

Abstract
A new protocol for the catalytic synthesis of cyclopropanes using electron‐deficient alkenes is presented, which is catalysed by a series of affordable, easy to synthesise and highly active substituted cobalt(II) tetraaza[14]annulenes. These catalysts are compatible with the use of sodium tosylhydrazone salts as precursors to diazo compounds in one‐pot catalytic transformations to afford the desired cyclopropanes in almost quantitative yields. The reaction takes advantage of the metalloradical character of the Co complexes to activate the diazo compounds. The reaction is practical and fast, and proceeds from readily available starting materials. It does not require the slow addition of diazo reagents or tosylhydrazone salts or heating and tolerates many solvents, which include protic ones such as MeOH. The CoII complexes derived from the tetramethyltetraaza[14]annulene ligand are easier to prepare than cobalt(II) porphyrins and present a similar catalytic carbene radical reactivity but are more active. The reaction proceeds at 20 °C in a matter of minutes and even at −78 °C in a few hours. The catalytic system is robust and can operate with either the alkene or the diazo reagent as the limiting reagent, which inhibits the dimerisation of diazo compounds totally. The protocol has been applied to synthesise a variety of substituted cyclopropanes. High yields and selectivities were achieved for various substrates with an intrinsic preference for trans cyclopropanes.
Keywords: alkenes, cobalt, macrocyclic ligands, phase-transfer catalysis, reaction mechanisms
Introduction
Cyclopropanes are fundamental building blocks in nature and part of the backbone of many biologically active synthetic compounds such as drugs and insecticides. Several methods are available for the synthesis of cyclopropanes, of which the transition‐metal‐catalysed cyclopropanation of olefins with diazo compounds is among the most robust and atom‐efficient methods. The first reports on asymmetric catalysed cyclopropanation were disclosed by Noyori and co‐workers in 1966.1 The catalyst they introduced is a Cu complex with a chiral chelating ligand. This discovery was followed by many others, most of which were based on Cu,2 Rh3 and Ru.4 Typically, these catalysts work well for the synthesis of chiral cyclopropanes derived from diazoacetates and electron‐rich olefins to give high yields and selectivities. However, these catalysts are typically poorly active for the cyclopropanation of electron‐deficient olefins because of the electrophilic nature of the metal carbene intermediates.5
A major drawback to obtain cyclopropanes by the reaction of electron‐deficient alkenes and diazo compounds is the competing un‐catalysed 1,3‐dipolar cycloaddition reaction to form pyrazolines (Scheme 1).6 If the dipolarophile is an α,β‐unsaturated ester, such as an acrylate, the reaction rate and selectivity are determined by the HOMO–LUMO interactions, so that the diazo compound tends to attack the β‐position of the unsaturated ester substrate. The larger the gap and the smaller the substituents, the more favourable the 1,3‐dipolar cycloaddition. In the first instance, the formed product is a 1‐pyrazoline, but because of its instability, isomerisation leads to the formation of 2‐pyrazolines.
Scheme 1.
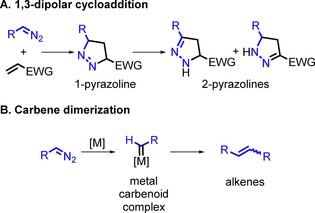
Side reactions during cyclopropanation. A) 1,3‐Dipolar cycloaddition between a diazo compound and an electron‐deficient alkene. B) Diazo compound dimerisation towards alkenes via metal carbenoids.7
Another competing pathway that leads to undesired side products is carbene–carbene dimerisation. This process occurs easily by the nucleophilic attack of the carbon atom of the diazo substrate at the prototypical electrophilic Fischer‐type metal carbene intermediates generated at the catalyst. To avoid the 1,3‐dipolar cycloaddition and form cyclopropanes instead, a catalyst must be used that has a high activity towards activation of diazo compounds and favours carbene formation and subsequent coupling to the alkene over the undesired cycloaddition or carbene–carbene dimerisation pathways. Such catalysts are still quite rare, and only a few examples have been reported, which are mostly based on CoII.8
In 1978, Nakamura and co‐workers introduced cobalt(II) dioximato complexes as enantioselective catalysts for the cyclopropanation of diazo compounds and alkenes.9 These catalysts produce cyclopropanes in high yields and with high enantioselectivities for specific substrates, but the yields were very low for alkenes that bear electron‐withdrawing substituents. Further investigation of these catalysts was also discouraged because of difficulties with catalyst homogeneity if chiral dioximato ligands are used. Chiral cobalt(II) salen complexes were later explored by Katsuki and co‐workers as cyclopropanation catalysts.10 Yamada et al.11 reported on the cyclopropanation of styrene derivatives with ethyl diazoacetate using similar complexes. However, again neither of these catalysts resulted in high yields, diastereoselectivities or enantioselectivities if electron‐deficient olefins were used. In 2003, Cenini and co‐workers and Zhang and co‐workers reported independently on the use of CoII catalysts with chiral porphyrins as ligands to produce cyclopropanes in high yields and selectivities from substituted styrenes and diazoacetates.12 Later, Zhang et al. demonstrated that these catalysts also allow the cyclopropanation of electron‐deficient alkenes (such as acrylates) in high yields, diastereomeric excess and enantiomeric excess.13 Several enantioselective protocols for the cyclopropanation of styrenes with cobalt porphyrins were also developed.14 The cyclopropanation reactivity of group 9 transition metal (Co, Rh, Ir) porphyrin complexes was reviewed recently by Gallo and co‐workers.15
This unique reactivity was explained by de Bruin et al. in 2010 through a detailed study of the cyclopropanation mechanism using DFT calculations and experimental studies (EPR spectroscopy and MS).16 During these investigations, they concluded that the mechanism of low‐spin planar CoII systems is a radical mechanism (Figure 1 A). The metalloradical activation of diazo compounds produces metal carbenoids with radical carbon character, which can best be described as one‐electron‐reduced Fischer‐type carbenes (Figure 1 B). Carbene generation at CoII leads to electron transfer from the CoII to the carbene moiety, which leads to a cobalt(III) carbene radical intermediate. The occupation of the carbenoid p orbital with an unpaired electron gives the cobalt carbenoid intermediate its unique radical‐type reactivity and makes it less electrophilic than common Fischer‐type carbenes. While in common Fischer‐type carbenes the (LUMO) carbenoid p orbital is completely empty, the respective carbon p orbital of the cobalt carbene radical intermediates shown in Figure 2 is half‐filled (singly occupied molecular orbital; SOMO). This explains the enhanced reactivity of the cobalt(III) carbene radical intermediates towards electron‐deficient olefins as well as their reduced tendency to undergo unwanted carbene–carbene dimerisation reactions.
