

Abstract
Helicobacter pylori (H. pylori) is a Gram-negative and motile bacterium that colonizes the hostile microniche of the human stomach, then persists for the host’s entire life, if not effectively treated. Clinically, H. pylori plays a causative role in the development of a wide spectrum of diseases including chronic active gastritis, peptic ulceration, gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue lymphoma. Due to the global distribution of H. pylori, it is no exaggeration to conclude that smart strategies are contributing to adaptation of the bacterium to its permanent host. Thirty-four years after the discovery of this bacterium, there are still many unanswered questions. For example, which strategies help the bacterium to survive in this inhospitable microniche? This question is slightly easier to answer if we presume the same clinical concept for both persistent infection and disease. Understanding the mechanisms governing H. pylori persistence will improve identification of the increased risk of diseases such as gastric cancer in patients infected with this bacterium. A well-defined and long-term equilibrium between the human host and H. pylori allows bacterial persistence in the gastric microniche; although this coexistence leads to a high risk of severe diseases such as gastric cancer. To escape the bactericidal activity of stomach acid, H. pylori secretes large amounts of surface-associated and cytosolic urease. The potential to avoid acidic conditions and immune evasion are discussed in order to explain the persistence of H. pylori colonization in the gastric mucosa, and data on bacterial genetic diversity are included. Information on the mechanisms related to H. pylori persistence can also provide the direction for future research concerning effective therapy and management of gastroduodenal disorders. The topics presented in the current review are important for elucidating the strategies used by H. pylori to help the bacterium persist in relation to the immune system and the many unfavorable features of living in the gastric microniche.
Keywords: Helicobacter pylori, Gastric microniche, Persistent infection, Gastric cancer
Core tip: Helicobacter pylori (H. pylori) infections are thought to cause severe gastroduodenal disorders which are the main outcome of this interaction between the host and this microorganism. The bacterium has developed interesting strategies which allow it to efficiently colonize the gastric mucosa leading to persistent infection. In this review, we aim to look at the five major reported strategies leading to the persistent survival of H. pylori within gastric epithelial cells.
INTRODUCTION
The groundbreaking discovery of a Gram-negative bacterium by Marshall and Warren changed the gastroenterologists’ view regarding the historic dogma that the stomach was a sterile organ[1]. Phylogenetic analysis emphasized the global distribution of the bacterium from East Africa to other continents thousands of years ago[2-5]. Helicobacter pylori (H. pylori) which was previously called Campylobacter pyloridis is mostly acquired during childhood and it has been genetically proved that it can be transmitted within families[4]. H. pylori is a ε-proteobacterium that colonizes the human stomach with patchy distribution and is mainly found in the antrum rather than the fundus[6]. Many published international guidelines recommend treatment of the bacterium with effective antibiotics[7-11]. Both in vitro and in vivo evidence in addition to epidemiologic data have confirmed that H. pylori is the main cause of both gastric and duodenal ulcers, gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue (MALT) lymphoma[12-15]. Due to the high prevalence of H. pylori worldwide and success of this long-term commensalism, it is no exaggeration to assume that complex strategies are involved in efficient colonization in the host. Virulence factors involved in the establishment of a persistent infection are shown in Table 1. As shown, an adhesion or secreted protein can be a determining factor in successful persistent colonization. This concept raises a challenging question concerning how H. pylori can persistently colonize the stomach in such unfavorable conditions. In the current review, we discuss the five observed strategies leading to persistent survival within gastric epithelial cells.
Table 1.
Major virulence factors involved in the persistence of Helicobacter pylori
| Virulence factors | Roles in persistent infection |
| OipA | Induces pro-inflammatory cytokines |
| In combination with CagA, causes disruption of cell tight junctions | |
| VacA | Increases IL8 expression and causes inflammation |
| Contributes to long-term colonization | |
| Cell proliferation and elongation | |
| Increases the level of MAPK signaling | |
| CagA | Stimulates the NF-κB pathway |
| Induces apoptosis | |
| Induces pro-inflammatory cytokines | |
| Disrupts cell tight junctions | |
| Increases mucosal inflammation | |
| BabA | Leads to effective cell adherence |
| Mediates in the effective interaction between H. pylori and epithelial cells | |
| DupA | Induces pro-inflammatory cytokines |
| Induces apoptosis | |
| GGT | Contributes to long-term gastric colonization |
| Induces immune response tolerance | |
| SabA | Mediates the effective interaction between H. pylori and epithelial cells |
MAPK: Mitogen-activated protein kinase; NF-κB: Nuclear factor-κB; H. pylori: Helicobacter pylori.
FIRST STRATEGY: ESCAPING GASTRIC ACIDITY
The human gastric lumen is known to consist of a large variety of physical and biochemical barriers to provide an unfavorable environment for all types of ingested microbes[16]. Successful gastric colonization by any microorganism in this microniche is due to the existence of various abilities to avoid hostile acidic conditions and the viscous mucosal layer[17,18]. To do this, H. pylori harbors specific features leading to successful colonization. In the following paragraphs, we discuss how this bacterium can cause persistent infection with a focus on acidity avoidance.
UREASE AS THE FIRST WEAPON AGAINST ACIDITY
The acidity of the stomach (pH 1-2) is the first danger to threaten bacterial survival in the stomach[14,17,19]. H. pylori is able to survive at approximately pH 5 which can kill many digested organisms within a few minutes after acid exposure. This is the main adaptive feature of this bacterium which facilitates its survival in the gastric microniche. H. pylori growth is limited in neutral pH, but can survive in acidic pH due to an increase in periplasmic pH and secretion of a large amount of urease enzyme[18,20]. Urease is a key feature in the capacity of H. pylori to avoid gastric acidity. To avoid the bactericidal activity of acid, H. pylori secretes large amounts of surface-associated and cytosolic urease[6]. However, hydrolysis of urea (occurring in the gastric lumen) produces large amounts of ammonia, which is then protonated to create ammonium, thus creating a neutral bacterial microniche[21]. In the presence of urea, pH is shifted to 2-6 and H. pylori can survive in the gastric microniche. In contrast, in the absence of urea, H. pylori can grow in the pH range of 6-8 and can even survive in the pH range of 4-8.5. Urea is imported through a specific channel, and with urease, ammonia will fill the entire cytosol and even bacterial periplasm[22,23]. H. pylori urease gene cluster consists of two major genes, ureA and ureB, and five accessory genes, ureI, ureE, ureF, ureG, and ureH. These accessory subunits are associated with urea absorption[16,18,21,24]. Urease is a nickel-binding hexamer of UreA and UreB. It has been reported that H. pylori can change the expression of certain genes (associated with the synthesis of ammonium ion) including arginase, urease and formamidase after exposure to hostile gastric acidic conditions[18]. The production of urease in 10% of bacterial proteomes enables it to hydrolyze large amounts of gastric urea to generate ammonia and CO2, causing a sharp increase in the pH around H. pylori[25]. However, in response to low pH by the ArsSR two-component system, production of ammonia can be continued by formamidase in addition to urease by the hydrolysis of short-chain amino acids, while arginase hydrolyzes L-arginine to L-ornithine and urea[26,27]. To establish persistent infection, after entering the gastric lumen, the bacterium should be able to penetrate and infiltrate the viscous mucosal layer[28,29]. Interestingly, mucosal viscosity is highly dependent on acidity. In general, in more acidic pH, mucus is stronger than at pH 4. Enzymatic activity governed by H. pylori facilitates bacterial penetration into the aggregated mucus. Subsequent diffusion of ammonia in the stomach provides an opportunity for H. pylori to penetrate deeply using corkscrew propulsion due to its flagella. The helical shape of the bacterium is another factor affecting its successful evasion of acid[30]. Due to low permeability of the mucosal layer, there is a scarcity of essential nutrients (for example Fe3+) in the stomach for all ingested microorganisms[13,16,18,21,22]. The release of large amounts of urease is a smart strategy adopted by H. pylori to weaken the integrity of the mucus to facilitate deep penetration to reach the nutrient enriched region. Moreover, moving into deeper layers gives the bacterium the opportunity to avoid the acidic conditions on the surface of the lumen; another strategy which increases the viability of this bacterium[6,13,31]. Thus, H. pylori would not survive in the stomach without its potential to escape high acidity which is not observed in any other microorganisms.
