

Abstract
Transcriptional activity of signal transducer and activator of transcription-3 (STAT-3) is a key element in the central regulation of appetite and energy homeostasis. Activation of hypothalamic STAT-3 has been attributed to cytokine-promoted phosphorylation at tyrosine-705 (Tyr-705). In nonhypothalamic cells, STAT-3 is also phosphorylated at serine-727 (Ser-727), but the functional significance of Ser-727 in the regulation of hypothalamic STAT-3 is not known. We used 2 hypothalamic cell lines and analyzed the effects of various hormones on STAT-3–dependent reporter gene activity and observed that IFN-γ, epidermal growth factor (EGF), and bradykinin (BK) induce similar STAT-3 reporter activation. EGF and BK solely increased Ser-727 and IFN-γ increased Tyr-705 phosphorylation of STAT-3. Specific inhibition of ERK-1/2 activity blocked EGF- and BK-induced STAT-3 activation and Ser-727 phosphorylation. BK-induced ERK-1/2 activation occurred via EGF receptor transactivation. Consequently, the BK-mediated effects on STAT-3 were blocked by a specific EGF receptor antagonist. Next, we analyzed the effects of IFN-γ and EGF on the expression of the STAT-3–dependent genes thyroliberin-releasing hormone and suppressors of cytokine signaling-3. EGF but not IFN-γ enhanced thyroliberin-releasing hormone expression via STAT-3. With regard to suppressors of cytokine signaling-3, we observed prolonged expression induced by IFN-γ and a transient effect of EGF that required coactivation of the activator protein-1. Thus, EGF-promoted Ser-727 phosphorylation by ERK-1/2 is not only sufficient to fully activate hypothalamic STAT-3, but, in terms of targeted genes and required cofactors, entails distinct modes of STAT-3 actions compared with IFN-γ–induced Tyr-705 phosphorylation.
Members of the signal transducer and activator of transcription (STAT) family are transcription factors originally discovered as regulators of gene expression in immune cells such as T lymphocytes (1, 2). It was soon discovered that STAT proteins also modulate gene expression in nonimmune cells and thus regulate a wide range of vital body functions (3–5). The STAT-3 subtype plays a pivotal role in the regulation of hypothalamic gene expression (6, 7). Neuronal disruption of the STAT-3 gene results in obesity and diabetes (8, 9); hence, hypothalamic STAT-3 plays an essential role in the central regulation of body weight and glucose homeostasis, and detailed insight into the molecular aspects of STAT-3 regulation in hypothalamic cells may help identify new therapeutic targets and improve current obesity and diabetes therapies.
Cytokine signaling is the major regulator of STAT-3 activity in immune cells and hypothalamic neurons. Cytokine receptors (CRs) are associated with Janus kinases (JAKs) that phosphorylate STAT-3 at Tyr-705 upon cytokine-induced receptor activation. Tyr-705 phosphorylation leads to STAT-3 dimerization, translocation to the nucleus, and increased DNA binding affinity (10–12). The adipocyte-derived cytokine leptin is the best-known stimulus to activate hypothalamic STAT-3 via JAK-mediated STAT-3 phosphorylation at Tyr-705 (13, 14). In fact, leptin is thought to be the strongest endogenous anorexigenic stimulus known so far whose central effects on body weight and energy homeostasis are mediated by STAT-3–dependent gene induction in hypothalamic neurons (7, 15–17). Consequently, STAT-3 expression is indispensable for physiological leptin actions, and STAT-3 dysfunction causes pathophysiological alterations in mice and humans (7–9, 18–20).
