

Abstract

The ToxCast EPA challenge was managed by TopCoder in Spring 2014. The goal of the challenge was to develop a model to predict the lowest effect level (LEL) concentration based on in vitro measurements and calculated in silico descriptors. This article summarizes the computational steps used to develop the Rank-I model, which calculated the lowest prediction error for the secret test data set of the challenge. The model was developed using the publicly available Online CHEmical database and Modeling environment (OCHEM), and it is freely available at http://ochem.eu/article/68104. Surprisingly, this model does not use any in vitro measurements. The logic of the decision steps used to develop the model and the reason to skip inclusion of in vitro measurements is described. We also show that inclusion of in vitro assays would not improve the accuracy of the model.
Introduction
The prediction of in vivo toxicity based on in vitro measurements is a challenging task, which is in the center of active development of modern computational toxicology.1−4
The TopCoder data science competition platform in collaboration with Environment Protection Agency (EPA) organized the ToxCast challenge in 2014.5 The target property was lowest effect level or LEL. LEL is defined as “the lowest dose that shows adverse effects in these animal toxicity tests. The LEL is then conservatively adjusted in different ways by regulators to derive a value that can be used by the Agency to set exposure limits that are expected to be tolerated by majority of the population.”5
The total ToxCast challenge included five consecutive subchallenges, which were executed over a seven month period and attracted 432 registrants from 32 countries. The first subchallenge was to identify software libraries and/or methods to describe the chemical structure of various compounds. The second subchallenge was about identification of a specific combination of in vitro assays, which could be used to predict the in vivo systemic LEL. The third subchallenge was executed privately and was entitled “Predictive Capability Tests”. The challenge described in this study was the fourth subchallenge. It had a goal “to build a prediction model (algorithm) using data from high-throughput in vitro assays, chemical properties, and chemical structural descriptors to quantitatively predict a chemical’s systemic LEL.”5 The final fifth subchallenge was about the documentation of the results of the models.
In this study, we present the results of the Rank-I model for the prediction challenge. According to the challenge rules, the participants were strictly forbidden to use any data other than the data that were provided in this competition. This was done in order to offer equal conditions to all participants as well as to better evaluate the performance of models developed using in vitro measurements. Indeed, the use of any information outside of the data provided within the challenge, e.g., information about toxicological chemical pathways, could potentially bias the comparison of algorithms. The summary of this prediction challenge was published by the EPA.6 Since September 2015, this information has been available from the Web archives7 and included as Supporting Information (see section “EPA ToxCast LELPredictor Marathon Match Results Summary”) to this study.
This article analyzes the steps that were taken to develop the Rank-I submission model by participant “novserj” (notice that the participant abbreviation is incorrectly reported as “noveserj” in the result table6). The Supporting Information also contains an extended technical description, which was submitted to TopCoder as part of the contest. Part of this description was also used by EPA in their report.6,7 Data, Rank-I model, and model predictions are available at http://ochem.eu/article/68104. We believe that this article will be interesting to both participants and organizers of the challenge and will help them to better understand and interpret the results of the challenge. It will also help other scientists in developing models with high prediction power. Moreover, the model reported in this study has the highest prediction ability for LEL end point as validated by the challenge organizers and thus can be of potential value for people working on the risk assessment of chemical compounds.
Data
Training and Test Data Sets
The in vitro measurements provided within the scope of the challenge included “a battery of more than 700 biochemical and cell-based in vitro assays to identify what proteins, pathways, and cellular processes these chemicals interact with and at what concentration they interact.”5
The total data set used during the challenge incorporated 1,854 molecules. The experimental LEL values were provided for 483 compounds that were used as the training set. The test set included LEL values for 143 chemicals, which were kept secret and were split into provisional (63) and final scoring (80) sets. During the challenge, TopCoder participants could submit predictions and receive the statistical results for the provisional set. Such results could be used to optimize the models during the submission stage. The 80 compounds from the final scoring set were used only once to rate the final model submissions.
The challenge organizers did not reveal information about the compounds, which were used as the test sets. After the challenge, the experimental values for 143 compounds from the test set were kindly released by the TopCoder organizers to the authors of the article. Both training and test set compounds are publicly available for download from http://ochem.eu/article/68104.
Methods
The detailed description of the Rank-I submission is provided in Supporting Information (see “Technical description” section). Below, we briefly recapitulate the main steps.
Descriptor Packages
Ten different descriptor packages implemented in the public platform OCHEM8 were used individually to create ten models for the resulting consensus. Four descriptor packages, E-state,9 QNPR,10 ISIDA fragmentor,11 and GSFrag,12 were based on the 2D representations of the chemical structures. The other six packages, Inductive,13 ChemAxon,14 Adriana,15 Mera/Mersy,16 CDK,17 and Dragon18 descriptors, were calculated using 3D representations of the chemical structures. The 3D structure representation was generated using Corina.19
Unsupervised Descriptor Selection
Within each individual model, the basic unsupervised descriptor selection procedure was performed. First, descriptors with constant values for the data set were removed. Next, duplicated descriptors with pairwise correlation of more than 0.95 were eliminated. Exactly the same procedure was used for all models, and thus, the same number of descriptors and molecules were utilized to develop each model with different machine learning methods.
The selected descriptors’ count for each model after the unsupervised filtering is shown in Table 1.