Figure 1.
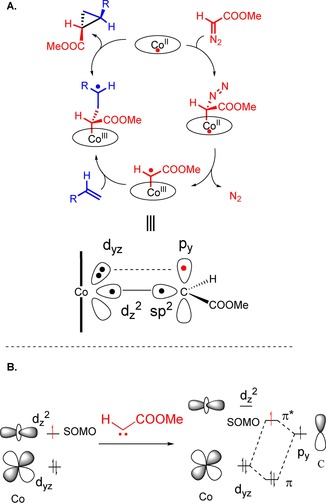
Mechanism of CoII‐catalysed cyclopropanation.
Figure 2.
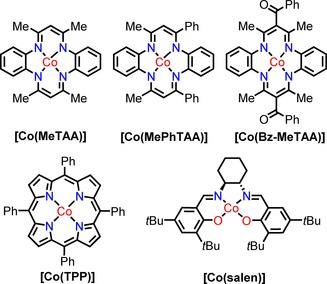
Structures of the cobalt(II) catalysts used in this study.
As a result of the synthetic challenges to prepare highly substituted CoII(porphyrin) catalysts that can activate diazo compounds efficiently via carbene radical intermediates, we decided to pursue the development of cheaper, easier to prepare and potentially more active low‐spin planar CoII catalysts. Based on our current understanding of the catalytic system (Figure 1), more‐electron‐rich complexes should facilitate electron transfer to the carbene in the diazo activation to create a more active catalyst. Therefore, we investigated the catalytic properties of cobalt(II) tetraaza[14]annulenes (Figure 2, top row) in alkene cyclopropanation reactions. They are cheap, easy to prepare and their macrocycle is smaller than that of the porphyrin analogue, which makes the metal centre more electron rich. These complexes were prepared and characterised in the late 1970s17 but their catalytic activity was never explored fully. The ligands have attracted renewed attention in view of their stronger electron‐donating properties and the higher reactivity of their coordination complexes compared to porphyrins,18 but the catalytic application of the Co complexes was thus far limited to a single example. Recently, we have shown that cobalt(II) tetramethyltetraaza[14]annulene [Co(MeTAA)] is able to activate diazo compounds and form substituted 1H‐indenes.19 In this study, we tested the performance of similar Co complexes for the cyclopropanation of electron‐deficient alkenes with (precursors of) diazo compounds using a metalloradical approach. In addition, we demonstrate an unprecedented one‐pot methodology for the cyclopropanation of electron‐deficient alkenes with tosylhydrazone salts as carbene precursors, which shows that the catalyst is fully compatible with the in situ generation of diazo compounds from tosylhydrazone salts in cyclopropanation reactions.
Results and Discussion
The straightforward synthesis of the cobalt(II) tetraaza[14]annulenes [Co(MeTAA)], [Co(MePhTAA)] (Co(II) 6,15‐dimethyl‐8,17‐diphenyltetraaza[14]annulene) and [Co(Bz‐MeTAA)] (Co(II) 7,16‐dibenzoyl‐6,8,15,17‐tetramethyltetraaza[14]annulene) is performed by a template condensation between 1,2‐diaminobenzene and a 2,4‐substituted diketone.20 The metal used for the template reaction can be either NiII or CoII. The NiII complex has the advantage that is diamagnetic, not sensitive to air and easy to demetalate to obtain the free ligand. The substitution of the methine carbon atom of the 2,4‐pentanediiminato ring is possible using aromatic acyl chlorides and is easier and higher yielding if we use [Ni(MeTAA)] or [Ni(MePhTAA)] rather than the metal‐free ligand. The heating of the metal‐free ligands to reflux in the presence of cobalt acetate yields the desired CoII complexes in high yields (Scheme 2).
Scheme 2.
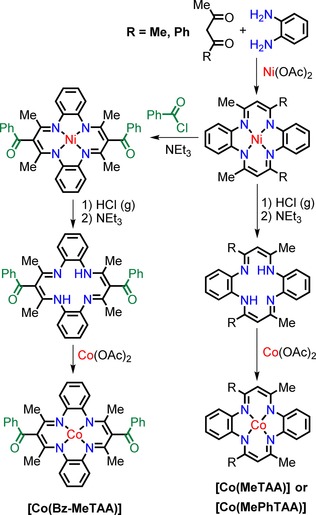
Synthesis of [Co(MeTAA)].