In nonhypothalamic cells, STAT-3 phosphorylation at Ser-727 by serine/threonine kinases such as protein kinase C, ERK-1/2, protein kinase B (AKT), c-Jun NH2-terminal kinase, or p38 kinase has been observed (21). It has been proposed that exclusive Ser-727 phosphorylation increases neither STAT-3 dimerization nor its affinity to DNA (22). Furthermore, Ser-727 phosphorylation has been shown to diminish Tyr-705 phosphorylation (23, 24). Thus, Ser-727 phosphorylation was considered a negative regulatory mechanism of STAT-3 activity (23–28). On the contrary, other researchers reported that both phosphorylation events are required for maximal STAT-3 activation (29–31) or that exclusive phosphorylation of STAT-3 at Ser-727 is sufficient for activation and induction of STAT-3-related biological functions (32–35). Different stimuli applied to induce STAT-3 phosphorylation or tissue and cell type-specific effects may have contributed to the aforementioned controversial findings. Despite the importance of STAT-3 in the central regulation of appetite and energy metabolism, no data about a potential role of Ser-727 in the regulation of hypothalamic STAT-3 are available at present. Therefore, it is not known whether hypothalamic STAT-3 signaling is subject to noncytokine cell surface receptors expressed in hypothalamic cells such as receptor tyrosine kinases (RTKs) and G protein–coupled receptors.
Herein, we used 2 recently established murine hypothalamic cell lines (mHypoA-2/10 and -2/12 cells) and analyzed STAT-3 activity in response to hormones that activate distinct classes of cell surface receptors. As expected, IFN-γ activated STAT-3 via Tyr-705 phosphorylation. Epidermal growth factor (EGF) also enhanced STAT-3 activity, however via Ser-727 without any Tyr-705 phosphorylation. Hence, regulation of hypothalamic STAT-3 signaling is not restricted to CRs but is also downstream of RTKs such as the epidermal growth factor receptor (EGFR). Therefore, manipulation of hypothalamic STAT-3 phosphorylation at Ser-727 may open up new avenues to interfere with metabolic disorders.
Materials and Methods
Materials
Cell culture reagents were obtained from Invitrogen, and TurboFect was from Fermentas. The anti-p-ERK-1/2 (E-4), anti-ERK-2 (C-14), anti-thyroliberin-releasing hormone (TRH) (M-166) antiserum, and 3,3′,5′-tri-iodo-l-thyronine (T3) were from Santa Cruz. The p-STAT-3-Ser-727 (no. 9134) and Tyr-705 (no. 9131) antisera were purchased from Cell Signaling, and pertussis toxin (PTX), AG-1478, AG-1296, and the peroxidase-conjugated anti-mouse and anti-rabbit antibodies, both raised in goat, were from Sigma-Aldrich. The firefly luciferase substrate was from Promega, and coelenterazine H was from Biaffin. Bradykinin (BK), PP-2, BIM-X, and MSH were purchased from Biotrend. Murine EGF was from Peprotech. PD-184352, genistein, and U-73122 were from Enzo Life Science, SR-11302, GM-6001, and neuropeptide Y (NPY) were from Tocris, STATTIC and S3I-201 were from Abcam, and IFN-γ was from Millipore. The STAT-3 reporter gene construct pGL4.47 (luc2P/SIE/Hygro) containing 3 times the STAT-3 binding site (5′-TTCCCGTCAA-3′) was purchased from Promega, the TRH reporter gene containing the human TRH promotor (product no. S713424) was from Switchgear Genomics, and the suppressor of cytokine signaling-3 (SOCS-3) reporter and its mutants as well as the cAMP response element (CRE) reporter, containing 6 5′-TGACCTCAC-3′ sites were reported previously (36, 37).
Cell culture and transfections
The adult mouse hypothalamic cell lines mHypoA-2/10 (Clu-176) and mHypoA-2/12 (CLU-177) were purchased from Cedarlane and cultured in DMEM (10% fetal bovine serum, 2 mM l-glutamine, and penicillin/streptomycin) at 37°C and 5% CO2. To obtain either cell line, hypothalamic neurons from 2-month-old male C57BL/6 mice were isolated, ciliary neurotrophic factor was used to trigger neurogenesis of hypothalamic primary cultures, and immortalization was induced by retroviral transfer of SV40 T antigen. To obtain mHypoA-2/10 cells stably expressing the CRE reporter, cells were cotransfected with the CRE reporter construct and an empty pcDNA4 vector containing the resistance gene for Zeocin. Transfected cells were then selected with 600 μg/mL Zeocin for 4 weeks, and positively tested cell pools were used for further studies.
