

Abstract
Deconjugation of the Atg8/LC3 protein family members from phosphatidylethanolamine (PE) by Atg4 proteases is essential for autophagy progression, but how this event is regulated remains to be understood. Here, we show that yeast Atg4 is recruited onto autophagosomal membranes by direct binding to Atg8 via two evolutionarily conserved Atg8 recognition sites, a classical LC3‐interacting region (LIR) at the C‐terminus of the protein and a novel motif at the N‐terminus. Although both sites are important for Atg4–Atg8 interaction in vivo, only the new N‐terminal motif, close to the catalytic center, plays a key role in Atg4 recruitment to autophagosomal membranes and specific Atg8 deconjugation. We thus propose a model where Atg4 activity on autophagosomal membranes depends on the cooperative action of at least two sites within Atg4, in which one functions as a constitutive Atg8 binding module, while the other has a preference toward PE‐bound Atg8.
Keywords: autophagosome, autophagy, deconjugation, LC3, phagophore assembly site
Subject Categories: Autophagy & Cell Death; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Autophagy is a highly conserved catabolic process that under stress conditions, such as nutrient starvation, allows the cell to degrade part of its content in a regulated manner in order to maintain cell homeostasis 1. It also allows the turnover of a large number of unwanted structures including defective proteins, dysfunctional organelles, and invading pathogens 2. As a result, impairments or defects in autophagy lead to different human diseases and disorders such as cancer and neurodegenerative disorders 3, 4.
Autophagy is characterized by the sequestration of structures targeted for destruction into autophagosomes, which fuse with lysosomes/vacuoles to expose their cargo to the degradative activity of the hydrolases present in their lumen 5, 6. Autophagosomes are formed by nucleation and subsequent expansion of a cistern known as the phagophore or isolation membrane. This event takes place at the so‐called phagophore assembly site or pre‐autophagosomal structure (PAS), as the result of the orchestrated action of the Atg proteins 5, 6.
One central component of the conserved core Atg machinery is the Atg8/LC3 ubiquitin‐like protein family with the sole yeast member Atg8 and seven mammalian orthologues 7. These molecules have an important role in the phagophore expansion and closure 8, 9, 10, 11. Atg8/LC3 associates to the outer and inner membrane of the phagophore 12. On the outer surface, Atg8/LC3 proteins appear to form a coat around the autophagosome 13. Within the inner surface, they play an important role in selecting cargoes targeted for degradation by binding them through specific receptors 14. The association of Atg8/LC3 proteins to autophagosomal membranes depends on their conjugation with phosphatidylethanolamine (PE), a molecular process that was initially and thoroughly characterized in yeast 15. The formation of Atg8‐PE requires the constitutive post‐translational priming of Atg8, which involves its C‐terminal processing by the Atg4 protease to expose a glycine residue 16, 17. Upon autophagy induction, the sequential action of the E1‐like enzyme Atg7 and the E2‐like enzyme Atg3 together with the Atg12–Atg5–Atg16 complex catalyzes the formation of an amide bond between the carboxyl group of the exposed glycine in Atg8 and the amino group of PE 18, 19. Once the autophagosome is completed, the Atg machinery, including Atg8, is released from its surface and recycled. As for the priming event, deconjugation of Atg8 from PE also relies on the catalytic activity of Atg4 13, 17. Atg4 also releases error‐prone conjugated Atg8 from various cellular membranes, which was shown to be essential for normal autophagy progression in yeast 20, 21. Similarly, human ATG4B and ATG4D activities are important for autophagosome fusion with degradative compartments in human erythroblasts during differentiation 22. Moreover, overexpression of human ATG4B reduces autophagy levels 23, 24. Altogether, these observations suggest that the regulation of Atg4 activity is likely to be an essential step for the control of autophagy, but the mechanism underlying this event is far from being understood.
Members of the Atg4 protein family have been exclusively detected in the cytoplasm, with the exception of yeast Atg4, which also localizes to the nucleus under autophagy‐inducing conditions 21, 25, 26. Surprisingly, Atg4 proteins have not been detected on forming and/or complete autophagosomes. As a result, the observed localization of Atg4 and its action on other cellular membranes has led to the hypothesis that the Atg8‐PE pool on forming autophagosomes is maintained by either an unbalanced Atg8‐PE conjugation/deconjugation kinetics and/or the presence of factor protecting Atg8‐PE from Atg4 activity 21, 27. In this study, we demonstrate that yeast Atg4 can be recruited to autophagosomal membranes during autophagy. This is mediated by its binding to Atg8 via conserved Atg8 recognition sites, which in turn play a role in the efficient deconjugation and release of Atg8 from lipid bilayers. Our results thus reveal the existence of a mechanism that regulates the association of Atg4 onto autophagosomal membranes that is essential for normal autophagy progression.
Results
Atg4 associates with the PAS
In order to better understand how Atg4 is regulated, we analyzed its subcellular localization. Under autophagy‐inducing conditions, endogenous Atg4‐GFP was mainly found in the cytoplasm and in the nucleus, but also rarely in punctate structures corresponding to the PAS highlighted with the specific marker proteins RFP‐Ape1 and mCherry‐Atg8 (Fig 1A–C). This observation is largely in agreement with a previous report 21, in which it was concluded that Atg4 can act on all intracellular membranes because of its dispersed localization. The sporadic localization of Atg4 at the PAS, however, could also be explained by this being a transient event that takes place at a precise time interval during autophagosome biogenesis. Indeed, time‐lapse live‐cell imaging showed that Atg4‐GFP is always recruited when or after the formation of mCherry‐Atg8‐positive PAS, and leaves this structure before its disappearance, what is probably when the fusion of the complete autophagosome with the vacuole happens (Fig EV1 and Movie EV1). In order to modulate such dynamic association and possibly detect a more pronounced population of Atg4 at the PAS, we decided to inspect the localization of this protease in cells lacking Atg1, one of the core Atg proteins to stall autophagosome formation at an early step 28. As shown in Fig 1A–C, Atg4‐GFP puncta formation and its colocalization with the PAS marker proteins RFP‐Ape1 and mCherry‐Atg8 significantly increased in atg1∆ cells. Altogether, these results show that Atg4 is recruited to the PAS.
Figure 1. Atg4 localizes to the PAS during autophagy.
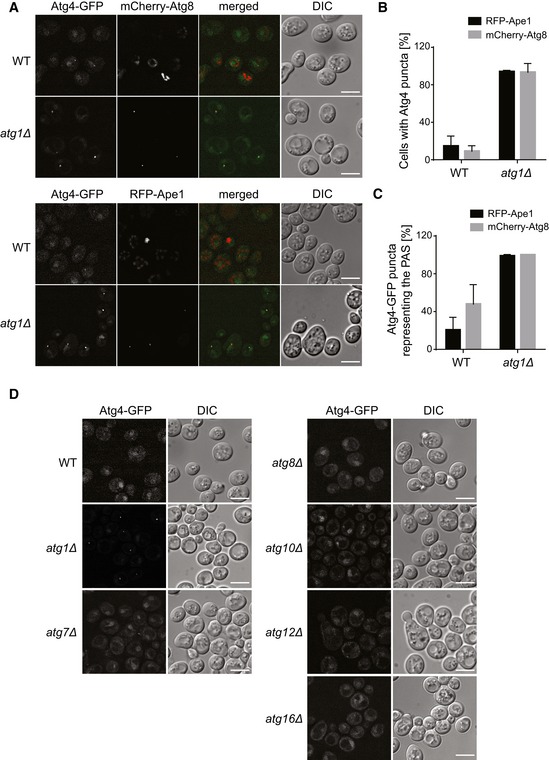
- Subcellular distribution of Atg4‐GFP under autophagy‐inducing conditions in WT and atg1∆ strains expressing the PAS marker proteins RFP‐Ape1 (SAY071 and SAY020) and mCherry‐Atg8 (MNY006 and SAY010 transformed with pCumCherryV5Atg8). DIC, differential interference contrast. Scale bars, 5 μm.
- Percentage of cells in which Atg4‐GFP is observed in a punctate structure in the experiments depicted in panel (A). Data represent the average of three independent experiments ± standard deviation (SD).
- Percentage of Atg4 puncta co‐localizing with the PAS marker proteins Atg8 and Ape1 in the experiments shown in panel (A). Data represent the average of three independent experiments ± SD.
- Localization of Atg4‐GFP in WT (MNY006), atg1∆ (SAY10), atg7∆ (SAY014), atg8∆ (SAY015), atg10∆ (SAY059), atg12∆ (SAY029), and atg16∆ (SAY064) mutant strains, under starvation. Cells were analyzed by fluorescence microscopy as in panel (A). DIC, differential interference contrast. Scale bars, 5 μm.
Figure EV1. Analysis of Atg4 association with the PAS by time‐lapse live‐cell imaging.