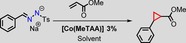
The catalytic activity of the thus obtained [Co(MeTAA)] was investigated using ethyl diazoacetate (EDA) and methyl acrylate as the model substrates initially. This reaction was performed at room temperature for 1 h in dichloromethane using 1.5 mol % catalyst, 1.0 equivalents of EDA and 3.0 equivalents of methyl acrylate to yield the desired cyclopropane product in 85 % isolated yield. For comparison, cobalt(II) meso‐tetraphenylporphyrin ([Co(TPP)]) was tested as a catalyst under identical conditions and yielded the cyclopropane in less than 30 % isolated yield after 1 h. Therefore, the reaction was continued for 20 h to yield 67 % of the cyclopropane product. Hence, [Co(MeTAA)] is much faster than [Co(TPP)]. It is clear from the results summarised in Table 1 that [Co(MeTAA)] led to higher isolated yields of the cyclopropane products in a shorter time, which reveals the higher activity of this catalyst for the cyclopropanation of methyl acrylate with EDA. Moreover, the trans/cis ratio of 93:7 is superior to that obtained with [Co(TPP)] (88:12). Entry 8 shows the reaction in which EDA is added in excess (relative to methyl acrylate, which is the limiting reagent) affords a similar yield of 87 %. Consequently, that both reagents, that is, either the diazo compound or the alkene, can be used interchangeably as limiting reagents without a noticeable effect on the obtained yields. This has a net advantage over the well‐established copper bisoxazoline catalysts, for which the alkene always has to be present in excess and the diazo compound has to be added slowly to the reaction mixture. Not even traces of dimerisation products (maleates or fumarates) were observed if [Co(MeTAA)] was used as the catalyst.
Table 1.
Screening of reaction conditions for the cyclopropanation of methylacrylate with EDA.[a]
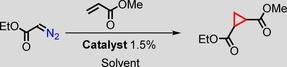
| Entry | Catalyst | Solvent | T [°C] | t [h] | Yield [%][b] | trans/cis [c] |
|---|---|---|---|---|---|---|
| 1 | – | CH2Cl2 | 20 | 1 | 0 | – |
| 2 | Co(OAc)2⋅4 H2O | CH2Cl2 | 20 | 1 | <5 | – |
| 3 | [Co(TPP)] | CH2Cl2 | 20 | 20 | 67 | 88:12 |
| 4 | [Co(TPP)] | CH2Cl2 | 10 | 20 | 58 | 88:12 |
| 5 | [Co(salen)] | CH2Cl2 | 20 | 18 | 43 | 70:30 |
| 6 | [Co(BzMeTAA)] | PhMe | 20 | 1 | 97 | 94:6 |
| 7 | [Co(MePhTAA)] | CH2Cl2 | 20 | 1 | 90 | 89:10 |
| 8[d] | [Co(MeTAA)] | CH2Cl2 | 20 | 1 | 87 | 93:7 |
| 9 | [Co(MeTAA)] | CH2Cl2 | 20 | 1 | 85 | 93:7 |
| 10 | [Co(MeTAA)] | THF | 20 | 1 | 72 | 94:6 |
| 11 | [Co(MeTAA)] | PhMe | 20 | 1 | 74 | 95:5 |
| 12 | [Co(MeTAA)] | PhCl | 20 | 1 | 69 | 93:7 |
| 13 | [Co(MeTAA)] | CH3CN: CH2Cl2 (4:1) | 20 | 1 | 80 | 93:7 |
| 14 | [Co(MeTAA)] | CH3OH: CH2Cl2 (4:1) | 20 | 1 | 84 | 94:6 |
| 15 | [Co(MeTAA)] | CH2Cl2 | 10 | 1 | 80 | 93:7 |
| 16 | [Co(MeTAA)] | CH2Cl2 | 0 | 2 | 74 | 97:3 |
| 17 | [Co(MeTAA)] | CH2Cl2 | −78 | 5 | 67 | 99:1 |
| 18[e] | [Co(MeTAA)] | PhMe | 10 | 1 1 2 6 | 85[f] 83[g] 75[h] 14[i] | 96:4 96:4 95:5 95:5 |
[a] Reactions were performed under N2 with 1.0 equiv. of EDA, 3.0 equiv. of methylacrylate and 1.5 mol % catalyst. Concentration: 1.9 mmol EDA/5 mL solvent. [b] Isolated yields. [c] Determined by using GC. [d] 1.0 equiv. methylacrylate, 1.2 equiv. EDA and 1.5 mol % catalyst. [e] Recyclability study performed starting with 5.0 mol % catalyst; [f] cycle 1; [g] cycle 2; [h] cycle 3; [i] cycle 4.
Several other CoII complexes were tested (Figure 2) to compare their activity to that of [Co(MeTAA)]. If we compare the activity of two other catalysts that belong to the tetraazaannulene family (Table 1, entries 6–7), it is clear that they are all active, and [Co(BzMeTAA)] (Table 1, entry 6) produces cyclopropanes in the highest isolated yield (97 %). This is possibly because the benzyl substituents slow down the catalyst deactivation pathways (e.g., 1,4‐addition of the alkene between the methine carbon atom of the 2,4‐pentanediiminato ring and the Co centre observed for the base catalyst).21 Other complexes, such as [Co(salen)], were much slower and gave lower yields under the same reaction conditions (Table 1, entry 5).
We decided to further explore the scope of the cyclopropanation reaction using the base catalyst [Co(MeTAA)] because it is simple, cheap and easier to synthesise in high quantities than [Co(Bz‐MeTAA)]. Furthermore, if nitrogen bases are used as additives, the activity of this catalyst can be further improved to reach high yields similar to those obtained with [Co(Bz‐MeTAA)] (vide infra), although the latter is slightly more active in the absence of additives.
Solvent screening (Table 1, entries 9–14) revealed that the reaction works well in both nonpolar solvents such as toluene (Table 1, entry 11) and polar solvents such as CH2Cl2, THF, chlorobenzene and CH3CN (Table 1, entries 9, 10, 12, 13). The catalyst even has an exceptionally high tolerance for protic solvents such as MeOH (Table 1, entry 14), which is rather unexpected for carbene‐transfer reactions. Mixtures of acetonitrile and methanol with dichloromethane were used to circumvent solubility issues with the catalyst. The reaction does not proceed without a catalyst (entry 1) or just with cobalt(II) acetate (entry 2). However, if [Co(MeTAA)] was used, cyclopropanation can be performed at 0 °C and even at −78 °C to obtain the products in good yields (Table 1, entries 16 and 17). Notably, [Co(TPP)] does not work below 10 °C in a reasonable amount of time (entry 4). A recyclability study was performed to test the stability of [Co(MeTAA)] (Table 1, entry 18). We started from a catalyst loading of 5.0 mol %, and the first three cycles achieve full conversion and high isolated yields. However, from the fourth cycle, the reaction becomes slower and the conversion does not exceed 20 % because of catalyst deactivation/poisoning. Further investigations into the stability and kinetics of the catalysis are currently underway.
