. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells expressing GFP‐USP26, GFP‐USP26 C/S, or control vector were treated overnight with TGF‐β. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected as indicated with GFP‐USP26 and Flag‐tagged SMAD1, SMAD2, SMAD3, SMAD4, SMAD6, and SMAD7. After 48 h, cells were lysed and immunoprecipitated with anti‐Flag affinity resin. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected as indicated with GFP‐USP26 and Flag‐tagged SMAD6 and SMAD7. After 48 h, cells were lysed and immunoprecipitated with anti‐GFP affinity resin. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected with GFP‐USP26 and cells were lysed and immunoprecipitated with anti‐GFP affinity resin. Whole‐cell extracts were probed with SMAD7.
- 293T cells were transfected with GFP‐USP26 and cells were lysed and immunoprecipitated with anti‐GFP affinity resin. Whole‐cell extracts were probed with SMAD6.
- 293T cells transfected with FL‐SMAD7, pRS‐USP26, control vector, and HA‐tagged ubiquitin. Following immunoprecipitation of SMAD7, lysates were resolved by SDS–PAGE and probed with the indicated antibodies.
- 293T cells transfected with FL‐SMAD7, pRS‐USP26, control vector, and wild‐type HA‐tagged ubiquitin or K48‐ubiquitin or K63‐ubiquitin mutants. Following immunoprecipitation of SMAD7, lysates were resolved by SDS–PAGE and probed with the indicated antibodies.
Source data are available online for this figure.
As USP26 appears to mediate SMAD2/3 phosphorylation and downstream TGF‐β activity, we reasoned that USP26 may function through the canonical TGF‐β pathway. To gain insight into the molecular mechanisms underlying USP26 function, we performed co‐immunoprecipitation assays with USP26 and Flag‐tagged SMADs. We found that immunoprecipitation of SMAD3, SMAD6, and SMAD7 from lysates of co‐transfected cells exhibited the greatest efficacy of USP26 binding (Figs 2C and EV2A). We also detected this interaction reciprocally by immunoprecipitating GFP‐tagged USP26 with a GFP antibody and probing the blotted precipitate with a Flag antibody (Fig 2D). Furthermore, ectopically expressed USP26 co‐immunoprecipitated with endogenous SMAD3, SMAD6, and SMAD7 (Figs 2E and F, and EV2B). These data suggest that USP26 can form a stable complex in vivo with SMAD3, SMAD6, and SMAD7. We decided to focus our attention on SMAD7.
Figure EV2. USP26 binds to and deubiquitinates SMAD7.
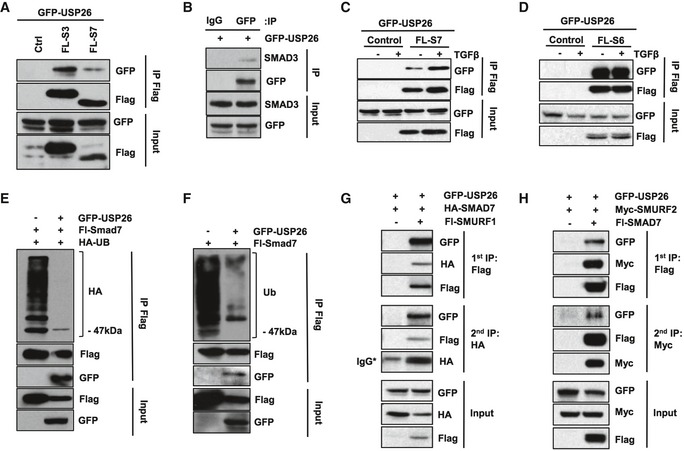
- 293T cells were transfected as indicated with GFP‐USP26 and Flag‐tagged SMAD3 and SMAD7. After 48 h, cells were lysed and immunoprecipitated with anti‐Flag affinity resin. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected with GFP‐USP26, and cells were lysed and immunoprecipitated with anti‐GFP affinity resin. Whole‐cell extracts were probed for SMAD3.
- 293T cells were transfected as indicated with GFP‐USP26 and Flag‐tagged SMAD7. After 48 h, cells were treated with TGF‐β for 1 h and lysed. Flag‐tagged proteins were immunoprecipitated with anti‐Flag affinity resin and whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected as indicated with GFP‐USP26 and Flag‐tagged SMAD6. After 48 h, cells were treated with TGF‐β and lysed. Flag‐tagged proteins were immunoprecipitated with anti‐Flag affinity resin and whole‐cell extracts were probed with the indicated antibodies.
- 293T cells transfected with FL‐SMAD7, GFP‐USP26, control vector, and HA‐tagged ubiquitin. Following immunoprecipitation of SMAD7, lysates were resolved by SDS–PAGE and probed with the indicated antibodies.
- Endogenous ubiquitination assay. 293T cells transfected with FL‐SMAD7, GFP‐USP26, or control vector. Following immunoprecipitation of SMAD7, lysates were resolved by SDS–PAGE and probed with the indicated antibodies.
- 293T cells were transfected as indicated with GFP‐USP26, Flag‐tagged SMAD7, and Myc‐tagged SMURF2. After 48 h, cells were lysed and immunoprecipitated with anti‐Flag affinity resin overnight, eluted with anti‐Flag peptide, and re‐immunoprecipitated with anti‐Myc affinity resin. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected as indicated with GFP‐USP26, HA‐tagged SMAD7, and Myc‐tagged SMURF1. After 48 h, cells were lysed and immunoprecipitated with anti‐Flag affinity resin overnight, cleaved with an anti‐Flag peptide, and re‐immunoprecipitated with anti‐Myc affinity resin. Whole‐cell extracts were probed with the indicated antibodies.
Source data are available online for this figure.
SMAD7 protein expression is tightly regulated by a number of post‐translational modifications including ubiquitylation. 26. Therefore, in light of our findings that both USP26 and SMAD7 are upregulated following stimulation with TGF‐β, we speculated that USP26 might form a complex with SMAD7 following TGF‐β exposure leading to USP26‐mediated deubiquitination. TGF‐β treatment enhanced the binding of USP26 to SMAD7 but not SMAD6 (Fig EV2C and D). To reveal ubiquitylated isoforms of SMAD7, we co‐transfected HEK293T cells with expression plasmids encoding Flag‐tagged SMAD7, HA‐tagged ubiquitin (Ub), and USP26. SMAD7 was affinity‐purified and its ubiquitylation pattern analyzed by immunoblotting with an HA antibody. Strikingly, the overall ubiquitylation pattern of SMAD7 appears to contain robust mono‐ and multimonoubiquitin bands as well as a polyubiquitin configuration (Figs 2G, and EV2E and F). Importantly, ectopic expression of USP26 completely abolished polyubiquitin chain linkages associated with SMAD7, while monoubiquitinated isoforms were still detected (Fig EV2E). Similar results were observed when we analyzed endogenous ubiquitylation levels of SMAD7 (Fig EV2F). In contrast, SMAD7 polyubiquitination was enhanced in HEK293T cells depleted for USP26 (Fig 2G).