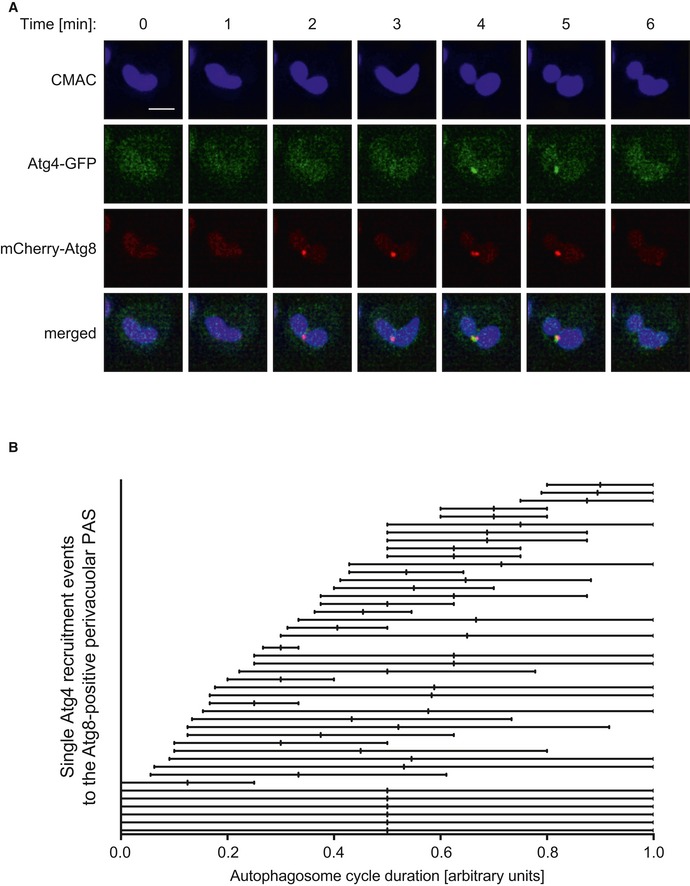
- Cells expressing Atg4‐GFP and mCherry‐V5‐Atg8 (RGY287) were starved in SD‐N medium for 30 min and incubated with CMAC for 10 min, before being examined by time‐lapse fluorescence microscopy. PAS were considered mCherry‐Atg8‐positive puncta adjacent to the vacuole, stained with CMAC. Images of the same cells were collected every 1 min for 15 min. For the complete movie, see the supplemental data (Movie EV1). Scale bar, 2 μm.
- Experiments as in panel (A) were quantified by normalizing the autophagosome cycle, defined as the interval of time form the appearance until disappearance of mCherry‐Atg8 puncta 49, to 1 and integrating Atg4‐GFP recruitment to the PAS overtime. Data are from four independent experiments where the PAS remained in the imaged focal planes over the course of the entire filming.
Atg8 is required for Atg4 association with the PAS
To determine how the recruitment of Atg4 to the PAS is mediated, we investigated the localization of this protein in cells lacking various Atg proteins belonging to different functional clusters. In absence of components that are part of the Atg1 complex (Atg13), the Atg9 trafficking system (Atg2, Atg9, and Atg18) and the phosphatidylinositol 3‐kinase complex (Atg6 and Atg14), Atg4‐GFP was detected in single puncta but the percentage of cells displaying this profile varied between the different knockout strains (Fig EV2A and B). In contrast, no Atg4‐GFP‐positive punctate structures were observed and Atg4 was exclusively nuclear and cytoplasmic, when proteins of the two ubiquitin‐like conjugation systems (Atg7, Atg8, Atg10, Atg12, and Atg16) that link Atg8 to PE were knocked out (Fig 1D). This result indicates that Atg8‐PE is very likely the factor involved in Atg4 recruitment to the PAS. To further confirm that this is indeed the case, we deleted components of the two ubiquitin‐like conjugation systems (i.e., Atg3, Atg7, Atg8, Atg10, Atg12, and Atg16) in the atg1∆ strain, in which we had observed the most pronounced association of Atg4 to the PAS (Fig 1A–C). In the resulting double knockout strains, Atg4‐GFP was not distributed in punctate structures, but instead it was exclusively cytosolic and nuclear (Fig EV2C), reinforcing the notion of a key role of Atg8‐PE in Atg4 recruitment to the PAS.
Figure EV2. Atg4 association with the PAS does not require components of the Atg1 complex, Atg9 cycling system and PI3K complex.
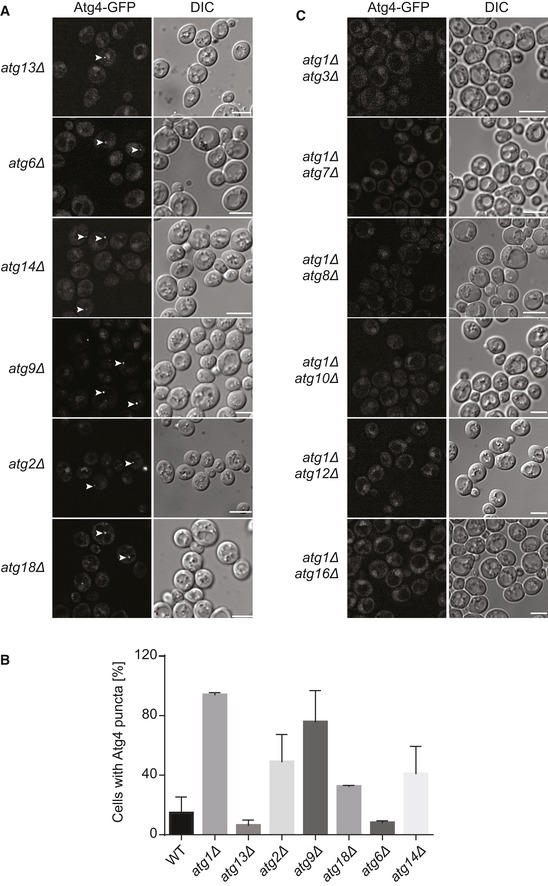
- Fluorescence microscopy images showing the subcellular localization of Atg4‐GFP in atg13∆ (SAY030), atg2∆ (SAY109), atg9∆ (SAY016), atg18∆ (SAY017), atg6∆ (SAY013), and atg14∆ (SAY110) strains analyzed as in Fig 1. White arrows highlight Atg4‐GFP puncta. DIC, differential interference contrast. Scale bars, 5 μm.
- Percentage of cells, in the experiments shown in panel (A), that display an Atg4‐GFP punctate structure. Data represent the average of three independent experiments ± SD.
- Subcellular localization of Atg4‐GFP in double knockout cells lacking Atg1 and components of the conjugation systems leading to the formation of Atg8‐PE: atg7∆ (SAY032), atg3∆ (SAY031), atg8∆ (SAY033), atg10∆ (SAY035), atg12∆ (SAY134), and atg16∆ (SAY135). Cells were grown and imaged as in Fig 1. DIC, differential interference contrast. Scale bars, 5 μm.
Atg4 contains several putative LIR motifs
Since Atg8 appeared to be responsible for the Atg4 association with the PAS, we investigated whether this event requires binding between these two proteins. It has been reported that recombinant Atg4 and Atg8 bind directly in vitro 29. To determine whether this interaction also takes place in vivo, as shown for ATG4B in mammalian cells 24, TAP‐tagged Atg4 was used to immunoprecipitate GFP‐Atg8 from cell lysates. As shown in Fig 2A, Atg4‐TAP was able to specifically pull down Atg8, showing that these two proteins interact in vivo as well.
Figure 2. Atg4 LIR motif at amino acid position F102 to I105 is essential for autophagy.
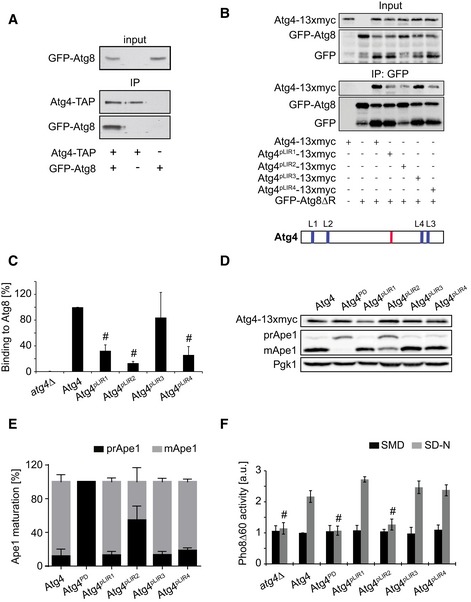
- Atg4‐TAP atg8∆ (yMS69) or atg8∆ (yCK765) strains carrying an empty plasmid (pRS416) or one expressing GFP‐Atg8 (pCK15) were grown to a log phase and exposed to 220 nM rapamycin for 1 h before preparing cell extracts. Atg4‐TAP was subsequently immunoprecipitated using IgG magnetic beads. Finally, immunoprecipitates were analyzed by Western blot for GFP and protein A.
- The atg4Δ (SAY084) or the atg4Δ (JAY151) strains carrying the integrative GFP‐ATG8ΔR plasmid were transformed with the centromeric plasmids expressing either Atg4‐13xmyc, Atg4pLIR1‐13xmyc, Atg4pLIR2‐13xmyc, Atg4pLIR3‐13xmyc, or Atg4pLIR4‐13xmyc. The strains were exponentially grown before being nitrogen starved in SD‐N medium for 1 h. Cell lysates were then subjected to pull‐down experiments using GFP‐trap agarose beads. Isolated proteins, 1% of cell lysate (input) or 50% of the pull‐down material (IP: GFP), were resolved by SDS–PAGE and analyzed by Western blot using either anti‐myc or anti‐GFP antibodies. A schematic view of the distribution of the putative LIR motif (blue), that is, LIR1 (L1), LIR2 (L2), LIR3 (L3), and LIR4 (L4), and the catalytic site (red) over within Atg4 is presented on the bottom of the panel.
- Quantification of the experiments shown in panel (B). Values are relative to WT Atg4 and represent the average of three independent experiments ± SD. Significant differences (P < 0.05) between cells expressing WT Atg4 were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the # symbol.