After we determined that [Co(MeTAA)] is able to catalyse cyclopropanation reactions in both polar and nonpolar solvents as well as at a wide range of temperatures, we decided to explore the effect of coordinating additives on the activity of the catalyst. Previous studies have shown that the axial coordination of nitrogen donors to cobalt(II) porphyrins increases the overall activity of the catalyst.12 Several additives, such as 4‐dimethylaminopyridine (DMAP) and N‐methylimidazole were tested (Table 2). With [Co(MeTAA)] as the catalyst, the yield increased from 78 % without an additive (Table 2, entry 1) to 94 % in the presence of N‐methylimidazole (1 equivalent with respect to the catalyst; Table 2, entry 2).
Table 2.
Additive screening for the cyclopropanation of acrylates with EDA and [Co(MeTAA)].[a]
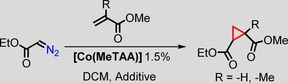
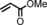
| Entry[a] | Olefin | Additive | Product | Yield [%][c] | trans/cis [d] |
|---|---|---|---|---|---|
| 1 |
|
– |
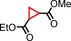
|
78 | 97:3 |
| 2 |
|
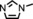
|
|
94 | 97:3 |
| 3 |
|
|
|
90 | 97:3 |
| 4 |
|
– |
|
52 | 93:7 |
| 5 |
|
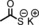
|
|
45 | 94:6 |
| 6[b] |
|
|
|
70 | 94:6 |
| 7 |
|
|
|
79 | 93:7 |
| 8[b] |
|
|
|
81 | 93:7 |
| 9 |
|
|
|
83 | 94:6 |
[a] Performed using 1.5 mol % [Co(MeTAA)] and 1.5 mol % additive under N2 with 1.0 equiv. of EDA (0.48 m) and 2 equiv. of olefin. [b] Performed using 1.5 mol % [Co(MeTAA)] and 25 mol % additive with 1.0 equiv. of EDA (0.38 m) and 3 equiv. of olefin. [c] Isolated yields. [d] Determined by using GC.
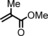
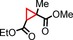
To better illustrate the influence of the additive, the alkene was changed to a more challenging one, methylmethacrylate (Table 2, entry 4), which only affords 52 % isolated yield without an additive. On addition of an equimolar amount of N‐methylimidazole with respect to the catalyst, the yield increased to 83 % (Table 2, entry 9). A positive effect is, therefore, observed upon the coordination of a nitrogen donor on one face of the catalyst. The addition of excess additive resulted in slightly reduced yields (Table 2, entries 6 and 8 vs. entries 7 and 9), which were, however, still higher than that without any additive. A partial blocking of both faces of the catalyst might play a role here. We hypothesise that the coordination of an additive to the CoII centre makes the catalyst more electron rich, thus more prone to carbene reduction and the formation of the cobalt(III) carbene radical intermediate during catalysis, which accelerates the reaction.
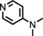
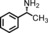
The increased yield of the cyclopropanation reaction caused by the coordination of additives to the catalyst led us to try several other ligands, which included chiral ones. These additives contain nitrogen donors, which can coordinate to Co, and −OH and −NH groups, which potentially stabilise intermediates during the catalytic cycle through hydrogen bonding interactions. We hoped this could lead to chirality transfer as well. Additives such as 1‐phenylethylamine, nicotine or ephedrine were good for the cyclopropanation of methyl acrylate with EDA, and the yield was increased to 99 % (Table 3, entries 2, 4 and 5). Bulkier additives such as di‐2‐naphthylprolinol in combination with [Co(MeTAA)] led to outstanding results in the cyclopropanation of styrene even at −78 °C (Table 3, entry 10), and the desired product was obtained almost quantitatively in a short reaction time. Although the additives have a beneficial effect on the overall yield and to decrease the reaction times, no chirality transfer was observed for any of the reactions in which a chiral additive was used. Even if we applied a high additive/catalyst ratio of 17:1 (25 mol % additive) the products were isolated as racemic mixtures. This implies that the catalyst itself requires a chiral backbone for efficient chirality transfer.
Table 3.
(Chiral) additive screening in the [Co(MeTAA)]‐catalysed cyclopropanation of methylacrylate and styrene.[a]

| Entry | R | Additive | T [°C] | t [h] | Yield [%][b] | trans/cis [c] |
|---|---|---|---|---|---|---|
| 1 | COOMe | – | 20 | 1 | 78 | 97:3 |
| 2 | COOMe |
|
20 | 1 | 99 | 94:6 |
| 3 | COOMe |
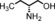
|
20 | 1 | 81 | 95:5 |
| 4 | COOMe |
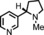
|
20 | 1 | 99 | 96:4 |
| 5 | COOMe |
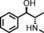
|
20 | 1 | 82 | 98:2 |
| 6 | COOMe |
|
0 | 2 | 83 | 97:3 |
| 7 | COOMe |
|
−78 | 5 | 65 | 99:1 |
| 8 | Ph | – | 20 | 1 | 85 | 85:15 |
| 9 | Ph |
|
0 | 2 | 92 | 87:13 |
| 10 | Ph |
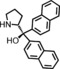
|
−78 | 5 | 98 | 89:11 |
| 11 | Ph |
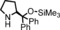
|
−78 | 5 | 93 | 89:11 |
[a] Performed using 1.5 mol % [Co(MeTAA)] and 25 mol % additive with 1.0 equiv. of EDA (0.38 m) and 3 equiv. of olefin. [b] Isolated yields. [c] Determined by using GC.