Intracellular or extracellular life
During the time research had been undertaken on H. pylori colonization in the human stomach, no clear evidence has been published to indicate that H. pylori is an invasive organism in the human gastric microniche[4,6,19,32]. Following the assessment of a large number of in vivo and in vitro studies, we suggest that H. pylori is a noninvasive gastric pathogen[33,34]. Interestingly, H. pylori is able to repopulate the extracellular environment following bacterial clearance (for example with effective antibiotics) of the gastric lumen, suggesting it may be better described as a facultative intracellular microbe[35]. To date, no direct evidence on H. pylori replication in gastric cells has been reported[36]. However, despite the progress already achieved, more in vitro experiments are required before we can conclude that the intracellular effect of H. pylori is a strategy involved in long-term persistence in gastric epithelial cells. However, it can be assumed that H. pylori is a facultative intracellular bacterium rather than an extracellular organism. As a temporary strategy to avoid chemical and physical barriers, H. pylori can transiently enter the intracellular space for a short time to reduce exposure duration to antibiotics, acidic conditions and the immune response[37]. Moreover, as H. pylori is optimally evolved to reduce its exposure to acidic gastric conditions, it can localize in mucus close to the epithelial surface where acidity is tolerable for longer survival. This strategy adopted by the microbe further highlights the importance of research in finding effective antibiotics to treat this organism.
Direct contact with host cells
By involving various surface proteins, H. pylori can bind to epithelial cells[38]. BabA is a major outer membrane protein and is one of these contributing proteins (Figures 1, 2 and 3)[39,40]. Interestingly, there are other adhesion molecules which facilitate attachment of the bacterium to epithelial cells[41]. Bacterial secreted lectin is another binding factor which binds to the sialic residues of laminin[42]. A cascade pathway allows the successful irreversible attachment of H. pylori to polarized epithelial cells (Figure 2)[43]. Small cytoplasmic changes can be observed following direct contact between H. pylori and host cells. Following first contact of the bacterium with the plasma membrane and rearrangement of the cellular cytoskeleton, an extension of the plasma membrane can engulf a part of the bacterium[38]. Undoubtedly, certain bacterial factors such as cagA and dupA are involved in the above processes; however, data regarding details of this contribution are lacking[44]. Interestingly, it was noted that only a small fraction of available H. pylori strains in the gastric mucosa can attach to the surface. Although H. pylori adherence is mediated by candidate surface proteins (e.g., AlpA, AlpB, DupA, BabA, OipA, SabA and HopZ), no specific molecule has been shown to be essential in adhesive mechanisms[45,46]. For example, H. pylori SabA supports the release of nutrients in the apical side of epithelial cells for bacterial survival in the stomach. This nutrient release is in parallel with translocated CagA protein which can induce inflammation in infected cells[47]. Different expression of adhesion molecules occurs among H. pylori strains suggesting various adaptations induced by genetic recombination, on/off switching of gene expression. It is clear that bacterial attachment is an inevitable step in the establishment of H. pylori infection, although determining the molecules involved is not easy. New technologies in the near future will reveal further details of the complex initial process in persistent H. pylori infection.
Figure 1.
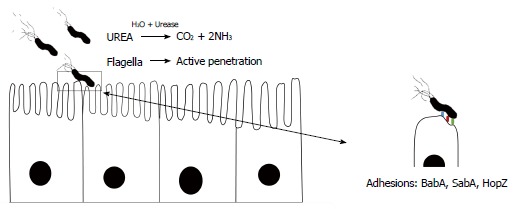
Basic overview of the events leading to the successful colonization of Helicobacter pylori.
Figure 2.
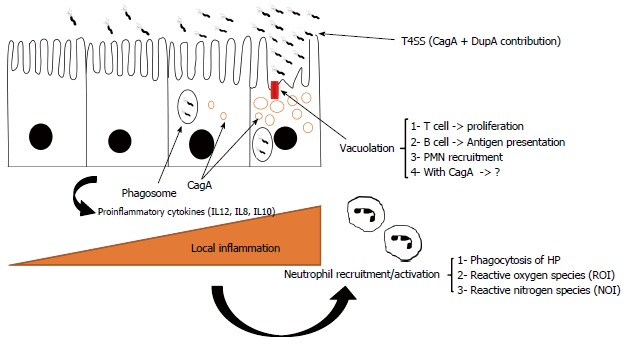
Subsequent activated events causing inflammation in chronic infection by Helicobacter pylori.
Figure 3.
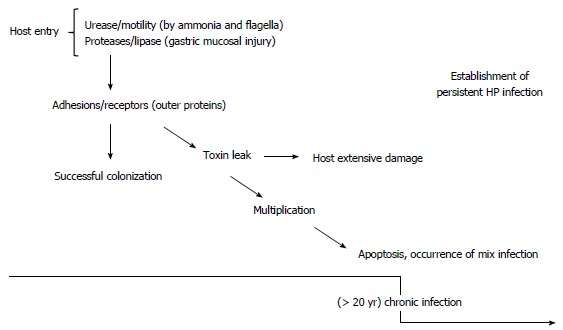
Flowchart of the strategies involved in persistent infection.
SECOND STRATEGY: HELICAL SHAPE
The helical shape of H. pylori has been suggested to provide a mechanical advantage for penetrating the viscous stomach mucus layer. Special cross-linkages in bacterial peptidoglycan can result in an efficient screw-like movement to facilitate H. pylori survival in the viscous environment of the stomach[48]. Consistent with this, H. pylori typically possesses a helical morphology and multiple uni-polar flagella. In an animal model, H. pylori cell shape mutants showed impaired stomach colonization indicating the importance of bacterial shape in motility and adherence[49]. The combination of a curved structure and cell elongation provides H. pylori with a suitable helical body shape. To date, our knowledge indicates that a certain number of genes such as Ccrp89, Ccrp58, Ccrp1142 and Ccrp1143 are involved in H. pylori morphology[48]. Thus, any mutations in these genes can cause a deficiency in bacterial motility and shape[50]. It should be noted that the helical shape in addition to flagella are a major part of the successful penetration process.
THIRD STRATEGY: THE PRESENCE OF FLAGELLA (MOTILITY)
To avoid elimination by regular gastric turnover, persistent infection is dependent on bacterial movement. H. pylori is mostly distributed within approximately 30 μm of mucosal epithelial cells and its histopathologic effects can be observed within the gastric crypts. During the last decade, H. pylori flagella have been widely investigated, and current knowledge demonstrates the critical role of these organelles in the colonization and establishment of persistent infection[43]. Extensive isogenic mutant studies have shown the role of intact flagellar apparatus in governing H. pylori infection, especially long-term infection[51]. H. pylori is an actively motile organism which has the ability to move between different regions of the stomach. Furthermore, flagella give the bacterium the potential for penetration which is necessary to avoid acidic pressure on the surface of the gastric mucosa[52]. Due to the combination of more than 39 genes in the H. pylori genome, 3-5 tuft-like flagella appear on the side of the bacterium. These flagella consist of a basal body, hook, and flagellar filament[53]. The deletion or any other changes in the genes related to the flagella can produce non-motile H. pylori which is unable to effectively colonize gastric mucosa[54]. In order to move both in counterclockwise (CCW) and clockwise (CW) directions, H. pylori requires a rotating movement around the filament[55]. The existence of polar flagella in H. pylori is a requirement for persistent infection. Following entry into the stomach, the bacterium has a durable acid-tolerance range[56]. H. pylori is tolerant to weakly acidic conditions in the stomach which occur after ingestion. Given that ammonia is produced by bacterial urease, the rheologic properties of the gastric mucin changes allowing penetration of H. pylori into the gastric mucosa using flagellated motility and its helical shape. The detailed mechanism by which H. pylori weakens mucus viscosity is not fully understood, but we now know that loss of tightness on the side of polarized epithelial cells may be involved leading to several gastroduodenal diseases. Thus, movement of H. pylori is necessary in order to attach to epithelial cells[57]. Taken together, these findings show that the presence of flagella is useful for H. pylori to produce infection in the gastric mucosa, and urease is an essential enzyme guaranteeing successful bacterial penetration.