Different ubiquitin chain topologies act as signals to regulate various substrate outcomes. Therefore, we analyzed whether loss of USP26 increases the levels of SMAD7 Lys48 or Lys63‐incorporated ubiquitin chains. HEK293T cells were transfected with Flag‐tagged SMAD7 and either wild‐type Ub or Lys48 or Lys63 Ub variants where all the lysine residues have been replaced with arginine except at the lysine 48 and lysine 63 residues, respectively. Among these mutants' depletion of USP26 significantly enhanced Lys48 ubiquitylation of SMAD7 while having no effect on Lys63‐ubiquitinated SMAD7 isoforms (Fig 2H). Taken together, these results suggest that USP26 acts as a SMAD7 Lys48 ubiquitin chain‐specific deubiquitylase.
Next, we sought to explore whether USP26 is present in the SMAD7:SMURF1 or SMAD7:SMURF2 complexes which target multiple components of the TGF‐β pathway for degradation. Indeed, USP26 interacts with both SMURF1 and SMURF2 when in complex with SMAD7 as demonstrated by sequential co‐immunoprecipitation (Fig EV2G and H). These results suggest that USP26 may potentially act in conjunction with SMAD7 and SMURF2 to downregulate canonical TGF‐β signaling.
USP26 regulates the stability of SMAD7 leading to TβR degradation
As ubiquitylation through Lys48 generally targets proteins for proteasome‐mediated degradation, our data strongly suggest that USP26 activity may regulate overall SMAD7 stability. In order to examine this possibility, we transfected HEK293T cells with Flag‐tagged SMAD7 and wild‐type USP26 or its catalytic dead variant. Overexpression of USP26, but not the catalytically inactive mutant USP26 C/S, markedly increased the expression of SMAD7 (Fig 3A). Furthermore, knockdown of USP26 significantly diminished both ectopically expressed and endogenous SMAD7 levels (Fig 3B and C). This effect could be rescued by the addition of the proteasome inhibitor, MG132 (Fig 3D and E).
Figure 3. USP26 stabilization of SMAD7 enhances TGF‐β receptor degradation.
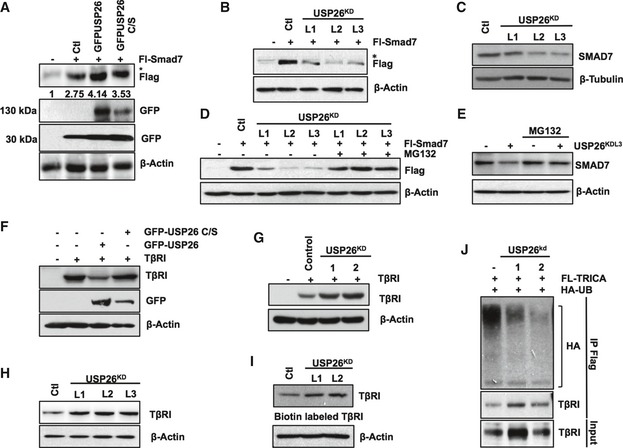
- 293T cells transfected with FL‐SMAD7 (1 μg), GFP‐USP26 (4 μg), GFP‐USP26 C/S (4 μg) or control vector. Whole‐cell extracts were probed with the indicated antibodies. * denotes background band.
- 293T cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were transfected with FL‐SMAD7 (1 μg). Whole‐cell extracts were probed with the indicated antibodies. * denotes background band.
- 293T cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were lysed, and whole‐cell extracts were probed with the indicated antibodies.
- 293T cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were transfected with FL‐SMAD7 (1 μg); the cells were treated with MG132 (5 μg/ml) as indicated overnight and subsequently lysed. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells stably expressing a USP26 knockdown vector or control vector were treated with MG132 (5 μg/ml) as indicated overnight and subsequently lysed. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells transfected with TβRI (1 μg), GFP‐USP26 (5 μg), GFP‐USP26 C/S (5 μg), or control vector. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells were transfected with TβRI (5 μg) and either control or knockdown vectors targeting USP26. Whole‐cell extracts were probed with the indicated antibodies.
- 293T cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were lysed, and whole‐cell extracts were probed with the indicated antibodies.
- Immunoblots of biotinylated TβRI in MDA‐MB‐231 stably expressing lentiviral knockdown vectors targeting USP26 or control vector.
- 293T cells transfected with FL‐TRICA and knockdown vectors targeting USP26, and HA‐tagged ubiquitin. Following immunoprecipitation of FL‐TRICA, lysates were resolved by SDS–PAGE and probed with the indicated antibodies.
Source data are available online for this figure.
Previous work has clearly established that SMAD7 acts as a scaffold protein to recruit SMURF2 to the TGF‐β receptor complex to facilitate receptor degradation and attenuate TGF‐β signaling 8. Therefore, in light of our recent results, we sought to address whether increased stabilization of SMAD7 by USP26 results in greater TGF‐β receptor degradation. Indeed, overexpression of USP26 significantly decreased the levels of co‐transfected TβRI. On the other hand, TβRI levels were relatively unchanged when co‐transfected with the catalytically inactive mutant USP26 C/S compared to controls (Fig 3F). In contrast, knockdown of USP26 significantly enhanced both ectopically expressed and endogenous levels of TβRI (Fig 3G and H). To further validate the relationship between USP26 and SMAD7 toward TβRI function, we determined the membrane localization of TβRI. Consistent with our previous results, USP26 depletion enhanced membrane‐associated TβRI (Fig 3I). Next we sought to address whether loss of USP26 and subsequent degradation of SMAD7 deregulates SMAD7/SMURF2‐mediated ubiquitylation of the TβR complex. To this end, we co‐transfected HEK293T cells with expression plasmids encoding a constitutively active TβR (TRICA) and HA‐tagged ubiquitin in the presence of shRNA vectors targeting USP26. Knockdown of USP26 caused a reduction in TβRI ubiquitylation compared to controls (Fig 3J). Taken together, these data indicate that USP26 inhibits TGF‐β pathway activity by limiting Lys48 ubiquitin‐mediated degradation of SMAD7 consequently resulting in enhanced ubiquitylation and degradation of the TβR complex.