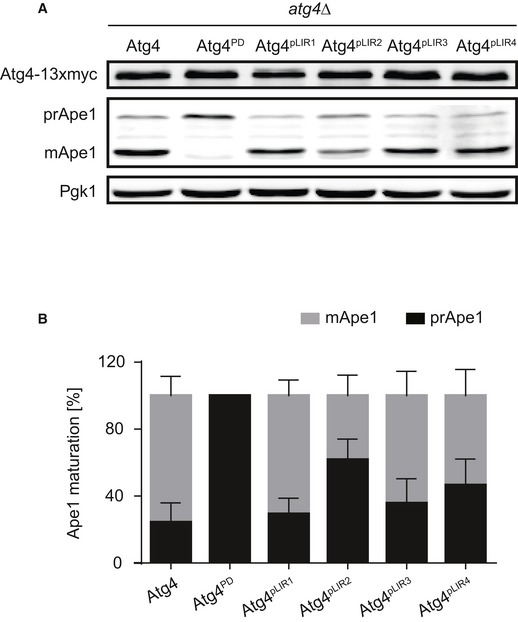
- The atg4Δ (SAY084) mutant was transformed with integrative vectors expressing 13xmyc‐tagged Atg4 (SAY173) or its mutant versions (Atg4PD, SAY174; Atg4pLIR1, SAY175; Atg4pLIR2, SAY176; Atg4pLIR3, SAY177; and Atg4pLIR4, SAY178). The resulting strains were grown to a log phase in SMD medium before being nitrogen starved in SD‐N medium for 3 h. Proteins were precipitated with 10% trichloroacetic acid (TCA) and analyzed by Western blot using the anti‐myc, anti‐Ape1, and anti‐Pgk1 antibodies (loading control).
- The percentages of prApe1 and mApe1 in the experiment shown in panel (D) were quantified, and values were plotted. Data represent the average of five independent experiments ± SD.
- The experiment described in panel (D) was repeated with the SAY130 strain (Pho8∆60 pho13∆ atg4∆) carrying an empty pRS416 vector (atg4∆) or plasmids expressing Atg4, Atg4PD, Atg4pLIR1, Atg4pLIR2, Atg4pLIR3, and Atg4pLIR4. Pho8∆60 activity was subsequently measured before (SMD) or after (SD‐N) the nitrogen starvation and expressed in arbitrary units (a.u.). Data represent the average of three independent experiments ± SD. Significant differences (P < 0.05) between cells expressing WT Atg4 were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the # symbol.
Most proteins known to bind Atg8 possess a so‐called LC3‐interacting region (LIR) or Atg8‐interacting motif characterized by a consensus amino acid sequence W/F/Y‐x‐x‐L/I/V (where x is any amino acid), which is often preceded by negatively charged amino acids 14, 30. Scrutiny of the amino acid sequence of Saccharomyces cerevisiae Atg4 revealed the presence of four putative LIR (pLIR) motifs, two of which are evolutionarily conserved, amino acids 102–105 (pLIR2) and 424–427 (pLIR4), and two being yeast‐specific, amino acids 36–39 (pLIR1) and 446–449 (pLIR3) (Fig EV3A and B). To determine which of these sequences might be a functional LIR motif essential for Atg4 recruitment to the PAS, we changed the key amino acids at position 1 and 4 (i.e., W/F/Y and L/I/V) of each putative LIR motif into alanines creating four Atg4 point mutants, namely Atg4pLIR1, Atg4pLIR2, Atg4pLIR3, and Atg4pLIR4. We then expressed the 13xmyc‐tagged Atg4 pLIR mutants in an atg4∆ strain carrying GFP‐Atg8∆R, a form of Atg8 already primed to exclude potential influences of a non‐identical first cleavage by the Atg4 variants, before generating cell extracts and immunoprecipitating GFP‐Atg8. As shown in Fig 2B and C, GFP‐Atg8∆R specifically pulled down wild‐type (WT) Atg4‐13xmyc and Atg4pLIR3‐13xmyc under starvation conditions. This co‐isolation, however, was strongly impaired in cells expressing Atg4pLIR2‐13xmyc but also in those carrying AtgpLIR1‐13xmyc and Atg4pLIR4‐13xmyc, indicating that the mutated domains in these constructs play a role in the interaction between Atg4 and Atg8 in vivo.
Figure EV3. Putative LIR (pLIR) motifs in S. cerevisiae Atg4 and their conservation among eukaryotes.
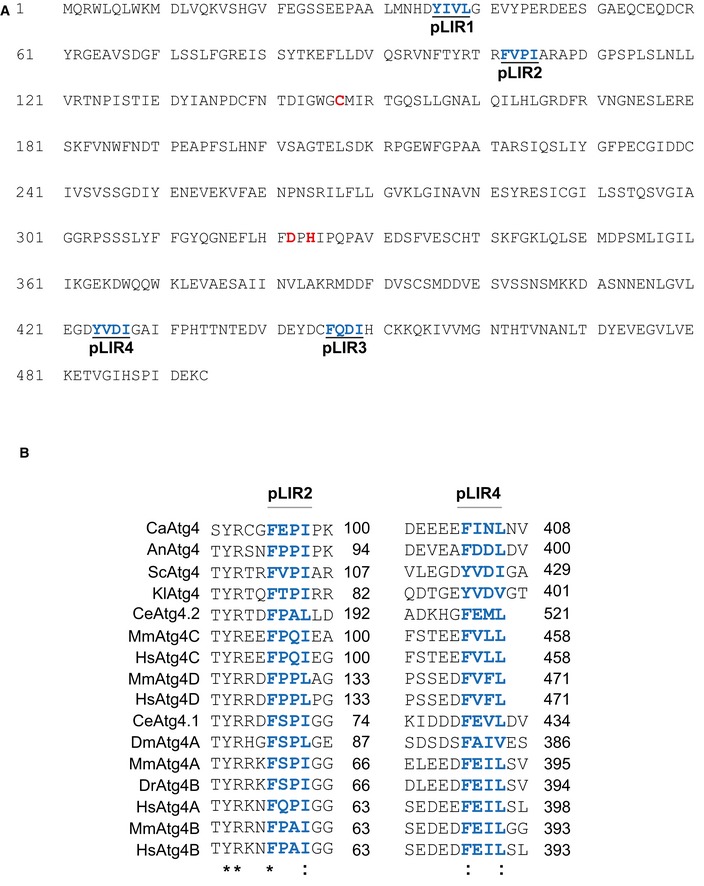
- Saccharomyces cerevisiae Atg4 amino acid sequence. The catalytic site (C147, D322, and H324) is highlighted in red, while the putative LIR motifs are indicated in blue.
- The amino acid sequence of the regions flanking the two conserved pLIR motifs of S. cerevisiae Atg4 (in blue), that is, F102 to I105 (pLIR2) and Y424 to I427 (pLIR4), were aligned with that of homologous proteins from different species using the Kalign alignment tool (http://www.ebi.ac.uk/Tools/msa/kalign/). UniprotKB accession numbers are C. albicans Atg4 (Q59UG3), A. nidulans Atg4 (Q5B7L0), S. cerevisiae Atg4 (P53867), K. lactis Atg4 (Q6CQ60), C. elegans Atg4.1 (Q9NA30), C. elegans Atg4.2 (Q9U1N6), D. melanogaster Atg4 (M9PBM3), D. rerio Atg4B (Q6DG88), M. musculus ATG4A (Q8C9S8), M. musculus ATG4B (Q8BGE6), M. musculus ATG4C (Q811C2), M. musculus ATG4D (Q8BGV9), H. sapiens ATG4A (Q8WYN0), H. sapiens ATG4B (Q9Y4P1), H. sapiens ATG4C (Q96DT6), and H. sapiens ATG4D (Q86TL0). The asterisk indicates conservation of the residue while two dots designate similarity.
The pLIR2 motif in Atg4 is essential for autophagy
We next investigated whether the mutations in the putative LIR motifs, causing the observed reduction in Atg8 association with Atg4, have any effect on autophagy. Therefore, the 13xmyc‐tagged Atg4 pLIR variants were expressed in cells lacking the endogenous ATG4 gene before measuring both the cytoplasm‐to‐vacuole targeting (Cvt) pathway progression, by assessment of Ape1 maturation 31, and autophagy, using the Pho8∆60 assay 32. Although cells expressing Atg4pLIR1, Atg4pLIR3, and Atg4pLIR4 did not display an evident defect in neither the Cvt pathway nor bulk autophagy, the strain carrying the Atg4pLIR2 mutant revealed an impairment in both pathways (Figs 2D–F and EV4). As a positive control, we used the catalytically inactive protease‐dead Atg4 (Atg4PD) mutant in which the key cysteine in the active site is mutated into a serine, making Atg4 unable to process Atg8 17. Importantly, Western blot analysis of protein extracts with an anti‐myc antibody showed that cellular levels of Atg4pLIR2 were identical to those of WT Atg4 demonstrating that the defects of Atg4pLIR2‐expressing cells were not due to a protein instability caused by the introduced mutations (Figs 2D and EV4A). We concluded that the conserved region of Atg4 comprised between amino acids 102 and 105 plays an important role in autophagy.
Figure EV4. Atg4 LIR2 motif is essential for the Cvt pathway.
- The strains described in Fig 2C were grown to an exponential log phase before proteins were precipitated with TCA and analyzed by Western blotting with anti‐myc, anti‐Ape1, and anti‐Pgk1 antibodies.
- The percentage of prApe1 and mApe1 in the experiment shown in panel (A) were quantified, and values were plotted. Data represent the average of five independent experiments ± SD.
The Atg4 pLIR2 plays a major role in Atg8‐PE deconjugation
Next, we explored whether the autophagy defect in cells expressing Atg4pLIR2 is caused by an impairment of either Atg4 dependent proteolytic priming of Atg8 in the cytosol or the deconjugation of Atg8‐PE from autophagosomal membranes.