FOURTH STRATEGY: H. PYLORI EVASION OF THE HUMAN IMMUNE SYSTEM
During the relatively long time that H. pylori has been recognized, it is clear that long-term bacterial persistence is necessary to maintain successful existence of H. pylori against both innate and adaptive immune responses (Figure 4)[57,58]. Following successful colonization of gastric mucosal cells by H. pylori, host immune responses (both innate and acquired branches) are stimulated generating specific antibodies and activated Th cells. Thus, H. pylori is required to avoid both branches of the immune system to colonize the stomach for several years[59]. In addition to long-term infection, other diseases are caused by this microorganism. Any digestive diseases related to prolonged colonization by H. pylori can be due to adaptation between the microbe and human host[60]. We now know that subsequent to colonization by H. pylori, a percentage of the infected population can develop autoimmunity or immune system abnormalities[61-64]. These findings show bilateral effects between H. pylori and the human immune system. During the thousands of years of commensalism between humans and H. pylori, the bacterium has learned to subvert the immune response and it may serve as an example of bacterial evolution in maintaining its micro-territory in humans[62]. Taken together, these findings suggest that we should use the word “equilibrium” to define this status. In this equilibrium, H. pylori induces immune cells which cause minimum cell damage to shed nutrients onto the surface of the gastric mucosa for survival[32]. Cell damage following local superficial gastritis occurs, but seems necessary for governing long-term colonization as the infection can cause minimum nutrient shedding which is required for bacterial survival in the harsh stomach environment. It should be noted that the microbe will be immediately eliminated by active immune cells[65]. In conclusion, bacterial persistence only occurs if H. pylori and the immune system co-adapt following successful immune evasion. In the next section, we briefly describe how H. pylori effectively avoids innate and adaptive immune responses.
Figure 4.
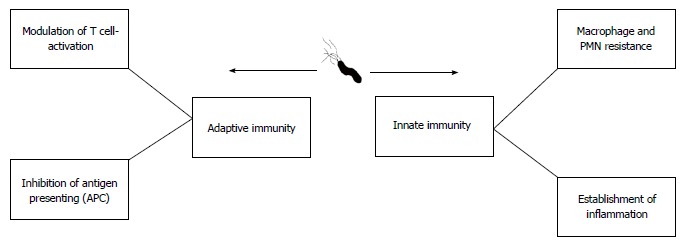
Different impacts of Helicobacter pylori on the two branches of the human immune system.
MECHANISMS OF IMMUNE EVASION BY H. PYLORI - INNATE IMMUNITY
Both components of the innate system (chemical and physical barriers) are involved in immune system responses against H. pylori in the stomach[66]. The gastric epithelial surface plays an active role in mucosal defense[67]. Immediately after entering the gastric epithelial cells, two major actions by the innate immune system work against H. pylori: (1) engulfment by phagocytes; and (2) chemical products of these cells include reactive nitrogen and oxygen[68,69]. Below, we focus on three different strategies adopted by H. pylori which facilitate establishment of persistent chronic infection in the gastric epithelium.
EVASION OF PHAGOCYTE KILLERS
Although H. pylori is usually phagocytosed by macrophages, in comparison to other Gram negative microbes, H. pylori is highly resistant to killing by these cells during phagocytosis[70-73]. A virulent H. pylori strain (type I: cagA + vacA+) is more resistant to phagocytosis than other strains[74-76]. Accumulation of the bacterium in vesicles which produce megasomes is the main evidence for delayed phagocytosis[77,78]. Macrophage apoptosis can result in H. pylori escaping phagosomes[79]. As mentioned previously, H. pylori induces inflammation to provide nutrients, thus inflammation may be present for months or years. Therefore, H. pylori needs to be well prepared to confront large numbers of phagocytes in the stomach due to the high rate of cell damage.
There are three proposed strategies which can help the bacterium to survive in this phagocyte-rich environment: (1) H. pylori induces mitochondrial-associated cell death in macrophages (mitochondrial dependent apoptosis)[80,81]; and (2) production of peroxiredoxin by AhpC gene actively defends the bacterium against NO products available in the gastric microniche[82-85].
Similarly, H. pylori arginase due to substrate competition can reduce NO or O2- radicals[86]. In vitro studies have shown that H. pylori activates macrophage nitric oxide synthase and thus apoptosis[87-89]. It has been shown that arginase-deficient bacteria are more sensitive to oxidative stress caused by macrophages[90,91]. Finally, H. pylori has adopted a smart strategy to inhibit polymorphonuclear neutrophil (PMN) and monocyte activation by involving CagA proteins (type IV secretion system)[92]. Furthermore, cagA positive strains produce more IL8, a trigger cytokine for inducing infiltration of inflammatory cells. Isogenic mutant experiments suggest the participation of VacA toxins in escape of the microbe from phagosomes[93]. Subsequent deactivation of PMNs and monocytes can cause interference in the innate immune response which is favorable for H. pylori[94]. An alteration in phagocyte function is the main mission of H. pylori to facilitate persistent infection. Therefore, there are undoubtedly other mechanisms involved in the persistence of H. pylori against phagocytosis[95,96]. Further in vitro and in vivo experiments are necessary to fully elucidate the complex interactions in chronic H. pylori infection.
ADAPTIVE IMMUNITY
A successful pathogenic organism which aims to overcome innate immunity to establish colonization in an ecological niche leading to chronic infection should possess strategies to overcome adaptive immunity. H. pylori modulates adaptive immunity by affecting T reg and T cell proliferation[97,98]. In the next paragraph, we describe the major reactions of H. pylori to adaptive immunity which include: (1) enhanced T and B cell functions; and (2) interference in the antigen presentation process.
EFFECTS ON B AND T CELLS AND THEIR PROLIFERATION
Recent data have demonstrated that H. pylori can modulate T cell responses[99-102]. Elegant in vivo experiments suggested that CD4 T lymphocytes are highly involved in the adaptive immune response against H. pylori, in contrast to the relatively inactive role of CD8 T cells. As shown previously, H. pylori stimulates the immune response using Th1[101,103]. The main interference due to H. pylori in the adaptive immune response is its inhibitory effect on T cell proliferation via three mechanisms: (1) binding the VacA toxin to the unknown surface ligand in T cells which results in actin rearrangement and then inhibition of cell proliferation[74,103]; (2) the VacA toxin is able to form an anion-selective channel in the host cell membrane. These specific channels can cause vacuoles in host cells leading to apoptosis[104]. This event starts following attachment of VacA to surface receptor alpha (RPTPα)[105]; and (3) VacA can bind to mitochondria and trigger the associated apoptotic pathway. This mechanism can induce inhibition of T cell proliferation. Moreover, VacA located in the membrane of anionic channels inhibits the release of cytochrome C, indicating the crucial role of channel formation in apoptosis induced by H. pylori VacA. The exact apoptotic role of VacA needs to be elucidated in the future. We noted that the antibody response is not sufficient to eliminate H. pylori infection in humans, as the bacterium has developed strategies to protect itself from the effects of antibodies[106]. Unfortunately, the data concerning the actual role of any specific bacterial product to inhibit B cell response are lacking although some experiments have shown that Janus kinase (JAK)-STAT (signal transducer and activator of transcription) suppression by CagA may be a major mechanism in reducing effective B cell response[107].
INTERFERENCE IN THE ANTIGEN PRESENTATION PROCESS
H. pylori has adopted various strategies to interrupt accurate antigen presentation in the host, which is the main method of interfering with late endocytic membrane trafficking. Inhibition of APC activation may be the key point in reducing B cell response to colonization of H. pylori in humans[108]. Inhibition of MHC-class II export to the cell surface is the main reason for impaired antigen presentation in dendritic cells[109,110]. In a novel study, the inhibitory effect of CagA on IL3-dependent B cell proliferation was reported to be via the JAK-STAT signaling pathway, which can cause ineffective antibody secretion in humans[111,112]. JAK/STAT signaling is an important immune signaling pathway. Certain inflammatory molecules can activate different types of JAK and STAT. Phosphorylated STAT generates dimers that translocate to the nucleus, thereafter its binding to the promoter of specific genes involved in immune inflammatory response can be stimulated[112,113]. As mentioned previously, data regarding the actual role of H. pylori in limiting B cell response are scarce and more detailed studies are required to elucidate the complex mechanism responsible for the establishment of persistent infection[114].