USP26 depletion enhances TGF‐β activity and TGF‐β biological responses in breast cancer and glioma
In certain advanced cancers including glioblastoma and breast cancer, the TGF‐β pathway is highly active and can act as an oncogenic factor driving cancer progression 3, 27. Furthermore, in patients with glioma, elevated TGF‐β activity has been demonstrated to correlate with poor overall survival 3. As USP26 regulates TGF‐β activity in the HEK293T cells, we sought to determine whether USP26 is also pertinent factor in glioma and breast cancer cell line models. Similar to HEK293T, depletion of USP26 expression in the breast cancer cell lines MCF7, MDA‐MB‐231, and the glioblastoma cell line U373 enhanced the levels of phosphorylated SMAD2 following exposure to TGF‐β (Fig EV3A–C). Given that the intrinsic properties of cultured cell lines tend to diverge from the original tumor from which they were originally derived, we confirmed our results in short‐term primary cultured tumor cells (PCTC) obtained directly from freshly resected GBM tumor samples 28. In line with our other models, knockdown of USP26 in this patient‐derived cell line enhanced TGF‐β activity as evidenced by increased levels of phospho‐SMAD2 and increased expression of TGF‐β target genes CTGF, LIF, and SMAD7 (Fig EV3D and E). In addition, depletion of USP26 significantly reduced the expression of SMAD7 in both the MDA‐MB‐231 and U373 cell lines (Fig EV3F and G). This finding confirms USP26 as a novel regulator of TGF‐β activity in breast cancer and glioma. Our previous results demonstrated that USP26 mRNA levels significantly increased following TGF‐β induction in HEK293T cells. To verify whether USP26 induction by TGF‐β is prevalent in other cell types, we analyzed the induction of USP26 and SMAD7 at early time points in the TGF‐β‐responsive breast cancer cell lines MCF7, MDA‐MB‐231, T47D, and CAL51 and glioma cell lines U373, PCTC, and A172 (Fig EV4A–G). In all cell lines tested, TGF‐β enhanced the mRNA expression levels of both USP26 and SMAD7. These data confirm that regulation of USP26 and SMAD7 mRNA by TGF‐β is a common occurrence in multiple types of cancer.
Figure EV3. Regulation of the TGF‐β by USP26 in breast cancer and GBM cell lines.
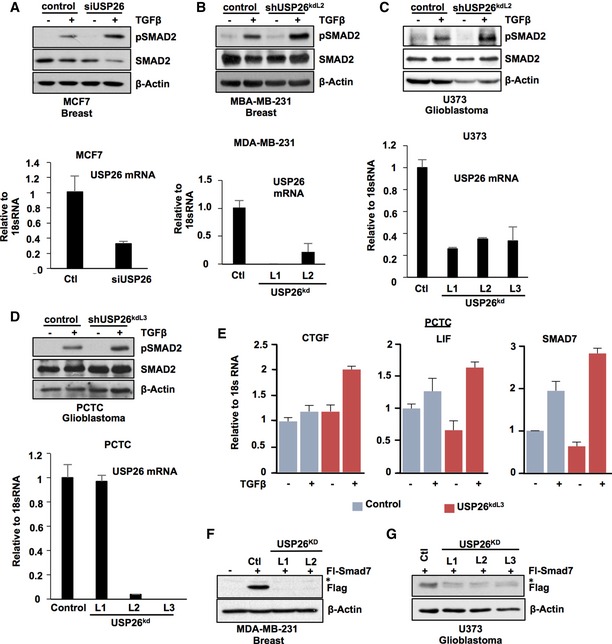
-
AMCF7 cells transfected with siRNA targeting USP26 or control vector were treated with TGF‐β overnight. Whole‐cell extracts were probed with the indicated antibodies (top panel). Corresponding USP26 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR (bottom panel). Data are mean ± SD of triplicate samples.
-
B–DMDA‐MB‐231 (B), U373 (C), and PCTC (D) cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were treated with TGF‐β overnight. Whole‐cell extracts were probed with the indicated antibodies (top panel). Corresponding USP26 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR (bottom panel). Data are mean ± SD of triplicate samples.
-
EPCTC cells stably transduced with a hairpin targeting USP26 or vector control were stimulated with TGF‐β for 3 h. CTGF, LIF, and SMAD7 mRNA levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are mean ± SD of triplicate samples.
-
FMDA‐MB‐231 cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were treated with transfected with FL‐SMAD7 (1 μg). Whole‐cell extracts were probed with the indicated antibodies. * denotes background band.
-
GU373 cells stably expressing lentiviral knockdown vectors targeting USP26 or control vector were treated with transfected with FL‐SMAD7 (1 μg). Whole‐cell extracts were probed with the indicated antibodies. * denotes background band.
Source data are available online for this figure.
Figure EV4. Expression of SMAD7 and USP26 following TGF‐β induction in breast cancer and GBM cell lines.
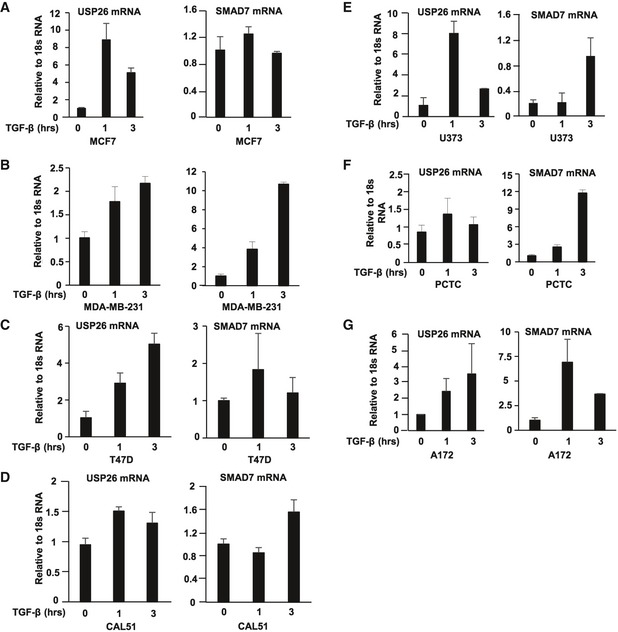
-
A–DThe breast cancer cell lines MCF7 (A), MDA‐MB‐231 (B), T47D (C), and CAL51 (D) were stimulated with TGF‐β for 1 and 3 h. USP26 mRNA (left panel) and SMAD7 (right panel) levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are mean ± SD of triplicate samples.
-
E–GThe glioblastoma cancer cell lines U373 (E), PCTC (F), and A172 (G) were stimulated with TGF‐β for 1 and 3 h. USP26 mRNA (left panel) and SMAD7 (right panel) levels relative to 18S are shown as evaluated by quantitative real‐time PCR. Data are mean ± SD of triplicate samples.
Depending on cellular context, the canonical TGF‐β signaling pathway can either induce growth arrest in early neoplasias or in advanced cancers promote migration and invasion. As knockdown of USP26 potently activated the TGF‐β pathway in breast cancer, we investigated whether depletion of USP26 in the TGF‐β‐responsive metastatic cell line MDA‐MB‐231 enhanced cellular motility and invasion. Stably infected MDA‐MB‐231 cells harboring knockdown vectors targeting USP26 or a control vector were utilized and migration and invasion were quantified using a scratch assay and transwell migration assay. Silencing of USP26 significantly enhanced TGF‐β‐induced migration and invasion (Fig 4A–D). In comparison, stable expression of USP26, but not the catalytically inactive mutant USP26 C/S, attenuated invasion of MDA‐MB‐231 cells following exposure to TGF‐β (Fig 4E–G). Taken together, these results show that USP26 is a critical regulator of the biological effects mediated by the TGF‐β pathway.