Table 1. Number of Descriptors and Models’ Accuracy for the Prediction of the Test Set Compounds.
| RMSE |
||||
|---|---|---|---|---|
| descriptor set | number of selected descriptors | whole test set (n = 143) | inside of ADa (n = 136) | outside of AD (n = 7) |
| CDK | 159 | 1.13 | 1.01 | 2.4 |
| Dragon | 1824 | 1.15 | 1.05 | 2.4 |
| Fragmentor | 631 | 1.18 | 1.04 | 2.7 |
| GSFrag | 202 | 1.1 | 0.97 | 2.5 |
| Mera, Mersy | 242 | 1.04 | 0.96 | 2.1 |
| Chemaxon | 97 | 1.16 | 1.06 | 2.4 |
| Inductive | 39 | 1.17 | 1.03 | 2.7 |
| Adriana | 133 | 1.14 | 1.01 | 2.5 |
| QNPR | 381 | 1.12 | 1.02 | 2.7 |
| E-state | 185 | 1.16 | 1 | 2.8 |
| in vitro | 143 | 1.21 | 1.11 | 2.5 |
| Consensus | 4036 | 1.08 | 0.96 | 2.5 |
Machine Learning Methods
The model used in the challenge was developed with Associative Neural Networks (ASNN). The ASNN exploits the idea of ensemble learning. It can be considered in a simplified way as a combination of k-nearest neighbor (kNN) method applied in the space of ensemble predictions. The models developed with the ASNN were top-ranked in several benchmarking studies3,20−29 and that is why this method was selected for the EPA challenge.
The default parameters for the ASNN algorithm, as optimized during previous studies and provided on the OCHEM Web site were used. They included 64 neural networks in ensemble, 3 neurons in a hidden layer trained by the SuperSAB30 algorithm.
In addition to ASNN, we also analyzed kNN, support vector machines (as implemented in LibSVM),31,32 and partial least squares33 methods. As with the ASNN method, the default parameters of these algorithms as provided on the OCHEM site were used.
Validation Protocols
The unbiased estimation of the models’ performance is critically important for selection and decision making for development of models. Two protocols, cross-validation, and bagging are frequently used to estimate validation accuracy for the training set. The cross-validation protocol splits the initial data set into n chunks. It uses n – 1 subsets as the training set and predicts the remaining chunk of the data.
Bootstrap aggregation (bagging) is another powerful approach to develop and validate models developed by Leo Breiman.34 It is based on the aggregation of models, each one of which is developed with its own training set (“bag” in the terminology of Breiman). Each bag is formed by random sampling with replacement from the initial training set and has the same size as the initial set. The molecules (on average 37%), which do not participate in the respective training set, are called “out-of-the-bag”. The predictions for these molecules are used to evaluate the predictive power of models. The bag size of 64 models was used.
Supervised Descriptor Selection Using Neural Network Pruning
In the 90s, there were several theoretical developments to identify the most significant descriptors for neural networks.35−39 Some of these methods calculate the sensitivity (importance) of input parameters according to derivatives of neural network weights with respect to the error function,35 while others provide such estimations based on the analysis of the magnitudes of the neural network weights.37 For this study, we used a method from the second group, which provided the best results in our previous studies.37−39 The sensitivity Si of a neuron i was calculated as
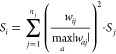 |
1 |
where wij were weights connecting neuron i and j, maxa was taken over all weights ending at the neuron j having sensitivity Sj, and summation was taken over all weights connecting the neuron i with the upper layer neurons. The sensitivity calculations were performed recursively starting from the last layer neuron, which had sensitivity of 1.
The pruning procedure started once neural network training was completed. On each step, the least significant descriptors with smallest Si were eliminated, and the models were retrained with the decreased set of descriptors. The sets of descriptors, which calculated the minimal errors, were considered as the optimal ones. In order to avoid overfitting and overtraining,40 the neural networks were trained using the efficient partitioning algorithm,41 which uses the early stopping procedure.40
Statistical Parameters
The root mean square error (RMSE) metric was used to score models. The RMSE is lower for models with higher performance. The challenge organizers used the following scoring function
| 1a |
to rank the models. As a result, models with lower RMSE got a higher score and higher rank among the others. In addition to RMSE, the organizers also reported Pearson correlation coefficients and AUC defined as “percentage of pairs where predicted1 < predicted2 among those where ground_truth1 < ground_truth2 (the higher the value, the better the result)”.6
Results
The workflow for the model development used in the challenge (see “Technical description” in the Supporting Information) was based on our previous expertise to develop recently published models.4,22,23,42 Final statistical results for the top-ranking models are summarized in Table 2. Below, we provide a detailed analysis of the steps, which were used to develop the model.
Table 2. Summary of the Performance of the Top-Ranked Models of the EPA ToxCast Challenge.
| test
set |
||||||||
|---|---|---|---|---|---|---|---|---|
| training
set (n = 483)a |
provisional
subset (n = 63) |
final
subset (n = 80) |
full, n = 143 | |||||
| model | RMSE | R2 | RMSE | rank | RMSE | R2 | rank | RMSE |
| novserj | 0.88 ± 0.04 | 0.27 ± 0.04 | 1.03 ± 0.08b | 8 | 1.12 ± 0.08b | 0.31 | 1 | 1.08 ± 0.07 |
| NobuMiu | 1.03 | 9 | 1.13 | 0.30 | 2 | 1.09 | ||
| a9108tc | 1.05 | 16 | 1.13 | 0.29 | 3 | 1.10 | ||
| klo86 min | 1.09 | 27 | 1.14 | 0.29 | 4 | 1.12 | ||
| in vitro assaysc | 0.97 ± 0.04 | 0.11 ± 0.03 | 1.24 ± 0.09 | |||||
| MW + NCd | 0.97 ± 0.04 | 0.11 ± 0.03 | 1.18 ± 0.08 | |||||
Prediction accuracy for the “out-of-the-bag” samples.