SUBVERSION OF PATTERN RECOGNITION
Different Toll-like receptors (TLRs) corresponding to microbial components called pathogen-associated microbial patterns (PAMPs) are the main components which provide a new strategy for H. pylori to avoid involvement with the human immune system. Any interruption of this complex process can hamper bacterial clearance by the immune system. We now know that successful recognition of pathogens by TLRs is involved in both the innate and adaptive systems[115-118]. Subversion of TLRs is dependent on recognition by H. pylori which helps the bacterium to survive in the strongly immune cell-rich gastric microniche[119-122]. TLR4-mediated inflammatory response to LPS in H. pylori is largely different to other organisms (almost 1000-fold less reactogenic than other Gram negative bacteria)[123-129]. Indeed, co-evolution with humans has resulted in a reduction in immunogenic ligands[130]. TLR-dependent immune subversion is a strategy that works in both innate and adaptive immune responses to H. pylori; therefore, this adopted strategy should have an effect in hiding the bacterium in the human gastric microniche.
FIFTH STRATEGY: H. PYLORI GENETIC DIVERSITY: FACILITATING PERSISTENT INFECTION
The bacterium is relatively competent in DNA uptake from other H. pylori strains. The diversity of the H. pylori strains even within a single host is impressive[131]. H. pylori is a highly heterogeneous bacterium with more than 1600 genes[12,58,68,69]. H. pylori is a highly competent at taking up both environmental DNA and DNA from other H. pylori strains. H. pylori was identified as being heterogeneous by a large amount of evidence from experiments investigating (1) allelic variation; (2) SNP; (3) genome size; and (4) bacterial recombination[21,132-134]. Genetic recombination is a very important factor involved in adaptation of H. pylori in the human stomach[135]. CagA is a classic virulence factor of the bacterium and has been isolated in different regions of the world (Figure 5). It can be seen from this figure that reported polymorphisms can directly affect its clinical importance. H. pylori is considered to be a genetically diverse organism infecting individuals around the world[19]. Its high potential for DNA uptake results in H. pylori having one of highest recombination rates reported in human pathogenic bacteria. This high rate of recombination causes high genomic variability among the bacterial strains[134]. Due to natural selection after these genetic changes, H. pylori is more persistent in the gastric microniche. However, it should be mentioned that in addition to genetic recombination, bacterial mutation can have a role in generating highly variable strains observed in persistent infections[136]. Almost 10% of its genome consists of specific genes for H. pylori[12]. Scientists have suggested that chronic persistent infection cannot occur independently. Comparative analyses of full genome sequences of H. pylori strains J99 and 26695 in addition to the large amount of data on new sequencing techniques provide new insight into bacterial diversity and its potential to infect humans life-long[137]. The H. pylori genome is not diverse, but various changes in the third base of codons has resulted in specific strains called “quasi-species”[133,138,139]. Currently, both antigenic and phase variation contribute to H. pylori persistence against immune responses[140,141]. Indeed, wide diversification in the H. pylori genome increases the chance of survival in the hostile gastric environment. In order to answer a direct question on the possible mechanism of bacterial diversity to establish chronic persistent infection we need to look at the biology of H. pylori. Clearly, bacteria such as H. pylori which is exposed to the human immune response and PMNs, it is crucial to promote strategies to avoid these unfavorable conditions. Apart from bacteria such as E. coli which have a large genome, H. pylori has half the genome of these bacteria. The main genetic deficiency in H. pylori is its two-component regulatory system[142]. Deficiencies in mismatch-repair systems of H. pylori can increase the frequency of genetic variation, and may facilitate minimal genetic diversity. Thus, H. pylori can take advantage of diversity in genetic sequences following selective pressure. To compensate for the lack of genetic contents in the regulatory systems, H. pylori has adopted the strategy of remaining life-long in the gastric microniche. This strategy means nonstop recombination and genetic change in the genome and then natural selection to choose the fittest organisms to adapt and live in the gastric environment. This adaptation can also be translated into successful bacterial immune evasion during H. pylori persistence.
Figure 5.
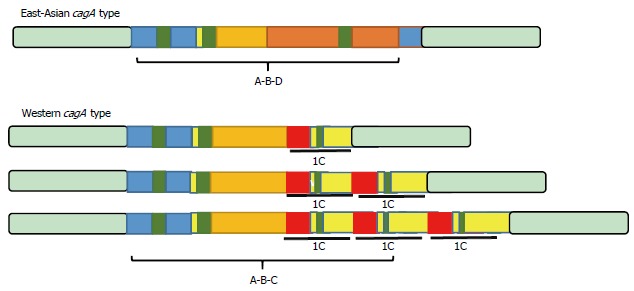
Polymorphism of the cagA gene reported in various regions of the world.
CONCLUSION
The beginning of the H. pylori era in 1984 was characterized by questions which require considerable attention; such as treatment, efficient vaccines, useful biomarkers and management of the infection[6,31,143]. For H. pylori, similar to most common gastric pathogens, we have information on its many molecular mechanisms, but its survival strategies in the human host are not well elucidated. The human stomach is a challenging niche for bacterial colonization; therefore, sophisticated strategies to aid survival of the bacterium would not be surprising. Bacterial genetic diversity and extensive polymorphism also facilitate the survival of H. pylori within the hostile gastric environment[14,63,144]. The potential for H. pylori to survive in the strong acidic conditions in the stomach is considered a major feature of bacterial persistence. Even with the five strategies described in this study, there is a big gap in the knowledge concerning the mechanisms involved in the persistent colonization of H. pylori. Initially, a useful buffering potential in addition to the presence of sufficient bacterial adhesion facilitate primary colonization and then continuous genetic diversification in parallel with immune evasion are the main strategies used for persistent infection. Despite intensive studies, the molecular host - H. pylori interactions, particularly in symptomatic patients colonized by persistent H. pylori are obscure. Understanding the details of H. pylori persistence and its contributing elements will help identify better strategies to treat the infection or may design better drug targets.
ACKNOWLEDGMENTS
The content of this review article is the sole responsibility of the author and necessarily represents a personal prospective. Moreover, the funding agencies had no role in the decision to publish, or preparation of the manuscript.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Iran
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: Author declares no conflict of interests for this article.