Figure 4. USP26 modulates TGF‐β‐mediated biological responses.
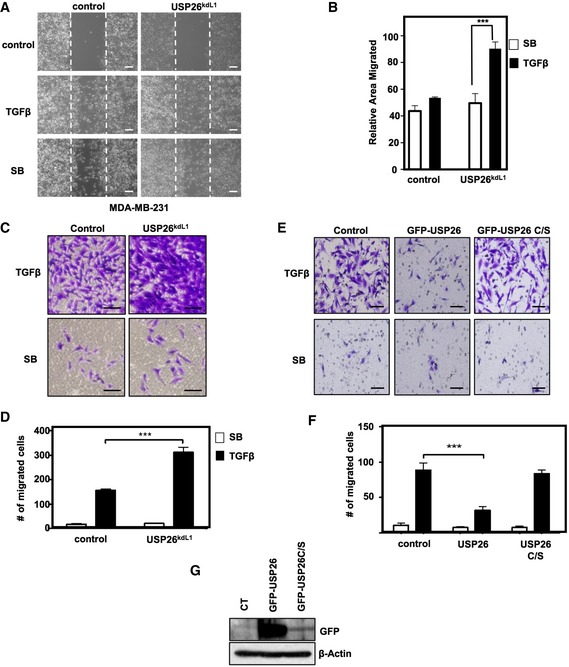
- MDA‐MB‐231 cells stably expressing a lentiviral hairpin targeting USP26 (L1) or relevant control vector were plated for scratch assay and treated with either SB431542 (5 μM) or TGF‐β (5 ng/ml); panels show migration at 0 and 24 h. Representative images are shown (scale bars, 50 μm).
- Percentage of migrated area within the white dotted lines was estimated with respect to control (0 h) and a graph was plotted. ***P < 0.001 using Student's t‐test. Data are mean ± SD of three random fields. Data are representative of two independent experiments with similar results.
- Transwell assay of MDA‐MB‐231 cells stably expressing a lentiviral hairpin targeting USP26 (L1) or relevant control vector treated with SB431542 (5 μM) or TGF‐β (5 ng/ml) for 16 h prior to fixation and crystal violet staining. Representative images are shown (scale bars, 100 μm).
- Graph represents average number of migrated cells taken from four different random fields from panel (C). Data are mean ± SD of triplicate samples from a representative experiment performed three times. ***P < 0.001 using Student's t‐test.
- Transwell assay of MDA‐MB‐231 cells stably expressing GFP‐USP26, GFP‐USP26 C/S, or relevant control vector treated with SB431542 (5 μM) or TGF‐β (5 ng/ml) for 16 h prior to fixation and crystal violet staining. Representative images are shown (scale bars, 100 μm).
- Graph represents average number of migrated cells taken from four different random fields from panel (E). Data are mean ± SD of triplicate samples from a representative experiment performed three times. ***P < 0.001 using Student's t‐test.
- Immunoblotting showing ectopically expressed GFP‐USP26, GFP‐USP26 C/S, or control vector in MDA‐MB‐231.
Source data are available online for this figure.
USP26 expression correlates with lower levels of p‐SMAD2 and patient survival in glioblastoma
TGF‐β activity has been demonstrated to play a pivotal role in glioblastoma pathogenesis 3. In light of our previous observations, we sought to further determine the relevance of USP26 on TGF‐β activity in glioblastoma patients. To this end, we performed immunohistochemical staining of USP26 and pSMAD2, with antibodies validated for immunohistochemistry, on glioblastoma tissue microarrays containing samples from 36 patients (Figs EV5A, and 5A and B). A significant negative correlation (P = −0.15, P = <0.0001) was observed between USP26 and pSMAD2 protein levels. Next, to determine the clinical significance of these findings, we probed The Cancer Genome Atlas (TCGA) database and performed copy number analyses on 577 glioblastoma patients. Indeed, USP26 copy number loss was observed in 17% of patients (Fig 4C) 29. Furthermore, using the REMBRANDT (Repository for Molecular BRAin Neoplasia DaTA) database, we found that glioblastoma patients with low levels of USP26 had a lower overall survival than patients with high levels of USP26 (Fig 4D). As loss of USP26 degrades SMAD7 leading to the stabilization of TβR, we evaluated the role of TβR and SMAD7 on clinical outcome. In line with the notion that high TGF‐β activity is a poor prognostic factor in glioblastoma, patients with high TβRI or TβRII, but not TβRIII, exhibited lower overall survival (Fig EV5B–D). Importantly, patients with low levels of SMAD7 also exhibited poorer overall survival then patients with higher levels of SMAD7 (Fig EV5E). Taken together, these findings confirmed our hypothesis that loss of USP26 enhances TGF‐β signaling and confers poor prognosis in glioblastoma patients.
Figure EV5. Correlation of TGF‐β pathway components with overall survival in GBM.
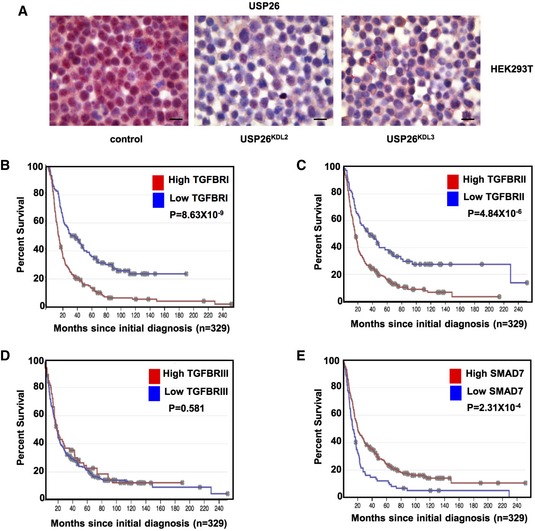
-
AValidation of the USP26 antibody for immunohistochemistry. 293T cells stably infected with knockdown vectors targeting USP26 were analyzed by immunohistochemistry for USP26 expression. Representative images are shown. To prepare sections, cell pellets were fixed in formalin and embedded in paraffin. Red staining indicates positive immunoreactivity. Scale bars: 50 μm.
-
B–EKaplan–Meier curves of glioblastoma patients (n = 329) with TGFBRI (B), TGFBRII (C), TGFBRIII (D), and SMAD7 (E) (REMBRANDT). P‐value was obtained by log‐rank test.
Figure 5. USP26 expression correlates with pSMAD2 levels in glioblastoma and poor overall survival.