After we had optimised the reaction conditions, we decided to explore the substrate scope. We tested the cyclopropanation of 13 alkenes, which included a variety of electron‐deficient acrylates (Table 4, entries 1–6), using a 1:1 mixture of [Co(MeTAA)] and N‐methylimidazole in CH2Cl2 at room temperature under conditions in which the alkene is the limiting reagent. Reasonable yields in the range of 72–94 % were obtained with good diastereoselectivities up to 97:3 in favour of the trans isomer. A somewhat lower trans/cis ratio of 69:31 was obtained for acrylonitrile, but this is known to be a more problematic substrate, and catalysts reported previously did not achieve a higher ratio.5 The yield could be increased significantly by using [Co(BzMeTAA)] instead of [Co(MeTAA)]. Substituted styrenes were also tested (Table 4, entries 7–10) and yielded around 80 % of the isolated product, and again the trans isomer was favoured. No general trend was observed for the substitution pattern of styrene, which is more consistent with a radical mechanism rather than a concerted one. As can be observed in entries 11–13, internal alkenes and electron‐rich alkenes were challenging substrates for [Co(MeTAA)].
Table 4.
Substrate scope with the variation of both the alkene and the diazo compound.[a]
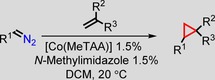
| Entry[a] | R1 | Olefin | Product | Yield [%][b] | trans/cis [c] |
|---|---|---|---|---|---|
| 1 | COOEt |
|
|
94 | 97:3 |
| 2 | COOEt |
|
|
83 | 94:6 |
| 3 | COOEt |
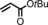
|
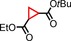
|
75 | 90:10 |
| 4 | COOEt |
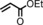
|
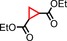
|
81 | 97:3 |
| 5 | COOEt |
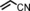
|
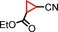
|
79 96[d] | 69:31 70:30 |
| 6 | COOEt |
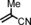
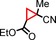
|
|
72 94[d] | 34:66 35:65 |
| 7 | COOEt |
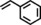
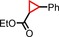
|
|
93 | 86:14 |
| 8 | COOEt |
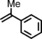
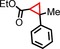
|
|
79 | 67:33 |
| 9 | COOEt |
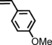
|
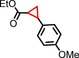
|
79 | 78:22 |
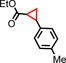
| 10 | COOEt |
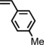
|
|
78 | 78:22 |
| 11 | COOEt |
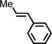
|
|
9 5[d] | 54:46 |
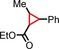
| 12 | COOEt |
|
|
<1 | n.d. |
| 13 | COOEt |
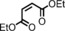
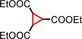
|
|
2 <1[d] | n.d. |
| 14 | COOtBu |
|
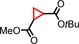
|
86 | 98:2 |
| 15 | COOBn |
|
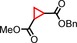
|
72 | 82:18 |
[a] Performed using 5 mol % [Co(MeTAA)] and 5 mol % N‐methylimidazole with 1.0 equiv. of olefin (0.47 m) and 1.2 equiv. of EDA. [b] Isolated yields. [c] Determined by using GC. [d] [Co(BzMeTAA)] was used instead of [Co(MeTAA)].
As a result of the intrinsic instability and potentially explosive nature of diazo compounds in the absence of an electron‐withdrawing substituent, commercially available diazo compounds are mainly ester‐substituted ones. As such, only a few diazo compounds could be screened conveniently for these experiments (Table 4, entries 1, 14, 15), which afforded moderate to good yields with a similar preference towards the trans cyclopropane as the base reaction with EDA. However, the substrate scope can be expanded by switching from the use of stable diazo compounds to tosylhydrazone salts as precursors for non‐stabilised diazo compounds. The thermal decomposition of tosylhydrazone salts was developed by Aggarwal et al. as an attractive methodology to prepare substituted diazo compounds in situ.22 They applied this method in the catalytic synthesis of epoxides from ketones. However, with the Rh and Fe catalysts tested, cyclopropane formation was much less successful using tosylhydrazone salts as carbene precursors and led to poor yields and selectivities. As such, we wondered if [Co(MeTAA)] was more tolerant to (reaction conditions associated with) the use of tosylhydrazone salts as precursors to diazo compounds in one‐pot catalytic transformations. Hence, we performed some screening experiments for the cyclopropanation of methyl acrylate with sodium benzyl tosylhydrazone salt catalysed by [Co(MeTAA)] at a temperature between 20 and 45 °C (Table 5).
Table 5.
Screening of the reaction conditions for the cyclopropanation of methylacrylate with sodium benzyl tosylhydrazone salt catalysed by [Co(MeTAA)].[a]
| Solvent | PTC[c] | Additive[d] | T [°C] | t [h] | Yield [%] | trans/cis | |
|---|---|---|---|---|---|---|---|
| 1 | CH3CN | – | – | 45 | 18 | 80 | 74:26 |
| 2 | THF | – | – | 45 | 18 | 67 | 76:24 |
| 3 | PhMe | – | – | 45 | 18 | 47 | 75:25 |
| 4 | PhCl | – | – | 45 | 18 | 52 | 76:24 |
| 5 | Dioxane | – | – | 45 | 18 | 13 | – |
| 6 | 1,2‐Dichloro ethane | – | – | 45 | 18 | 38 | 74:26 |
| 7 | DMF | – | – | 45 | 18 | 64 | 75:25 |
| 8[b] | MeOH | – | – | 45 | 18 | – | – |
| 9 | THF | Aliquat® 336 | – | 45 | 18 | >99 | 76:24 |
| 10 | THF | Aliquat® 336 | – | 45 | 4 | >99 | 76:24 |
| 11 | THF | Aliquat® 336 | – | 45 | 2 | >99 | 76:24 |
| 12 | PhMe | Aliquat® 336 | – | 45 | 18 | >99 | 76:24 |
| 13 | PhCl | Aliquat® 336 | – | 45 | 18 | 94 | 75:25 |
| 14 | THF | Aliquat® 336 | – | 20 | 18 | 85c | 76:24 |
| 15 | CH2Cl2 | Aliquat® 336 | – | 20 | 18 | 16c | – |
| 16 | CH2Cl2 | – | – | 20 | 18 | – | – |
| 17 | THF | – | 1‐methyl‐ imidazole | 45 | 18 | 96 | 75:25 |
| 18 | THF | – | DMAP | 45 | 18 | 86 | 74:26 |
| 19 | THF | – | pyridine | 45 | 18 | >99 | 76:24 |
| 20 | THF | – | 2,4,6‐trimethyl pyridine | 45 | 18 | >99 | 76:24 |
[a] Reactions were performed under N2 with 1.0 equiv. of sodium benzyl tosylhydrazone, 3.0 equiv. of methylacrylate and 3 mol % [Co(MeTAA)]. Concentration: 0.675 mmol sodium benzyl tosylhydrazone/5 mL solvent; [b] methyl 3‐methoxypropanoate is obtained as the sole product; [c] 0.15 equiv. Aliquat®336; [d] 0.5 equiv. additive. [f] Aliquat ® 336 is a quaternary ammonium salt that contains a mixture of C8 (octyl) and C10 (decyl) chains (predominantly C8) and a chloride counter ion.