Peer-review started: December 6, 2016
First decision: January 10, 2017
Article in press: February 16, 2017
P- Reviewer: Bernal G, Manguso F, Koch TR S- Editor: Ma YJ L- Editor: Webster JR E- Editor: Wang CH
References
- 1.Abadi ATB, Ierardi E, Lee YY. Why do we still have Helicobacter Pylori in our Stomachs? Malays J Med Sci. 2015;22:70–75. [PMC free article] [PubMed] [Google Scholar]
- 2.Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlebusch CM, Skoglund P, Sjödin P, Gattepaille LM, Hernandez D, Jay F, Li S, De Jongh M, Singleton A, Blum MG, et al. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science. 2012;338:374–379. doi: 10.1126/science.1227721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Didelot X, Nell S, Yang I, Woltemate S, van der Merwe S, Suerbaum S. Genomic evolution and transmission of Helicobacter pylori in two South African families. Proc Natl Acad Sci USA. 2013;110:13880–13885. doi: 10.1073/pnas.1304681110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malfertheiner P, Mégraud F, O’Morain C, Hungin AP, Jones R, Axon A, Graham DY, Tytgat G. Current concepts in the management of Helicobacter pylori infection--the Maastricht 2-2000 Consensus Report. Aliment Pharmacol Ther. 2002;16:167–180. doi: 10.1046/j.1365-2036.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- 8.Malfertheiner P, Megraud F, O’Morain C, Bazzoli F, El-Omar E, Graham D, Hunt R, Rokkas T, Vakil N, Kuipers EJ. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut. 2007;56:772–781. doi: 10.1136/gut.2006.101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malfertheiner P, Megraud F, O’Morain CA, Atherton J, Axon AT, Bazzoli F, Gensini GF, Gisbert JP, Graham DY, Rokkas T, et al. Management of Helicobacter pylori infection--the Maastricht IV/ Florence Consensus Report. Gut. 2012;61:646–664. doi: 10.1136/gutjnl-2012-302084. [DOI] [PubMed] [Google Scholar]
- 10.Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808–1825. doi: 10.1111/j.1572-0241.2007.01393.x. [DOI] [PubMed] [Google Scholar]
- 11.Talebi Bezmin Abadi A. Helicobacter pylori treatment: New perspectives using current experience. J Glob Antimicrob Resist. 2017;8:123–130. doi: 10.1016/j.jgar.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 12.Yamaoka Y. Pathogenesis of Helicobacter pylori-Related Gastroduodenal Diseases from Molecular Epidemiological Studies. Gastroenterol Res Pract. 2012;2012:371503. doi: 10.1155/2012/371503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basso D, Plebani M, Kusters JG. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2010;15 Suppl 1:14–20. doi: 10.1111/j.1523-5378.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- 14.Abadi AT, Kusters JG. Management of Helicobacter pylori infections. BMC Gastroenterol. 2016;16:94. doi: 10.1186/s12876-016-0496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer F, Robbe-Saule M, Turlin E, Mancuso F, Michel V, Richaud P, Veyrier FJ, De Reuse H, Vinella D. Characterization in Helicobacter pylori of a Nickel Transporter Essential for Colonization That Was Acquired during Evolution by Gastric Helicobacter Species. PLoS Pathog. 2016;12:e1006018. doi: 10.1371/journal.ppat.1006018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. Acid-adaptive genes of Helicobacter pylori. Infect Immun. 2003;71:5921–5939. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson K, Letley DP, Kaneko K. The Human Stomach in Health and Disease: Infection Strategies by Helicobacter pylori. Curr Top Microbiol Immunol. 2017;400:1–26. doi: 10.1007/978-3-319-50520-6_1. [DOI] [PubMed] [Google Scholar]
- 18.Marcus EA, Scott DR. Gastric Colonization by H. pylori. Springer: 2016. pp. 23–34. [Google Scholar]
- 19.Israel DA, Peek RM. The role of persistence in Helicobacter pylori pathogenesis. Curr Opin Gastroenterol. 2006;22:3–7. doi: 10.1097/01.mog.0000194790.51714.f0. [DOI] [PubMed] [Google Scholar]
- 20.van Vliet AH. Use of pan-genome analysis for the identification of lineage-specific genes of Helicobacter pylori. FEMS Microbiol Lett. 2017;364 doi: 10.1093/femsle/fnw296. [DOI] [PubMed] [Google Scholar]
- 21.Sachs G, Wen Y, Scott DR. Gastric infection by Helicobacter pylori. Curr Gastroenterol Rep. 2009;11:455–461. doi: 10.1007/s11894-009-0070-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauerfeind P, Garner R, Dunn BE, Mobley HL. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut. 1997;40:25–30. doi: 10.1136/gut.40.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stingl K, De Reuse H. Staying alive overdosed: how does Helicobacter pylori control urease activity? Int J Med Microbiol. 2005;295:307–315. doi: 10.1016/j.ijmm.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Huang JY, Goers Sweeney E, Guillemin K, Amieva MR. Multiple Acid Sensors Control Helicobacter pylori Colonization of the Stomach. PLoS Pathog. 2017;13:e1006118. doi: 10.1371/journal.ppat.1006118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed J. 2016;39:14–23. doi: 10.1016/j.bj.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcus EA, Sachs G, Wen Y, Scott DR. Phosphorylation-dependent and Phosphorylation-independent Regulation of Helicobacter pylori Acid Acclimation by the ArsRS Two-component System. Helicobacter. 2016;21:69–81. doi: 10.1111/hel.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Servetas SL, Carpenter BM, Haley KP, Gilbreath JJ, Gaddy JA, Merrell DS. Characterization of Key Helicobacter pylori Regulators Identifies a Role for ArsRS in Biofilm Formation. J Bacteriol. 2016;198:2536–2548. doi: 10.1128/JB.00324-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carpenter BM, West AL, Gancz H, Servetas SL, Pich OQ, Gilbreath JJ, Hallinger DR, Forsyth MH, Merrell DS, Michel SL. Crosstalk between the HpArsRS two-component system and HpNikR is necessary for maximal activation of urease transcription. Front Microbiol. 2015;6:558. doi: 10.3389/fmicb.2015.00558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott DR, Marcus EA, Wen Y, Oh J, Sachs G. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc Natl Acad Sci USA. 2007;104:7235–7240. doi: 10.1073/pnas.0702300104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishna U, Romero-Gallo J, Suarez G, Azah A, Krezel AM, Varga MG, Forsyth MH, Peek RM. Genetic Evolution of a Helicobacter pylori Acid-Sensing Histidine Kinase and Gastric Disease. J Infect Dis. 2016;214:644–648. doi: 10.1093/infdis/jiw189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talebi Bezmin Abadi A. Helicobacter pylori and Gastric Cancer. Front Med (Lausanne) 2016;3:36. doi: 10.3389/fmed.2016.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talebi Bezmin Abadi A. Helicobacter pylori: Emergence of a Superbug. Front Med (Lausanne) 2014;1:34. doi: 10.3389/fmed.2014.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudley J, Wieczorek T, Selig M, Cheung H, Shen J, Odze R, Deshpande V, Zukerberg L. Clinicopathological characteristics of invasive gastric Helicobacter pylori. Hum Pathol. 2017;61:19–25 [10.1016/j.humpath.2016.09.029]. doi: 10.1016/j.humpath.2016.09.029. [DOI] [PubMed] [Google Scholar]
- 34.Backert S, Schmidt TP, Harrer A, Wessler S. Exploiting the Gastric Epithelial Barrier: Helicobacter pylori’s Attack on Tight and Adherens Junctions. Curr Top Microbiol Immunol. 2017;400:195–226. doi: 10.1007/978-3-319-50520-6_9. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y, Wang QL, Cheng DD, Xu WT, Lu NH. Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter pylori. Front Cell Infect Microbiol. 2016;6:159. doi: 10.3389/fcimb.2016.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amieva MR, Salama NR, Tompkins LS, Falkow S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol. 2002;4:677–690. doi: 10.1046/j.1462-5822.2002.00222.x. [DOI] [PubMed] [Google Scholar]