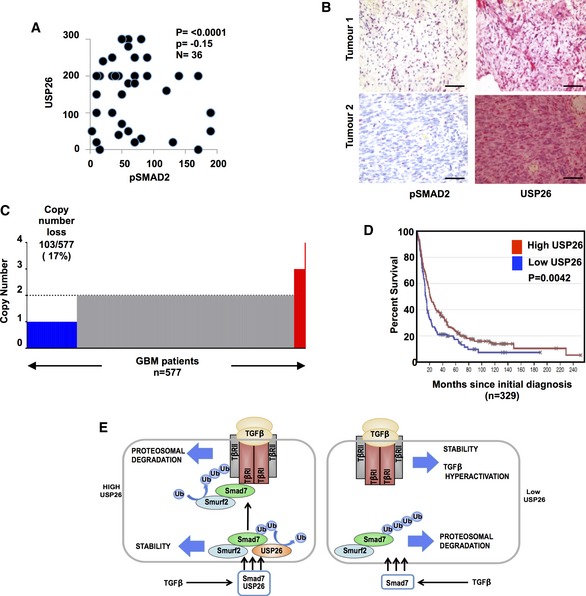
- Scatter plot showing correlation with USP26 levels and pSMAD2 in tissue microarrays from human glioblastoma patients (n = 36). A Spearman's test was used to determine correlation between nuclear USP26 and nuclear pSMAD2 staining (P = −0.15). Statistical significance was determined by two‐tailed test (P < 0.0001).
- Immunohistochemical staining of USP26 and pSMAD2 in representative glioblastoma specimens from the GL806D (Biomax) tissue microarray. Red staining indicates positive immunoreactivity. Scale bars: 50 μm.
- Bar plots of USP26 gene level copy number (y‐axis) from TCGA glioblastoma (n = 577). Color code: copy number loss, blue; copy number gain, red; and copy number normal, gray.
- Kaplan–Meier curves showing that glioblastoma patients with low levels of USP26 have a significantly lower overall survival than patients with high levels of USP26 (P = 0.0042) (REMBRANDT). P‐value was obtained by log‐rank test.
- Schematic overview of USP26 converging with the SMAD7 complex to regulate SMAD7 stability and downregulate TGF‐β activity.
Discussion
Targeting the TGF‐β pathway is emerging as a promising strategy for certain cancers 30. However, due to its dichotomous role in cancer acting as both an oncogene and tumor suppressor, systemic inhibition of TGF‐β signaling by TGF‐β pathway inhibitors may confer a number of undesirable side effects. Therefore, the identification of reliable biomarkers correlated with high TGF‐β activity may lead to the identification of patients most likely to benefit from these compounds.
Extensive studies have shown that SMAD7 functions as a scaffold protein recruiting a number of HECT type E3 ligases (SMURF2, NEDD4‐2, and WWP1) to induce polyubiquitination of the receptor and channeling the TβR‐SMAD7‐SMURF2 complex for degradative endocytosis 7. Pathway regulation by these ligases is countered upon by an apparent disproportionate number of deubiquitinating enzymes, the majority of which require SMAD7 as an adapter either to deubiquitinate complex bound E3 ligases or ubiquitylated TGF‐β receptors. On the other hand, very little is known regarding the mechanistic details that underlie the post‐translational modifications of SMAD7 itself. SMAD7 is targeted for ubiquitylation and degradation by the E3 ligases ARKADIA, ITCH, and JAB1 7. Furthermore, SMAD7 is acetylated at two N‐terminal lysine residues by the acetyltransferase p300 26. Acetylation of SMAD7 inhibits the ubiquitylation and degradation of SMAD7. Thus, there exists an interplay between acetylation and ubiquitylation in the regulation of overall SMAD7 protein levels. Both deacetylating enzymes HDAC1 and SIRT1 have been reported to deacetylate SMAD7; however, no deubiquitinating enzyme for SMAD7 has been identified 7, 31.
In this report, we identify USP26 as a novel regulator of TGF‐β activity. Our data suggest that as part of a negative feedback loop, TGF‐β enhances the expression of not only SMAD7 but also USP26, which acts to deubiquitinate and stabilize SMAD7. This then permits SMAD7 to remain in stable conformation with SMURF2, permitting SMURF2 recruitment to the TGF‐β receptor complex potentiating complex degradation. In contrast, loss of USP26 rapidly degrades SMAD7 stabilizing the TGF‐β receptors leading to enhanced TGF‐β activity as observed by increased levels of phosphorylated SMAD2. Further experimentation will be required to elucidate the precise mechanism of how TGF‐β enhances USP26 expression.
As previously discussed, the interaction between SMURF2 and SMAD7 serves a number of purposes including mediating the shuttling of the SMAD7‐SMURF2 complex from the nucleus to the cytoplasm and facilitating the interaction of SMURF2 with the TβR complex 8. Furthermore, to prevent autoubiquitination and degradation, SMURF2 exists in a closed conformation with the C2 domain of the protein coming into close association with the catalytic HECT domain 32. SMAD7 binding alleviates this closed conformation allowing SMURF2 catalysis and appropriate substrate targeting. However, SMAD7 itself does not appear to be a substrate for SMURF2 9. Furthermore, NEDD4 family members, including SMURF2, predominantly act as Lys63 ubiquitin ligases 33. As USP26 acts as SMAD7 Lys48‐specific deubiquitinase, we postulate that USP26 counteracts the ubiquitylation and degradation of SMAD7 by another E3 ligase in the cytoplasm while in complex with SMURF2. SMAD7 has also been described to regulate the activity of a number of other pathways and it would be interesting to determine what effect USP26 has on SMAD7 function in these non‐TGF‐β settings 7.
We also validated these novel mechanistic findings in breast cancer and glioblastoma cell line models, two types of cancer where TGF‐β has been described to act as an oncogene in certain scenarios. Two of the most prominent pro‐tumoral effects mediated by the TGF‐β pathway include invasion and migration. We observed that depletion of USP26 enhanced both of these TGF‐β‐induced oncogenic effects in the TGF‐β‐responsive breast cancer cell line MDA‐MB‐231. These results identify USP26 as a bona fide regulator of the TGF‐β pathway.
USP26 has been described to be predominantly expressed in testis where it is required for androgen receptor hormone‐induced activation in spermatogenesis, with various reports indicating an association with USP26 mutations and human male infertility in these settings 34, 35. Furthermore, USP26 has been shown to stabilize the androgen receptor in prostate cancer cell lines leading to androgen receptor transcriptional activity 24. Besides acting as downstream modulator of testosterone‐mediated transcription, the androgen receptor can additionally bind to SMAD3, constraining the DNA binding ability of SMAD3 and consequently limiting TGF‐β‐mediated transcriptional responses 36. As part of this study, we demonstrate that USP26 binds to SMAD3, suggesting that USP26 may also regulate the TGF‐β pathway by enhancing androgen receptor‐mediated repression of SMAD3 transcriptional targets. This is similar to the role observed by other DUBs which act on multiple nodes in the TGF‐β pathway to regulate overall TGF‐β output 21, 37, 38, 39. Further work will be required to analyze the potential role of USP26 on androgen receptor stability and TGF‐β activity.
More recently, it was found that USP26 regulates homologous recombination partly by counteracting RNF‐168 ubiquitylation 40. Interestingly, the authors found that USP26 expression was lost or amplified in 1,457 and 309 cancer cell lines, respectively, strongly suggesting that the function of USP26 in the TGF‐β pathway may be observed in other cancers besides glioblastoma and breast cancer.