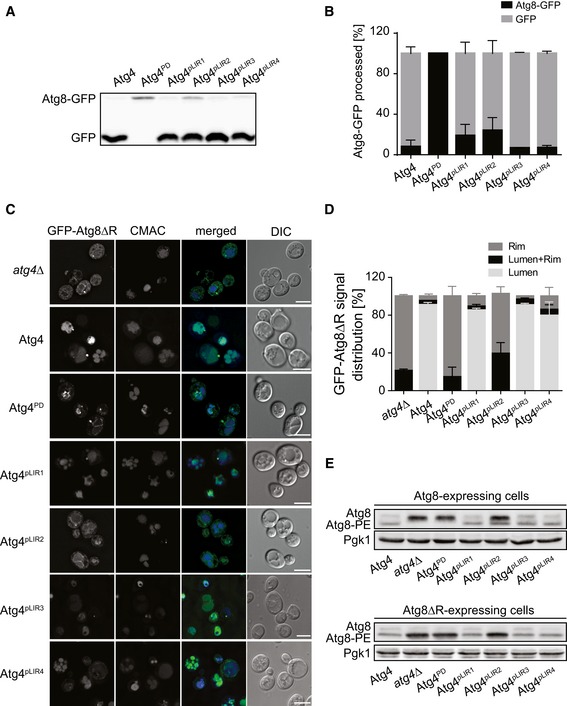
First, we assessed the initial post‐translational C‐terminal cleavage of Atg8 by Atg4 using the Atg8‐GFP chimera 16 under autophagy‐inducing conditions. As expected, Atg8‐GFP was effectively processed in atg4∆ cells expressing WT Atg4 whereas it remained intact in those carrying Atg4PD (Fig 3A and B). Importantly, all the Atg4 mutant proteins had normal proteolytic cleavage of Atg8‐GFP except Atg4pLIR2, which displayed a very slight defect.
Figure 3. Atg4 pLIR2 motif is essential for Atg8‐PE deconjugation.
- The atg4Δ Atg8‐GFP mutant (SAY113) was transformed with integration vectors expressing 13xmyc‐tagged Atg4 (SAY173) or its mutant variants (Atg4PD, SAY174; Atg4pLIR1, SAY175; Atg4pLIR2, SAY176; Atg4pLIR3, SAY177; and Atg4pLIR4, SAY178). Proteins were TCA‐precipitated and analyzed by Western blot using the anti‐GFP antibody.
- The percentages of Atg8‐GFP processed in the experiment shown in panel (A) were quantified and values were plotted. Data represent the average of three independent experiments ± SD.
- The atg4Δ strain carrying the integration plasmid pCuGFPAtg8ΔR(305) (JSY151) and an empty vector (atg4Δ) or plasmids expressing 13xmyc‐tagged Atg4 variants (Atg4, Atg4PD, Atg4pLIR1, Atg4pLIR2, Atg4pLIR3, and Atg4pLIR4) were grown in SMD, labeled with the vacuole‐specific dye CMAC, nitrogen starved in SD‐N for 3 h, and imaged. DIC, differential interference contrast. Scale bars, 5 μm.
- Quantification of GFP‐Atg8 distribution in cells imaged in panel (C): vacuole lumen, vacuole rim, or both localizations (lumen + rim). Data represent the average of three independent experiments ± SD.
- The atg4∆ (SAY084, top) and atg4∆ atg8∆ ATG8∆R (RHY012, bottom) mutants were transformed with an empty vector or plasmids expressing the 13xmyc‐tagged Atg4 variants (Atg4, Atg4PD, Atg4pLIR1, Atg4pLIR2, Atg4pLIR3, and Atg4pLIR4), and the resulting strains were grown to a log phase in SMD medium before being nitrogen starved in SD‐N medium for 3 h. Proteins were precipitated with TCA and analyzed by Western blot using the anti‐Atg8 and anti‐Pgk1 antibodies.
Subsequently, we specifically analyzed the Atg8‐PE deconjugating activity of the different Atg4 mutants in vivo by examining the distribution of GFP‐Atg8∆R in an atg4∆ background 20, 21. As reported, PE‐anchored GFP‐Atg8 failed to be released from the surface of autophagosomes before fusion with the vacuole in atg4∆ cells carrying either an empty vector or Atg4PD, and thus, the fluorescence signal mainly localized to the vacuolar limiting membrane (Fig 3C and D) 20, 21. Complementation of the atg4∆ mutant with Atg4, Atg4pLIR1, Atg4pLIR3, or Atg4pLIR4 led to normal GFP‐Atg8 recycling and delivery into the vacuolar lumen 20, 21. Crucially, Atg4pLIR2‐expressing atg4∆ cells showed a GFP‐Atg8∆R distribution pattern identical to the one of the strain carrying either an empty plasmid or Atg4PD, revealing a defect of Atg4pLIR2 in cleaving Atg8 from its PE anchor (Fig 3C and D). The fluorescence microscopy observations were confirmed by Western blot analysis after separation of free Atg8 from its PE‐conjugated form in either Atg8 or Atg8∆R expressing cells, which revealed a marked accumulation of Atg8‐PE in atg4∆ cells expressing Atg4pLIR2 (Fig 3E).
We wondered whether the impairment of recycling Atg8 from its lipid bound form seen in the pLIR2 mutant variant is a result of its inefficient recruitment to the PAS. GFP‐tagged Atg4, Atg4pLIR1, Atg4pLIR2, Atg4pLIR3, and Atg4pLIR4 were expressed in an atg1∆ mutant background, where endogenous Atg4‐GFP was shown to be enriched at the PAS (Fig 1). The recruitment of Atg4pLIR2‐GFP to the PAS was significantly reduced compared to WT Atg4‐GFP and the other analyzed fluorescent chimeras (Fig 4A–C). This reduction was very similar to the one observed in the Atg4PD‐GFP strain, where the defect in Atg8 conjugation to PE does not allow Atg4 association with the PAS (Fig 4A–C). We concluded from these results that the conserved sequence within Atg4 between positions 102 and 105 is important for the Atg4 recruitment to autophagosomal membranes and subsequent Atg8‐PE deconjugation.
Figure 4. Atg4p LIR 2 mutant recruitment to the PAS is reduced.
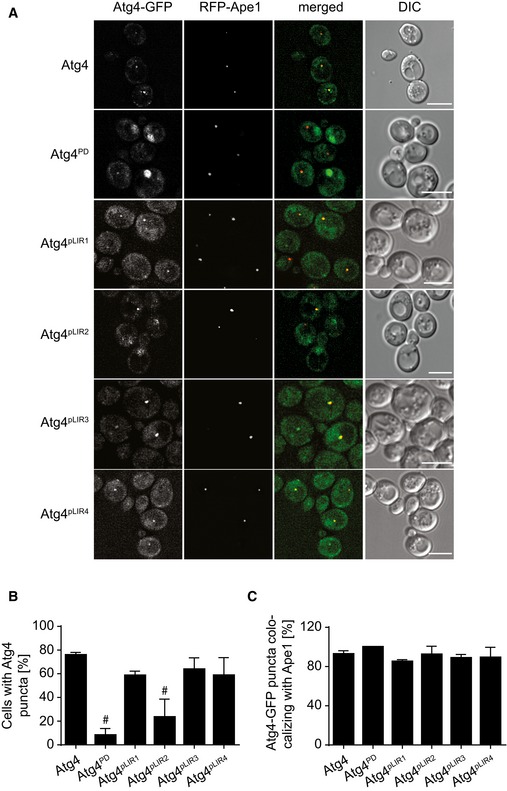
- The atg1∆ cells expressing integrated RFP‐Ape1 and Atg4‐GFP (SAY136) Atg4PD‐GFP (SAY137), Atg4pLIR1‐GFP (RHY016), Atg4pLIR2‐GFP (SAY139), Atg4pLIR3‐GFP (RHY017), or Atg4pLIR4‐GFP (RHY018) were grown in YPD to an early log phase and then starved for 3 h in SD‐N medium before imaging. Scale bars, 5 μm.
- Percentage of cells in which Atg4‐GFP is observed in a punctate structure in the experiments depicted in panel (A). Data represent the average of three independent experiments ± SD. Significant differences (P < 0.05) between the Atg4 mutants and the WT were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the symbol #.
- Percentage of Atg4 puncta co‐localizing with the PAS marker proteins Ape1 in panel (A). Data represent the average of three independent experiments ± SD.
Impairment of Atg8 deconjugation is reflected in autophagosome size
Next, we assessed the consequence of the observed defects caused by the Atg4 mutant variants on autophagosome biogenesis. Hence, we examined the accumulation of autophagic bodies (AB) in cells lacking the major vacuolar protease Pep4 by electron microscopy 33. The atg4Δ pep4Δ strain expressing Atg4pLIR2 exhibited a severe decrease in the number of AB in comparison to WT Atg4 and was similar to the one observed in the strains carrying Atg4PD or an empty vector (Fig 5A and B). Moreover, the average diameter of the AB observed in the Atg4pLIR2 mutant was smaller than in atg4∆ cells expressing Atg4, Atg4pLIR1, Atg4pLIR3 and Atg4pLIR4 (Fig 5C). This result shows that the failure of the Atg4pLIR2 mutant in recycling Atg8 from Atg8‐PE leads to a defect in autophagosome biogenesis.
Figure 5. The pLIR2 sequence is new motif involved in the specific recognition of Atg8‐PE.
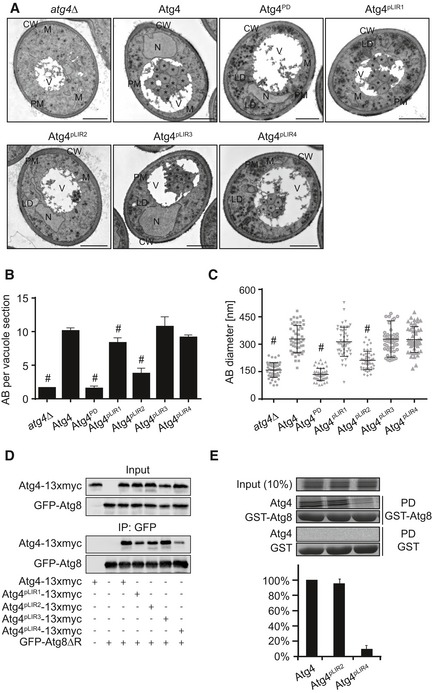
- The atg4Δ pep4Δ strain transformed with integration plasmids expressing Atg4 (SAY144), Atg4PD (SAY145), Atg4pLIR1 (SAY146), Atg4pLIR2 (SAY147), Atg4pLIR3 (RHY009), and Atg4pLIR4 (RHY010) or an empty plasmid (JSY163) was grown and starved as in Fig 1D before being processed for EM. Autophagic bodies (AB) are highlighted in the EM micrographs with asterisks. CW, cell wall; LD, lipid droplet; M, mitochondria; N, nucleus; PM, plasma membrane; V, vacuole. Scale bars, 1 μm.