- 37.Deen NS, Huang SJ, Gong L, Kwok T, Devenish RJ. The impact of autophagic processes on the intracellular fate of Helicobacter pylori: more tricks from an enigmatic pathogen? Autophagy. 2013;9:639–652. doi: 10.4161/auto.23782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 39.Talebi Bezmin Abadi A, Taghvaei T, Mohabbati Mobarez A, Vaira G, Vaira D. High correlation of babA 2-positive strains of Helicobacter pylori with the presence of gastric cancer. Intern Emerg Med. 2013;8:497–501. doi: 10.1007/s11739-011-0631-6. [DOI] [PubMed] [Google Scholar]
- 40.Appelmelk BJ, Monteiro MA, Martin SL, Moran AP, Vandenbroucke-Grauls CM. Why Helicobacter pylori has Lewis antigens. Trends Microbiol. 2000;8:565–570. doi: 10.1016/s0966-842x(00)01875-8. [DOI] [PubMed] [Google Scholar]
- 41.Edwards NJ, Monteiro MA, Faller G, Walsh EJ, Moran AP, Roberts IS, High NJ. Lewis X structures in the O antigen side-chain promote adhesion of Helicobacter pylori to the gastric epithelium. Mol Microbiol. 2000;35:1530–1539. doi: 10.1046/j.1365-2958.2000.01823.x. [DOI] [PubMed] [Google Scholar]
- 42.Valkonen KH, Wadström T, Moran AP. Identification of the N-acetylneuraminyllactose-specific laminin-binding protein of Helicobacter pylori. Infect Immun. 1997;65:916–923. doi: 10.1128/iai.65.3.916-923.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kavermann H, Burns BP, Angermuller K, Odenbreit S, Fischer W, Melchers K, Haas R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003;197:813–822. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003;22:515–528. doi: 10.1093/emboj/cdg050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oleastro M, Ménard A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology (Basel) 2013;2:1110–1134. doi: 10.3390/biology2031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Talebi Bezmin Abadi A, Perez-Perez G. Role of dupA in virulence of Helicobacter pylori. World J Gastroenterol. 2016;22:10118–10123. doi: 10.3748/wjg.v22.i46.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol. 2003;161:249–255. doi: 10.1083/jcb.200208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sycuro LK, Pincus Z, Gutierrez KD, Biboy J, Stern CA, Vollmer W, Salama NR. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell. 2010;141:822–833. doi: 10.1016/j.cell.2010.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bansil R, Celli JP, Hardcastle JM, Turner BS. The Influence of Mucus Microstructure and Rheology in Helicobacter pylori Infection. Front Immunol. 2013;4:310. doi: 10.3389/fimmu.2013.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sycuro LK, Rule CS, Petersen TW, Wyckoff TJ, Sessler T, Nagarkar DB, Khalid F, Pincus Z, Biboy J, Vollmer W, et al. Flow cytometry-based enrichment for cell shape mutants identifies multiple genes that influence Helicobacter pylori morphology. Mol Microbiol. 2013;90:869–883. doi: 10.1111/mmi.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim JS, Chang JH, Chung SI, Yum JS. Molecular cloning and characterization of the Helicobacter pylori fliD gene, an essential factor in flagellar structure and motility. J Bacteriol. 1999;181:6969–6976. doi: 10.1128/jb.181.22.6969-6976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Celli JP, Turner BS, Afdhal NH, Ewoldt RH, McKinley GH, Bansil R, Erramilli S. Rheology of gastric mucin exhibits a pH-dependent sol-gel transition. Biomacromolecules. 2007;8:1580–1586. doi: 10.1021/bm0609691. [DOI] [PubMed] [Google Scholar]
- 53.Ottemann KM, Lowenthal AC. Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect Immun. 2002;70:1984–1990. doi: 10.1128/IAI.70.4.1984-1990.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoenhofen IC, Lunin VV, Julien JP, Li Y, Ajamian E, Matte A, Cygler M, Brisson JR, Aubry A, Logan SM, et al. Structural and functional characterization of PseC, an aminotransferase involved in the biosynthesis of pseudaminic acid, an essential flagellar modification in Helicobacter pylori. J Biol Chem. 2006;281:8907–8916. doi: 10.1074/jbc.M512987200. [DOI] [PubMed] [Google Scholar]
- 55.Rhee KH, Park JS, Cho MJ. Helicobacter pylori: bacterial strategy for incipient stage and persistent colonization in human gastric niches. Yonsei Med J. 2014;55:1453–1466. doi: 10.3349/ymj.2014.55.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGowan CC, Cover TL, Blaser MJ. Helicobacter pylori and gastric acid: biological and therapeutic implications. Gastroenterology. 1996;110:926–938. doi: 10.1053/gast.1996.v110.pm8608904. [DOI] [PubMed] [Google Scholar]
- 57.Robinson K, Argent RH, Atherton JC. The inflammatory and immune response to Helicobacter pylori infection. Best Pract Res Clin Gastroenterol. 2007;21:237–259. doi: 10.1016/j.bpg.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 58.Baldari CT, Lanzavecchia A, Telford JL. Immune subversion by Helicobacter pylori. Trends Immunol. 2005;26:199–207. doi: 10.1016/j.it.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 59.Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001;107:767–773. doi: 10.1172/JCI12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graham DY. History of Helicobacter pylori, duodenal ulcer, gastric ulcer and gastric cancer. World J Gastroenterol. 2014;20:5191–5204. doi: 10.3748/wjg.v20.i18.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Müller A, Solnick JV. Inflammation, immunity, and vaccine development for Helicobacter pylori. Helicobacter. 2011;16 Suppl 1:26–32. doi: 10.1111/j.1523-5378.2011.00877.x. [DOI] [PubMed] [Google Scholar]
- 62.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Backert S, Neddermann M, Maubach G, Naumann M. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2016;21 Suppl 1:19–25. doi: 10.1111/hel.12335. [DOI] [PubMed] [Google Scholar]
- 64.Mejías-Luqu R, Gerhard M. Immune Evasion Strategies and Persistence of Helicobacter pylori. Molecular Pathogenesis and Signal Transduction by Helicobacter pylori. Springer: 2017. pp. 53–71. [DOI] [PubMed] [Google Scholar]
- 65.Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, Zähringer U, Mollenkopf HJ, Heinz E, Meyer TF. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med. 2006;12:1030–1038. doi: 10.1038/nm1480. [DOI] [PubMed] [Google Scholar]
- 66.Aebischer T, Laforsch S, Hurwitz R, Brombacher F, Meyer TF. Immunity against Helicobacter pylori: significance of interleukin-4 receptor alpha chain status and gender of infected mice. Infect Immun. 2001;69:556–558. doi: 10.1128/IAI.69.1.556-558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crabtree JE. Immune and inflammatory responses to Helicobacter pylori infection. Scand J Gastroenterol Suppl. 1996;215:3–10. [PubMed] [Google Scholar]
- 68.D’Elios MM, Andersen LP. Helicobacter pylori inflammation, immunity, and vaccines. Helicobacter. 2007;12 Suppl 1:15–19. doi: 10.1111/j.1523-5378.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 69.Michetti P, Svennerholm AM. Helicobacter pylori- inflammation, immunity and vaccines. Helicobacter. 2003;8 Suppl 1:31–35. doi: 10.1046/j.1523-5378.2003.00164.x. [DOI] [PubMed] [Google Scholar]
- 70.Allen LA. The role of the neutrophil and phagocytosis in infection caused by Helicobacter pylori. Curr Opin Infect Dis. 2001;14:273–277. doi: 10.1097/00001432-200106000-00005. [DOI] [PubMed] [Google Scholar]
- 71.Allen LA. Phagocytosis and persistence of Helicobacter pylori. Cell Microbiol. 2007;9:817–828. doi: 10.1111/j.1462-5822.2007.00906.x. [DOI] [PubMed] [Google Scholar]
- 72.Allen LA. Rate and extent of Helicobacter pylori phagocytosis. Methods Mol Biol. 2008;431:147–157. doi: 10.1007/978-1-60327-032-8_12. [DOI] [PubMed] [Google Scholar]
- 73.Deen NS, Gong L, Naderer T, Devenish RJ, Kwok T. Analysis of the Relative Contribution of Phagocytosis, LC3-Associated Phagocytosis, and Canonical Autophagy During Helicobacter pylori Infection of Macrophages. Helicobacter. 2015;20:449–459. doi: 10.1111/hel.12223. [DOI] [PubMed] [Google Scholar]
- 74.Abadi AT, Lee YY. Helicobacter pylori vacA as marker for gastric cancer and gastroduodenal diseases: one but not the only factor. J Clin Microbiol. 2014;52:4451. doi: 10.1128/JCM.02640-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blaser MJ. Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. J Infect Dis. 1990;161:626–633. doi: 10.1093/infdis/161.4.626. [DOI] [PubMed] [Google Scholar]