It has recently been suggested that the role of TGF‐β inhibitors in cancer function primarily by targeting the tumor microenvironment as TGF‐β inhibitors have demonstrated minimal effect on cancer cell proliferation in vitro. However, it has clearly been demonstrated that effective blocking of the TGF‐β signaling in the tumor cells thwarts the ability of TGF‐β to induce tumorigenesis and invasion. This is especially true in settings where accurate biomarkers are available which correspond with hyperactivation of the TGF‐β pathway. Our results indicate that low levels of USP26 may be considered as a potential biomarker for response to TGF‐β inhibitors in glioblastoma.
Materials and Methods
Western blotting and quantification
Cells were lysed in solubilizing buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP‐40, 0.5% deoxycholic acid, 0.1% SDS, 1 mM sodium vanadate, 1 mM pyrophosphate, 50 mM sodium fluoride, 100 mM β‐glycerol phosphate), supplemented with protease inhibitors (Complete; Roche). Whole‐cell extracts were then separated on 7–12% SDS–PAGE gels and transferred to polyvinylidene difluoride membranes (Millipore). Membranes were blocked with bovine serum albumin for all antibodies except phospho‐SMAD2, which was blocked in milk and probed with specific antibodies. Blots were then incubated with an HRP‐linked secondary antibody and resolved with chemiluminescence (Pierce).
Plasmids and antibodies
The DUB knockdown library vectors were generated by annealing the individual oligonucleotide primer pairs and cloning them into pSuper as described in 41. The bacterial colonies of each DUB hairpin were then pooled and used for plasmid preparation. For USP26 knockdown, pSuper sequences are as follows (A): GATATCCTGGCTCCACACA; (B): TGGCTTGTTTATTGAAGGA 24. Short interfering RNA (siRNA) targeting USP26 was purchased from Dharmacon. Lentiviral knockdown vectors targeting USP26 were purchased from Transomic. Lentiviral sequences are as follows #1: CAGATTGTTCGAGGTGTGTAA; #2. GAGAGAAACAATTGAAGTTAA; and #3 CCGGCTAAGTGATAATATTCAA. Human GFP USP26 was a kind gift from Rene Bernards. Flag‐tagged USP26 was purchased from MRC Protein phosphorylation and Ubiquitylation unit (83844). Generation of catalytically inactive USP26 mutant (USP15 C/S) was generated by site‐directed mutagenesis as described in Papa et al 25. Flag‐SMAD1, 2, 3, 4, 6, 7 were kind gifts from Joan Seoane. HA‐ubiquitin and respective HA‐tagged ubiquitin mutants were purchased from Addgene 42. Additional cloning information will be given upon request. Antibodies anti‐p‐SMAD2 (3108), anti‐SMAD2 (3103), anti‐tubulin (2128) were from Cell Signaling; anti‐SMAD4 (sc‐7966), anti‐SMAD7 (sc‐7004), anti‐HA (Y‐11), anti‐GFP (sc‐57592), anti‐MYC (sc‐789), and anti‐TβRI [V‐22 (sc‐398); R‐20 (sc‐399)] were from Santa Cruz; anti‐β‐actin (A1978) and anti‐Flag (F7425) were from Sigma. Anti‐USP26 (ab101650) was purchased from Abcam.
Cell culture and transient transfections
HEK293T, MCF7, MDA‐MB‐231, U373, PCTC, A172, T47D, and CAL51 cells were cultured in Dulbecco's modified Eagle's medium (DMEM (Hyclone)) supplemented with 10% fetal calf serum, d‐glutamate, and penicillin/streptomycin (Gibco). HEK293T cells were divided into 10‐cm dishes 1 day prior to transfection. Sub‐confluent cells were transfected using the calcium phosphate transfection method 43. Cells were incubated overnight and washed twice in PBS. Lysates were collected 48–72 h post‐transfection. When appropriate, TGF‐β (100 pM; R&D), SB431542 (5 μM; Tocris) or MG132 (5 μM; Sigma) was added. PCTCs were generated as described previously 3, 21, 44.
Luciferase assays
Luciferase assays were performed using the Dual luciferase system (Promega). CAGA‐luciferase vector (300 ng) was transfected in the presence of CMV‐USP26 (1 μg), CMV‐USP26 C/S (1 μg), or a control vector and CMV‐Renilla (0.25 μg). For loss‐of‐function experiments, CAGA‐luciferase vector (300 ng) and CMV‐Renilla (0.25 μg) were co‐transfected with 1.5 μg of relevant pSuper vector or pSuper USP26 knockdown vectors. After 72 h, 100 pM TGF‐β was added in the presence of DMEM (0% FCS) and luciferase counts were measured 12 h later using a Sirius Luminometer (Berthold).
Biotin labeling of TGF‐β receptor
Cell surface biotinylation experiment was performed using Pierce Cell surface protein isolation kit (catalog no. 89881) according to the manufacturer's instructions. MDA‐MB‐231 cells either control, USP26 shRNA#1, or USP26 shRNA#2 were grown on 15‐cm dishes, labeled with biotin, and lysed. Protein estimation was performed, and equal amount of protein lysates was incubated with streptavidin beads. The beads were boiled in sample buffer with 50 mM DTT and loaded onto an SDS–PAGE gel.
Immunoprecipitation and in vivo deubiquitination assay
For co‐immunoprecipitation experiments, cells were lysed in ELB [0.25 M NaCl, 0.1% NP‐40, 50 mM HEPES (pH 7.3)] supplemented with proteasome inhibitors. Cell lystates (500 μg to 1 mg) were incubated for 2 h to overnight with 2 μg of the indicated antibodies conjugated to protein A or protein G Sepharose beads (GE Healthcare), washed three times in ELB buffer, and separated out on SDS–PAGE gels. When appropriate, cell lysates were immunoprecipitated with ANTI‐Flag M2 affinity resin (Sigma). For in vivo ubiquitination experiments, Flag‐tagged SMAD7 (2 μg) was co‐transfected with HA‐ubiquitin (5 μg) and CMV‐USP26 (5 μg), CMV‐USP26 C/S (5 μg), or a control vector. For loss‐of‐function experiments, Flag‐tagged SMAD7 (5 μg) or Flag‐TRICA (2 μg) was co‐transfected with HA‐ubiquitin (5 μg) and pSuper USP26 (10 μg) or control vector. After 72 h, MG132 (5 μM) was added and incubated overnight, and cells were lysed in ELB buffer.