Whole-cell ELISA
Cells were seeded in 24-well plates (∼25,000/well) 2 days before the experiment. After 20 hours of serum starvation, untreated or inhibitor- or dimethylsulfoxide (DMSO)-pretreated cells were stimulated with BK, placed on ice, and washed with ice-cold PBS. After the cells were fixed for 15 minutes with 4% paraformaldehyde, they were permeabilized with ice-cold methanol-acetone (50:50) for 5 minutes. Next, cells were washed with PBS and then incubated with 0.5% of BSA in PBS for 15 minutes at 37°C to block unspecific binding sites. After an additional washing step, cells were incubated for 30 minutes at 37°C with phospho (p)-ERK-1/2 (1:2,500) in PBS/BSA. First, antibodies were removed, and cells were washed and incubated with the horseradish peroxidase–conjugated goat anti-rabbit secondary antibody (1:4000) in PBS/BSA for 30 minutes at 37°C. After several washing steps 200 μL of 1-Step-Ultra-TMB-ELISA substrate (Thermo Scientific) was added to the cells, and the solution was incubated for 15 minutes at room temperature. The reaction was stopped by addition of 50 mL of 1 M H2SO4, 200 μL was transferred to 96-well plates with a clear bottom, and OD450 was measured in a FLUOstar Omega plate reader.
Western blotting
Cells were seeded on 6-well plates (∼100 000/well), cultured for 1 day, serum starved for 20 hours the next day, stimulated for the indicated period of time with IFN-γ, EGF, MSH, or BK, and then lysed in Laemmli buffer. Lysates were subjected to SDS-PAGE, and protein was transferred to nitrocellulose by Western blotting. For detection of TRH expression, blots were separated in 2 parts, by a horizontal cut at 35 kDa. The lower part was used to analyze TRH expression (30 kDa) with a TRH-specific antiserum (1:2000), and the upper part was used to detect total ERK-2 (42 kDa) proteins (1:10 000) as a loading control. For detection of STAT-3 phosphorylation, blots were separated by a horizontal cut at 50 kDa. The upper part was used to analyze STAT-3 (68 kDa) phosphorylation with phospho-specific STAT-3 (Tyr-705, 1:5000; and Ser-727, 1:2000) antisera, and the lower part was used to detect total ERK-2 (42 kDa) proteins (1:10 000) as a loading control. Immune reactivity was quantified by densitometry, ratios between TRH or p-STAT-3 and total ERK-2 signals were calculated, and ligand-induced TRH expression or STAT-3 phosphorylation was normalized to that of unstimulated cells.
Reporter gene assays
Cells were seeded on 12-well plates (∼20 000/well) and transfected with various luciferase reporter genes constructs using the TurboFect reagent (R0531; Thermo Scientific) on the next day, according to the manufacturer's protocol. After serum starvation for 20 hours, cells were stimulated on the following day for 4 to 6 hours in serum-free medium. Small molecule inhibitors (concentration as indicated) and the respective carrier control were preincubated for 30 to 60 minutes before addition of agonist and were present during ligand stimulation. After stimulation, cells were lysed (25 mM Tris-HCl [pH 7.4], 4 mM EGTA, 8 mM MgCl2, 1 mM dithiothreitol, and 1% Triton-X-100), and luciferase activity was measured in white-bottom 96-well plates after automatic injection of luciferase substrate (beetle luciferin in the case of the STAT-3, SOCS-3, or CRE reporter and coelenterazine H in the case of the TRH reporter). The resulting total light emission was detected every second for 10 seconds after injection in a FLUOstar Omega plate reader. Maximal light emission during this period was recorded and is indicated relative to that of unstimulated cells.
cAMP accumulation
To determine agonist-induced cAMP accumulation, 200 000 cells were seeded in 12-well dishes 24 hours before the experiment and labeled in serum-free DMEM containing 2 μCi/mL of [3H]adenine for 4 hours. Cells were stimulated for 30 minutes at 37°C in DMEM containing 1 μM 3-isobutyl-1-methylxanthine along with MSH or forskolin (FSK) and NPY. The reaction was terminated by removal of the medium and addition of ice-cold 5% trichloroacetic acid to the cells. [3H]ATP and [3H]cAMP were then purified by sequential chromatography (Dowex resin/aluminum oxide columns), and the accumulation of [3H]cAMP was expressed as the ratio of [3H]cAMP to ([3H]cAMP + [3H]ATP).