The catalyst is indeed tolerant to these reaction conditions, and this approach broadened the substrate scope substantially. Extended solvent screening was performed, in which eight different solvents were tested (Table 5, entries 1–8). The highest yields were obtained in acetonitrile (80 %) and THF (67 %), whereas 1,2‐dichloroethane (38 %) and dioxane (13 %) led to poor yields. Tosylhydrazone salts are insoluble in non‐polar solvents such as toluene and, therefore, are known to be troublesome for in situ diazo formation.22 This was confirmed in our studies. In toluene, the reaction afforded only 47 % of the desired cyclopropane. However, phase‐transfer catalysts (PTCs) are known to enhance the rate of the conversion of the tosylhydrazone salts into diazo compounds, even in non‐polar solvents such as toluene.23 Therefore, we tested Aliquat®336 in combination with several solvents (Table 5, entries 9–15).
The effect is remarkable, and the yield increased to 99 % (entry 9) even upon drastic reduction of the reaction time (entries 10–11) or decrease of the reaction temperature to 20 °C (entry 14). Interestingly, solvents that led to poor yields in the absence of this PTC, such as toluene, become viable solvents for this reaction in the presence of Aliquat®336 to afford almost quantitative yields of the desired cyclopropane product (entry 12).
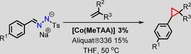
As nitrogen‐donor additives have a positive influence on the activity of [Co(MeTAA)] (vide supra), we decided to test their effect in combination with tosylhydrazone salts as well (Table 5, entries 17–20), this time in the absence of Aliquat®336 as a PCT. 1‐Methylimidazole again performs better than DMAP with a yield of 96 versus 86 %. The best results were obtained with pyridine and 2,4,6‐trimethylpyridine, both of which led to the almost quantitative formation of the desired cyclopropane with a negligible effect on the diastereoselectivity. As a result of the high yields obtained using either a PTC or a nitrogen‐donor ligand additive, the use of both would be redundant for these specific reactions. However, the mechanisms of action of the quaternary ammonium salt Aliquat®336 and nitrogen‐donor ligand additives are totally different, even it might seem that they lead to similar results. The PTC favours the in situ generation of the diazo compound, whereas the nitrogen donor additive enhances the electronics of the Co catalyst. Therefore, we decided to screen the substrate scope without the addition of nitrogen‐donor ligands but in the presence of Aliquat®336 as a PTC. However, it might be beneficial to use a combination of both for more challenging substrates. Five substituted sodium tosylhydrazone salts were used in the cyclopropanation of methyl acrylate. Tosylhydrazone salts that contain electron‐donating substituents afforded the desired products in high yields of 94–97 % (Table 6, entries 1,4 and 5). Tosylhydrazone salts that contain electron‐withdrawing substituents resulted in somewhat lower yields (Table 6, entries 2 and 3). The reaction of the iPr‐substituted tosylhydrazone salt and four electron‐deficient alkenes, from the acrylate family, were tested and afforded the desired cyclopropanes in yields up to 97 % (Table 6, entries 5–8). Remarkably, acrylonitrile yielded the substituted cyclopropane in 96 % yield with a 3:1 trans/cis selectivity (entry 7). Substituted styrenes (entries 9–11) are also cyclopropanated easily using [Co(MeTAA)] and tosylhydrazone salts.
Table 6.
Substrate scope with the variation of both the tosylhydrazone salt and the alkene.[a]
| Entry | R1 | Alkene | Product | Yield [%] | trans/cis |
|---|---|---|---|---|---|
| 1 | Ph |
|
|
94 | 78:22 |
| 2 | CN |
|
|
68 | 70:30 |
| 3 | NO2 |
|
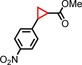
|
63 | 72:28 |
| 4 | OMe |
|
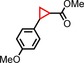
|
94 | 74:26 |
| 5 | iPr |
|
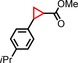
|
97 | 75:25 |
| 6 | iPr |
|
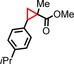
|
93 | 73:26 |
| 7 | iPr |
|
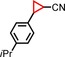
|
96 | 75:25 |
| 8 | iPr |
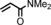
|
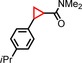
|
77 | 62:38 |
| 9 | iPr |
|
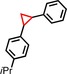
|
89 | 89:11 |
| 10 | iPr |
|
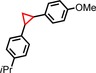
|
90 | 86:14 |
| 11 | iPr |
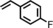
|
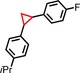
|
85 | 90:10 |
[a] Reactions were performed under N2 with 1.0 equiv. of sodium tosylhydrazone, 3.0 equiv. alkene, 0.15 equiv. Aliquat®336 and 3 mol % [Co(MeTAA)]. Concentration: 0.675 mmol sodium tosylhydrazone/5 mL THF solvent.