Wound‐healing and Transwell migration assays
About 2 × 105 MDA‐MB‐231 cells expressing either control vector, or hairpins targeting USP26 were seeded per well in a six‐well plate. The cells were serum‐starved for 24 h before a scratch was produced, and cells were washed with 1× PBS and replenished with 0.5% serum‐containing media together with either 5 μM SB431542 or TGF‐β (5 ng/ml) for 24 h. Images were captured immediately after producing the scratch and at 24 h. A 10 × 10 grid was used to quantify migration efficiency with each square either being scored positive or negative. For transwell migration assay, MDA‐MB‐231 cells were grown in 10‐cm dishes to 80% confluency and serum‐starved for 24 h, and about 50,000 cells were seeded on each transwell migration chamber and treated with either 5 μM SB431542 or 10 ng/ml of TGF‐β for 16 h. The cells were fixed in ice‐cold methanol for 10 min and stained with crystal violet solution. Migrated cells were then visualized through brightfield microscope, and pictures were taken at four random sites and quantified.
Quantitative real‐time PCR
Cells were collected and washed twice in PBS and RNA was isolated using GeneJet RNA extraction kit (Thermo Scientific). qRT–PCR was performed using TaqMan probes (USP26) from Applied Biosystems according to the manufacturer's recommendations. Reactions were carried out on a ABI 7900 or 7500 FAST sequence detector (Perkin Elmer). Relative mRNA values are calculated by the ∆∆C t method. GAPDH or 18S were used as internal normalization controls where specified. The following qRT–PCR primers were used: SMAD7: 5′‐AAA CAG GGG GAA CGA ATT ATC‐3′, 5′‐ACC ACG CAC CAG TGT GAC‐3′; PAI1: 5′‐AAG GCA CCT CTG AGA ACT TCA‐3′, 5′‐CCC AGG ACT AGG CAG GTG‐3′; CDKN1A (p21): 5′‐CCG AAG TCA GTT CCT TGT GG‐3′, 5′‐CAT GGG TTC TGA CGG ACA T‐3′; CTGF: 5′‐CCT GCA GGG TAG AGA AGC AG‐3′, 5′‐TGG AGA TTT TGG GAG TAC GG‐3′; LIF: 5′‐TGC CAA TGC CCT CTT TAT TC‐3′, 5′‐GTC CAG GTT GTT GGG GAA‐3′; and GAPDH: 5′‐AAC AGC GAC ACC CAC TCC TC‐3′, 5′‐CAT ACC AGG AAA TGA GCT TGA C‐3′.
Glioblastoma copy number analysis
TCGA glioblastoma copy number analysis data of USP26 gene are obtained from Broad GDAC website, version 2016_01_28.
Immunohistochemical staining and evaluation
Immunohistochemistry (IHC) staining on the respective formalin‐fixed, paraffin‐embedded tissue sections was performed using the Leica BOND‐MAX and Ventana Benchmark XT autostainers according to the conditions stated in Table 1. Tissue sections underwent automated deparaffinization followed by incubation with their optimized antigen retrieval solutions. Slides were then incubated with antibody as indicated in Table 1. Detection of antibody staining was carried out according to the manufacturer's protocol for the detection kits used with an extension of hematoxylin counterstain extended to 10 min to ensure for a defined stain. Slides were rinsed with deionized water followed by manual mounting of coverslips. Positive and negative controls were included in each run, consisting of tissue with known expression and tissue stained without primary antibody, respectively. Quantification was assessed double‐blind by a trained pathologist (BP) and expressed as an H‐score. The H‐score was determined by the formula 3 × percentage of strongly staining cells, 2 × percentage of moderately staining cells, and 1 × percentage of weak staining cells, giving a range of 0–300 for the H‐score.
Table 1.
Immunohistochemistry protocols for USP26 and pSMAD2
| Antibody | Manufacturer | Autostainer | Dilution | Antigen retrieval | Block (min) | Antibody incubation | Detection kit used |
|---|---|---|---|---|---|---|---|
| USP26 (ab101650) | Abcam | BOND‐MAX | 1/50 | pH 9, 20 min | None | 15 min | Bond Polymer Refine Red Detection Kit |
| pSMAD2 (3108) | Cell Signaling | BOND‐MAX | 1/100 | pH 9, 20 min | None | 15 min | Bond Polymer Refine Red Detection Kit |
Immunohistochemistry protocols for USP26 and pSMAD2 detection in glioblastoma samples.
Author contributions
SKLL, PVI, PJ, and ZFBAI performed the experiments. BP participated in data analysis. TZT performed the bioinformatics analysis. PJAE conceived the project, interpreted the results, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
This work was in part supported by the by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative, Koninklijke Philips N.V, and the Ministry of Education Academic Research Fund Tier 1 grants (T1‐2013 Sep‐10) and (T1‐2014 Oct‐08).
EMBO Reports (2017) 18: 797–808
References
- 1. Seoane J (2008) The TGFBeta pathway as a therapeutic target in cancer. Clin Transl Oncol 10: 14–19 [DOI] [PubMed] [Google Scholar]
- 2. Rodon J (2015) First‐in‐human dose study of the novel transforming growth factor‐β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res 21: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J et al (2007) High TGFbeta‐Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF‐B gene. Cancer Cell 11: 147–160 [DOI] [PubMed] [Google Scholar]
- 4. Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19: 2783–2810 [DOI] [PubMed] [Google Scholar]
- 5. Massague J (2008) TGFbeta in cancer. Cell 134: 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iyengar PV, Eichhorn PJ (2013) (De)‐Ubiquitination in the TGF‐B pathway. Cancer Res Ther Oncol 1: 1–6 [Google Scholar]
- 7. Yan X, Liu Z, Chen Y (2009) Regulation of TGF‐beta signaling by Smad7. Acta Biochim Biophys Sin 41: 263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6: 1365–1375 [DOI] [PubMed] [Google Scholar]
- 9. Ogunjimi AA, Briant DJ, Pece‐Barbara N, Le Roy C, Di Guglielmo GM, Kavsak P, Rasmussen RK, Seet BT, Sicheri F, Wrana JL (2005) Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell 19: 297–308 [DOI] [PubMed] [Google Scholar]
- 10. Komuro A, Imamura T, Saitoh M, Yoshida Y, Yamori T, Miyazono K, Miyazawa K (2004) Negative regulation of transforming growth factor‐beta (TGF‐beta) signaling by WW domain‐containing protein 1 (WWP1). Oncogene 23: 6914–6923 [DOI] [PubMed] [Google Scholar]