- Quantification of the autophagic bodies. Average number of autophagic bodies (AB) per 50 vacuole sections ± SD. Significant differences (P < 0.05) between the Atg4 mutants and the WT were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the symbol #.
- Quantification of the diameter of the AB. Average diameter of the AB in 50 vacuole sections ± SD. Significant differences (P < 0.05) between the various Atg4 mutants and the WT were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the symbol #.
- The atg4Δ atg3Δ (FKY428) or the atg4Δ atg3Δ (FKY437) strains carrying the integrative GFP‐ATG8ΔR plasmid were transformed with the centromeric plasmids expressing either Atg4‐13xmyc, Atg4pLIR1‐13xmyc, Atg4pLIR2‐13xmyc, Atg4pLIR3‐13xmyc, or and Atg4pLIR4‐13xmyc. Strains were processed for pull‐down experiments as in Fig 2B. Quantification is shown in Fig 6A.
- Atg4, Atg4pLIR2, and Atg4pLIR4 were translated and radiolabeled in vitro as described in the Materials and Methods section before pull‐down (PD) with GST or GST‐Atg8 immobilized on glutathione‐beads. Beads were successively washed, and the eluted material was resolved by SDS–PAGE. Atg4 was visualized by autoradiographs, while GST and GST‐Atg8 amounts were assessed by Coomassie brilliant blue staining of the SDS–PAGE gel. The graph is the quantification of three independent experiments ± SD, where the binding of Atg4 is set as 100%.
The pLIR2 motif is a novel Atg8 recognition site specific for membrane bound Atg8
Even though several mutations within Atg4 cause a decreased association between Atg4 and Atg8, our results indicated that only mutation of the pLIR2 amino acid sequence has a negative impact on autophagy mainly due to an impairment of Atg8‐PE deconjugation. We therefore reasoned that the area around pLIR2 could be important to specifically recognize conjugated Atg8 as a substrate. To confirm our hypothesis, we repeated the immunoprecipitation experiment with GFP‐Atg8∆R but in atg4Δ atg3Δ cells in which Atg8 cannot be conjugated to PE due to the lack of the E2‐like conjugation enzyme Atg3. Our results revealed that Atg4pLIR2 has no Atg8 binding defect in this strain background anymore and behaves like WT Atg4, while Atg4pLIR1 and Atg4pLIR4 still displayed a decrease in Atg8 association with various extents (Figs 5D and 6A).
Figure 6. The double Atg4APEAR , cLIR mutant leads to enhanced in vivo defects compared to the single APEAR and cLIR mutants.
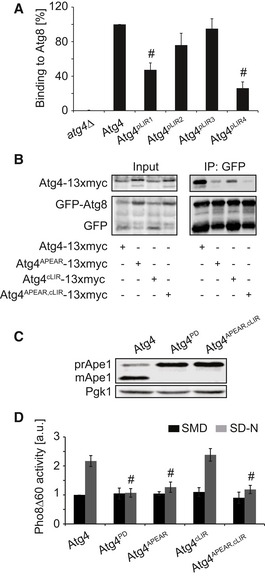
- Quantification of the experiments shown in Fig 5D. Values are relative to WT Atg4 and represent the average of three independent experiments ± SD. Significant differences (P < 0.05) between cells expressing WT Atg4 were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the # symbol.
- The atg4Δ (JSY151) strains carrying the integrative GFP‐ATG8ΔR plasmid were transformed with the centromeric plasmids expressing either Atg4‐13xmyc, Atg4APEAR‐13xmyc, Atg4cLIR‐13xmyc, or Atg4APEAR,cLIR‐13xmyc. Pull‐down experiments were carried out as in Fig 2B. 2% of cell lysate (input) or 50% of the pull‐down material (IP: GFP) were resolved by SDS–PAGE and analyzed by Western blot using either anti‐myc or anti‐GFP antibodies.
- The atg4Δ (SAY084) mutant was transformed with plasmids expressing 13xmyc‐tagged Atg4, Atg4PD, and Atg4APEAR,cLIR before being grown to a log phase in SMD medium. Proteins were precipitated with TCA and subsequently analyzed by Western blot using the anti‐Ape1 and anti‐Pgk1 antibodies (loading control).
- The SAY130 strain (Pho8∆60 pho13∆ atg4∆) carrying plasmids expressing Atg4, Atg4PD, Atg4APEAR, Atg4cLIR, and Atg4APEAR,cLIR was analyzed as in Fig 2E. Data represent the average of three independent experiments ± SD. Significant differences (P < 0.05) between cells expressing WT Atg4 were calculated using the paired two‐tailed Student's t‐test, and they are indicated with the # symbol.
To acquire additional insights into the pLIR2 binding mode, we analyzed the interaction between Atg4pLIR2 and GST‐ATG8 in vitro. In this experimental setup, Atg8 is present in its non‐lipidated form. As a control, we used Atg4pLIR4 since it is the only other evolutionarily conserved motif in Atg4 and very recent work has shown that this area is a LIR motif important for efficient cleavage of LC3/GABARAP proteins by ATG4B 34. As shown in Fig 5E, in vitro translated and radiolabeled Atg4 and Atg4pLIR2 specifically bound to Atg8, while Atg4pLIR4 lost its association with Atg8 almost completely.
Based on these experiments and our in vivo data, we concluded that Atg4 pLIR4 is a bona fide LIR motif that constitutively binds Atg8. Therefore, we named it cLIR (C‐terminal LIR). In contrast, pLIR2 is a novel Atg8 recognition site with a specificity for Atg8‐PE rather than non‐lipidated Atg8. We thus renamed it ATG8‐PE association region (APEAR).
APEAR and cLIR are cooperative Atg8 binding sites
Next, we wondered whether the two evolutionarily conserved APEAR and cLIR motifs in Atg4 function together. To investigate their impact on Atg8 binding, we revisited the association between Atg4 and Atg8 when both sites are mutated. As depicted in Fig 6B, the double mutant of Atg4, that is, Atg4APEAR,cLIR, reduces the interaction between the two proteins further compared to the single mutants, which suggests that APEAR and cLIR bind cooperatively. This notion was also supported by the fact that the strain carrying Atg4APEAR,cLIR displayed a defect in the Cvt pathway much more pronounced than the one observed for the single mutants (Fig 6C vs. Fig 2D and E). We further found that autophagy is similarly blocked in the double Atg4APEAR,cLIR mutant as in Atg4PD and Atg4APEAR (Fig 6D).
When we examined the priming of Atg8 using Atg8‐GFP, the double Atg4 mutant showed an enhanced impairment in cleaving GFP from Atg8 than the single Atg4 mutants (Fig 7A). Furthermore, deconjugation of Atg8‐PE from the autophagosomal membrane also proved to be almost completely abolished in an atg4∆ strain carrying the Atg4APEAR,cLIR mutant when analyzing the cellular distribution of GFP‐Atg8∆R by fluorescence microscopy and Atg8‐PE levels by Western blot (Fig 7B–D). Finally, we tested the ability of the different Atg4 mutants to deconjugate Atg8‐PE in vitro. Atg4 and Atg4cLIR were deconjugating Atg8‐PE on liposomes with very similar kinetics (Fig 7E). In contrast, Atg4APEAR and Atg4APEAR,cLIR displayed a complete defect in releasing Atg8 from its lipid anchor.
Figure 7. Atg4APEAR , cLIR displays similar Atg8‐PE deconjugation defect as Atg4APEAR .
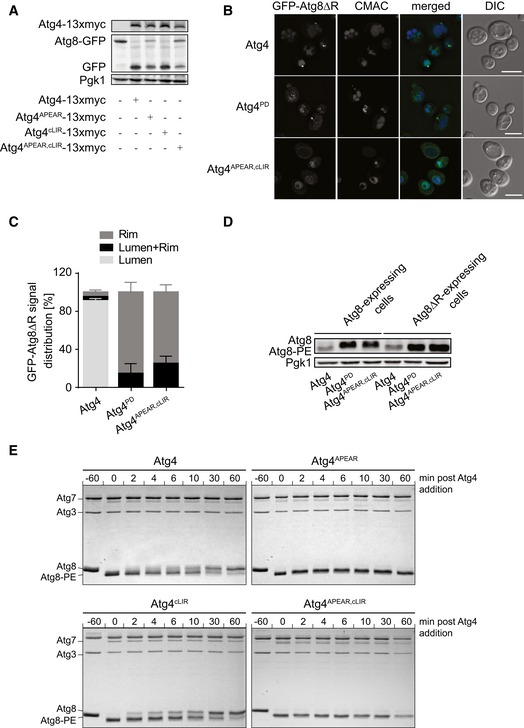
- The atg4Δ Atg8‐GFP mutant (SAY113) was transformed with an empty vector or plasmids expressing 13xmyc‐tagged Atg4, Atg4LIR2, Atg4LIR4, and Atg4APEAR,cLIR. Proteins from exponentially growing cells were TCA‐precipitated and analyzed by Western blot using the anti‐GFP antibody.