Confidence intervals were estimated using the subsets, which were sampled from the training set, and each had the same size as the respective test set (see for more details ref (23)).
Best model based on the in vitro assay descriptors developed using the LibSVM method (see also Table S1).
Model based on molecular weight (MW) and number of carbon atoms (NC) developed using the same approach as the above in vitro model.
Failed Molecules
There were 37 molecules, including 11 molecules from the training set, for which descriptor generation failed for different packages. CDK descriptor package does not support inorganic elements such as [Sn], [Hg], [B], and [As]. The failed molecules either included unsupported atoms for CDK or some groups, e.g., [N3+]. Several other molecules, e.g., rifampicin, α-cyclodextrin, milbemectin, emamectin benzoate, etc. were large chemical structures and failed either due to time-out or structure conversion problems. According to the challenge rules, the participants were required to submit predictions for all molecules. Therefore, we had to submit some values. As a simple solution, we used an average value of all training set molecules, logLEL = −3.2602 log(M), as the predicted values for the failed molecules
Scoring of Models: How Useful Is the Provisional Test Statistics?
The challenge organizers offered a provisional set of N = 63 compounds for the purpose of model analysis and selection. However, we decided to skip the testing on this set for the following considerations.
The provisional test set was much smaller than the training set. Thus, an attempt to rely on the models’ performance for this set by, e.g., submission of multiple predictions and selection of a “best” model using it, could contribute a higher uncertainty and result in the selection of a nonoptimal model for the final set.
Indeed, the final model RMSE was 0.88 ± 0.04 for N = 472 training set molecules. The provisional set was not available, and thus, we were unable to calculate the confidence intervals for it. We estimated the intervals by random sampling of N = 63 molecules from the training set, for each of which we calculated the intervals. The confidence intervals for these sets were about 2-fold larger ±0.08. Thus, selecting the best model based on the performance for the provisional test set is about twice as uncertain compared to selecting based on the training set. Therefore, instead of relying on the accuracy of models for the provisional test set, a strategy to rely on the estimated validated results for the training set is more reliable. An even better strategy could be to select a model based on the combined accuracies of the provisional and training sets, but such an analysis was not implemented.
The confidence intervals for N = 80 molecules were about the same as that for N = 63 molecules. The wide confidence intervals for both provisional and final test sets might have contributed to the fluctuations of ranks of challenge models for both sets. For example, the top final scoring model was only ranked #8 for the provisional submission, while the fourth model was ranked #27. Vice versa, the models ranked top #1 and #4 for provisional submissions were ranked as #9 and #34 (out of 47 participants) for the final test set.5 Thus, indeed, the provisional ranking score was not strongly predictive of the final one: provisional and final models’ ranks were correlated only with correlation coefficient R = 0.76.
The RMSE of the eight top-ranked final models were in the range 1.12 to 1.16 and thus were within the confidence intervals of the winning model. Thus, statistically speaking, these models had the same performance, and their differences in performance were due to chance.
Analysis of the Machine Learning Methods
The model submitted to the TopCoder challenge was a consensus of the bagging models developed with the ASNN method. In this section, we briefly describe the considerations that were used to develop and select this model for the challenge. The OCHEM Web site provides several machine learning methods and descriptors. Below, we compare the performance of different methods, which are described in the Methods section.
Bagging vs cross-validation was compared. Table S1 in Supporting Information demonstrates that models developed using bagging had consistently smaller validation RMSEs as compared to the cross-validation results. This result is in agreement with our previous observations.4,21,22,24 Therefore, the bagging approach was used.
Comparison of different machine learning methods (Table S1) shows that combinations of machine learning methods and descriptors provided quite similar performances with RMSE ranging from 0.9 to 1.2 log units. Considering that 95% confidence intervals of RMSE were ±0.4 log units, the majority of these models were not significantly different.
The LibSVM approach resulted in the lowest RMSE for individual models. The highest RMSE = 1.2 (i.e., the lowest performance) was calculated using the PLS method for Mera/Mersy descriptors. Actually, the failure of this method was due to several outlying molecules that had predictions far beyond the range of the training set values. They may be due to the sparseness of descriptors used to develop models and insufficient number of data points used to fit the coefficients in PLS. If we limited the predicted values for all compounds to the range of the training set values, the results of PLS models became similar to those of other methods.
It is interesting that the model developed using in vitro assay measurements consistently provided the lowest accuracy compared to the other descriptors.
Development of the Rank-I Submission Model
Considering that models developed with different descriptor sets had approximately similar performance, we decided to build our consensus models using a simple average of individual models. For each machine learning approach, a consensus model was built for all descriptor packages. Since the individual models were calculated using the bagging approach, the developed consensus models were also validated using the same protocol. The model based on the ASNN method calculated the lowest RMSE error compared to consensus models developed using other machine learning approaches.