- 11. Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K, Imamura T (2005) NEDD4‐2 (neural precursor cell expressed, developmentally down‐regulated 4‐2) negatively regulates TGF‐beta (transforming growth factor‐beta) signalling by inducing ubiquitin‐mediated degradation of Smad2 and TGF‐beta type I receptor. Biochem J 386: 461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aragon E, Goerner N, Xi Q, Gomes T, Gao S, Massague J, Macias MJ (2012) Structural basis for the versatile interactions of Smad7 with regulator WW domains in TGF‐beta Pathways. Structure 20: 1726–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park SH, Jung EH, Kim GY, Kim BC, Lim JH, Woo CH (2015) Itch E3 ubiquitin ligase positively regulates TGF‐beta signaling to EMT via Smad7 ubiquitination. Mol Cells 38: 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koinuma D, Shinozaki M, Komuro A, Goto K, Saitoh M, Hanyu A, Ebina M, Nukiwa T, Miyazawa K, Imamura T et al (2003) Arkadia amplifies TGF‐beta superfamily signalling through degradation of Smad7. EMBO J 22: 6458–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim BC, Lee HJ, Park SH, Lee SR, Karpova TS, McNally JG, Felici A, Lee DK, Kim SJ (2004) Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor beta signaling by binding to Smad7 and promoting its degradation. Mol Cell Biol 24: 2251–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81: 203–229 [DOI] [PubMed] [Google Scholar]
- 17. Nijman SM, Luna‐Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123: 773–786 [DOI] [PubMed] [Google Scholar]
- 18. Komander D, Clague MJ, Urbe S (2009) Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10: 550–563 [DOI] [PubMed] [Google Scholar]
- 19. Zhang L, Zhou F, Drabsch Y, Gao R, Snaar‐Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX et al (2012) USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF‐beta type I receptor. Nat Cell Biol 14: 717–726 [DOI] [PubMed] [Google Scholar]
- 20. Al‐Salihi MA, Herhaus L, Macartney T, Sapkota GP (2012) USP11 augments TGFbeta signalling by deubiquitylating ALK5. Open Biol 2: 120063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eichhorn PJ, Rodon L, Gonzalez‐Junca A, Dirac A, Gili M, Martinez‐Saez E, Aura C, Barba I, Peg V, Prat A et al (2012) USP15 stabilizes TGF‐beta receptor I and promotes oncogenesis through the activation of TGF‐beta signaling in glioblastoma. Nat Med 18: 429–435 [DOI] [PubMed] [Google Scholar]
- 22. Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A (2005) The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF‐beta signalling. Oncogene 24: 8080–8084 [DOI] [PubMed] [Google Scholar]
- 23. Iyengar PV, Jaynes P, Rodon L, Lama D, Law KP, Lim YP, Verma C, Seoane J, Eichhorn PJ (2015) USP15 regulates SMURF2 kinetics through C‐lobe mediated deubiquitination. Sci Rep 5: 14733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dirac AM, Bernards R (2010) The deubiquitinating enzyme USP26 is a regulator of androgen receptor signaling. Mol Cancer Res 8: 844–854 [DOI] [PubMed] [Google Scholar]
- 25. Papa FR, Hochstrasser M (1993) The yeast DOA4 gene encodes a deubiquitinating enzyme related to a product of the human tre‐2 oncogene. Nature 366: 313–319 [DOI] [PubMed] [Google Scholar]
- 26. Gronroos E, Hellman U, Heldin CH, Ericsson J (2002) Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 10: 483–493 [DOI] [PubMed] [Google Scholar]
- 27. Teicher BA (2001) Malignant cells, directors of the malignant process: role of transforming growth factor‐beta. Cancer Metastasis Rev 20: 133–143 [DOI] [PubMed] [Google Scholar]
- 28. Penuelas S, Anido J, Prieto‐Sanchez RM, Folch G, Barba I, Cuartas I, Garcia‐Dorado D, Poca MA, Sahuquillo J, Baselga J et al (2009) TGF‐beta increases glioma‐initiating cell self‐renewal through the induction of LIF in human glioblastoma. Cancer Cell 15: 315–327 [DOI] [PubMed] [Google Scholar]
- 29. Broad Institute TCGA Genome Data Analysis Center (2016) SNP6 Copy number analysis (GISTIC2). The Reference Broad Institute of MIT Harvard. doi:10.7908/C1HD7V19 [Google Scholar]
- 30. Neuzillet C, Tijeras‐Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A (2015) Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther 147: 22–31 [DOI] [PubMed] [Google Scholar]
- 31. Simonsson M, Heldin CH, Ericsson J, Gronroos E (2005) The balance between acetylation and deacetylation controls Smad7 stability. J Biol Chem 280: 21797–21803 [DOI] [PubMed] [Google Scholar]
- 32. Wiesner S, Ogunjimi AA, Wang HR, Rotin D, Sicheri F, Wrana JL, Forman‐Kay JD (2007) Autoinhibition of the HECT‐type ubiquitin ligase Smurf2 through its C2 domain. Cell 130: 651–662 [DOI] [PubMed] [Google Scholar]
- 33. Maspero E, Valentini E, Mari S, Cecatiello V, Soffientini P, Pasqualato S, Polo S (2013) Structure of a ubiquitin‐loaded HECT ligase reveals the molecular basis for catalytic priming. Nat Struct Mol Biol 20: 696–701 [DOI] [PubMed] [Google Scholar]
- 34. Wosnitzer MS, Mielnik A, Dabaja A, Robinson B, Schlegel PN, Paduch DA (2014) Ubiquitin Specific Protease 26 (USP26) expression analysis in human testicular and extragonadal tissues indicates diverse action of USP26 in cell differentiation and tumorigenesis. PLoS One 9: e98638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ribarski I, Lehavi O, Yogev L, Hauser R, Bar‐Shira Maymon B, Botchan A, Paz G, Yavetz H, Kleiman SE (2009) USP26 gene variations in fertile and infertile men. Hum Reprod 24: 477–484 [DOI] [PubMed] [Google Scholar]
- 36. Chipuk JE, Cornelius SC, Pultz NJ, Jorgensen JS, Bonham MJ, Kim SJ, Danielpour D (2002) The androgen receptor represses transforming growth factor‐beta signaling through interaction with Smad3. J Biol Chem 277: 1240–1248 [DOI] [PubMed] [Google Scholar]
- 37. Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, Enzo E, Moro S, Polo S, Dupont S et al (2011) USP15 is a deubiquitylating enzyme for receptor‐activated SMADs. Nat Cell Biol 13: 1368–1375 [DOI] [PubMed] [Google Scholar]
- 38. Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L et al (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 136: 123–135 [DOI] [PubMed] [Google Scholar]
- 39. Xie Y, Avello M, Schirle M, McWhinnie E, Feng Y, Bric‐Furlong E, Wilson C, Nathans R, Zhang J, Kirschner MW et al (2013) Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity‐dependent self‐degradation. J Biol Chem 288: 2976–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Typas D, Luijsterburg MS, Wiegant WW, Diakatou M, Helfricht A, Thijssen PE, van de Broek B, Mullenders LH, van Attikum H (2015) The de‐ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Res 43: 6919–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R (2003) Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF‐kappaB. Nature 424: 797–801 [DOI] [PubMed] [Google Scholar]
- 42. Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA et al (2005) Parkin mediates nonclassical, proteasomal‐independent ubiquitination of synphilin‐1: implications for Lewy body formation. J Neurosci 25: 2002–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van der Eb AJ, Graham FL (1980) Assay of transforming activity of tumor virus DNA. Methods Enzymol 65: 826–839 [DOI] [PubMed] [Google Scholar]
- 44. Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, Modrusan Z, Meissner H, Westphal M, Lamszus K (2008) Glioblastoma‐derived stem cell‐enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 27: 2897–2909 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4