Conclusions
We have shown that cobalt(II) tetraaza[14]annulene complexes are highly active in the catalytic cyclopropanation of electron‐deficient alkenes. These low‐spin CoII catalysts are cheap, easy to synthesise, afford high cyclopropanation yields and their activity is superior to that of cobalt(II) meso‐tetraphenylporphyrin. Fast and selective carbene‐transfer reactions are achieved by taking advantage of the radical mechanism that involves discrete cobalt(III) carbene radical species, and unwanted carbene–carbene dimerisation and 1,3‐dipolar addition reactions between the electron deficient alkene and the diazo reagent were almost completely suppressed. In addition, a new one‐pot protocol was presented that uses in situ generated diazo compounds. This method expands the substrate scope substantially by taking advantage of the compatibility between the catalyst and the thermal decomposition of tosylhydrazone salts in one‐pot transformations. This method yields substituted cyclopropanes in almost quantitative yields using relatively low catalyst loadings. All the reactions described in this paper can be performed in a one‐pot fashion, are practical and fast, tolerant to many solvents, can be performed in a broad temperature range and do not require the slow addition of any of the components. The reaction is diastereoselective, and the trans isomer is favoured in all cases. The formation of byproducts is kept to a minimum. To elucidate the mechanism of the cobalt(II) tetramethyltetraaza[14]annulene complex during radical catalysis fully, in depth kinetic studies and DFT investigations are required. These studies are underway and will be reported in a follow‐up paper.
Experimental Section
General considerations
All manipulations were performed under a nitrogen atmosphere using standard Schlenk techniques. All solvents used for catalysis were dried over and distilled from sodium (toluene, tetrahydrofuran, diethyl ether) or CaH2 (dichloromethane, methanol, acetonitrile). Diazo compounds and alkenes were degassed using the freeze–thaw–pump method. Acrylates were passed through basic alumina before use to remove radical scavengers. All other chemicals were purchased from commercial suppliers (Sigma Aldrich, Acros or Strem) and used without further purification. NMR spectra (1H, and 13C{1H}) were measured by using a Bruker AV400, AV300, DRX 500 or DRX 300 spectrometer. Unless noted otherwise, the NMR spectra were measured in CDCl3. Individual peaks are reported as: multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet), integration, coupling constant [Hz]. Mass spectra of the synthesised compounds were recorded by using an Agilent‐5973 GC–MS system, and the corresponding HRMS data were recorded by using a JEOL AccuTOF 4G with a direct injection probe using either EI or ESI. The GC used for isomer separation was a Shimadzu 17A with a Supelco SPB TM‐1 Fused Silica Capillary Column with a length of 30 m, a diameter of 0.32 mm and a film thickness of 2.0 μm.
Catalyst preparation
[Co(MeTAA)], [Co(MePhTAA)] and [Co(Bz‐MeTAA)] were synthesised according to reported procedures.20 [Co(TPP)] and [Co(salen)] were purchased from Sigma–Aldrich and used without further purification.
General procedure for cyclopropanation using diazo compounds
Under a N2 atmosphere, catalyst (0.05 equiv.) was added to a flame‐dried Schlenk tube. The tube was capped, evacuated and backfilled with N2. The solid was dissolved, and the additive (0.05 equiv.) and alkene (1.0 equiv., 0.48 m) were added. Then, the diazo compound (1.2 equiv.) was added, and the solution was stirred for 1 h. The resulting mixture was concentrated, and the residue was purified by flash chromatography (silica gel) or extracted into pentane.
General procedure for the synthesis of the N‐tosylhydrazone salts24
An equimolar mixture of the corresponding aldehyde and N‐tosylhydrazide was placed in a round‐bottomed flask and dissolved in methanol (2 mL mmol−1). The reaction mixture was stirred overnight at RT. The white precipitate was collected by filtration and washed with cold methanol and hexane to obtain the pure product. The formed N‐tosylhydrazone was then deprotonated in methanol with NaOMe (1 equiv.). After the evaporation of methanol, the pure product was obtained as a white powder.
General procedure for cyclopropanation using N‐tosylhydrazone salts
Under a N2 atmosphere, the respective N‐tosylhydrazone salt (1.0 equiv.), catalyst (0.03 equiv.) and Aliquat®336 (0.15 equiv.) were added to a flame‐dried Schlenk tube in a glovebox. The tube was capped, evacuated and backfilled with N2. Then, alkene (3.0 equiv., 0.405 m) and solvent were added. The Schlenk tube was then placed in an oil bath and heated to 50 °C under N2 for a set time. The resulting mixture was concentrated, and the residue was purified by flash chromatography (silica gel) or extracted into pentane.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Ed Zuidinga for MS measurements, Jan Meine Ernsting for NMR advice and Daan den Boer for help with experiments. Financial support from the Netherlands Organization for Scientific Research (NWO‐CW VICI project 016.122.613) and the University of Amsterdam (Research Priority Area Sustainable Chemistry) is gratefully acknowledged.