- The atg4Δ strain carrying the integration plasmid pCuGFPAtg8ΔR(305) (JSY151) and a plasmid expressing 13xmyc‐tagged Atg4, Atg4PD, or Atg4APEAR,cLIR was processed and analyzed as in Fig 3C. DIC, differential interference contrast. Scale bars, 5 μm.
- Quantification of GFP‐Atg8 distribution in cells imaged in (B): vacuole lumen, vacuole rim, or both localizations (lumen + rim). Error bars represent the SD of three independent experiments.
- The atg4∆ (SAY084) and atg4∆ ATG8∆R (RHY012) mutants were transformed with plasmids expressing 13xmyc‐tagged Atg4, Atg4PD, and Atg4APEAR,cLIR, and the resulting strains were processed as in Fig 3E.
- Recombinant GST‐tagged Atg4, Atg4APEAR, Atg4cLIR, and Atg4APEAR,cLIR were added to Atg8‐PE‐containing liposomes, and their deconjugation activity was assessed over time as described in the Materials and Methods section.
From these results, we draw the conclusion that Atg4APEAR and Atg4cLIR associate with Atg8 cooperatively and that both sites are important for Atg4 function.
Discussion
Atg8‐PE deconjugation by Atg4 on autophagosomal membranes, but also other organelles, is critical for autophagy progression because it allows reusing Atg8 for autophagosome biogenesis 11, 20, 21, 35. Our study is in line with these findings and confirms the relevance of Atg8‐PE deconjugation in autophagy. Additionally, it reveals that Atg4 can associate with autophagosomal membranes and that this recruitment is essential for Atg8‐PE deconjugation at this location. Our data further suggest that an evolutionarily conserved amino acid motif plays an important role in Atg4 recognition of Atg8‐PE and we have named it APEAR (Atg8‐PE association region). We further define another conserved Atg8 recognition site at the C‐terminus of Atg4, that is, cLIR, which, also in accordance with a recent publication 34, is a LIR motif. Together with APEAR, cLIR is relevant for Atg4 function.
The predicted protein structure of yeast Atg4 shows that APEAR is in close proximity to the catalytic site (Fig 8A) and hence could negatively affect its conformation. However, Western blot analysis shows that Atg4APEAR is stable. Moreover, we can conclude that the active site of the Atg4APEAR variant is still functional since priming of newly synthesized Atg8 is largely unaffected in cells expressing this mutant protein (Figs 2B and D, and 3A and B). Instead, the proteolytic defect appears to be specific for Atg8‐PE. This notion is supported by our finding that the interaction between Atg8 and Atg4APEAR is only impaired when Atg8 is conjugated to PE (in the cells employed for the pull‐down experiment shown in Fig 2B, Atg8 is mainly lipidated, Fig 3E) and explains why no binding defect is detected in our in vivo and in vitro binding experiments with non‐lipidated Atg8 (Figs 2B and C, 5D and E, and 6A). In contrast, mutation of the C‐terminal Atg4 cLIR impairs Atg8‐Atg4 interaction both in our in vitro and in vivo analyses. Very recent work has shown that a C‐terminal LIR motif in human ATG4B, which may functionally correspond to cLIR, is important for binding and efficient cleavage of LC3/GABARAP proteins 34. Our data are consistent with this report but also shows that cLIR is not key in Atg8‐PE deconjugation neither in vivo nor in vitro (Figs 3C–E and 7E), but it is rather a constitutive Atg8‐binding site that appears to indistinctively participate to both Atg8 priming and Atg8‐PE deconjugation. cLIR relevance, however, surfaces in combination with other Atg4 mutations and redundancies with other sites in promoting Atg8‐Atg4 interaction might be possible, such as the catalytic site or pLIR1. During the revision of this manuscript, a structural study describing the mechanism of Legionella RavZ‐mediated LC3‐PE C‐terminal processing was published 36. One of the hypotheses of the authors is that the N‐terminal and C‐terminal LIR motifs of RavZ are essential to bind the substrate simultaneously and open the catalytic groove to allow the access to the bond that has to be cleaved. Thus, the non‐conserved pLIR1, which plays a role in the in vivo interaction between Atg4 and Atg8 (Fig 2B), could functionally correspond to the N‐terminal LIR in RavZ and/or in human ATG4B, which together with the catalytic site is required for the proteolytic priming of LC3 36, 37, 38. Future investigations are needed to determine whether pLIR1 is indeed a LIR motif and whether it has any functional relationship with APEAR and/or cLIR.
Figure 8. Models for the structure of yeast Atg4 catalytic site and for Atg4 deconjugation activity on autophagosomal membranes.
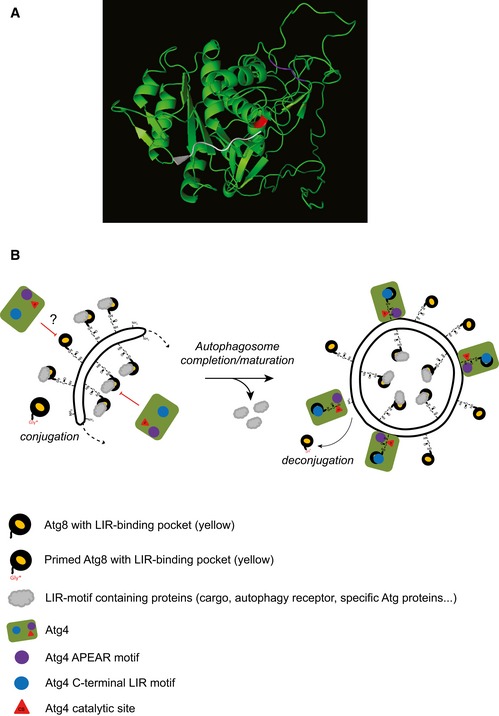
- Three‐dimensional model predicting yeast Atg4 structure generated using the RaptorX online program (http://raptorx.uchicago.edu/StructurePrediction/predict/) 55, which covered Atg4 sequence from aa 1–403 leaving C‐terminal region with 91 aa unpredicted. The catalytic site (C147) of Atg4 is highlighted in red while APEAR is colored in purple. The last eight C‐terminal amino acids of LC3B, in white, were positioned into the catalytic site as shown for ATG4B 37.
- After proteolytic priming, the C‐terminal glycine of Atg8 gets activated via the ubiquitin‐like conjugation system and linked through an amide bond to the PE present on autophagosomal membranes. Atg8‐PE is involved in autophagosome biogenesis at the PAS (dotted arrow). At this location, Atg8‐PE also associates with various proteins (gray clouds), such as cargo receptors, by binding their LIR motifs through a defined structural pocket (yellow). Atg4 binding to Atg8‐PE has to be controlled to avoid premature deconjugation. Occupation of the LIR motif‐binding pocket by other factors and/or other regulatory mechanisms such as post‐translational modifications (depicted with a question mark) could inhibit Atg4 action on autophagosomal membranes. As soon as the autophagosome is completed, the Atg machinery, including the shielding factors, is released allowing Atg4 access to autophagosomal membranes. This latter event involves both Atg4 binding to Atg8 via cLIR (blue circle) and association of the APEAR motif (purple circle) possibly with the C‐terminal region of Atg8‐PE, which allows the correct positioning of the C‐terminus into the catalytic site (red triangle) of Atg4.
Based on our data on the two evolutionarily conserved Atg8 recognition sites APEAR and cLIR, we propose the following model for the timely regulation of Atg8‐PE deconjugation by Atg4 (Fig 8B). During autophagosome biogenesis, the LIR‐binding pocket in Atg8 is shielded to prevent premature interaction between Atg4 and Atg8, which is in agreement with the observation that Atg4 is rarely detected at the PAS (Figs 1 and EV1). The formation of the autophagosomes in tight contact with cargoes, which is guaranteed by a LIR‐mediated interaction between Atg8‐PE and the autophagy receptors 14, 30, 39, could sterically and/or competitively impede Atg4 binding to the inner autophagosomal pool of Atg8‐PE. The outer pool, in contrast, would be protected by the LIR‐mediated association with components of the autophagy machinery such as the Atg3 and the Atg12‐Atg5‐Atg16 complex that are on the surface of nascent autophagosomes 13, 40. In addition or alternatively, post‐translational modifications like oxidoreduction 41, 42 or phosphorylation could contribute to modulate Atg4 activity and/or eventually its interaction with Atg8‐PE. The existence of one or more other mechanisms that protect the Atg8‐PE pool at the PAS from Atg4 proteolytic activity 21, 27 cannot be excluded and the accumulation of lipidated Atg8 at the PAS in knockout strains such as atg1∆, where also Atg4 is present at this location suggests this scenario (Figs 1A–C and EV2A and B) 43. Thus, different recognition determinants for Atg8 processing could be dictated by either post‐translational modifications or differences in structural conformations acquired by Atg8 before and after conjugation to PE. For example, Atg8‐PE appears to form multimers 8, 13, and this could influence the type of Atg4‐Atg8 interaction. Finally, once an autophagosome is completed, we propose that the release of the Atg machinery free the LIR‐binding pocket of Atg8 allowing Atg4 to interact via its cLIR (Fig 8B). Subsequent positioning of Atg4 APEAR, which we hypothesize is involved in the specific recognition of the C‐terminal part of PE‐conjugated Atg8, as also suggested by Atg4 structural modeling (Fig 8A), will permit proteolytic processing.
Altogether, our investigations have revealed the existence of a regulated recruitment of Atg4 onto autophagosomal membranes to deconjugate Atg8‐PE. Future studies are necessary to further dissect the function of all identified motifs and to understand how the deconjugation activity of Atg4 is timely orchestrated with the rest of the mechanisms that lead to the biogenesis of autophagosomes.