- 76.Ramarao N, Meyer TF. Helicobacter pylori resists phagocytosis by macrophages: quantitative assessment by confocal microscopy and fluorescence-activated cell sorting. Infect Immun. 2001;69:2604–2611. doi: 10.1128/IAI.69.4.2604-2611.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Demiray E, Bekmen N. [Helicobacter pylori infection and phagocytosis] Mikrobiyol Bul. 2008;42:177–184. [PubMed] [Google Scholar]
- 78.Nazligul Y, Aslan M, Horoz M, Celik Y, Dulger AC, Celik H, Erel O. The effect on serum myeloperoxidase activity and oxidative status of eradication treatment in patients Helicobacter pylori infected. Clin Biochem. 2011;44:647–649. doi: 10.1016/j.clinbiochem.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 79.Borlace GN, Butler RN, Brooks DA. Monocyte and macrophage killing of helicobacter pylori: relationship to bacterial virulence factors. Helicobacter. 2008;13:380–387. doi: 10.1111/j.1523-5378.2008.00625.x. [DOI] [PubMed] [Google Scholar]
- 80.Calvino-Fernández M, Benito-Martínez S, Parra-Cid T. Oxidative stress by Helicobacter pylori causes apoptosis through mitochondrial pathway in gastric epithelial cells. Apoptosis. 2008;13:1267–1280. doi: 10.1007/s10495-008-0255-0. [DOI] [PubMed] [Google Scholar]
- 81.Chaturvedi R, Cheng Y, Asim M, Bussière FI, Xu H, Gobert AP, Hacker A, Casero RA, Wilson KT. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem. 2004;279:40161–40173. doi: 10.1074/jbc.M401370200. [DOI] [PubMed] [Google Scholar]
- 82.Cross RK, Wilson KT. Nitric oxide in inflammatory bowel disease. Inflamm Bowel Dis. 2003;9:179–189. doi: 10.1097/00054725-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 83.Lundström AM, Sundaeus V, Bölin I. The 26-kilodalton, AhpC homologue, of Helicobacter pylori is also produced by other Helicobacter species. Helicobacter. 2001;6:44–54. doi: 10.1046/j.1523-5378.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- 84.Papinutto E, Windle HJ, Cendron L, Battistutta R, Kelleher D, Zanotti G. Crystal structure of alkyl hydroperoxide-reductase (AhpC) from Helicobacter pylori. Biochim Biophys Acta. 2005;1753:240–246. doi: 10.1016/j.bbapap.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 85.Zhang B, Li HL, Fan Q, Guo F, Ren XY, Zhou HB, Zhu JW, Zhao YS, Tian WJ. Serum Helicobacter pylori KatA and AhpC antibodies as novel biomarkers for gastric cancer. World J Gastroenterol. 2016;22:5060–5067. doi: 10.3748/wjg.v22.i21.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Augusto AC, Miguel F, Mendonça S, Pedrazzoli J, Gurgueira SA. Oxidative stress expression status associated to Helicobacter pylori virulence in gastric diseases. Clin Biochem. 2007;40:615–622. doi: 10.1016/j.clinbiochem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 87.Gobert AP, Cheng Y, Wang JY, Boucher JL, Iyer RK, Cederbaum SD, Casero RA, Newton JC, Wilson KT. Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol. 2002;168:4692–4700. doi: 10.4049/jimmunol.168.9.4692. [DOI] [PubMed] [Google Scholar]
- 88.El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, König T, Schleicher U, Koo MS, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. 2008;9:1399–1406. doi: 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 90.Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, Mobley HL, Wilson KT. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci USA. 2001;98:13844–13849. doi: 10.1073/pnas.241443798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rogers PA, Eide L, Klungland A, Ding H. Reversible inactivation of E. coli endonuclease III via modification of its [4Fe-4S] cluster by nitric oxide. DNA Repair (Amst) 2003;2:809–817. doi: 10.1016/s1568-7864(03)00065-x. [DOI] [PubMed] [Google Scholar]
- 92.Rittig MG, Shaw B, Letley DP, Thomas RJ, Argent RH, Atherton JC. Helicobacter pylori-induced homotypic phagosome fusion in human monocytes is independent of the bacterial vacA and cag status. Cell Microbiol. 2003;5:887–899. doi: 10.1046/j.1462-5822.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 93.Zheng PY, Jones NL. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol. 2003;5:25–40. doi: 10.1046/j.1462-5822.2003.00250.x. [DOI] [PubMed] [Google Scholar]
- 94.Borody T, Ren Z, Pang G, Clancy R. Impaired host immunity contributes to Helicobacter pylori eradication failure. Am J Gastroenterol. 2002;97:3032–3037. doi: 10.1111/j.1572-0241.2002.07121.x. [DOI] [PubMed] [Google Scholar]
- 95.Del Giudice G, Michetti P. Inflammation, immunity and vaccines for Helicobacter pylori. Helicobacter. 2004;9 Suppl 1:23–28. doi: 10.1111/j.1083-4389.2004.00245.x. [DOI] [PubMed] [Google Scholar]
- 96.Permin H, Andersen LP. Inflammation, immunity, and vaccines for Helicobacter infection. Helicobacter. 2005;10 Suppl 1:21–25. doi: 10.1111/j.1523-5378.2005.00337.x. [DOI] [PubMed] [Google Scholar]
- 97.Raghavan S, Quiding-Järbrink M. Immune modulation by regulatory T cells in Helicobacter pylori-associated diseases. Endocr Metab Immune Disord Drug Targets. 2012;12:71–85. doi: 10.2174/187153012799278974. [DOI] [PubMed] [Google Scholar]
- 98.Gray BM, Fontaine CA, Poe SA, Eaton KA. Complex T cell interactions contribute to Helicobacter pylori gastritis in mice. Infect Immun. 2013;81:740–752. doi: 10.1128/IAI.01269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R, Lee CK, Kleanthous H, Monath TP. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J Exp Med. 1998;188:2277–2288. doi: 10.1084/jem.188.12.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thamphiwatana S, Gao W, Obonyo M, Zhang L. In vivo treatment of Helicobacter pylori infection with liposomal linolenic acid reduces colonization and ameliorates inflammation. Proc Natl Acad Sci USA. 2014;111:17600–17605. doi: 10.1073/pnas.1418230111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ding H, Nedrud JG, Blanchard TG, Zagorski BM, Li G, Shiu J, Xu J, Czinn SJ. Th1-mediated immunity against Helicobacter pylori can compensate for lack of Th17 cells and can protect mice in the absence of immunization. PLoS One. 2013;8:e69384. doi: 10.1371/journal.pone.0069384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beigier-Bompadre M, Moos V, Belogolova E, Allers K, Schneider T, Churin Y, Ignatius R, Meyer TF, Aebischer T. Modulation of the CD4+ T-cell response by Helicobacter pylori depends on known virulence factors and bacterial cholesterol and cholesterol α-glucoside content. J Infect Dis. 2011;204:1339–1348. doi: 10.1093/infdis/jir547. [DOI] [PubMed] [Google Scholar]
- 103.Ernst PB, Pappo J. T-cell-mediated mucosal immunity in the absence of antibody: lessons from Helicobacter pylori infection. Acta Odontol Scand. 2001;59:216–221. doi: 10.1080/00016350152509238. [DOI] [PubMed] [Google Scholar]
- 104.Yahiro K, Akazawa Y, Nakano M, Suzuki H, Hisatune J, Isomoto H, Sap J, Noda M, Moss J, Hirayama T. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Discov. 2015;1:15035. doi: 10.1038/cddiscovery.2015.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Radin JN, González-Rivera C, Frick-Cheng AE, Sheng J, Gaddy JA, Rubin DH, Algood HM, McClain MS, Cover TL. Role of connexin 43 in Helicobacter pylori VacA-induced cell death. Infect Immun. 2014;82:423–432. doi: 10.1128/IAI.00827-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yokota Si, Amano Ki, Hayashi S, Kubota T, Fujii N, Yokochi T. Human antibody response to Helicobacter pylori lipopolysaccharide: presence of an immunodominant epitope in the polysaccharide chain of lipopolysaccharide. Infect Immun. 1998;66:3006–3011. doi: 10.1128/iai.66.6.3006-3011.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Engstrand L, Gustavsson S, Schwan A, Scheynius A. Local and systemic immune response in Helicobacter pylori-associated chronic gastritis before and after treatment. Scand J Gastroenterol. 1993;28:1105–1111. doi: 10.3109/00365529309098317. [DOI] [PubMed] [Google Scholar]