The exclusion of the model based on in vitro descriptors did not change the accuracy of the ASNN consensus model. The model based on a combination of both in silico and in vitro descriptors requires both sets of descriptors. This limits its application to compounds for which in vitro measurements are present, while the model based exclusively on in silico descriptors can be applied to any new compounds. Therefore, we decided to submit the model developed using only in silico descriptors to the challenge.
The model development steps were based on simple decisions, which followed “Occam’s razor” principle. First, we found that the models developed on the training set have large validation RMSE and that the provisional set statistics had a limited value for model selection. Therefore, we followed the model development steps, which were successful in our previous studies.20,21,23−27,42 This strategy allowed us to develop the Rank-I model.
Comparison with a Simple Two-Descriptors Based Model
Did the complexity of the final model (a consensus of several individual models, each of which uses a different descriptor set and is a bootstrap aggregation of multiple neural network submodels) add any value, or could we get some similar results using a simpler approach? In order to answer this question, we developed models using just two descriptors: molecular weight and number of carbon atoms using linear regression. The RMSE of this model on the training set was 1.0 ± 0.04 log unit. The use of the same descriptors for the bagging approach decreased RMSE to 0.97 ± 0.04 log unit for the LibSVM method (Table 2). This error was significantly higher than that of the Rank-I model. Interestingly, the best model calculated based on the in vitro assay measurements had exactly the same accuracy (RMSE = 0.97 ± 0.04).
Analysis of the Test Set Compounds
The TopCoder organizers kindly released information about the experimental values for the N = 143 test set molecules. It allowed us to provide an additional analysis of the results for this set and to better evaluate the influence of the in vitro descriptors on the prediction accuracy.
Analysis of Several Models Involving in Vitro Descriptors
The model developed using in vitro descriptors (see Table 2) had higher RMSE = 1.24 for the test set as compared to that of the consensus model, RMSE = 1.08, based on in silico descriptors. The extension of the consensus model by inclusion of the model based on in vitro descriptors increased the RMSE of the new consensus model to 1.10 log units. We also explored whether extension of in silico descriptors with in vitro descriptors can provide better prediction accuracy. For this study, we developed models using combinations of each descriptor set with in vitro descriptors. The RMSE of the models developed with in silico + in vitro sets were changed in the range of −0.02 to 0.01 log units compared to RMSEs of models calculated using only in silico descriptors. The RMSE of the consensus model based on in silico + in vitro sets was 1.09, i.e., 0.01 log units higher compared to that of the model based only on in silico descriptors.
Chemical Diversity
The RMSE calculated for the test set was significantly higher than that for the training set compounds (Table 2). An analysis of extended functional groups (EFG)43 was done to identify whether both sets contained chemically different compounds. The EFG consists of 583 manually curated functional groups, which provide comprehensive coverage of heterocyclic compounds and are relevant for medicinal chemistry. The SetCompare tool23 was used to determine statistically significantly overrepresented chemical groups in training and in test sets using hypergeometric distribution. It was found that hydroxy compounds, amines, saturated six-membered heterocycles containing one heteroatom, etc., were overrepresented in the test set, while pnictogens, thiophosphoric acid esters, halogen derivatives, etc. were overrepresented in the training set. The full list of overrepresented groups is available at http://ochem.eu/article/68104. Thus, the chemical diversity of molecules in both sets may have contributed to the observed differences in the RMSEs for the training and test sets.
Analysis of Compounds Predicted with Large Errors
The EFG and SetCompare tools were also used to analyze which chemical features contributed to predictions with high errors. A difference of 1.5 log units between predicted and experimental values was used to identify N = 62 compounds with high prediction errors. Most of these compounds had extreme LEL values, i.e., either low or high values, and only 7 compounds (10%) were within [−2, −4] log(M) interval as compared to 420 (75%) of compounds that were within this region for the remaining group. Thus, the model had difficulties in predicting highly toxic and nontoxic compounds.
Applicability Domain
According to the TopCoder rules, the submitted results were scored using predictions for all test set compounds. However, of course, some of the compounds from the test set could be outside of the applicability domain (AD) and have lower prediction accuracy. OCHEM uses standard deviation of the predictions of models, which contribute to the consensus model, as distance to the model (abbreviated as STD-CONS).20 STD-CONS was found as the best definition of the distance to the model.20,27 OCHEM defines AD of a model as the value of STD-CONS that covers 95% predictions from the training set. In the previous benchmarking study of 12 definitions of distances to the model applied to 11 models, we found that STD-CONS provided the best separation of molecules with large and low prediction errors notwithstanding the used model.20 Therefore, we concluded that AD of models is determined by the composition of the training set of molecules rather than by the used descriptors or machine learning methods. In the current study, seven compounds from the test set were outside of the AD of the consensus model. The consensus model as well as individual submodels calculated significantly higher RMSEs, which were in the range of 2.1 to 2.7 log units, for these seven compounds (Table 1). This result supports the previous conclusions about the universal nature of the AD of models and good discriminating power of STD-CONS distance to the model. It also indicates that taking into consideration the AD of the models is important to avoid predictions with high errors.
It is interesting that four out of seven compounds had LEL < 3, while other three compounds had LEL > 5.5. Thus, the used AD has identified compounds that had experimental toxicity values in the ranges that are difficult to predict.