Materials and Methods
Yeast strains and media
The S. cerevisiae strains used in this study are listed in Table EV1. Genes were knocked out by homologous recombination using PCR fragments amplified with primers containing 60 bases identical to the flanking regions of the gene open reading frames 44, 45 and were replaced with URA3 or LEU2 from K. lactis, TRP1 from S. cerevisiae, HIS5 from S. pombe, kanamycin‐resistance gene (kanMX), or hygromycin‐resistance gene (hphNT1). ATG4 was tagged on its chromosomal locus by integration of GFP at the 3′ end. The PCR product used for the integration was obtained by PCR amplification of the GFP ORF and the TRP1 marker using pFA6a‐GFP(S65T)‐TRP1 as template plasmid 44. The expression of tagged Atg4‐GFP and Atg4‐13xmyc was analyzed by light microscopy and Western blot using an anti‐GFP and anti‐myc antibodies, and Ape1 processing was used to test the functionality of the protein fusion. Gene disruptions were confirmed by PCR and Ape1 processing analysis.
Cells were grown in rich medium (YPD: 1% yeast extract, 2% peptone, and 2% glucose) or synthetic minimal medium (SMD: 0.67% yeast nitrogen base, 2% glucose, and amino acids and vitamins as needed). To induce autophagy, cells were grown to a logarithmic (log) phase in YPD or SMD medium and then transferred into a medium lacking nitrogen (SD‐N; 0.17% yeast nitrogen base without amino acids and ammonium sulfate, and 2% glucose) for 3 h.
Plasmids
The pATG4GFP(416) was created by PCR amplification of the ATG4 promoter and the ATG4‐GFP fusion from the genome of the MNY006 strain. Used primers contained overhanging ends generating XhoI and SacI restriction sites in order to clone the PCR fragment into the pRS416 vector 46. The pATG413xmyc(416) plasmid was obtained by swapping the sequence coding for GFP from the pATG4GFP(416) with the one for the 13xmyc tag obtained from pFA6a13xmycTRP1 plasmid 44 using PacI and SacI. The pRS416 backbone of pATG4GFP(416) was then replaced with the one from pRS406 46, using KpnI and SacI, in order to create pATG4GFP(406) and all other integrative ATG4 constructs, that is, pLIR1 (Y36A, L39A), pLIR2/APEAR (F102A, I105A), pLIR3 (F446A, I449A), and pLIR4/cLIR (Y424A, I427A).
The protease‐dead (C147S), that is, Atg4PD, and LIR mutant versions of Atg4 were created in the pATG413xmyc(416) and pATG4GFP(406) constructs using the site‐directed mutagenesis kit (Stratagene). The introduced mutations were verified by DNA sequencing. These plasmids were also used as templates to amplify by PCR the ATG4 promoter and the sequence coding for the various Atg4 forms, form, and clone them back in the same vectors as KpnI‐PacI fragments. This approach was taken to generate plasmids expressing untagged versions the different Atg4 constructs under the control of the authentic promoter.
To generate the pCuGFPATG8ΔR(305) and pCuATG8GFP(403) plasmids, we replaced the vector backbone of the pCuGFPATG8ΔR(406) 20 and pAUT7GFP(416) plasmids 16 with the one from pRS305 and pRS403 vectors 46, respectively, using SacI and XhoI. The promATG8ΔR(404) integration plasmid was created by PCR amplification of ATG8ΔR and 470 bp of ATG8 promoter from genomic DNA and clone it as a KpnI/AscI fragment upstream of the ADH1 promoter in a pRS404 vector.
ATG4 was subcloned into the pENTR3C vector before generating the Atg4pLIR2, Atg4pLIR4 and Atg4pLIR2,4 mutants using the QuikChange site‐directed mutagenesis kit (Agilent Technologies, 210515). pDONR201‐ATG8 (Harvard PlasmID Repository, ScCD00011665) and the pENTR‐based Atg constructs were subsequently inserted into the Gateway destination vectors pDEST15 (Thermo Fisher Scientific) and pDESTmyc (mammalian expression of N‐terminal myc‐tagged proteins), respectively, to create the pDESTmyc‐Atg4, pDESTmyc‐Atg4pLIR2, pDESTmyc‐Atg4pLIR4, and pDESTmyc‐Atg4pLIR2,4 plasmids.
Plasmids pCumCherryV5ATG8(415), pCK15 (centromeric plasmid carrying GFP‐Atg8 under control of the authentic promoter), and pPS128 (for the integration of RFP‐APE1 in the LEU2 locus) have been described elsewhere 43, 47, 48.
Antibodies and reagents
Western blot membranes were probed with monoclonal anti‐GFP (Roche, cat# 11814460001) and anti‐myc (Santa Cruz, cat# sc‐40) antibodies, or anti‐Ape1 47 and anti‐Pgk1 antisera (Sánchez‐Wandelmer et al, in revision). The anti‐Atg8 antiserum was generated by immunization of New Zealand White rabbits by injection of recombinant Atg8 obtained from Escherichia coli (New England Peptides). Secondary antibodies were Alexa‐680 conjugated anti‐rabbit or anti‐mouse IgG (Life Technologies).
Fluorescence microscopy
Fluorescence signals were visualized with a DeltaVision RT fluorescence microscope (Applied Precision) equipped with a CoolSNAP HQ camera (Photometrix). Images were generated by collecting a stack of 20 pictures with focal planes 0.20 μm apart to cover the entire volume of a yeast cell and subsequently deconvolved using the SoftWoRx software (Applied Precision). Where indicated, the CellTracker™ Blue 7‐amino‐4‐chloromethylcoumarin (CMAC) dye (Invitrogen) was used to specifically stain the vacuolar lumen. A single focal plane is shown at each time point. The number of Atg4‐GFP‐ and GFP‐Atg8∆R‐positive puncta per cell was counted in 50 cells from at least two independent experiments.
For time‐lapse imaging experiments, cells nitrogen starved in SD‐N medium and stained with CMAC for 10 min were imaged every 1 min, collecting a Z‐stack of six pictures with focal planes 0.30 μm apart. Images were deconvolved and mounted into movies before measuring the life of mCherry‐Atg8 and Atg4‐GFP puncta using the SoftWoRx software. The time point at which mCherry‐Atg8 appeared as a punctate structure was considered as time 0. Because of the variation in the life of mCherry‐Atg8‐positive puncta between cells, that is, 5–8 min as previously documented 49, the autophagosome cycle duration was normalized from 0 to 1. Atg4‐GFP puncta appearance was normalized identically, and the graph shows the relative period of time where Atg4‐GFP colocalizes with mCherry‐Atg8.
Immunoprecipitations
Yeast cell cultures were grown to a log phase in SMD medium and then for 5 h in YPD medium before adding 220 nM of rapamycin. Cells were harvested by centrifugation and washed in PBS with 2% glucose. Cells were then resuspended in a pellet volume of lysis buffer (PBS, 10% glycerol, 0.5% Tween‐20) supplemented with 1 mM NaF, 1 mM PMSF, 1 mM Na3VO4 and the complete protease inhibitors (Roche) and frozen in droplets in liquid nitrogen. After cell disruption with a cryomill (6770, Spex SamplePrep), extracts were cleared by centrifugation twice at 13,000 g for 10 min. For protein A immunoprecipitations (Fig 2A), Dyna epoxy magnetic beads (Invitrogen) were coupled with rabbit IgG according to manufacturer's protocol. Approximately 200 mg of cleared yeast extracts was incubated with 5 μl coupled IgG beads for 1 h at 4°C. The beads were washed three times in lysis buffer and resuspended in 15 μl urea loading buffer.
Alternatively (Fig 2E), equivalents of 100 OD600 of growing cells were transferred into SD‐N medium for 1 h, harvested by centrifugation, and resuspended in 500 μl of lysis buffer (45 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Tween‐20) supplemented with 10 mM NaF, 1 mM Na3VO4, 10 mM β‐glycerophosphate, 1 mM PMSF, and Complete protease inhibitors (Roche). Cells were lysed by vortexing at 4°C for 5 min in presence of glass beads, and lysates were subsequently cleared by centrifugation at 15,000 g for 5 min at 4°C. The supernatants were incubated with 25 μl of pre‐washed GFP‐Trap agarose beads (ChromoTek) on a rotating wheel for 1.5 h at 4°C. Beads were then washed three times with 500 μl of lysis buffer without supplements and immuno‐isolates were resuspended in loading buffer by boiling. Samples were then analyzed by SDS–PAGE followed by Western blot.
Electron microscopy
Cells were processed for EM as described previously 50. To determine the number of AB per vacuole and their diameter, three different grids with sections obtained from the same preparation were analyzed. In each grid, AB number and diameter were determined from 60 cells with vacuoles. Diameter was calculated from the AB area, measured using ImageJ software (https://imagej.nih.gov/ij/). Error bars represent the standard deviation from the values of the three grids.
GST pull‐down experiments
GST‐Atg8 was expressed in Escherichia coli BL21 (DE3) and affinity‐purified on glutathione‐Sepharose 4 Fast Flow beads (GE Healthcare, 17513201) followed by washing with NET‐N buffer (100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P‐40, 50 mM Tris–HCl pH 8) supplemented with the Complete EDTA‐free protease inhibitors (Roche). GST pull‐down assays were performed with [35S]‐labeled myc‐tagged Atg4 constructs co‐transcribed/translated using the TnT Coupled Reticulocyte Lysate System (Promega, L4610) as described previously 51, using pDESTmyc‐Atg4, pDESTmyc‐Atg4pLIR2, pDESTmyc‐Atg4pLIR4, and pDESTmyc‐Atg4pLIR2,4 plasmids as templates. For quantifications, gels were vacuum dried and [35S]‐labeled proteins detected on a Fujifilm bioimaging analyzer BAS‐5000 (Fujifilm, Tokyo, Japan).
In vitro deconjugation of Atg8
The proteins used for deconjugation assay (Atg3, Atg7, Atg8∆R, and Atg12–Atg5) were purified as described 52, and GST‐Atg4, GST‐Atg4APEAR, GST‐Atg4cLIR, and GST‐Atg4APEAR,cLIR were also purified as described 52, with the exception that the fusion proteins were not cleaved with thrombin but eluted using a buffer containing 20 mM glutathione. The in vitro conjugation was conducted also as described 52, with the following modification. The deconjugation reactions were carried out with 10 nM of GST‐Atg4 (or its mutant versions) in 137 mM NaCl, 25 mM HEPES, 2.7 mM KCl, 1 mM DTT for the indicated time points, and the proteins were separated on 13.5% polyacrylamide gels containing 4.8 M urea.
Miscellaneous reagents and procedures
The alkaline phosphatase activity, Ape1 maturation, and GFP‐Atg8 processing were measured as previously described 53. Protein isolation and Western blot analyses were conducted as explained 54. Detection of proteins by Western blot was done using an Odyssey system (LiCor Biosciences) and quantifications with the ImageJ software.
Author contributions
SA, FK, and FR conceived the study. SA, FK, MP, TJ, CK, SM, and FR designed the experiments. SA, FK, RG‐S, RSG, MM, JS‐W, MS, MSR, BZ, and RH realized the experiments. SA, FK, and FR wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Movie EV1
Review Process File
Acknowledgements
The authors thank Daniel Klionsky for reagents. F.R. is supported by ALW Open Program (822.02.014), DFG‐NWO Cooperation (DN82‐303), SNF Sinergia (CRSII3_154421), and ZonMW VICI (016.130.606) grants. M.P. is also a recipient of the SNF Sinergia grant (CRSII3_154421) and is supported by the ETH and an ERC advanced fellowship. C.K. is supported by a grant from the Vienna Science and Technology Fund (WWTF, VRG10‐001), the Austrian Science Fund (FWF, P25522‐B20 and P28113‐B28), and the EMBO YIP Program. S.M. is supported by an ERC grant (No. 646653), by the Austrian Science Fund (FWF, No. P25546‐B20) and by the EMBO Young Investigator Program. T.J. is funded by grants from the FRIBIOMED program of the Norwegian Research Council (grant number 214448), and the Norwegian Cancer Society (grant number 71043‐PR‐797 2006‐0320). S.A. is a recipient of a FCT Fellowship (SFRH/BD/95013/2013). R.G.‐S. is supported by a Marie Sklodowska‐Curie Individual Fellowship (IF‐EF) from the European Commission.
EMBO Reports (2017) 18: 765–780
References
- 1. Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sica V, Galluzzi L, Bravo‐San Pedro JM, Izzo V, Maiuri MC, Kroemer G (2015) Organelle‐specific initiation of autophagy. Mol Cell 59: 522–539 [DOI] [PubMed] [Google Scholar]
- 3. Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368: 651–662 [DOI] [PubMed] [Google Scholar]
- 4. Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self‐digestion. Nature 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774 [DOI] [PubMed] [Google Scholar]
- 6. Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- 7. Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) Atg8: an autophagy‐related ubiquitin‐like protein family. Genome Biol 12: 226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakatogawa H, Ichimura Y, Ohsumi Y (2007) Atg8, a ubiquitin‐like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130: 165–178 [DOI] [PubMed] [Google Scholar]
- 9. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE‐16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, et al (2011) SNARE proteins are required for macroautophagy. Cell 146: 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xie Z, Nair U, Klionsky DJ (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19: 3290–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaufmann A, Beier V, Franquelim HG, Wollert T (2014) Molecular mechanism of autophagic membrane‐scaffold assembly and disassembly. Cell 156: 469–481 [DOI] [PubMed] [Google Scholar]
- 14. Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- 15. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al (2000) A ubiquitin‐like system mediates protein lipidation. Nature 408: 488–492 [DOI] [PubMed] [Google Scholar]
- 16. Kim J, Huang W‐P, Klionsky DJ (2001) Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol 152: 51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y (2000) The reversible modification regulates the membrane‐binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 151: 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuma A, Mizushima N, Ishihara N, Ohsumi Y (2002) Formation of the approximately 350‐kDa Apg12‐Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277: 18619–18625 [DOI] [PubMed] [Google Scholar]
- 19. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y (1998) A protein conjugation system essential for autophagy. Nature 395: 395–398 [DOI] [PubMed] [Google Scholar]
- 20. Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, Reggiori F, Klionsky DJ (2012) A role for Atg8‐PE deconjugation in autophagosome biogenesis. Autophagy 8: 780–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakatogawa H, Ishii J, Asai E, Ohsumi Y (2012) Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 8: 177–186 [DOI] [PubMed] [Google Scholar]
- 22. Betin VM, Singleton BK, Parsons SF, Anstee DJ, Lane JD (2013) Autophagy facilitates organelle clearance during differentiation of human erythroblasts: evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy 9: 881–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanida I, Sou YS, Ezaki J, Minematsu‐Ikeguchi N, Ueno T, Kominami E (2004) HsAtg4B/HsApg4B/autophagin‐1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule‐associated protein light chain 3‐ and GABAA receptor‐associated protein‐phospholipid conjugates. J Biol Chem 279: 36268–36276 [DOI] [PubMed] [Google Scholar]
- 24. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19: 2092–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Betin VM, MacVicar TD, Parsons SF, Anstee DJ, Lane JD (2012) A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy 8: 664–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Betin VM, Lane JD (2009) Caspase cleavage of Atg4D stimulates GABARAP‐L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci 122: 2554–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nair U, Cao Y, Xie Z, Klionsky DJ (2010) Roles of the lipid‐binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J Biol Chem 285: 11476–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suzuki K, Kubota Y, Sekito T, Ohsumi Y (2007) Hierarchy of Atg proteins in pre‐autophagosomal structure organization. Genes Cells 12: 209–218 [DOI] [PubMed] [Google Scholar]
- 29. Lang T, Schaeffeler E, Bernreuther D, Bredschneider M, Wolf DH, Thumm M (1998) Aut2p and Aut7p, two novel microtubule‐associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J 17: 3597–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif – crucial for selective autophagy. J Cell Sci 126: 3237–3247 [DOI] [PubMed] [Google Scholar]
- 31. Lynch‐Day MA, Klionsky DJ (2010) The Cvt pathway as a model for selective autophagy. FEBS Lett 584: 1359–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noda T, Matsuura A, Wada Y, Ohsumi Y (1995) Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae . Biochem Biophys Res Commun 210: 126–132 [DOI] [PubMed] [Google Scholar]
- 33. Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y (1992) Autophagy in yeast demonstrated with proteinase‐deficient mutants and conditions for its induction. J Cell Biol 119: 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rasmussen MS, Mouilleron S, Kumar Shrestha B, Wirth M, Lee R, Bowitz Larsen K, Princely YA, O'Reilly N, Sjøttem E, Tooze SA, et al (2017) ATG4B contains a C‐terminal LIR motif important for binding and efficient cleavage of ATG8 family proteins. Autophagy doi: 10.1080/15548627.2017.1287651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, Chen Y, Zheng XL, Klionsky DJ, Liang Y, et al (2012) Dual roles of Atg8‐PE deconjugation by Atg4 in autophagy. Autophagy 8: 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kwon DH, Kim S, Jung YO, Roh KH, Kim L, Kim BW, Hong SB, Lee IY, Song JH, Lee WC, et al (2016) The 1:2 complex between RavZ and LC3 reveals a mechanism for deconjugation of LC3 on the phagophore membrane. Autophagy 13: 70–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F (2009) The structure of Atg4B‐LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J 28: 1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sugawara K, Suzuki NN, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F (2004) The crystal structure of microtubule‐associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes Cells 9: 611–618 [DOI] [PubMed] [Google Scholar]
- 39. Sawa‐Makarska J, Abert C, Romanov J, Zens B, Ibiricu I, Martens S (2014) Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane‐cargo apposition during selective autophagy. Nat Cell Biol 16: 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamaguchi M, Noda NN, Nakatogawa H, Kumeta H, Ohsumi Y, Inagaki F (2010) Autophagy‐related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3‐Atg8 interaction and is crucial for the cytoplasm‐to‐vacuole targeting pathway. J Biol Chem 285: 29599–29607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perez‐Perez ME, Zaffagnini M, Marchand CH, Crespo JL, Lemaire SD (2014) The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy 10: 1953–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scherz‐Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26: 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y (2001) The pre‐autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 20: 5971–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- 45. Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre‐mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F (2010) An Atg9‐containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 190: 1005–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stromhaug PE, Reggiori F, Guan J, Wang CW, Klionsky DJ (2004) Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell 15: 3553–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geng J, Baba M, Nair U, Klionsky DJ (2008) Quantitative analysis of autophagy‐related protein stoichiometry by fluorescence microscopy. J Cell Biol 182: 129–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffith J, Mari M, De Maziere A, Reggiori F (2008) A cryosectioning procedure for the ultrastructural analysis and the immunogold labelling of yeast Saccharomyces cerevisiae . Traffic 9: 1060–1072 [DOI] [PubMed] [Google Scholar]
- 51. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- 52. Fracchiolla D, Zens B, Martens S (2017) In vitro reconstitution of Atg8 conjugation and deconjugation. Methods Enzymol 587: 377–390 [DOI] [PubMed] [Google Scholar]
- 53. Guimaraes RS, Delorme‐Axford E, Klionsky DJ, Reggiori F (2015) Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae . Methods 75: 141–150 [DOI] [PubMed] [Google Scholar]
- 54. Reggiori F, Wang C‐W, Stromhaug PE, Shintani T, Klionsky DJ (2003) Vps51 is part of the yeast Vps fifty‐three tethering complex essential for retrograde traffic from the early endosome and Cvt vesicle completion. J Biol Chem 278: 5009–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J (2012) Template‐based protein structure modeling using the RaptorX web server. Nat Protoc 7: 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Movie EV1
Review Process File