- 109.Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS One. 2010;5:e10844. doi: 10.1371/journal.pone.0010844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kranzer K, Eckhardt A, Aigner M, Knoll G, Deml L, Speth C, Lehn N, Rehli M, Schneider-Brachert W. Induction of maturation and cytokine release of human dendritic cells by Helicobacter pylori. Infect Immun. 2004;72:4416–4423. doi: 10.1128/IAI.72.8.4416-4423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Umehara S, Higashi H, Ohnishi N, Asaka M, Hatakeyama M. Effects of Helicobacter pylori CagA protein on the growth and survival of B lymphocytes, the origin of MALT lymphoma. Oncogene. 2003;22:8337–8342. doi: 10.1038/sj.onc.1207028. [DOI] [PubMed] [Google Scholar]
- 112.Leonard WJ. The Jak-STAT pathway. Hormone Signaling. 2002;7:103–120. [Google Scholar]
- 113.Igaz P, Tóth S, Falus A. Biological and clinical significance of the JAK-STAT pathway; lessons from knockout mice. Inflamm Res. 2001;50:435–441. doi: 10.1007/PL00000267. [DOI] [PubMed] [Google Scholar]
- 114.Kim N. Immunological Reactions on H. pylori Infection. Helicobacter pylori: Springer; 2016. pp. 35–52. [DOI] [Google Scholar]
- 115.West AC, Jenkins BJ. Investigating the Role of Toll-Like Receptors in Mouse Models of Gastric Cancer. Methods Mol Biol. 2016;1390:427–449. doi: 10.1007/978-1-4939-3335-8_25. [DOI] [PubMed] [Google Scholar]
- 116.Hammond CE, Beeson C, Suarez G, Peek RM, Backert S, Smolka AJ. Helicobacter pylori virulence factors affecting gastric proton pump expression and acid secretion. Am J Physiol Gastrointest Liver Physiol. 2015;309:G193–G201. doi: 10.1152/ajpgi.00099.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hossain MJ, Tanasescu R, Gran B. Innate immune regulation of autoimmunity in multiple sclerosis: Focus on the role of Toll-like receptor 2. J Neuroimmunol. 2017;304:11–20. doi: 10.1016/j.jneuroim.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 118.Hu Y, Liu JP, Zhu Y, Lu NH. The Importance of Toll-like Receptors in NF-κB Signaling Pathway Activation by Helicobacter pylori Infection and the Regulators of this Response. Helicobacter. 2016;21:428–440. doi: 10.1111/hel.12292. [DOI] [PubMed] [Google Scholar]
- 119.Djekic A, Müller A. The Immunomodulator VacA Promotes Immune Tolerance and Persistent Helicobacter pylori Infection through Its Activities on T-Cells and Antigen-Presenting Cells. Toxins (Basel) 2016;8 doi: 10.3390/toxins8060187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kivrak Salim D, Sahin M, Köksoy S, Adanir H, Süleymanlar I. Local Immune Response in Helicobacter pylori Infection. Medicine (Baltimore) 2016;95:e3713. doi: 10.1097/MD.0000000000003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sjökvist Ottsjö L, Jeverstam F, Yrlid L, Wenzel AU, Walduck AK, Raghavan S. Induction of mucosal immune responses against Helicobacter pylori infection after sublingual and intragastric route of immunization. Immunology. 2017;150:172–183. doi: 10.1111/imm.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith SM, Freeley M, Moynagh PN, Kelleher DP. Differential modulation of Helicobacter pylori lipopolysaccharide-mediated TLR2 signaling by individual Pellino proteins. Helicobacter. 2017;22 doi: 10.1111/hel.12325. [DOI] [PubMed] [Google Scholar]
- 123.Mai UE, Perez-Perez GI, Wahl LM, Wahl SM, Blaser MJ, Smith PD. Soluble surface proteins from Helicobacter pylori activate monocytes/macrophages by lipopolysaccharide-independent mechanism. J Clin Invest. 1991;87:894–900. doi: 10.1172/JCI115095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kidd M, Miu K, Tang LH, Perez-Perez GI, Blaser MJ, Sandor A, Modlin IM. Helicobacter pylori lipopolysaccharide stimulates histamine release and DNA synthesis in rat enterochromaffin-like cells. Gastroenterology. 1997;113:1110–1117. doi: 10.1053/gast.1997.v113.pm9322505. [DOI] [PubMed] [Google Scholar]
- 125.Li H, Liao T, Debowski AW, Tang H, Nilsson HO, Stubbs KA, Marshall BJ, Benghezal M. Lipopolysaccharide Structure and Biosynthesis in Helicobacter pylori. Helicobacter. 2016;21:445–461. doi: 10.1111/hel.12301. [DOI] [PubMed] [Google Scholar]
- 126.Pachathundikandi SK, Müller A, Backert S. Inflammasome Activation by Helicobacter pylori and Its Implications for Persistence and Immunity. Curr Top Microbiol Immunol. 2016;397:117–131. doi: 10.1007/978-3-319-41171-2_6. [DOI] [PubMed] [Google Scholar]
- 127.Naumann M, Sokolova O, Tegtmeyer N, Backert S. Helicobacter pylori: A Paradigm Pathogen for Subverting Host Cell Signal Transmission. Trends Microbiol. 2017;25:316–328. doi: 10.1016/j.tim.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 128.Koch M, Mollenkopf HJ, Meyer TF. Macrophages recognize the Helicobacter pylori type IV secretion system in the absence of toll-like receptor signalling. Cell Microbiol. 2016;18:137–147. doi: 10.1111/cmi.12492. [DOI] [PubMed] [Google Scholar]
- 129.Li S, Cao M, Song L, Qi P, Chen C, Wang X, Li N, Peng J, Wu D, Hu G, et al. The contribution of toll-like receptor 2 on Helicobacter pylori activation of the nuclear factor-kappa B signaling pathway in gastric epithelial cells. Microb Pathog. 2016;98:63–68. doi: 10.1016/j.micpath.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 130.de Jonge R, Kusters JG, Timmer MS, Gimmel V, Appelmelk BJ, Bereswill S, van Vliet AH, Meuwissen SG, Kist M, Vandenbroucke-Grauls CM, et al. The role of Helicobacter pylori virulence factors in interleukin production by monocytic cells. FEMS Microbiol Lett. 2001;196:235–238. doi: 10.1111/j.1574-6968.2001.tb10570.x. [DOI] [PubMed] [Google Scholar]
- 131.Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc Natl Acad Sci USA. 2000;97:14668–14673. doi: 10.1073/pnas.97.26.14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol. 1999;32:459–470. doi: 10.1046/j.1365-2958.1999.01382.x. [DOI] [PubMed] [Google Scholar]
- 133.Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. doi: 10.1038/nrmicro1658. [DOI] [PubMed] [Google Scholar]
- 134.Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. Free recombination within Helicobacter pylori. Proc Natl Acad Sci USA. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Falush D, Kraft C, Taylor NS, Correa P, Fox JG, Achtman M, Suerbaum S. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc Natl Acad Sci USA. 2001;98:15056–15061. doi: 10.1073/pnas.251396098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang G, Humayun MZ, Taylor DE. Mutation as an origin of genetic variability in Helicobacter pylori. Trends Microbiol. 1999;7:488–493. doi: 10.1016/s0966-842x(99)01632-7. [DOI] [PubMed] [Google Scholar]
- 137.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 138.Kuipers EJ, Israel DA, Kusters JG, Gerrits MM, Weel J, van Der Ende A, van Der Hulst RW, Wirth HP, Höök-Nikanne J, Thompson SA, et al. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J Infect Dis. 2000;181:273–282. doi: 10.1086/315173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Covacci A, Rappuoli R. Helicobacter pylori: molecular evolution of a bacterial quasi-species. Curr Opin Microbiol. 1998;1:96–102. doi: 10.1016/s1369-5274(98)80148-3. [DOI] [PubMed] [Google Scholar]
- 140.Portal-Celhay C, Perez-Perez GI. Immune responses to Helicobacter pylori colonization: mechanisms and clinical outcomes. Clin Sci (Lond) 2006;110:305–314. doi: 10.1042/CS20050232. [DOI] [PubMed] [Google Scholar]
- 141.Bergman M, Del Prete G, van Kooyk Y, Appelmelk B. Helicobacter pylori phase variation, immune modulation and gastric autoimmunity. Nat Rev Microbiol. 2006;4:151–159. doi: 10.1038/nrmicro1344. [DOI] [PubMed] [Google Scholar]
- 142.Beier D, Frank R. Molecular characterization of two-component systems of Helicobacter pylori. J Bacteriol. 2000;182:2068–2076. doi: 10.1128/jb.182.8.2068-2076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abadi AT. Vaccine against Helicobacter pylori: Inevitable approach. World J Gastroenterol. 2016;22:3150–3157. doi: 10.3748/wjg.v22.i11.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shafiee A, Amini M, Emamirad H, Talebi Bezmin Abadi A. Recombination and phenotype evolution dynamic of Helicobacter pylori in colonized hosts. Int J Syst Evol Microbiol. 2016 doi: 10.1099/ijsem.0.001072. Epub ahead of print. [DOI] [PubMed] [Google Scholar]