A. Chirila, B. Gopal Das, N. D. Paul, B. de Bruin, ChemCatChem 2017, 9, 1413.
References
- 1. Nozaki H., Moriuti S., Takaya H., Noyori R., Tetrahedron Lett. 1965, 6, 2563–2567. [Google Scholar]
- 2. Lo M., Fu G. C., J. Am. Chem. Soc. 1998, 120, 10270–10271. [Google Scholar]
- 3. Davies H. M. L., Bruzinski P. R., Lake D. H., Kong N., Fall M. J., J. Am. Chem. Soc. 1996, 118, 6897–6907. [Google Scholar]
- 4. Che C., Huang J., Lee F., Li Y., Lai T., Kwong H., Teng P., Lee W., Lo W., Peng S., Zhou Z., J. Am. Chem. Soc. 2001, 123, 4119–4129. [DOI] [PubMed] [Google Scholar]
- 5. Chen Y., Ruppel J. V., Zhang X. P., J. Am. Chem. Soc. 2007, 129, 12074–12075. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Simovic D., Di M., Marks V., Chatfield D. C., Rein K. S., J. Org. Chem. 2007, 72, 650; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Novikov R. A., Platonov D. N., Dokichev V. A., Tomilov Y. V., Nefedov O. M., Russ. Chem. Bull. 2010, 59, 984. [Google Scholar]
- 7. Baratta W., Del Zotto A., Rigo P., Chem. Commun. 1997, 2163–2164. [Google Scholar]
- 8. Doyle M. P., Angew. Chem. Int. Ed. 2009, 48, 850–852; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 864–866. [Google Scholar]
- 9. Nakamura A., Konishi A., Tatsuno Y., Otsuka S., J. Am. Chem. Soc. 1978, 100, 3443–3448. [Google Scholar]
- 10. Niimi T., Uchida T., Irie R., Katsuki T., Adv. Synth. Catal. 2001, 343, 79–88. [Google Scholar]
- 11. Ikeno T., Sato M. I., Sekino H., Nishizuka A., Yamada T., Bull. Chem. Soc. Jpn. 2001, 74, 2139–2150. [Google Scholar]
- 12.
- 12a. Huang L., Chen Y., Gao G., Zhang X. P., J. Org. Chem. 2003, 68, 8179–8184; [DOI] [PubMed] [Google Scholar]
- 12b. Penoni A., Wanke R., Tollari S., Gallo E., Musella D., Ragaini F., Demartin F., Cenini S., Eur. J. Inorg. Chem. 2003, 1452–1460. [Google Scholar]
- 13.
- 13a. Zhu S., Perman J. A., Zhang X. P., Angew. Chem. Int. Ed. 2008, 47, 8460–8463; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8588–8591; [Google Scholar]
- 13b. Zhu S. F., Xu X., Perman J. A., Zhang X. P., J. Am. Chem. Soc. 2010, 132, 12796–12799. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Caselli A., Gallo E., Ragaini F., Ricatto F., Abbiati G., Cenini S., Inorg. Chim. Acta 2006, 359, 2924–2932; [Google Scholar]
- 14b. Fantauzzi S., Gallo E., Rose E., Raoul N., Caselli A., Issa S., Ragaini F., Cenini S., Organometallics 2008, 27, 6143–6151; [Google Scholar]
- 14c. Rose E., Gallo E., Raoul N., Bouche L., Pille A., Caselli A., Lequin O., J. Porphyrins Phthalocyanines 2010, 14, 646–659. [Google Scholar]
- 15. Intrieri D., Caselli A., Gallo E., Eur. J. Inorg. Chem. 2011, 5071–5081. [Google Scholar]
- 16.
- 16a. Dzik W. I., Xu X., Zhang X. P., Reek J. N. H., de Bruin B., J. Am. Chem. Soc. 2010, 132, 10891–10902; [DOI] [PubMed] [Google Scholar]
- 16b. Lu H., Dzik W. I., Xu X., Wojtas L., de Bruin B., Zhang X. P., J. Am. Chem. Soc. 2011, 133, 8518–8521; [DOI] [PubMed] [Google Scholar]
- 16c. Dzik W. I., Zhang X. P., de Bruin B., Inorg. Chem. 2011, 50, 9896–9903. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Cotton F. A., Czuchajowska J., Polyhedron 1990, 9, 2553; [Google Scholar]
- 17b. Mountford P., Chem. Soc. Rev. 1998, 27, 105. [Google Scholar]
- 18.
- 18a. Imler G. H., Bhagan S., Coffin V. L., Wayland B. B., Inorg. Chem. 2012, 51, 3352; [DOI] [PubMed] [Google Scholar]
- 18b. Imler G. H., Zdil-la M. J., Wayland B. B., Inorg. Chem. 2013, 52, 11509; [DOI] [PubMed] [Google Scholar]
- 18c. Imler G. H., Zdilla M. J., Wayland B. B., J. Am. Chem. Soc. 2014, 136, 5856; [DOI] [PubMed] [Google Scholar]
- 18d. Imler G. H., Peters G. M., Zdilla M. J., Wayland B. B., Inorg. Chem. 2015, 54, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Das B. G., Chirila A., Tromp M., Reek J. N. H., de Bruin B., J. Am. Chem. Soc. 2016, 138, 8968. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Eilmes J., Polyhedron 1991, 10, 1779; [Google Scholar]
- 20b. Dzugan S. J., Busch D. H., Inorg. Chem. 1990, 29, 2528; [Google Scholar]
- 20c. Eilmes J., Polyhedron 1985, 4, 943. [Google Scholar]
- 21.
- 21a. Weiss M. C., Goedken V. L., J. Am. Chem. Soc. 1976, 98, 3389; [Google Scholar]
- 21b. Weiss M. C., Gordon G. C., Goedken V. L., J. Am. Chem. Soc. 1979, 101, 857. [Google Scholar]
- 22.
- 22a. Aggarwal V. K., Alonso E., Bae I., Hynd G., Lydon K. M., Palmer M. J., Patel M., Porcelloni M., Richardson J., Stenson R. A., Studley J. R., Vasse J.-L., Winn C. L., J. Am. Chem. Soc. 2003, 125, 10926; [DOI] [PubMed] [Google Scholar]
- 22b. Fulton J. R., Aggarwal V. K., De Vicente J., Eur. J. Org. Chem. 2005, 1479; [Google Scholar]
- 22c. Adams L. A., Aggarwal V. K., Bonnert R. V., Bressel B., Cox R. J., Shepherd J., De Vicente J., Walter M., Whittingham W. G., Winn C. L., J. Org. Chem. 2003, 68, 9433. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Z., Liu Y., Ling L., Li Y., Dong Y., Gong M., Zhao X., Zhang Y., Wang J., J. Am. Chem. Soc. 2011, 133, 4330. [DOI] [PubMed] [Google Scholar]
- 24. Arunprasath D., Muthupandi P., Sekar G., Org. Lett. 2015, 17, 5448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary