

Abstract
The activins were discovered and named based on their abilities to stimulate FSH secretion and FSHβ (Fshb) subunit expression by pituitary gonadotrope cells. According to subsequent in vitro observations, activins also stimulate the transcription of the GnRH receptor (Gnrhr) and the activin antagonist, follistatin (Fst). Thus, not only do activins stimulate FSH directly, they have the potential to regulate both FSH and LH indirectly by modulating gonadotrope sensitivity to hypothalamic GnRH. Moreover, activins may negatively regulate their own actions by stimulating the production of one of their principal antagonists. Here, we describe our current understanding of the mechanisms through which activins regulate Fshb, Gnrhr, and Fst transcription in vitro. The activin signaling molecules SMAD3 and SMAD4 appear to partner with the winged-helix/forkhead transcription factor, forkhead box L2 (FOXL2), to regulate expression of all 3 genes. However, in vivo data paint a different picture. Although conditional deletion of Foxl2 and/or Smad4 in murine gonadotropes produces impairments in FSH synthesis and secretion as well as in pituitary Fst expression, Gnrhr mRNA levels are either unperturbed or increased in these animals. Surprisingly, gonadotrope-specific deletion of Smad3 alone or with Smad2 does not impair FSH production or fertility; however, mice harboring these mutations may express a DNA binding-deficient, but otherwise functional, SMAD3 protein. Collectively, the available data firmly establish roles for FOXL2 and SMAD4 in Fshb and Fst expression in gonadotrope cells, whereas SMAD3's role requires further investigation. Gnrhr expression, in contrast, appears to be FOXL2, SMAD4, and, perhaps, activin independent in vivo.
The activins are pleiotropic members of the TGFβ superfamily but were discovered and named based on their effects on FSH synthesis by pituitary gonadotrope cells. Research groups led by Vale and Guillemin, fresh on the heels of their successful purification of the inhibins from porcine follicular fluid, identified 2 novel ligands that we now call activin A and activin AB (1–3). These proteins proved to be homo- or heterodimers of the newly identified inhibin β-subunits. Despite (or perhaps because of) their structural similarity, activins and inhibins revealed themselves to be functionally antagonistic. That is, when applied to rat pituitaries dispersed in culture, activins selectively stimulated FSH release with no effects on LH, whereas inhibins suppressed FSH (2–5). Research over the next decade established that activins produce their effects, in large part, by stimulating expression of the FSHβ (Fshb) subunit gene (6–9). Although inhibins function as bona fide endocrine hormones, activins appear to act in autocrine or paracrine fashion, like most TGFβ superfamily members.
Activin B emerged as the gonadotrope cell-derived ligand most likely to stimulate FSH synthesis (10–12). It was also quickly appreciated that activins' actions in gonadotropes were not limited to FSH synthesis. In particular, activins stimulated GnRH receptor (Gnrhr) expression (13, 14), suggesting a role for autocrine/paracrine regulation of gonadotrope cell responsiveness to hypothalamic GnRH. At around the same time, activins were observed to stimulate pituitary synthesis and secretion of follistatin (Fst) (15, 16). Follistatins bind and bioneutralize activins (17, 18). Therefore, activins can stimulate the production of their own endogenous antagonists via a short feedback loop.
Research by many groups, predominantly using in vitro approaches, has uncovered candidate mechanisms through which activins regulate transcription of the Fshb, Gnrhr, and Fst genes. In this minireview, we first summarize these proposed mechanisms, particularly in rodents (Figure 1). Next, we discuss more recent in vivo data from genetically modified mice that both support and challenge current models of activin action in gonadotropes. Finally, we highlight new questions inspired by these and other in vivo observations.
Figure 1. Schematic representation of activin signaling in murine gonadotrope cells.

Activin B (or a related ligand) produced by gonadotropes acts in autocrine or paracrine fashion to stimulate Fshb, Gnrhr, and Fst transcription. The dimeric ligand binds complexes of type I/type II serine/threonine receptor kinases. ACVR2 and perhaps BMPR2 are the most likely type II receptors in this system. ACVR1B (also known as ALK4) and ACVR1C (ALK7) are the candidate type I receptors. Upon ligand binding, the type II receptors phosphorylate the type I receptors. The type I receptors then phosphorylate the signaling molecules SMAD2 (S2) and SMAD3 (S3). These receptor-regulated SMADs then partner with the common partner SMAD, SMAD4 (S4), and accumulate in the nucleus. According to in vitro models (pictured at the left of the nucleus), these SMAD complexes partner with FOXL2 (F2) to regulate all 3 genes. Left top, In the murine Fshb promoter, SMAD3 partners with FOXL2 at a distal FBE (FBE1); with SMAD2 and SMAD4 at a composite 8-bp SBE (SBEx2); with SMAD4 and FOXL2 at a proximal SBE/FBE3 composite element; and perhaps with paired-like homeodomain transcription factors (PITX or P) at a proximal PITX-binding site. Also, pictured is the location of a composite FBE2/SBE element, which may be unique to the porcine Fshb promoter (see Figure 2). Left middle, SMAD3 partners with DNA-bound SMAD4 and FOXL2 at a composite response element referred to as GRAS to stimulate the murine Gnrhr promoter. An adjacent response element called DARE, which binds LHXs, also mediates activin responsiveness but through currently unknown mechanisms. Left bottom, SMAD3 partners with FOXL2 at an intronic enhancer in the rat Fst gene to mediate its activin induction. SMAD4's role in the response is poorly defined. At the right, The models have been revised to reflect the results of in vivo mouse knockout experiments. Right top, The model of Fshb regulation is largely unchanged, with 2 exceptions: 1) SMAD2 has been removed, and 2) SMAD3 does not need to bind DNA to produce its effects, casting doubt on a role for the 8-bp SBE (SBEx2). Right middle, SMAD4 and FOXL2 do not positively regulate murine Gnrhr. Whether the gene is activin regulated via alternative mechanisms is unclear but appears doubtful. Right bottom, Both FOXL2 and SMAD4 regulate Fst transcription in gonadotropes. The model has been revised to suggest that SMAD4 rather than SMAD3 binds the SBE in the enhancer and the relative location of the FBE is proposed to be directly adjacent to, but 5′ of the SBE (see Figure 3).
Activin Signaling In Vitro
Like other TGFβ superfamily members, activins bind and signal through heterotetrameric complexes of transmembrane type I and 2 type II serine/threonine kinase receptors (eg, Figure 1) (19, 20). Once activated (phosphorylated) by their type II partners, type I receptors phosphorylate effector proteins in the homologs of Drosophila mothers against decapentaplegic (SMAD) family. These so-called receptor-regulated SMADs then complex with the common partner SMAD, SMAD4, and accumulate in the nucleus, where they act as transcription factors, most often in association with DNA binding partners (21, 22). As reviewed in the next sections, this general model seems to apply to activin regulation of Fshb, Gnrhr, and Fst in immortalized gonadotrope-like cells. Moreover, the same signaling proteins and DNA binding cofactor appear to mediate activin-induction of all 3 genes, at least in vitro.
Activin induction of Fshb transcription
Activins can signal via either of 2 type II receptors, activin receptor type IIA (ACVR2) or bone morphogenetic protein receptor type II (BMPR2), to stimulate Fshb transcription in the immortalized murine gonadotrope-like cell line, LβT2 (23, 24). In contrast, the other canonical activin type II receptor, ACVR2B, appears dispensable for FSH synthesis (23, 24). It should be noted that LβT2 cells are the only differentiated gonadotrope cell line available in any species (25) and have therefore provided the model for most research on Fshb transcriptional regulation. Studies of Gnrhr and Fst have employed both this and a second, less mature, murine gonadotrope-like cell line, αT3-1 (25). Consistent with the cell line data, FSH levels are reduced in Acvr2 knockout mice (26), whereas BMPR2's role in FSH synthesis in vivo has not yet been reported.
Downstream of ACVR2A (and BMPR2?), activins can signal through either of 2 type I receptors, ACVR1B or ACVR1C, to regulate Fshb transcription (27). The relative importance of these receptors is presently unclear and may be ligand specific, because activin B and AB can engage both receptors, whereas activin A only signals via ACVR1B (27, 28). Notably, activin A or B induction of FSH secretion is preserved in cultured pituitaries of Acvr1c knockout mice, suggesting that ACVR1B may be the preferred type I receptor, or that it can compensate for the loss of ACVR1C (29).
SMAD2 and SMAD3 are the canonical receptor-regulated SMADs downstream of activin type I receptors (22), and SMAD3 appears most critical for regulation of rodent, porcine, and ovine Fshb promoter reporters in immortalized gonadotropes (30–38). SMAD2's role is less well defined (30, 32, 34, 35). Phosphorylated SMAD3 typically partners with SMAD4 and accumulates in the nucleus to regulate transcription (22). As revealed by RNA interference-mediated knockdown studies, SMAD4 is required for activin induction of the murine and ovine Fshb promoters (38, 39). Thus, the available in vitro evidence indicates that activins stimulate Fshb gene expression through a canonical type I/II receptor-SMAD3/4-mediated signaling cascade (Figure 1).
Once in the nucleus, SMAD proteins recognize specific, but low affinity 4-bp SMAD-binding elements (SBEs) (GTCT) in target gene promoters (22). A rarer, but higher affinity site is formed by a palindromic arrangement of 2 minimal SBEs (GTCTAGAC), which can accommodate the simultaneous binding of 2 SMAD proteins (40, 41). The proximal Fshb promoters in rats and mice contain this 8-bp SBE (labeled SBEx2 in Figure 1), but it is notably absent in the orthologous promoters in humans, sheep, and pigs. This suggests a species-specific role for this cis-element in rodents. Indeed, activin induction of rodent Fshb promoters is reduced (by ∼50%), but not lost, when this site is mutated (33, 35, 42). Therefore, whereas the 8-bp SBE may play a role in the overall amplitude of the promoter response in rodents, it is not necessary for activin-mediated transcription in any mammalian species investigated to date.
These observations motivated the search for additional response elements conferring activin sensitivity (32–35, 42, 43). As reviewed previously (44), a major breakthrough came through the comparison of activin responsiveness of the human and porcine Fshb promoters. In brief, the porcine promoter contains a high-affinity binding element, hereafter forkhead-binding element (FBE), for the transcription factor forkhead box L2 (FOXL2), and this site is adjacent to a 4-bp SBE (43). This particular FBE (labeled FBE2 in Figure 1), which is unique to the porcine Fshb promoter (Figure 2), is required for its pronounced activin sensitivity (43). Nonetheless, FOXL2 knockdown experiments in LβT2 cells indicate a role for the protein in activin's regulation of murine Fshb (43). In silico analysis identified a second, more proximal FBE (labeled FBE3 in Figure 1). Importantly, this element is conserved in Fshb promoters of pigs, mice, rats, and humans and, in all cases, is 5′ flanked by a minimal SBE. According to the current model (Figure 1), this composite SBE/FBE3 binds SMAD4 and FOXL2, which are linked through their shared physical interactions with SMAD3 (37, 45, 46). Although only explored thus far in the porcine and murine Fshb promoters, it seems likely that this represents a common mechanism of activin-regulated Fshb transcription across mammalian species. However, the relative spacing of the SBE and FBE3 may contribute to interspecies differences in activin sensitivity (43) as might the presence of additional nonconserved FBEs that have thus far been described in the human, murine, and porcine promoters (Figures 1 and 2) (36, 37, 43, 46–48; reviewed in Ref. 44).
Figure 2. The high-affinity FBE2 is unique to the porcine Fshb promoter.

Bottom, Fshb/FSHB promoter sequences from pig, human, sheep, and mouse were aligned to show the similarities and differences in the composite FBE2/SBE response element. There is a single base pair difference at the first position in pig (T) relative to the other species (C). As described in Ref. 43, this enables high-affinity binding of FOXL2 to the porcine, but not human, promoter. Top, Replacement of the C with a T (C→T) in human, ovine, and murine FSHB/Fshb promoter reporters confers enhanced activin sensitivity in LβT2 cells. The murine promoter differs at 2 bp from that of the pig in FBE2. Introduction of the porcine base pairs into the murine promoter (CA→TG) confers even greater activin sensitivity than the C→T base pair change alone. The data reflect the means (±SD or SEM) of 2 (human and sheep; n = 2) or 3 (mouse; n = 3) independent experiments, with treatments performed in duplicate or triplicate. Note the break in the y-axis.
In addition to FBE and SBE, a conserved binding site for paired-like homeodomain transcription factors, such as PITX1 and PITX2, is present in the Fshb/FSHB promoters of several species, including rodents and humans (Figure 1) (49, 50). Mutations that block PITX1 and PITX2 protein binding impair both basal and activin-stimulated Fshb transcription (35, 49–51). PITX1/2 and SMAD proteins physically interact and cooperatively activate the Fshb promoter. Moreover, depletion of endogenous PITX proteins impairs activin-stimulated Fshb transcription in LβT2 cells (35, 37, 50, 52). Nevertheless, it is presently unclear whether or how functional PITX/SMAD complexes are formed on the Fshb promoter as there are no SBEs in immediate proximity to the PITX-binding site.
Overall, signaling through SMAD/FOXL2 complexes and their cognate binding sites appears to represent a fundamental mechanism underlying activin induced Fshb transcription. The contributions, if any, of PITX proteins and perhaps other factors (53–55) require more investigation.
Activin induction of murine Gnrhr transcription
GnRH is a necessary and potent regulator of gonadotropin synthesis and secretion (eg, Refs. 56–59). Gonadotrope cell responsiveness to GnRH, in turn, is regulated by relative GnRHR expression, which varies under a variety of physiological conditions (eg, Refs. 60, 61). The main drivers of Gnrhr expression are GnRH itself (62–64), steroid hormones (64, 65), and activins (14). GnRH and activin regulation of GnRHR protein expression in primary rat pituitary cultures appears to involve distinct, but complementary, mechanisms (13, 66, 67). This has been substantiated by mechanistic analyses of the murine Gnrhr promoter in αT3-1 and LβT2 cells (7, 65, 66, 68–71).
A tripartite enhancer in the murine Gnrhr 5′-flanking region is critical for transcriptional activity in gonadotrope-like cells (69). The enhancer includes binding sites for steroidogenic factor 1 (also known as NR5A1) and activator protein-1 (AP-1) complexes (69, 72). Gnrhr mRNA levels are markedly reduced in Nr5a1 knockout mice (73) as well as in mice harboring a mutation in the AP-1 site (74). Therefore, the extant data confirm roles for steroidogenic factor 1 and AP-1 proteins (or at least the AP-1-binding site) in Gnrhr expression in vivo. The third essential element of the enhancer is referred to as GnRHR-activating sequence (GRAS) (5′-CTAGTCACAACA-3′) (Figure 1), which mediates activin induction of the murine Gnrhr promoter in vitro (67). Its role in activin regulation of Gnrhr in other species is less clear (75).
SMAD3 and SMAD4 associate with the 5′-end of GRAS with SMAD4 brokering direct DNA binding to the sequence CTAGTC (Figure 1) (71). The latter 4 bp (underlined) resemble the reverse-complement of a consensus SBE (AGAC). According to crystallographic data, the base pair at the third position (T in GRAS and A in the consensus sequence) does not contribute to SMAD binding (40, 76) and therefore may tolerate sequence variation. Activin and GnRH synergistically regulate Gnrhr expression (66, 70, 71), and their cooperation depends on an AP-1 site in GRAS (not shown in Figure 1 and distinct from the AP-1 site described above), which partly overlaps with the SBE (70, 71). Remarkably, the 3′-end of GRAS binds FOXL2. Moreover, SMAD3 induction of a GRAS reporter requires FOXL2 binding (71). The putative FOXL2-binding site (CACAACA), which has not yet been thoroughly characterized, resembles FBE3 in the proximal Fshb promoter (CTAAACA; base pair differences are underlined). Therefore, in both Gnrhr and Fshb, composite SBE/FBE sites in the 5′-flanking region mediate activin responsiveness. In both cases, SMAD4 and FOXL2 are predicted to bind DNA directly, with SMAD3 mediating their interaction.
As mentioned, GRAS contains an AP-1 element between the SBE and FBE that plays a role in activin/GnRH synergism. No such AP-1 site has been described within the SBE/FBE3 of Fshb, but the promoter is similarly synergistically regulated by activin and GnRH (eg, Refs. 7, 32, 77, 78). In the case of the human FSHB promoter, this involves cooperation between SMAD and AP-1 proteins acting via an AP-1-binding site just 3′ of SBE/FBE3 (78). A definitive role for the FOXL2 protein or FBE3 in activin or activin/GnRH induction of human FSHB promoter activity has yet to be established (43, 47). However, SMADs, FOXL2, and AP-1 proteins, acting at least in part via the SBE/FBE3, may mediate activin/GnRH synergistic regulation of murine Fshb (46). Thus, there appear to be similar mechanisms of activin action on the murine Fshb and Gnrhr promoters.
Nonetheless, there are notable differences. Activins regulate murine Fshb via an 8-bp SBE as well as a more distal FBE (FBE1 in Figure 1), which lacks an adjacent SBE (36, 46, 47). Moreover, although PITX proteins also regulate Gnrhr gene transcription (79, 80), they have not been implicated in activin induction. The murine Gnrhr promoter contains an additional cis-element called downstream activin regulatory element (DARE), which is located 16-bp 3′ of GRAS (Figure 1). DARE is necessary for activin induction of Gnrhr transcription in αT3-1 cells (75). DARE contains 2 TAAT motifs, which can bind LIM-homeodomain proteins (LHXs) 2 and 3 (LHX2/3) (81). At present, it is unclear whether or how LHX proteins mediate activin responses. Interestingly, LHX3 can also regulate the Fshb/FSHB promoters in pigs and humans, but its binding sites are not required for activin induction of porcine Fshb (82).
Collectively, the data suggest that activin regulates murine Gnrhr promoter activity in αT3-1 cells via at least 2 promoter elements, GRAS and DARE (Figure 1). GRAS binds complexes of SMAD3, SMAD4, and FOXL2, whereas the specifics of DARE's role in activin responsiveness are less clear. It is noteworthy that neither GRAS nor DARE is conserved in other species, including rat. Furthermore, activin negatively regulates Gnrhr expression in primary ovine pituitary cells (83–85). Therefore, the effects of activins on Gnrhr expression and their underlying regulatory mechanisms may be species specific.
Activin induction of rat Fst transcription
In the anterior pituitary, Fst is principally expressed by 2 cell types, gonadotropes and folliculostellate cells. Activin A stimulates Fst mRNA expression in cultured rat pituitary cells and this effect appears to be specific to the gonadotrope cell population (86, 87). Consistent with this idea, activins stimulate Fst transcription in αT3-1 cells (88). This model has therefore been exploited to decipher the underlying molecular mechanisms. Unlike Fshb and Gnrhr, the primary activin-responsive element in rat Fst, at least in the gonadotrope cell context, maps to an enhancer at the 3′-end of intron 1 rather than in the proximal promoter (Figure 1) (88, 89). Importantly, in nongonadotropes (HepG2 and HEK293T cells), activin A induces human FST and rat Fst transcription via the proximal promoter (45, 90). Therefore, activin's actions on Fst in gonadotropes may be via cell type-specific mechanisms.
These mechanisms once again appear to involve SMADs and FOXL2. The rat Fst intronic enhancer contains an SBE (88) and a nearby FBE (Figure 1) (45). The precise location and nature of the latter has not yet been characterized, but activin stimulates the recruitment of both SMAD3 and FOXL2 to the enhancer in αT3-1 cells (45, 88). According to both knockdown and overexpression analyses, SMAD3 but not SMAD2, mediates activin's actions on Fst transcription. SMAD4's role, if any, has not been investigated thoroughly in vitro (Figure 1) (45). Although mechanisms of murine Fst transcription have not been reported, the high sequence identity of the intronic enhancer suggests that they are likely to be conserved with those of rat.
In the Fshb and Gnrhr promoters, the SBEs and FBEs are in close proximity. In contrast, the SBE is 17- to 24-bp 5′ of the putative FBE (ACATTGAT) in the Fst intronic enhancer. This FBE was mapped computationally (rather than experimentally) based on its apparent similarity to a recently described “consensus” FOXL2-binding site (GT[c/g]AAGG) (91). This site bears little resemblance to the FBEs in Fshb or Gnrhr, nor to other known forkhead factor binding sequences. Mutations in the putative FBE in Fst attenuate but do not block activin-induced reporter activity in αT3-1 cells (45). Given the incomplete effects of these mutations, the relative distance between the SBE and the putative FBE, and concerns about the veracity of the consensus FOXL2-binding site (91), we examined FOXL2 binding to the putative Fst FBE by electrophoretic mobility shift assays. As shown in Figure 3, a probe containing the consensus FOXL2-binding site from Ref (91). was unable to compete for FOXL2 binding to a probe containing FBE2 from the porcine Fshb promoter (lanes 14–17) (43). This contrasts with porcine and murine Fshb probes containing their respective FBE3 sequences (lanes 6–13); a probe containing the FBE in GRAS (of murine Gnrhr, lanes 21–24); and a probe containing the activin responsive region of the rat Fst intron 1 (lanes 25–28), which could all dose dependently compete for FOXL2 binding (Figure 3). A mutation (Mut#2 from Ref. 45) that impairs activin responsiveness and is presumed to block FOXL2 binding had no effect on the Fst probe's ability to compete for binding to FOXL2 (lanes 33–36). This suggests that FOXL2 binds the Fst enhancer, but perhaps not via the sequence presumed to be the FBE. We noted that the sequence immediately 5′ of the SBE, TGTTGTG, resembles FBE3 in the Fshb promoter, TGTTTAG (reverse complement of CTAAACA), and therefore questioned whether it might actually mediate FOXL2 binding. Indeed, introduction of a mutation to the first position (T to C) impaired competition and therefore FOXL2 binding (Figure 3, lanes 29–32). Clearly, more work is needed to characterize the mechanisms through which FOXL2 binds and regulates the Fst intronic enhancer, but the early indications are that it too may contain a composite FBE/SBE element.
Figure 3. FOXL2 binds regulatory sequences in Fshb, Gnrhr, and Fst.

Nuclear extracts from heterologous Chinese hamster ovary cells expressing murine FLAG-FOXL2 were incubated with a radiolabeled double-stranded probe corresponding to −185/−145 of the porcine Fshb promoter, which contains the high-affinity FBE2 (see Figure 2). As reported previously (43), FOXL2 forms a specific complex with the probe when resolved on a nondenaturing polyacrylamide gel (lane 1; labeled at extreme right). The complex contains FLAG-FOXL2 as indicated by the supershifted complexes observed when antibodies against the FLAG tag (lane 18) or FOXL2 (lane 19) are included in the mix (labeled at right). Binding is dose dependently competed when an unlabeled form of the same probe is included at 50-, 100-, 500-, or 1000-fold molar excess relative to the radiolabeled probe (lanes 2–5). Similarly, unlabeled double-stranded DNA probes containing FBE3 in pig (lanes 6–9) or mouse (lanes 10–13), GRAS from murine Gnrhr (lanes 21–24), the rat Fst intronic enhancer (lanes 25–28), or the Fst enhancer containing mutations in the putative FBE (lanes 33–36) also compete for binding, indicating that they too contain intact FOXL2-binding sites. A probe containing the putative consensus FOXL2-binding site (lanes 14–17) (Ref. 91) fails to compete for binding, suggesting that it lacks a true FOXL2 binding sequence. A Fst enhancer probe containing a mutation in what we predict to be the actual FBE is impaired in its ability to compete for binding (lanes 29–32). Probe sequences (sense strand only) are shown, putative FBEs are underlined, and mutated base pairs are shown in lower case: porcine FBE2, TTATTTTTCCTGTTCCACTGTGTTTAGACTACTTTAGTAAG; porcine FBE3, CCTGTCTATCTAAACACTGATTCACTTACAG; murine FBE3, GCTTGATCTCCCTGTCCGTCTAAACAATGATTCCCTTTCAG; consensus FBE, CCTGTCACGGTCAAGGTCACTATCACTCAC; GRAS, TTTTGTATCTGTCTAGTCACAACAGTTTTT; Fst, GCTGCACGTGTTGTGTCTGGGTCACTGGTAACTGACATTGATATGGCTAG; Fst T->C, GCTGCACGcGTTGTGTCTGGGTCACTGGTAACTGACATTGATATGGCTAG; and Fst FBEmut, GCTGCACGTGTTGTGTCTGGGTCACTGGTAACTGACAcaGcTATGGCTAG.
FOXL2/SMAD Regulation of Activin Target Genes in Mice
The 3 classic targets of activin signaling in gonadotropes (Fshb, Gnrhr, and Fst) appear to share the same transcriptional regulators, in particular FOXL2, SMAD4, and SMAD3 (Figure 1). It is important to note, however, that the above described in vitro analyses were conducted principally in 2 cell lines, αT3-1 and LβT2, using promoters or enhancers from a relatively small number of mammalian species. Although these reagents have enabled detailed analyses, their limited number and variety raises legitimate questions about how accurately they model transcriptional mechanisms in nontransformed gonadotropes (eg, Refs. 92–96). Although not unique to this system, there are also concerns that cultured cells might not accurately model cell function in the context of intact tissues and physiological systems (77, 97, 98). It is therefore imperative to probe the involvement of FOXL2 and SMAD proteins in gonadotrope function in vivo.
FOXL2 regulates Fshb and Fst, but not Gnrhr, expression in vivo
Humans harboring inactivating mutations in the FOXL2 gene suffer from blepharophimosis-ptosis-epicanthus-inversus syndrome (BPES), which is characterized by craniofacial defects with (type I) or without (type II) premature ovarian failure (99, 100). Although FOXL2 is expressed in human gonadotrope cells (101, 102), women with BPES type I typically exhibit elevated FSH (eg, Refs. 100, 103). Although this might suggest that FOXL2 is dispensable for FSH synthesis in humans, it is important to note that only a single FOXL2 allele is affected in these patients. Therefore, the loss of both alleles might be required to observe the predicted declines in FSH synthesis. To our knowledge, only 2 families harboring homozygous FOXL2 mutations have been described and the particular mutations do not cause a complete loss of protein function (104–106). Therefore, in the absence of human clinical insight into FOXL2's role in FSH synthesis in vivo, investigators have employed genetic mouse models. These mice not only provide the opportunity to assess effects of loss of Foxl2 (and other gene) function on circulating FSH levels but also permit the investigation of the corresponding consequences for pituitary Fshb, Gnrhr, and Fst mRNA expression.
The single exon Foxl2 gene has been deleted in mice using conventional and conditional gene targeting approaches. Homozygous Foxl2 knockout mice die soon after birth (107, 108). Those that survive are generally unhealthy, rarely live beyond 5 weeks postnatally, and exhibit ovarian dysgenesis. Nonetheless, at embryonic day 18.5, pituitary Fshb mRNA levels are significantly reduced in knockouts relative to age-matched controls (46). There is also a nonsignificant trend for reduced Gnrhr expression in these animals (Fst data were not reported). At 3 weeks of age, female Foxl2 knockouts exhibit significant reductions in pituitary expression of Fshb, Gnrhr, and Fst (109). However, they also show profound defects in Gh and prolactin (Prl) expression. As neither somatotrope nor lactotrope cells express Foxl2, at least some pituitary phenotypes in these mice may be noncell autonomous.
Indeed, the selective deletion of Foxl2 in gonadotropes using a Cre/lox approach causes reductions in Fshb and Fst, but not Gnrhr, Gh, or Prl, mRNA levels in both adult males and females (110). These conditional knockout (cKO) mice are subfertile, with females producing smaller litters at reduced frequencies relative to controls. Circulating FSH levels are markedly reduced in both male and female Foxl2 cKOs, whereas LH is reduced in males and increased in females. Increased LH secretion in females and normal Lhb expression in both sexes are consistent with intact GnRH signaling in these mice. FSH and LH secretion appear normal in mice in which only a single Foxl2 allele is deleted in gonadotropes (Figure 4). These observations are consistent with the absence of FSH deficiency in BPES patients who, as noted above, usually carry only 1 mutated FOXL2 allele.
Figure 4. Serum FSH is reduced in male mice lacking 2, but not 1, Foxl2 allele in gonadotrope cells.
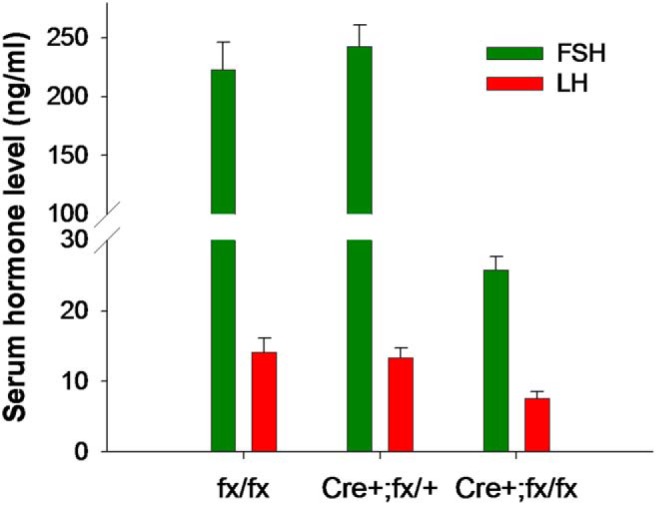
Blood was collected from 8- to 10-week-old males with 2 intact (floxed) Foxl2 alleles (fx/fx, n = 8), or 1 (Cre+;fx/+, n = 5) or 2 (Cre+;fx/fx, n = 10) Foxl2 alleles selectively deleted in gonadotrope cells. GRIC mice were used as Cre drivers (142). Serum FSH (green) and LH (red) were measured by ELISA at the UVA Ligand Assay Core. Data are mean ± SEM. Note the break in the y-axis.
Collectively, these data indicate that FOXL2 is required for Fshb and Fst mRNA expression in murine gonadotropes in vivo. A single functional copy of the gene is sufficient to maintain FSH synthesis in mice (Gnrhr and Fst were not measured in these animals). This might explain the absence of FSH deficiency in humans with loss of function mutations in only 1 FOXL2 allele. In contrast to the in vitro data (see above), gonadotrope Gnrhr expression may be FOXL2 independent in vivo.
SMAD4 is required for Fshb and Fst, but not Gnrhr, expression in vivo
As reviewed above, SMAD4 mediates activin-regulated Fshb and Gnrhr, but perhaps not Fst, transcription in vitro. Data from mice with a conditional deletion of Smad4 specifically in gonadotropes (Smad4 cKO) both support and challenge these observations (111). Smad4 cKO mice are hypogonadal and females are subfertile. Both male and female cKOs exhibit markedly reduced circulating FSH levels and pituitary Fshb mRNA expression (111). Serum LH is reduced in Smad4 cKO males, but not females, which is similar to the phenotype of Foxl2 cKO mice. Counter to the a priori prediction, pituitary Gnrhr mRNA levels were actually increased in Smad4 cKOs relative to controls. Originally, we reported no effect of the Smad4 deletion on Fst expression in pituitaries of males and only a small decrease in females. Although these observations were seemingly consistent with the in vitro data, we subsequently discovered a technical problem with the Fst primers used in the mRNA analysis. When the assays were repeated with new primers and archival material, we observed marked reductions in Fst mRNA expression in both male and female Smad4 cKO mice relative to controls. Therefore, loss of SMAD4 impairs both Fshb and Fst, but not Gnrhr, mRNA expression in vivo.
At present, the mechanisms mediating the nearly 2-fold increase in pituitary Gnrhr mRNA levels in Smad4 cKO mice are unclear. It seems unlikely that SMAD4 actively represses Gnrhr expression in vivo, although no existing data formally reject this possibility. Perhaps increased Gnrhr reflects enhanced GnRH release and signaling due to reduced steroid negative feedback. Female Smad4 cKOs show impaired ovarian follicle development. Because FSH stimulates granulosa cell proliferation and estradiol production, one might expect these animals to be hypoestrogenemic. However, neither uterine weights nor serum estradiol levels (on metestrus/diestrus) differ between cKOs and controls. Perhaps analyses on other days of the estrous cycle, particularly when estradiol levels are normally elevated (afternoon of proestrus) might reveal estrogen deficiency in these animals.
The data in males are more difficult to reconcile at present. Both serum testosterone and seminal vesicle weights appear normal in Smad4 cKOs relative to controls. Therefore, it is not clear that a loss of testosterone negative feedback on GnRH secretion explains the increase in Gnrhr expression. However, endocrine regulatory mechanisms are clearly altered in these mice. First, testosterone levels are normal in the face of reduced serum LH. Second, LH secretion is reduced, even though Gnrhr expression (and presumably GnRH signaling?) is increased. Male, but not female, Smad4 cKO mice exhibit reduced gonadotropin α-subunit (Cga) expression, which could explain their reduced LH secretion. SMAD4's role in gonadotrope Cga expression has not been explored to our knowledge; however, activin A suppresses murine Cga promoter activity in αT3-1 cells (112). If this reflects activin action in vivo, then it would appear to be independent of, if not impaired by, SMAD4. Along these lines, SMAD3 potentiates the actions of the androgen receptor (AR) in prostate cancer cells, but this synergism is antagonized by SMAD4 coexpression (113). Androgens also repress CGA transcription in αT3-1 cells (114). It is therefore possible that SMAD3 and the AR cooperatively inhibit Cga expression and that this activity is normally attenuated by SMAD4. In the absence of SMAD4, SMAD3/AR could repress Cga expression more readily. The effect might be sex specific, in turn, because of higher androgen levels in males than females. Although such a mechanism might explain Cga suppression in male Smad4 cKO mice, the mechanism of their enhanced Gnrhr remains a mystery.
The combined activities of FOXL2 and SMAD4 are required for pituitary Fshb, but not Gnrhr, expression in vivo
Based on in vitro analyses of activin-regulated Fshb, Gnrhr, and Fst, the phenotypes of gonadotrope-specific Foxl2 or Smad4 knockout mice have been eye opening. First, Foxl2 cKO and Smad4 cKO mice have greatly reduced FSH as expected, but they do not lack FSH entirely and females remain (sub)fertile in most cases. Second, Smad4 cKO mice have reduced Fst expression, although SMAD4 was not previously implicated in activin regulation of this gene in gonadotrope-like cell lines. Third, neither the loss of Foxl2 nor Smad4 causes decreases in Gnrhr expression, as would be expected based on their cooperative actions at the GRAS element in vitro (Figure 1). In the case of Fst, more mechanistic research will be needed to identify how and where SMAD4 produces its action(s). For both Fshb and Gnrhr, residual or maintained expression could reflect compensatory mechanisms. That is, in the murine Fshb promoter, there are cis-elements where FOXL2 can act independently of SMAD4 (FBE1 in Figure 1) and at least 1 where SMAD4 might function independently of FOXL2 (SBEx2 in Figure 1). These residual actions may be sufficient to maintain some activin-stimulated Fshb expression. In the case of the Gnrhr promoter, it is possible that SMAD3 might compensate for the absence of SMAD4 at the 5′-end of GRAS in Smad4 cKO mice. In Foxl2 cKO mice, SMADs and AP-1 proteins might compensate for the absence of FOXL2 binding at the 3′-end of GRAS. The simultaneous deletion of Foxl2 and Smad4 in gonadotropes should, therefore, preclude these compensatory/redundant mechanisms.
Indeed, the reproductive phenotype of Smad4/Foxl2 cKO mice is more dramatic than that of either Smad4 cKO or Foxl2 cKO mice (111). In many ways, these mice resemble Fshb knockouts (115). Female Smad4/Foxl2 cKOs are sterile, lack estrous cyclicity, and exhibit an arrest in ovarian follicle development at the early antral stage. Their FSH deficiency is also more pronounced than in the single knockout mice lines. Once again, however, these animals fail to show any impairment in Gnrhr mRNA levels, raising serious doubts about a role for GRAS in receptor expression. A more definitive conclusion regarding GRAS's role in vivo could be obtained by disrupting the element in transgenic mice.
Notably, the double knockout mice have a pituitary Gnrhr phenotype (ie, no change relative to control) more similar to single Foxl2 than Smad4 cKOs. Smad4/Foxl2 cKO females have thread-like uteri suggestive of hypoestrogenemia. They also do not ovulate and lack corpora lutea, suggesting that their progesterone levels are also low. In mice, GnRHR levels depend on both GnRH and gonadal steroids (62, 63, 116). Therefore, loss of steroid negative feedback and consequent increased GnRH release will not lead to up-regulated GnRHR levels (as they do in rats), if steroid levels are below a certain threshold level. Therefore, we postulate that in Smad4 cKO mice, steroid levels are reduced, but sufficient to synergize with enhanced GnRH release to drive increases in Gnrhr expression. In Foxl2 or Foxl2/Smad4 cKO mice, steroid levels may be below the threshold required for these synergistic actions. The increased Lhb mRNA expression in Smad4/Foxl2 cKOs, however, is consistent with enhanced GnRH secretion. We acknowledge that these explanations are speculative and that other possibilities must also be considered.
An uncertain role for SMAD3 in Fshb expression in vivo
SMAD3 is arguably the linchpin in activin-regulated gene expression in gonadotropes. It is the protein that most clearly links activin receptor binding to transcriptional activation. SMAD3 is phosphorylated by the type I receptor and then accumulates in the nucleus where it binds DNA either directly (eg, at the 8-bp SBE in the Fshb promoter) or via its interactions with SMAD4 and/or FOXL2 (in Fshb, GRAS in Gnrhr, and the Fst intronic enhancer) (Figure 1). Without SMAD3, there is no (obvious) link between receptor complex activation and SMAD4 or FOXL2, because there is no evidence that either protein is directly regulated by activin receptors. Therefore, a priori, one would predict that the loss of Smad3 in gonadotropes should produce effects comparable with, if not more extreme than, the combined deletion of Smad4 and Foxl2.
Surprisingly, however, circulating FSH levels, pituitary Fshb expression, and fertility are unimpaired in mice with a targeted deletion of Smad3 in gonadotropes, either alone or in combination with Smad2 (Smad2/3 cKO) (117). Cre-mediated recombination removes exons 2 and 3 of the 9 exon Smad3 gene in these mice. However, they exhibit a 2-fold up-regulation of a Smad3 transcript, which contains exon 1 spliced to exons 4 through 9. Although translation starting in exon 1 should cause a frame-shift mutation and the absence of most of the SMAD3 protein, alternative translation can commence from a consensus Kozak sequence in exon 4. The resulting protein lacks the N-terminal MAD homology domain (MH)1, which binds DNA, but contains the entirety of the C-terminal MH2 domain. The SMAD3 MH2 domain is phosphorylated by the type I receptor (118) and mediates the interactions with SMAD4 (119–121) and FOXL2 (45, 71). Although we do not yet known whether a truncated SMAD3 protein is expressed in gonadotropes of these mice or whether it actually reflects a naturally occurring variant of the protein (122), it can activate the Fshb promoter equivalently to full-length SMAD3 in vitro (117). These data indicate that, in mice, SMAD2 is dispensable for FSH synthesis and that, if required, SMAD3 does not need to bind DNA directly to regulate Fshb transcription. This result is somewhat surprising, as stimulation of the murine Fshb promoter through the 8-bp SBE requires direct binding of both SMAD3 and SMAD4 and this element contributes to overall activin sensitivity in vitro (33). In contrast, regulation through the composite SBE/FBE3, which preferentially binds SMAD4, should be maintained in Smad2/3 cKO mice, as the truncated SMAD3 would still be activated by the type I receptor and interact with SMAD4 and FOXL2.
Neither pituitary Fst nor Gnrhr expression were measured in Smad2/3 cKO mice. According to in vitro observations, however, one would predict impaired Fst expression, as SMAD3 binds the intronic enhancer. Regardless, Fst deficiency in Smad4 cKO mice suggests that SMAD4 might be the preferred DNA binding partner for FOXL2 in Fst as it is in Fshb. Here too, the truncated SMAD3 could provide a bridge between the DNA-bound SMAD4 and FOXL2. This idea can be tested readily in vitro (45), although it would also be valuable to assess Fst expression in pituitaries of these mice. It seems unlikely that Gnrhr expression is impaired in Smad2/3 cKOs in light of the observations in Foxl2 and Smad4 cKOs. Moreover, LH secretion is normal in Smad2/3 cKO females and is rapidly inhibited by a GnRHR antagonist (Figure 5A), indicating intact GnRHR signaling.
Figure 5. GnRHR signaling and expression are activin independent.
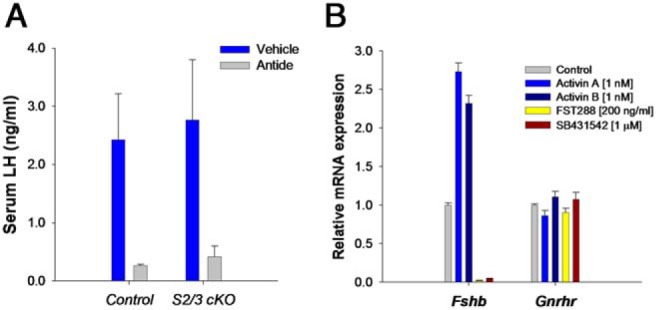
A, Intact adult female wild-type or gonadotrope-specific Smad2/3 knockout (S2/3 cKO) mice were treated sc with 3-mg/kg body weight of the GnRHR antagonist, Antide, or with vehicle. After 8 hours, blood was collected and serum LH measured by ELISA. Antide potently suppressed LH release in both genotypes. B, Pituitaries from adult wild-type male and female mice were dispersed and cultured. The next day, cells were treated with the indicated ligands or inhibitors in low serum (2%) medium overnight. RNA was collected; Fshb and Gnrhr mRNA concentrations were determined by RT-qPCR. Data are from a single experiment with treatments performed in quadruplicate (mean ± SEM). Male and female data were comparable and therefore pooled for analysis. Both exogenous activin A and B stimulated Fshb mRNA levels, whereas follistatin-288 and the type I receptor inhibitor, SB431542, abolished Fshb expression. The latter results suggest that an endogenous TGFβ superfamily ligand that signals via ACVR1B, ACVR1C, or TGFBR1 is the main driver of Fshb expression in cultured murine gonadotropes. None of the treatments affected Gnrhr mRNA levels, suggesting that receptor expression is independent of activin signaling, at least in culture and under these experimental conditions.
Collectively, these data show that SMAD2 is dispensable for murine Fshb expression in vivo, contrary to earlier in vitro results (30). Moreover, if SMAD3 does mediate activin induction of the Fshb and Fst genes in gonadotropes, it can do so via protein-protein interactions rather than through direct DNA binding. This too is inconsistent with current in vitro models. It seems unlikely that SMAD2 or SMAD3 plays a significant role, if any, in pituitary Gnrhr expression. Definitive conclusions regarding SMAD3 function in gonadotropes must await the development of a new model that effectively and completely silences the gene.
Summary and Future Directions: The Known Knowns and Known Unknowns
As summarized in Figure 1, the results of recent in vivo experiments in genetically modified mice require us to modify existing models of activin signaling in gonadotrope cells. The extant data allow us to reach the following firm conclusions: 1) FOXL2 and SMAD4 are essential regulators of Fshb and Fst, but not Gnrhr, expression by gonadotropes; 2) SMAD2 is dispensable for Fshb, Gnrhr, and Fst expression by gonadotropes; 3) if SMAD3 regulates Fshb, Gnrhr, or Fst expression in vivo, it can do so independently of its DNA binding activity; 4) at least 1 TGFβ superfamily ligand (activin B?) is an essential regulator of FSH synthesis in mice; and 5) this ligand may play a more fundamental role than GnRH in FSH regulation. Below, we briefly elaborate on a few of these conclusions and consider some additional questions they raise.
Do FOXL2 and SMAD4 play necessary roles in activin signaling in vivo?
Although it is clear that Fshb and Fst expression are impaired in gonadotropes of Foxl2 or Smad4 cKO mice, these results raise more questions than they answer. For example, can we assume that the observed results derive from specific impairments in activin-induced gene transcription? It is important to remember that both genes were deleted during embryonic life, which could have affected gonadotrope development in ways we do not currently appreciate. Although conditional deletion of the genes in adulthood should address this concern, it remains possible (if not probable) that FOXL2 and SMAD4 directly and indirectly regulate Fshb and Fst expression via mechanisms we have yet to discover. Systems level analyses (eg, chromatin immunoprecipitation-sequencing) could undercover novel binding sites through which the proteins independently and cooperatively regulate transcription of these or upstream genes. Such analyses may also reveal how SMAD4 regulates the Fst gene, if not through the intronic enhancer. Combined with expression analyses (123), we are poised to identify networks of activin-, FOXL2-, and SMAD4-regulated genes that go well beyond the currently appreciated short list of targets in gonadotropes.
Is Gnrhr an activin target gene in gonadotropes?
Although in vitro data indicate that murine Gnrhr is a bona fide activin target gene, the in vitro data suggest otherwise. At a minimum, the normal, if not enhanced, Gnrhr expression in Foxl2- and Smad4-deficient mice indicate that the GRAS regulatory sequence is unlikely to play a major role in transcription of this gene. Although activins might signal independently of FOXL2 or SMAD4, this possibility is undermined by the phenotype of Acvr2 knockout mice, which show reduced Fshb but normal Gnrhr expression (124). Moreover, in cultured murine pituitary cells, activin B or a related ligand is the principal driver of Fshb expression. As we showed previously (24), antagonism of this ligand with either follistatin-288 or an inhibitor of activin type I receptors, SB431542 (125), abolishes Fshb expression (Figure 5B). The same treatments, however, have no effect on Gnrhr expression. Exogenous activin A or activin B, at concentrations sufficient to stimulate Fshb, also fail to alter Gnrhr expression in these cultures (Figure 5B). These latter experiments, which were performed on pituitaries of wild-type mice, rule out compensatory mechanisms that might occur in the existing knockout models. Therefore, we conclude that Gnrhr is not an activin response gene in murine gonadotropes.
Is activin B the relevant TGFβ ligand regulating Fshb (and Fst) in gonadotropes?
FSH deficiency in Acvr2 (26) and gonadotrope-specific Smad4 knockout mice (111) suggests an essential role for TGFβ superfamily signaling in Fshb expression. These results alone, however, do not implicate activin B as the relevant ligand. 1) Several TGFβ ligands bind ACVR2 (eg, Refs. 126–128), and SMAD4 mediates the actions of all ligands in the family. 2) Mice deficient in activin B have increased, rather than reduced FSH levels (129). 3) And pituitaries from mice lacking 1 of the 2 activin type I receptors, ACVR1C, show normal basal and activin B-stimulated FSH release in culture (29). Thus, either activin B is not the primary ligand driving FSH synthesis in vivo, or there are additional ligands that compensate in its absence or act redundantly. The identities of these ligands (and their type I receptor[s]) are presently unknown; however, they would clearly depend on ACVR2 and SMAD4 to mediate their actions. The results of experiments such as those in Figure 5B also indicate that the ligand(s) can be antagonized by follistatin-288 and signal(s) through one (or more) of type I receptors blocked by SB431542. An important challenge for the field will be to identify the relevant ligand(s) and corresponding type I receptor(s).
What are the relative roles of activins (or activin-like ligands) and GnRH in FSH synthesis?
The recent cKO data in mice force us to revisit questions regarding the relative roles of TGFβ superfamily and GnRH signaling in FSH synthesis. Hpg mice (which lack GnRH) and Gnrhr knockout mice are gonadotropin deficient (130–132), clearly indicating that GnRH signaling is required for FSH synthesis in this species. At the same time, FSH deficiency in Smad4/Foxl2 cKO mice suggests a vital role for activin (TGFβ superfamily) signaling (110, 111). How can we reconcile these observations? There is no clear answer at present. It is possible that GnRH signaling is critical during reproductive development, but FSH becomes more dependent on activin-like signaling later in life. Consistent with this idea, GnRHR antagonists are more effective in inhibiting LH than FSH secretion in adulthood (eg, Refs. 133, 134). Another possibility is that GnRH regulates Fshb transcription via FOXL2 and SMAD4. To our knowledge, however, there is no evidence that GnRH signals directly to the Fshb promoter via either of these proteins (39, 43). This said, both newer and older data suggest that GnRH may stimulate FSH indirectly via regulation of TGFβ superfamily ligands and/or their antagonists (135–139). Future efforts, particularly in vivo, should be committed to determining the extent to which GnRH regulation of FSH depends on activins or other members of the TGFβ superfamily.
Overall, accumulating evidence from experiments in model cell lines and genetically modified mice indicate a prominent role for activin-like signaling in the regulation of FSH synthesis and fertility. Assuming the underlying mechanisms are conserved in humans, it is tempting to speculate that components of the activin signaling cascade may represent promising targets for the selective regulation of FSH in the treatment of reproductive disorders, including anovulation and polycystic ovary syndrome (140, 141).
Acknowledgments
We thank Dr Ulrich Boehm, Dr Mathias Treier, Dr Chu-Xia Deng, Dr Jonathan Graff, and Dr Michael Weinstein for their collaboration on the various conditional knockout studies.
This work was supported by Canadian Institutes of Health Research Operating Grants MOP-89991, MOP-123447, and MOP-238760 (to D.J.B.).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by Canadian Institutes of Health Research Operating Grants MOP-89991, MOP-123447, and MOP-238760 (to D.J.B.).
Footnotes
- ACVR2
- activin receptor type IIA
- AP-1
- activator protein-1
- AR
- androgen receptor
- BMPR2
- bone morphogenetic protein receptor type II
- BPES
- blepharophimosis-ptosis-epicanthus-inversus syndrome
- cKO
- conditional knockout
- DARE
- downstream activin regulatory element
- FBE
- forkhead-binding element
- FOXL2
- forkhead box L2
- Fshb
- FSHβ
- Fst
- follistatin
- Gnrhr
- GnRH receptor
- GRAS
- GnRHR-activating sequence
- LHX
- LIM-homeodomain protein
- MH
- MAD homology domain
- PTIX
- paired-like homeodomain transcription factor
- SBE
- SMAD-binding element.
References
- 1. Ling N, Ying SY, Ueno N, et al. . A homodimer of the β-subunits of inhibin A stimulates the secretion of pituitary follicle stimulating hormone. Biochem Biophys Res Commun. 1986;138:1129–1137. [DOI] [PubMed] [Google Scholar]
- 2. Ling N, Ying SY, Ueno N, et al. . Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature. 1986;321:779–782. [DOI] [PubMed] [Google Scholar]
- 3. Vale W, Rivier J, Vaughan J, et al. . Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature. 1986;321:776–779. [DOI] [PubMed] [Google Scholar]
- 4. Ling N, Ying SY, Ueno N, Esch F, Denoroy L, Guillemin R. Isolation and partial characterization of a Mr 32,000 protein with inhibin activity from porcine follicular fluid. Proc Natl Acad Sci USA. 1985;82:7217–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rivier J, Spiess J, McClintock R, Vaughan J, Vale W. Purification and partial characterization of inhibin from porcine follicular fluid. Biochem Biophys Res Commun. 1985;133:120–127. [DOI] [PubMed] [Google Scholar]
- 6. Weiss J, Guendner MJ, Halvorson LM, Jameson JL. Transcriptional activation of the follicle-stimulating hormone β-subunit gene by activin. Endocrinology. 1995;136:1885–1891. [DOI] [PubMed] [Google Scholar]
- 7. Pernasetti F, Vasilyev VV, Rosenberg SB, et al. . Cell-specific transcriptional regulation of follicle-stimulating hormone-β by activin and gonadotropin-releasing hormone in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. [DOI] [PubMed] [Google Scholar]
- 8. Attardi B, Miklos J. Rapid stimulatory effect of activin-A on messenger RNA encoding the follicle-stimulating hormone β-subunit in rat pituitary cell cultures. Mol Endocrinol. 1990;4:721–726. [DOI] [PubMed] [Google Scholar]
- 9. Carroll RS, Corrigan AZ, Vale W, Chin WW. Activin stabilizes follicle-stimulating hormone-β messenger ribonucleic acid levels. Endocrinology. 1991;129:1721–1726. [DOI] [PubMed] [Google Scholar]
- 10. Corrigan AZ, Bilezikjian LM, Carroll RS, et al. . Evidence for an autocrine role of activin B within rat anterior pituitary cultures. Endocrinology. 1991;128:1682–1684. [DOI] [PubMed] [Google Scholar]
- 11. Roberts V, Meunier H, Vaughan J, et al. . Production and regulation of inhibin subunits in pituitary gonadotropes. Endocrinology. 1989;124:552–554. [DOI] [PubMed] [Google Scholar]
- 12. DePaolo LV, Bald LN, Fendly BM. Passive immunoneutralization with a monoclonal antibody reveals a role for endogenous activin-B in mediating FSH hypersecretion during estrus and following ovariectomy of hypophysectomized, pituitary-grafted rats. Endocrinology. 1992;130:1741–1743. [DOI] [PubMed] [Google Scholar]
- 13. Braden TD, Conn PM. Activin-A stimulates the synthesis of gonadotropin-releasing hormone receptors. Endocrinology. 1992;130:2101–2105. [DOI] [PubMed] [Google Scholar]
- 14. Fernández-Vázquez G, Kaiser UB, Albarracin CT, Chin WW. Transcriptional activation of the gonadotropin-releasing hormone receptor gene by activin A. Mol Endocrinol. 1996;10:356–366. [DOI] [PubMed] [Google Scholar]
- 15. Bilezikjian LM, Corrigan AZ, Vaughan JM, Vale WM. Activin-A regulates follistatin secretion from cultured rat anterior pituitary cells. Endocrinology. 1993;133:2554–2560. [DOI] [PubMed] [Google Scholar]
- 16. DePaolo LV, Mercado M, Guo Y, Ling N. Increased follistatin (activin-binding protein) gene expression in rat anterior pituitary tissue after ovariectomy may be mediated by pituitary activin. Endocrinology. 1993;132:2221–2228. [DOI] [PubMed] [Google Scholar]
- 17. Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H. Activin-binding protein from rat ovary is follistatin. Science. 1990;247:836–838. [DOI] [PubMed] [Google Scholar]
- 18. Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell. 2005;9:535–543. [DOI] [PubMed] [Google Scholar]
- 19. Wrana JL, Attisano L, Cárcamo J, et al. . TGF β signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. [DOI] [PubMed] [Google Scholar]
- 20. Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. [DOI] [PubMed] [Google Scholar]
- 21. Chen Y, Lebrun JJ, Vale W. Regulation of transforming growth factor β- and activin-induced transcription by mammalian Mad proteins. Proc Natl Acad Sci USA. 1996;93:12992–12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 23. Rejon CA, Hancock MA, Li YN, Thompson TB, Hébert TE, Bernard DJ. Activins bind and signal via bone morphogenetic protein receptor type II (BMPR2) in immortalized gonadotrope-like cells. Cell Signal. 2013;25:2717–2726. [DOI] [PubMed] [Google Scholar]
- 24. Rejon CA, Ho CC, Wang Y, Zhou X, Bernard DJ, Hébert TE. Cycloheximide inhibits follicle-stimulating hormone β subunit transcription by blocking de novo synthesis of the labile activin type II receptor in gonadotrope cells. Cell Signal. 2013;25:1403–1412. [DOI] [PubMed] [Google Scholar]
- 25. Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. 1996;122:3319–3329. [DOI] [PubMed] [Google Scholar]
- 26. Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–360. [DOI] [PubMed] [Google Scholar]
- 27. Bernard DJ, Lee KB, Santos MM. Activin B can signal through both ALK4 and ALK7 in gonadotrope cells. Reprod Biol Endocrinol. 2006;4:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsuchida K, Nakatani M, Yamakawa N, Hashimoto O, Hasegawa Y, Sugino H. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol Cell Endocrinol. 2004;220:59–65. [DOI] [PubMed] [Google Scholar]
- 29. Sandoval-Guzmán T, Göngrich C, Moliner A, et al. . Neuroendocrine control of female reproductive function by the activin receptor ALK7. FASEB J. 2012;26:4966–4976. [DOI] [PubMed] [Google Scholar]
- 30. Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. [DOI] [PubMed] [Google Scholar]
- 31. Dupont J, McNeilly J, Vaiman A, Canepa S, Combarnous Y, Taragnat C. Activin signaling pathways in ovine pituitary and LβT2 gonadotrope cells. Biol Reprod. 2003;68:1877–1887. [DOI] [PubMed] [Google Scholar]
- 32. Gregory SJ, Lacza CT, Detz AA, Xu S, Petrillo LA, Kaiser UB. Synergy between activin A and gonadotropin-releasing hormone in transcriptional activation of the rat follicle-stimulating hormone-β gene. Mol Endocrinol. 2005;19:237–254. [DOI] [PubMed] [Google Scholar]
- 33. Lamba P, Santos MM, Philips DP, Bernard DJ. Acute regulation of murine follicle-stimulating hormone β subunit transcription by activin A. J Mol Endocrinol. 2006;36:201–220. [DOI] [PubMed] [Google Scholar]
- 34. Suszko MI, Balkin DM, Chen Y, Woodruff TK. Smad3 mediates activin-induced transcription of follicle-stimulating hormone β-subunit gene. Mol Endocrinol. 2005;19:1849–1858. [DOI] [PubMed] [Google Scholar]
- 35. Suszko MI, Lo DJ, Suh H, Camper SA, Woodruff TK. Regulation of the rat follicle-stimulating hormone β-subunit promoter by activin. Mol Endocrinol. 2003;17:318–332. [DOI] [PubMed] [Google Scholar]
- 36. Tran S, Lamba P, Wang Y, Bernard DJ. SMADs and FOXL2 synergistically regulate murine FSHβ transcription via a conserved proximal promoter element. Mol Endocrinol. 2011;25:1170–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lamba P, Wang Y, Tran S, et al. . Activin A regulates porcine follicle-stimulating hormone β-subunit transcription via cooperative actions of SMADs and FOXL2. Endocrinology. 2010;151:5456–5467. [DOI] [PubMed] [Google Scholar]
- 38. Wang Y, Bernard DJ. Activin A induction of murine and ovine follicle-stimulating hormone β transcription is SMAD-dependent and TAK1 (MAP3K7)/p38 MAPK-independent in gonadotrope-like cells. Cell Signal. 2012;24:1632–1640. [DOI] [PubMed] [Google Scholar]
- 39. Wang Y, Libasci V, Bernard DJ. Activin A induction of FSHβ subunit transcription requires SMAD4 in immortalized gonadotropes. J Mol Endocrinol. 2010;44:349–362. [DOI] [PubMed] [Google Scholar]
- 40. Shi Y, Wang YF, Jayaraman L, Yang H, Massagué J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell. 1998;94:585–594. [DOI] [PubMed] [Google Scholar]
- 41. Zawel L, Dai JL, Buckhaults P, et al. . Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. [DOI] [PubMed] [Google Scholar]
- 42. McGillivray SM, Thackray VG, Coss D, Mellon PL. Activin and glucocorticoids synergistically activate follicle-stimulating hormone β-subunit gene expression in the immortalized LβT2 gonadotrope cell line. Endocrinology. 2007;148:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lamba P, Fortin J, Tran S, Wang Y, Bernard DJ. A novel role for the forkhead transcription factor FOXL2 in activin A-regulated follicle-stimulating hormone β subunit transcription. Mol Endocrinol. 2009;23:1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernard DJ, Tran S. Mechanisms of activin-stimulated FSH synthesis: the story of a pig and a FOX. Biol Reprod. 2013;88:78. [DOI] [PubMed] [Google Scholar]
- 45. Blount AL, Schmidt K, Justice NJ, Vale WW, Fischer WH, Bilezikjian LM. FoxL2 and Smad3 coordinately regulate follistatin gene transcription. J Biol Chem. 2009;284:7631–7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roybal LL, Hambarchyan A, Meadows JD, et al. . Roles of binding elements, FOXL2 domains, and interactions with cJUN and SMADs in regulation of FSHβ. Mol Endocrinol. 2014;28:1640–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Corpuz PS, Lindaman LL, Mellon PL, Coss D. FoxL2 Is required for activin induction of the mouse and human follicle-stimulating hormone β-subunit genes. Mol Endocrinol. 2010;24:1037–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghochani Y, Saini JK, Mellon PL, Thackray VG. FOXL2 is involved in the synergy between activin and progestins on the follicle-stimulating hormone β-subunit promoter. Endocrinology. 2012;153:2023–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lamba P, Khivansara V, D'Alessio AC, Santos MM, Bernard DJ. Paired-like homeodomain transcription factors 1 and 2 regulate follicle-stimulating hormone β-subunit transcription through a conserved cis-element. Endocrinology. 2008;149:3095–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zakaria MM, Jeong KH, Lacza C, Kaiser UB. Pituitary homeobox 1 activates the rat FSHβ (rFSHβ) gene through both direct and indirect interactions with the rFSHβ gene promoter. Mol Endocrinol. 2002;16:1840–1852. [DOI] [PubMed] [Google Scholar]
- 51. Han SO, Miller WL. Activin A induces ovine follicle stimulating hormone β using −169/−58 bp of its promoter and a simple TATA box. Reprod Biol Endocrinol. 2009;7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suszko MI, Antenos M, Balkin DM, Woodruff TK. Smad3 and Pitx2 cooperate in stimulation of FSHβ gene transcription. Mol Cell Endocrinol. 2008;281:27–36. [DOI] [PubMed] [Google Scholar]
- 53. Charles MA, Saunders TL, Wood WM, et al. . Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol. 2006;20:1366–1377. [DOI] [PubMed] [Google Scholar]
- 54. Jacobs SB, Coss D, McGillivray SM, Mellon PL. Nuclear factor Y and steroidogenic factor 1 physically and functionally interact to contribute to cell-specific expression of the mouse follicle-stimulating hormone-β gene. Mol Endocrinol. 2003;17:1470–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bailey JS, Rave-Harel N, McGillivray SM, Coss D, Mellon PL. Activin regulation of the follicle-stimulating hormone β-subunit gene involves Smads and the TALE homeodomain proteins Pbx1 and Prep1. Mol Endocrinol. 2004;18:1158–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cattanach BM, Iddon CA, Charlton HM, Chiappa SA, Fink G. Gonadotrophin-releasing hormone deficiency in a mutant mouse with hypogonadism. Nature. 1977;269:338–340. [DOI] [PubMed] [Google Scholar]
- 57. Mason AJ, Hayflick JS, Zoeller RT, et al. . A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science. 1986;234:1366–1371. [DOI] [PubMed] [Google Scholar]
- 58. Chan YM, de Guillebon A, Lang-Muritano M, et al. . GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 2009;106:11703–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bouligand J, Ghervan C, Tello JA, et al. . Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–2748. [DOI] [PubMed] [Google Scholar]
- 60. Savoy-Moore RT, Schwartz NB, Duncan JA, Marshall JC. Pituitary gonadotropin-releasing hormone receptors during the rat estrous cycle. Science. 1980;209:942–944. [DOI] [PubMed] [Google Scholar]
- 61. Bauer-Dantoin AC, Hollenberg AN, Jameson JL. Dynamic regulation of gonadotropin-releasing hormone receptor mRNA levels in the anterior pituitary gland during the rat estrous cycle. Endocrinology. 1993;133:1911–1914. [DOI] [PubMed] [Google Scholar]
- 62. Naik SI, Saade G, Detta A, Clayton RN. Homologous ligand regulation of gonadotrophin-releasing hormone receptors in vivo: relationship to gonadotrophin secretion and gonadal steroids. J Endocrinol. 1985;107:41–47. [DOI] [PubMed] [Google Scholar]
- 63. Naik SI, Young LS, Saade G, Kujore A, Charlton HM, Clayton RN. Role of GnRH in the regulation of pituitary GnRH receptors in female mice. J Reprod Fertil. 1985;74:605–614. [DOI] [PubMed] [Google Scholar]
- 64. Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Regulation of rat pituitary gonadotropin-releasing hormone receptor mRNA levels in vivo and in vitro. Endocrinology. 1993;133:931–934. [DOI] [PubMed] [Google Scholar]
- 65. Seong JY, Kang SS, Kam K, et al. . Differential regulation of gonadotropin-releasing hormone (GnRH) receptor expression in the posterior mediobasal hypothalamus by steroid hormones: implication of GnRH neuronal activity. Brain Res Mol Brain Res. 1998;53:226–235. [DOI] [PubMed] [Google Scholar]
- 66. Norwitz ER, Xu S, Jeong KH, et al. . Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology. 2002;143:985–997. [DOI] [PubMed] [Google Scholar]
- 67. Duval DL, Ellsworth BS, Clay CM. Is gonadotrope expression of the gonadotropin releasing hormone receptor gene mediated by autocrine/paracrine stimulation of an activin response element? Endocrinology. 1999;140:1949–1952. [DOI] [PubMed] [Google Scholar]
- 68. Albarracin CT, Kaiser UB, Chin WW. Isolation and characterization of the 5′-flanking region of the mouse gonadotropin-releasing hormone receptor gene. Endocrinology. 1994;135:2300–2306. [DOI] [PubMed] [Google Scholar]
- 69. Duval DL, Nelson SE, Clay CM. The tripartite basal enhancer of the gonadotropin-releasing hormone (GnRH) receptor gene promoter regulates cell-specific expression through a novel GnRH receptor activating sequence. Mol Endocrinol. 1997;11:1814–1821. [DOI] [PubMed] [Google Scholar]
- 70. Norwitz ER, Xu S, Xu J, et al. . Direct binding of AP-1 (Fos/Jun) proteins to a SMAD binding element facilitates both gonadotropin-releasing hormone (GnRH)- and activin-mediated transcriptional activation of the mouse GnRH receptor gene. J Biol Chem. 2002;277:37469–37478. [DOI] [PubMed] [Google Scholar]
- 71. Ellsworth BS, Burns AT, Escudero KW, Duval DL, Nelson SE, Clay CM. The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol Cell Endocrinol. 2003;206:93–111. [DOI] [PubMed] [Google Scholar]
- 72. Norwitz ER, Cardona GR, Jeong KH, Chin WW. Identification and characterization of the gonadotropin-releasing hormone response elements in the mouse gonadotropin-releasing hormone receptor gene. J Biol Chem. 1999;274:867–880. [DOI] [PubMed] [Google Scholar]
- 73. Ingraham HA, Lala DS, Ikeda Y, et al. . The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes Dev. 1994;8:2302–2312. [DOI] [PubMed] [Google Scholar]
- 74. Noel SD, Martin C, Muyide T, et al. . A proximal AP-1 site in the Gnrhr gene promoter is critical for normal pubertal and reproductive development in female mice. ICE/ENDO2014, Chicago, IL, 2014;pp OR30–5. [Google Scholar]
- 75. Cherrington BD, Farmerie TA, Lents CA, Cantlon JD, Roberson MS, Clay CM. Activin responsiveness of the murine gonadotropin-releasing hormone receptor gene is mediated by a composite enhancer containing spatially distinct regulatory elements. Mol Endocrinol. 2005;19:898–912. [DOI] [PubMed] [Google Scholar]
- 76. Chai J, Wu JW, Yan N, Massagué J, Pavletich NP, Shi Y. Features of a Smad3 MH1-DNA complex. Roles of water and zinc in DNA binding. J Biol Chem. 2003;278:20327–20331. [DOI] [PubMed] [Google Scholar]
- 77. Huang HJ, Sebastian J, Strahl BD, Wu JC, Miller WL. Transcriptional regulation of the ovine follicle-stimulating hormone-β gene by activin and gonadotropin-releasing hormone (GnRH): involvement of two proximal activator protein-1 sites for GnRH stimulation. Endocrinology. 2001;142:2267–2274. [DOI] [PubMed] [Google Scholar]
- 78. Wang Y, Fortin J, Lamba P, et al. . Activator protein-1 and smad proteins synergistically regulate human follicle-stimulating hormone β-promoter activity. Endocrinology. 2008;149:5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jeong KH, Chin WW, Kaiser UB. Essential role of the homeodomain for pituitary homeobox 1 activation of mouse gonadotropin-releasing hormone receptor gene expression through interactions with c-Jun and DNA. Mol Cell Biol. 2004;24:6127–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tremblay JJ, Goodyer CG, Drouin J. Transcriptional properties of Ptx1 and Ptx2 isoforms. Neuroendocrinology. 2000;71:277–286. [DOI] [PubMed] [Google Scholar]
- 81. McGillivray SM, Bailey JS, Ramezani R, Kirkwood BJ, Mellon PL. Mouse GnRH receptor gene expression is mediated by the LHX3 homeodomain protein. Endocrinology. 2005;146:2180–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. West BE, Parker GE, Savage JJ, et al. . Regulation of the follicle-stimulating hormone β gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology. 2004;145:4866–4879. [DOI] [PubMed] [Google Scholar]
- 83. Wu JC, Sealfon SC, Miller WL. Gonadal hormones and gonadotropin-releasing hormone (GnRH) alter messenger ribonucleic acid levels for GnRH receptors in sheep. Endocrinology. 1994;134:1846–1850. [DOI] [PubMed] [Google Scholar]
- 84. Sealfon SC, Laws SC, Wu JC, Gillo B, Miller WL. Hormonal regulation of gonadotropin-releasing hormone receptors and messenger RNA activity in ovine pituitary culture. Mol Endocrinol. 1990;4:1980–1987. [DOI] [PubMed] [Google Scholar]
- 85. Gregg DW, Schwall RH, Nett TM. Regulation of gonadotropin secretion and number of gonadotropin-releasing hormone receptors by inhibin, activin-A, and estradiol. Biol Reprod. 1991;44:725–732. [DOI] [PubMed] [Google Scholar]
- 86. Prendergast KA, Burger LL, Aylor KW, Haisenleder DJ, Dalkin AC, Marshall JC. Pituitary follistatin gene expression in female rats: evidence that inhibin regulates transcription. Biol Reprod. 2004;70:364–370. [DOI] [PubMed] [Google Scholar]
- 87. Bilezikjian LM, Leal AM, Blount AL, Corrigan AZ, Turnbull AV, Vale WW. Rat anterior pituitary folliculostellate cells are targets of interleukin-1β and a major source of intrapituitary follistatin. Endocrinology. 2003;144:732–740. [DOI] [PubMed] [Google Scholar]
- 88. Blount AL, Vaughan JM, Vale WW, Bilezikjian LM. A Smad-binding element in intron 1 participates in activin-dependent regulation of the follistatin gene. J Biol Chem. 2008;283:7016–7026. [DOI] [PubMed] [Google Scholar]
- 89. Winters SJ, Dalkin AC, Tsujii T. Evidence that pituitary adenylate cyclase activating polypeptide suppresses follicle-stimulating hormone-β messenger ribonucleic acid levels by stimulating follistatin gene transcription. Endocrinology. 1997;138:4324–4329. [DOI] [PubMed] [Google Scholar]
- 90. Bartholin L, Maguer-Satta V, Hayette S, et al. . Transcription activation of FLRG and follistatin by activin A, through Smad proteins, participates in a negative feedback loop to modulate activin A function. Oncogene. 2002;21:2227–2235. [DOI] [PubMed] [Google Scholar]
- 91. Benayoun BA, Caburet S, Dipietromaria A, et al. . The identification and characterization of a FOXL2 response element provides insights into the pathogenesis of mutant alleles. Hum Mol Genet. 2008;17:3118–3127. [DOI] [PubMed] [Google Scholar]
- 92. Boerboom D, Kumar V, Boyer A, et al. . β-Catenin stabilization in gonadotropes impairs follicle-stimulating hormone synthesis in male mice in vivo. Endocrinology. 2014;en20141296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fortin J, Kumar V, Zhou X, et al. . NR5A2 regulates Lhb and Fshb transcription in gonadotrope-like cells in vitro, but is dispensable for gonadotropin synthesis and fertility in vivo. PLoS One. 2013;8:e59058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang Q, Chikina M, Zaslavsky E, Pincas H, Sealfon SC. β-Catenin regulates GnRH-induced FSHβ gene expression. Mol Endocrinol. 2013;27:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Salisbury TB, Binder AK, Nilson JH. Welcoming β-catenin to the gonadotropin-releasing hormone transcriptional network in gonadotropes. Mol Endocrinol. 2008;22:1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zheng W, Yang J, Jiang Q, He Z, Halvorson LM. Liver receptor homologue-1 regulates gonadotrope function. J Mol Endocrinol. 2007;38:207–219. [DOI] [PubMed] [Google Scholar]
- 97. Strahl BD, Huang HJ, Pedersen NR, Wu JC, Ghosh BR, Miller WL. Two proximal activating protein-1-binding sites are sufficient to stimulate transcription of the ovine follicle-stimulating hormone-β gene. Endocrinology. 1997;138:2621–2631. [DOI] [PubMed] [Google Scholar]
- 98. Strahl BD, Huang HJ, Sebastian J, Ghosh BR, Miller WL. Transcriptional activation of the ovine follicle-stimulating hormone β-subunit gene by gonadotropin-releasing hormone: involvement of two activating protein-1-binding sites and protein kinase C. Endocrinology. 1998;139:4455–4465. [DOI] [PubMed] [Google Scholar]
- 99. Strømme P, Sandboe F. Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES). Acta Ophthalmol Scand. 1996;74:45–47. [DOI] [PubMed] [Google Scholar]
- 100. Crisponi L, Deiana M, Loi A, et al. . The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159–166. [DOI] [PubMed] [Google Scholar]
- 101. Chesnokova V, Zonis S, Wawrowsky K, et al. . Clusterin and FOXL2 act concordantly to regulate pituitary gonadotroph adenoma growth. Mol Endocrinol. 2012;26:2092–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Egashira N, Takekoshi S, Takei M, Teramoto A, Osamura RY. Expression of FOXL2 in human normal pituitaries and pituitary adenomas. Mod Pathol. 2011;24:765–773. [DOI] [PubMed] [Google Scholar]
- 103. Amati P, Gasparini P, Zlotogora J, et al. . A gene for premature ovarian failure associated with eyelid malformation maps to chromosome 3q22–q23. Am J Hum Genet. 1996;58:1089–1092. [PMC free article] [PubMed] [Google Scholar]
- 104. Nallathambi J, Moumné L, De Baere E, et al. . A novel polyalanine expansion in FOXL2: the first evidence for a recessive form of the blepharophimosis syndrome (BPES) associated with ovarian dysfunction. Hum Genet. 2007;121:107–112. [DOI] [PubMed] [Google Scholar]
- 105. Kaur I, Hussain A, Naik MN, Murthy R, Honavar SG. Mutation spectrum of fork-head transcriptional factor gene (FOXL2) in Indian Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) patients. Br J Ophthalmol. 2011;95:881–886. [DOI] [PubMed] [Google Scholar]
- 106. Dipietromaria A, Benayoun BA, Todeschini AL, Rivals I, Bazin C, Veitia RA. Towards a functional classification of pathogenic FOXL2 mutations using transactivation reporter systems. Hum Mol Genet. 2009;18:3324–3333. [DOI] [PubMed] [Google Scholar]
- 107. Schmidt D, Ovitt CE, Anlag K, et al. . The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131:933–942. [DOI] [PubMed] [Google Scholar]
- 108. Uda M, Ottolenghi C, Crisponi L, et al. . Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum Mol Genet. 2004;13:1171–1181. [DOI] [PubMed] [Google Scholar]
- 109. Justice NJ, Blount AL, Pelosi E, Schlessinger D, Vale W, Bilezikjian LM. Impaired FSHβ expression in the pituitaries of Foxl2 mutant animals. Mol Endocrinol. 2011;25:1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tran S, Zhou X, Lafleur C, et al. . Impaired fertility and FSH synthesis in gonadotrope-specific Foxl2 knockout mice. Mol Endocrinol. 2013;27:407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Fortin J, Boehm U, Deng CX, Treier M, Bernard DJ. Follicle-stimulating hormone synthesis and fertility depend on SMAD4 and FOXL2. FASEB J. 2014;28:3396–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Attardi B, Klatt B, Little G. Repression of glycoprotein hormone α-subunit gene expression and secretion by activin in α T3-1 cells. Mol Endocrinol. 1995;9:1737–1749. [DOI] [PubMed] [Google Scholar]
- 113. Kang HY, Huang KE, Chang SY, Ma WL, Lin WJ, Chang C. Differential modulation of androgen receptor-mediated transactivation by Smad3 and tumor suppressor Smad4. J Biol Chem. 2002;277:43749–43756. [DOI] [PubMed] [Google Scholar]
- 114. Heckert LL, Wilson EM, Nilson JH. Transcriptional repression of the α-subunit gene by androgen receptor occurs independently of DNA binding but requires the DNA-binding and ligand-binding domains of the receptor. Mol Endocrinol. 1997;11:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204. [DOI] [PubMed] [Google Scholar]
- 116. Naik SI, Young LS, Charlton HM, Clayton RN. Evidence for a pituitary site of gonadal steroid stimulation of GnRH receptors in female mice. J Reprod Fertil. 1985;74:615–624. [DOI] [PubMed] [Google Scholar]
- 117. Fortin J, Boehm U, Weinstein MB, Graff JM, Bernard DJ. Follicle-stimulating hormone synthesis and fertility are intact in mice lacking SMAD3 DNA binding activity and SMAD2 in gonadotrope cells. FASEB J. 2014;28:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci USA. 1997;94:10669–10674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chacko BM, Qin B, Correia JJ, Lam SS, de Caestecker MP, Lin K. The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat Struct Biol. 2001;8:248–253. [DOI] [PubMed] [Google Scholar]
- 120. Chacko BM, Qin BY, Tiwari A, et al. . Structural basis of heteromeric smad protein assembly in TGF-β signaling. Mol Cell. 2004;15:813–823. [DOI] [PubMed] [Google Scholar]
- 121. Correia JJ, Chacko BM, Lam SS, Lin K. Sedimentation studies reveal a direct role of phosphorylation in Smad3:Smad4 homo- and hetero-trimerization. Biochemistry. 2001;40:1473–1482. [DOI] [PubMed] [Google Scholar]
- 122. Kim SY, Zhu J, Woodruff TK. A truncated, activin-induced Smad3 isoform acts as a transcriptional repressor of FSHβ expression in mouse pituitary. Mol Cell Endocrinol. 2011;342:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang H, Bailey JS, Coss D, et al. . Activin modulates the transcriptional response of LβT2 cells to gonadotropin-releasing hormone and alters cellular proliferation. Mol Endocrinol. 2006;20:2909–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kumar TR, Agno J, Janovick JA, Conn PM, Matzuk MM. Regulation of FSHβ and GnRH receptor gene expression in activin receptor II knockout male mice. Mol Cell Endocrinol. 2003;212:19–27. [DOI] [PubMed] [Google Scholar]
- 125. Inman GJ, Nicolás FJ, Callahan JF, et al. . SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. [DOI] [PubMed] [Google Scholar]
- 126. Nishitoh H, Ichijo H, Kimura M, et al. . Identification of type I and type II serine/threonine kinase receptors for growth/differentiation factor-5. J Biol Chem. 1996;271:21345–21352. [DOI] [PubMed] [Google Scholar]
- 127. Yamashita H, ten Dijke P, Huylebroeck D, et al. . Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. J Cell Biol. 1995;130:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Vitt UA, Mazerbourg S, Klein C, Hsueh AJ. Bone morphogenetic protein receptor type II is a receptor for growth differentiation factor-9. Biol Reprod. 2002;67:473–480. [DOI] [PubMed] [Google Scholar]
- 129. Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin β B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994;8:414–427. [DOI] [PubMed] [Google Scholar]
- 130. McDowell IF, Morris JF, Charlton HM, Fink G. Effects of luteinizing hormone releasing hormone on the gonadotrophs of hypogonadal (hpg) mice. J Endocrinol. 1982;95:331–340. [DOI] [PubMed] [Google Scholar]
- 131. Charlton HM, Halpin DM, Iddon C, et al. . The effects of daily administration of single and multiple injections of gonadotropin-releasing hormone on pituitary and gonadal function in the hypogonadal (hpg) mouse. Endocrinology. 1983;113:535–544. [DOI] [PubMed] [Google Scholar]
- 132. Wu S, Wilson MD, Busby ER, Isaac ER, Sherwood NM. Disruption of the single copy gonadotropin-releasing hormone receptor in mice by gene trap: severe reduction of reproductive organs and functions in developing and adult mice. Endocrinology. 2010;151:1142–1152. [DOI] [PubMed] [Google Scholar]
- 133. Grady RR, Shin L, Charlesworth MC, et al. . Differential suppression of follicle-stimulating hormone and luteinizing hormone secretion in vivo by a gonadotropin-releasing hormone antagonist. Neuroendocrinology. 1985;40:246–252. [DOI] [PubMed] [Google Scholar]
- 134. Kartun K, Schwartz NB. Effects of a potent antagonist to gonadotropin-releasing hormone on male rats: luteinizing hormone is suppressed more than follicle-stimulating hormone. Biol Reprod. 1987;36:103–108. [DOI] [PubMed] [Google Scholar]
- 135. Besecke LM, Guendner MJ, Schneyer AL, Bauer-Dantoin AC, Jameson JL, Weiss J. Gonadotropin-releasing hormone regulates follicle-stimulating hormone-β gene expression through an activin/follistatin autocrine or paracrine loop. Endocrinology. 1996;137:3667–3673. [DOI] [PubMed] [Google Scholar]
- 136. Choi SG, Jia J, Pfeffer RL, Sealfon SC. G proteins and autocrine signaling differentially regulate gonadotropin subunit expression in pituitary gonadotrope. J Biol Chem. 2012;287:21550–21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Choi SG, Wang Q, Jia J, Pincas H, Turgeon JL, Sealfon SC. Growth differentiation factor 9 (GDF9) forms an incoherent feed-forward loop modulating follicle-stimulating hormone β-subunit (FSHβ) gene expression. J Biol Chem. 2014;289:16164–16175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pincas H, Choi SG, Wang Q, Jia J, Turgeon JL, Sealfon SC. Outside the box signaling: secreted factors modulate GnRH receptor-mediated gonadotropin regulation. Mol Cell Endocrinol. 2014;385:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Dalkin AC, Haisenleder DJ, Gilrain JT, Aylor K, Yasin M, Marshall JC. Gonadotropin-releasing hormone regulation of gonadotropin subunit gene expression in female rats: actions on follicle-stimulating hormone β messenger ribonucleic acid (mRNA) involve differential expression of pituitary activin (β-B) and follistatin mRNAs. Endocrinology. 1999;140:903–908. [DOI] [PubMed] [Google Scholar]
- 140. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7:219–231. [DOI] [PubMed] [Google Scholar]
- 141. Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57:1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wen S, Schwarz JR, Niculescu D, et al. . Functional characterization of genetically labeled gonadotropes. Endocrinology. 2008;149:2701–2711. [DOI] [PubMed] [Google Scholar]