Development of Models Using Descriptors Optimized with Pruning
The final consensus model was based on 10 submodels, which were developed with N = 4036 descriptors (Table 3). These descriptors were selected from the initial set following the unsupervised filtering procedure. We explored whether the performance of this model can be further improved by using a supervised descriptor selection procedure based on neural network pruning of the least sensitive descriptors. The application of this procedure decreased the numbers of descriptors in 5 to 100-fold (Table 3). Models developed using these descriptors had on average lower training set RMSEs as compared to those based on descriptors selected by the unsupervised filtering, while the opposite result was calculated for the test set RMSEs (Table 3). Thus, selection of descriptors optimal for the training set introduced variable selection bias.44 Indeed, during supervised selection of descriptors we evaluated the performance of models for the training set molecules multiple times. This resulted in selection of descriptors with improved fit for this set but at the same time decreased the prediction accuracy for test set compounds, which have different chemical diversities. The neural networks are very efficient methods to work with high-dimensional data45 and can be also efficiently used without a need of supervised variable selection.
Table 3. Performances of Models Developed Using Different Descriptor Selection Proceduresa.
| unsupervised
selection |
neural
network pruning |
|||||
|---|---|---|---|---|---|---|
| RMSE |
RMSE |
|||||
| descriptor set | N | training | test | N | training | test |
| CDK | 159 | 0.93 | 1.13 | 6 | 0.89 | 1.2 |
| Dragon | 1824 | 0.93 | 1.15 | 18 | 0.87 | 1.19 |
| Fragmentor | 631 | 0.98 | 1.18 | 12 | 0.92 | 1.21 |
| GSFrag | 202 | 0.97 | 1.1 | 24 | 0.97 | 1.18 |
| Mera, Mersy | 242 | 0.93 | 1.04 | 10 | 0.93 | 1.18 |
| Chemaxon | 97 | 0.93 | 1.16 | 11 | 0.92 | 1.16 |
| Inductive | 39 | 0.94 | 1.17 | 21 | 0.93 | 1.16 |
| Adriana | 133 | 0.93 | 1.14 | 8 | 0.92 | 1.1 |
| QNPR | 381 | 0.95 | 1.12 | 74 | 0.89 | 1.13 |
| E-state | 185 | 0.96 | 1.16 | 11 | 0.9 | 1.24 |
| Consensus | 4036 | 0.88 | 1.08 | 186 | 0.85 | 1.13 |
N is the number of descriptors selected to develop the respective model. RMSE is the root mean squared error calculated for the training (n = 483) and full test set (n = 143).
Discussion
In this study, we highlighted the steps used to develop the Rank-I submission for the EPA ToxCast challenge, which was organized by the TopCoder community. We have shown how to consider limitations of the training and test data sets and that following “Occam’s razor” principle helps to provide a top-entry to the challenge. This conclusion is supported by other studies. A similar consensus approach was used to achieve the overall best balanced accuracy for 12 end points for another ToxCast challenge3 organized by NIH.46 The consensus modeling was also successfully used in the CERAPP project to identify potential endocrine disruptors.47
It is rather surprising that the Rank-I model did not involve the in vitro descriptors. This can be attributed to several factors.
Poor Definition of the Predicted End Point
The LEL is defined as lowest effect level dose across multiple animal studies. This can contribute to considerable differences in the determined quantitative toxicity thresholds due to interspecies variations as well as differences in the experimental protocols. These factors could contribute to the biological noise of the measured values and make their prediction a difficult task.
Lack of Domain-Specific Modeling Approaches
The relatively weak performance of this model and all others in the challenge can also point out the limitation of the brute-force machine learning approach to this problem. The in vitro assay data may need to be treated as more than just a table of numbers, and one will need to incorporate biological knowledge into the structure of the model. Indeed, pharmacokinetic and pharmacodynamics properties of the analyzed molecules could be essential for their toxicity. Thus, we can expect that the use of systems biology methods can contribute to more accurate predictions of the LEL. It should be mentioned that the use of external data was explicitly forbidden for the purpose of the ToxCast challenge.
Insufficiency of Used in Vitro Assays
We cannot exclude the possibility that some of the currently used in vitro assays could be insufficient for the analyzed end point. For example, if toxicity is caused by metabolites of the analyzed compound the in vitro assays ignoring metabolic activation may not correctly report toxicity. Currently, it is not clear whether such problems frequently occur, but recent studies suggest that taking into consideration the metabolic activation was an important factor for prioritization of potentially emerging contaminants.21 Of course, the same problem can also contribute to difficulties with prediction of toxicity based on in silico descriptors.
Which of these factors contributed to the low accuracy of the model? Such analysis is beyond the scope of the article and will hopefully be answered in the future with new computational studies by the scientific community. Importantly, the public availability of the test set compounds released in this article will help other users to develop and benchmark new approaches to predict LEL and benchmark their results against the Rank I model of the ToxCast challenge. Moreover, since the model is publicly available and does not use in vitro descriptors, it can be used to predict the LEL of new compounds in prospective studies and can be benchmarked using new measurements, which may be available in the future. We believe publishing models online in a usable and reproducible manner will become an integral part of future computational chemistry.48
In summary, we have described the protocol for developing the Rank-I model of the EPA ToxCast challenge. The model is based only on in silico descriptors, and we were not able to increase its prediction ability using in vitro measurements in a postmarathon study presented in this article. The relatively low accuracy of this model indicates high complexity of the LEL and suggests that pure brute-force machine-learning approaches may not be sufficient to accurately predict such a complex biological end point. Possibly, systems biology approaches can help to develop better models for the prediction of LEL using the available in vitro measurements. At the same time, we cannot exclude the possibility that the currently used in vitro assays may not be sufficient to correctly characterize this end point. The developed model and used data are publicly available at http://ochem.eu/article/68104 and can be used by interested users to answer these questions as well as to benchmark new ideas, methods, or approaches.
Acknowledgments
We thank the organizers of the challenge for providing the test set data. We thank ChemAxon (http://www.chemaxon.com), Molecular Networks GmbH (http://www.molecular-networks.com), Kode srl (http://www.kode-solutions.net), and ChemoSophia (http://www.chemosophia.com) for contributing their software tools used in this study. We also thank TopCoder and EPA for their permission to use “EPA ToxCast LELPredictor marathon match results summary” in the Supporting Information of this article.
Glossary
Abbreviations
- AD
applicability domain
- ASNN
associative neural network
- CDK
chemistry development kit
- CEO
chief executive officer
- CERAPP
Collaborative Estrogen Receptor Activity Prediction Project
- EFG
extended functional groups
- EPA
Environmental Protection Agency
- kNN
k nearest neighbors
- LEL
lowest effect level
- LibSVM
Library of Support Vector Methods
- MW
molecular weight
- NC
number of carbon atoms
- NIH
National Institutes of Health
- OCHEM
Online Chemical Database and Modeling Environment, http://ochem.eu
- PLS
partial least squares
- QNPR
quantitative name property relationship
- R2
square of correlation coefficient
- RMSE
root mean squared error
- STD-CONS
standard deviation of predictions of models, which contribute to the consensus model
- SVM
support vector machines
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.5b00481.
EPA ToxCast LELPredictor marathon match results summary (reprinted with permission from the U.S. Environmental Protection Agency), technical description of the consensus model for the prediction of lowest effect level (LEL) concentrations, which was submitted to TopCoder as part of the contest, and Table S1 with statistical parameters of models (PDF)
Author Present Address
# S.N.: Facebook U.K. Ltd., London, UK.
Author Present Address
○ Y.S.: Google Inc., Munich, Germany.
Author Present Address
∇ R.K.: OSB AG, Regensburg, Germany.
All research was performed at eADMET GmbH.
The authors declare the following competing financial interest(s): Dr. Igor Tetko is CEO and founder of BigChem GmbH, which licenses OCHEM software. The other authors declared that they have no actual or potential competing financial interests.
Supplementary Material
References
- Judson R. S.; Houck K. A.; Kavlock R. J.; Knudsen T. B.; Martin M. T.; Mortensen H. M.; Reif D. M.; Rotroff D. M.; Shah I.; Richard A. M.; Dix D. J. (2010) In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environ. Health Perspect. 118, 485–492. 10.1289/ehp.0901392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock R.; Dix D. (2010) Computational toxicology as implemented by the U.S. EPA: providing high throughput decision support tools for screening and assessing chemical exposure, hazard and risk. J. Toxicol. Environ. Health, Part B 13, 197–217. 10.1080/10937404.2010.483935. [DOI] [PubMed] [Google Scholar]
- Abdelaziz A.; Spahn-Langguth H.; Werner-Schramm K.; Tetko I. V. (2016) Consensus Modeling for HTS Assays Using In silico Descriptors Calculates the Best Balanced Accuracy in Tox21 Challenge. Front. Environ. Sci. 4, E2. 10.3389/fenvs.2016.00002. [DOI] [Google Scholar]
- Abdelaziz A.; Sushko Y.; Novotarskyi S.; Korner R.; Brandmaier S.; Tetko I. V. (2015) Using Online Tool (iPrior) for Modeling ToxCast Assays Towards Prioritization of Animal Toxicity Testing. Comb. Chem. High Throughput Screening 18, 420–438. 10.2174/1386207318666150305155255. [DOI] [PubMed] [Google Scholar]
- TopCoder Data Science Competition Platform. http://www.topcoder.com/epa/toxcast/ (March 6, 2016).
- Prediction challenge. http://www.epa.gov/ncct/challenges.html (March 6, 2016).
- Archives of the challenge. http://web.archive.org/web/20150616141428/http://www.epa.gov/ncct/challenges.html (March 6, 2016).
- Sushko I.; Novotarskyi S.; Korner R.; Pandey A. K.; Rupp M.; Teetz W.; Brandmaier S.; Abdelaziz A.; Prokopenko V. V.; Tanchuk V. Y.; Todeschini R.; Varnek A.; Marcou G.; Ertl P.; Potemkin V.; Grishina M.; Gasteiger J.; Schwab C.; Baskin I. I.; Palyulin V. A.; Radchenko E. V.; Welsh W. J.; Kholodovych V.; Chekmarev D.; Cherkasov A.; Aires-de-Sousa J.; Zhang Q. Y.; Bender A.; Nigsch F.; Patiny L.; Williams A.; Tkachenko V.; Tetko I. V. (2011) Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J. Comput.-Aided Mol. Des. 25, 533–554. 10.1007/s10822-011-9440-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall L. H.; Kier L. B. (1995) Electrotopological State Indexes for Atom Types - a Novel Combination of Electronic, Topological, and Valence State Information. J. Chem. Inf. Model. 35, 1039–1045. 10.1021/ci00028a014. [DOI] [Google Scholar]
- Vidal D.; Thormann M.; Pons M. (2005) LINGO, an efficient holographic text based method to calculate biophysical properties and intermolecular similarities. J. Chem. Inf. Model. 45, 386–393. 10.1021/ci0496797. [DOI] [PubMed] [Google Scholar]
- Varnek A.; Fourches D.; Horvath D.; Klimchuk O.; Gaudin C.; Vayer P.; Solov’ev V.; Hoonakker F.; Tetko I. V.; Marcou G. (2008) ISIDA - Platform for virtual screening based on fragment and pharmacophoric descriptors. Curr. Comput.-Aided Drug Des. 4, 191–198. 10.2174/157340908785747465. [DOI] [Google Scholar]
- Skvortsova M. I.; Baskin I. I.; Skvortsov L. A.; Palyulin V. A.; Zefirov N. S.; Stankevich I. V. (1999) Chemical graphs and their basis invariants. J. Mol. Struct.: THEOCHEM 466, 211–217. 10.1016/S0166-1280(98)00467-9. [DOI] [Google Scholar]
- Cherkasov A. (2005) ’Inductive’ Descriptors: 10 Successful Years in QSAR. Curr. Comput.-Aided Drug Des. 1, 21–42. 10.2174/1573409052952288. [DOI] [Google Scholar]
- OCHEM User’s Manual. http://docs.ochem.eu/display/MAN/OCHEM+Introduction (March 6, 2016).
- Gasteiger J. (2006) Of molecules and humans. J. Med. Chem. 49, 6429–6434. 10.1021/jm0608964. [DOI] [PubMed] [Google Scholar]
- Potemkin V. A.; Grishina M. A. (2008) A new paradigm for pattern recognition of drugs. J. Comput.-Aided Mol. Des. 22, 489–505. 10.1007/s10822-008-9203-x. [DOI] [PubMed] [Google Scholar]
- Steinbeck C.; Han Y.; Kuhn S.; Horlacher O.; Luttmann E.; Willighagen E. (2003) The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 43, 493–500. 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todeschini R., and Consonni V. (2000) Handbook of Molecular Descriptors, WILEY-VCH, Weinheim, Germany. [Google Scholar]
- Sadowski J.; Gasteiger J.; Klebe G. (1994) Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Model. 34, 1000–1008. 10.1021/ci00020a039. [DOI] [Google Scholar]
- Tetko I. V.; Sushko I.; Pandey A. K.; Zhu H.; Tropsha A.; Papa E.; Oberg T.; Todeschini R.; Fourches D.; Varnek A. (2008) Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 48, 1733–1746. 10.1021/ci800151m. [DOI] [PubMed] [Google Scholar]
- Rybacka A.; Ruden C.; Tetko I. V.; Andersson P. L. (2015) Identifying potential endocrine disruptors among industrial chemicals and their metabolites - development and evaluation of in silico tools. Chemosphere 139, 372–378. 10.1016/j.chemosphere.2015.07.036. [DOI] [PubMed] [Google Scholar]
- Nizami B.; Tetko I. V.; Koorbanally N. A.; Honarparvar B. (2015) QSAR models and scaffold-based analysis of non-nucleoside HIV RT inhibitors. Chemom. Intell. Lab. Syst. 148, 134–144. 10.1016/j.chemolab.2015.09.011. [DOI] [Google Scholar]
- Vorberg S.; Tetko I. V. (2014) Modeling the Biodegradability of Chemical Compounds Using the Online CHEmical Modeling Environment (OCHEM). Mol. Inf. 33, 73–85. 10.1002/minf.201300030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko I. V.; Novotarskyi S.; Sushko I.; Ivanov V.; Petrenko A. E.; Dieden R.; Lebon F.; Mathieu B. (2013) Development of dimethyl sulfoxide solubility models using 163 000 molecules: using a domain applicability metric to select more reliable predictions. J. Chem. Inf. Model. 53, 1990–2000. 10.1021/ci400213d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotarskyi S.; Sushko I.; Korner R.; Pandey A. K.; Tetko I. V. (2011) A comparison of different QSAR approaches to modeling CYP450 1A2 inhibition. J. Chem. Inf. Model. 51, 1271–1280. 10.1021/ci200091h. [DOI] [PubMed] [Google Scholar]
- Sushko I.; Novotarskyi S.; Körner R.; Pandey A. K.; Kovalishyn V. V.; Prokopenko V. V.; Tetko I. V. (2010) Applicability domain for in silico models to achieve accuracy of experimental measurements. J. Chemom. 24, 202–208. 10.1002/cem.1296. [DOI] [Google Scholar]
- Sushko I.; Novotarskyi S.; Korner R.; Pandey A. K.; Cherkasov A.; Li J.; Gramatica P.; Hansen K.; Schroeter T.; Muller K. R.; Xi L.; Liu H.; Yao X.; Oberg T.; Hormozdiari F.; Dao P.; Sahinalp C.; Todeschini R.; Polishchuk P.; Artemenko A.; Kuz’min V.; Martin T. M.; Young D. M.; Fourches D.; Muratov E.; Tropsha A.; Baskin I.; Horvath D.; Marcou G.; Muller C.; Varnek A.; Prokopenko V. V.; Tetko I. V. (2010) Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model. 50, 2094–2111. 10.1021/ci100253r. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Varbanov H. P.; Galanski M.; Talmaciu M.; Platts J. A.; Ravera M.; Gabano E. (2016) Prediction of logP for Pt(II) and Pt(IV) complexes: Comparison of statistical and quantum-chemistry based approaches. J. Inorg. Biochem. 156, 1–13. 10.1016/j.jinorgbio.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Jaroszewicz I.; Platts J. A.; Kuduk-Jaworska J. (2008) Calculation of lipophilicity for Pt(II) complexes: Experimental comparison of several methods. J. Inorg. Biochem. 102, 1424–1437. 10.1016/j.jinorgbio.2007.12.029. [DOI] [PubMed] [Google Scholar]
- Tollenaere T. (1990) SuperSAB: Fast adaptive back propagation with good scaling properties. Neural Netw. 3, 561–573. 10.1016/0893-6080(90)90006-7. [DOI] [Google Scholar]
- Cortes C.; Vapnik V. (1995) Support-vector networks. Machine Learn. 20, 273–297. 10.1007/BF00994018. [DOI] [Google Scholar]
- Chang C.-C.; Lin C.-J. (2011) LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2 2 (3), 1–27. 10.1145/1961189.1961199. [DOI] [Google Scholar]
- Eriksson L., Johansson E., Kettaneh-Wold N., and Wold S. (2001) Multi- and Megavariate Data Analysis: Principles and Applications, Umetrics, Umeå, Sweden. [Google Scholar]
- Breiman L. (1996) Bagging Predictors. Machine Learn. 24, 123–140. 10.1007/BF00058655. [DOI] [Google Scholar]
- Reed R. (1993) Pruning algorithms-a survey. IEEE Transactions on Neural Networks 4, 740–747. 10.1109/72.248452. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Tanchuk V. Y.; Chentsova N. P.; Antonenko S. V.; Poda G. I.; Kukhar V. P.; Luik A. I. (1994) HIV-1 reverse transcriptase inhibitor design using artificial neural networks. J. Med. Chem. 37, 2520–2526. 10.1021/jm00042a005. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Villa A. E.; Livingstone D. J. (1996) Neural network studies . 2. Variable selection. J. Chem. Inf. Comput. Sci. 36, 794–803. 10.1021/ci950204c. [DOI] [PubMed] [Google Scholar]
- Kovalishyn V. V.; Tetko I. V.; Luik A. I.; Kholodovych V. V.; Villa A. E. P.; Livingstone D. J. (1998) Neural network studies. 3. Variable selection in the cascade-correlation learning architecture. J. Chem. Inf. Comput. Sci. 38, 651–659. 10.1021/ci980325n. [DOI] [Google Scholar]
- Tetko I. V.; Villa A. E.; Aksenova T. I.; Zielinski W. L.; Brower J.; Collantes E. R.; Welsh W. J. (1998) Application of a pruning algorithm to optimize artificial neural networks for pharmaceutical fingerprinting. J. Chem. Inf. Comput. Sci. 38, 660–668. 10.1021/ci970439j. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Livingstone D. J.; Luik A. I. (1995) Neural network studies. 1. Comparison of overfitting and overtraining. J. Chem. Inf. Model. 35, 826–833. 10.1021/ci00027a006. [DOI] [Google Scholar]
- Tetko I. V.; Villa A. E. P. (1997) Efficient partition of learning data sets for neural network training. Neural Netw 10, 1361–1374. 10.1016/S0893-6080(97)00005-1. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Sushko Y.; Novotarskyi S.; Patiny L.; Kondratov I.; Petrenko A. E.; Charochkina L.; Asiri A. M. (2014) How Accurately Can We Predict the Melting Points of Drug-like Compounds?. J. Chem. Inf. Model. 54, 3320–3329. 10.1021/ci5005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmina E. S.; Haider N.; Tetko I. V. (2016) Extended Functional Groups (EFG): An Efficient Set for Chemical Characterization and Structure-Activity Relationship Studies of Chemical Compounds. Molecules 21, E1. 10.3390/molecules21010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko I. V., Baskin I. I., and Varnek A. (2008) Tutorial 2b. Descriptor Selection Bias, Strasbourg Summer School on Chemoinformatics: CheminfoS3, Obernai, France. [Google Scholar]
- Baskin I. I.; Winkler D. A.; Tetko I. V.. A renaissance of neural networks in drug discovery. Expert Opin. Drug Discovery 2016, In press [DOI] [PubMed]
- ToxCast challenge. http://tripod.nih.gov/tox21/challenge (March 6, 2016).
- Mansouri K.; Abdelaziz A.; Rybacka A.; Roncaglioni A.; Tropsha A.; Varnek A.; Zakharov A.; Worth A.; Richard A. M.; Grulke C. M.; Trisciuzzi D.; Fourches D.; Horvath D.; Benfenati E.; Muratov E.; Wedebye E. B.; Grisoni F.; Mangiatordi G. F.; Incisivo G. M.; Hong H.; Ng H. W.; Tetko I. V.; Balabin I.; Kancherla J.; Shen J.; Burton J.; Nicklaus M.; Cassotti M.; Nikolov N. G.; Nicolotti O.; Andersson P. L.; Zang Q.; Politi R.; Beger R. D.; Todeschini R.; Huang R.; Farag S.; Rosenberg S. A.; Slavov S.; Hu X.; Judson R. S. (2016) CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect. 10.1289/ehp.1510267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko I. V. (2012) The perspectives of computational chemistry modeling. J. Comput.-Aided Mol. Des. 26, 135–136. 10.1007/s10822-011-9513-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.