RT-PCR
Cells were seeded on 6-well plates (∼100 000/well) and serum starved for 20 hours on the following day. Afterward, cells were stimulated for the indicated period of time in serum-free medium. Total RNA was isolated using TRI Reagent (Sigma-Aldrich), according to the manufacturer's instructions. First-strand synthesis was performed with oligo(dT)18 primer using 2 μg of total RNA and the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas). RT-PCR was performed using an advanced primus 25 cycler from Peqlab, DreamTaq polymerase (Fermentas), 0.08 μL of the first-strand synthesis reaction, and 1 μM concentrations of the following primer pairs: β-actin (forward 5′-CCAACCGCGAGAAGATGA-3′ and reverse 5′-CCAGAGGCGTACAGGGATAG-3′), SOCS-3 (forward 5′-CACACAAGGAGCCAAACACA-3′ and reverse 5′-TAGCCACCTGGGTGAATCC-3′), and c-fos (forward 5′-GGGACAGCCTTTCCTACT-3′ and reverse 5′-GATCTGCGCAAAAGTCCT-3′). The following conditions were using for amplification: initial denaturation for 15 minutes at 94°C, 30 cycles (β-actin) or 40 cycles (SOCS-3 and c-fos) of 94°C for 10 seconds, 55°C for 10 seconds, and 72°C for 10 seconds.
Data analysis
Data were analyzed using Prism 4.0 (GraphPad Software Inc). The statistical significance of differences was assessed by one- or two-sample Student t tests. Asterisks (***, P < .001; **, P < .01; and *, P < .05) in the case of the one-sample test and hash signs (###, P < .001; ##, P < .01; and #, P < .05) in the case of the 2-sample test were used to indicate significant differences.
Results
mHypoA-2/10 and mHypoA-2/12 cells resemble MSH/NPY-sensitive, hypothalamic neurons
The adult mouse hypothalamic cell lines mHypoA-2/10 and -2/12 were immortalized from 2-month-old male mice and express a wide range of hypothalamic markers (38–43). The hypothalamus contains distinct areas of specialized neurons such as the paraventricular nucleus (PVN) or the dorsomedial hypothalamic nucleus (DMH). Neurons of these areas have been reported to enhance or suppress TRH expression when challenged with MSH or NPY, respectively (15, 44, 45). Thus, in a first set of experiments, we investigated whether mHypoA-2/10 and -2/12 cells respond to both peptides. As seen in Figure 1A, both cell lines strongly reacted to MSH with cAMP accumulation indicating expression of Gs-coupled MSH receptors. NPY receptors antagonize MSH action in hypothalamic neurons by activating Gi/o proteins and thus inhibit cAMP signals (46). Accordingly, NPY significantly inhibited FSK-induced cAMP production (Figure 1B), revealing that mHypoA-2/10 and -2/12 cells respond to MSH and NPY on the level of cAMP. MSH has been shown to enhance and NPY to suppress TRH expression in hypothalamic neurons of the PVN or DMH (47). Furthermore, the thyroid hormone T3 has been shown to depress TRH expression in hypothalamic neurons by a negative-feedback mechanism (48, 49). Thus, we stimulated mHypoA-2/10 and -2/12 cells with MSH for different time periods and analyzed TRH expression by Western analysis. As shown in Figure 1, C and D, MSH significantly induced TRH expression in both cell lines after 2 hours of stimulation, an effect that lasted for additional 2 hours. Furthermore, MSH-induced activation and NPY or T3 suppression of TRH-promoter reporter activity in mHypoA-2/10 cells (Figure 1E), strongly indicating that mHypoA-2/10 and -2/12 cells resemble hypothalamic neurons with regard to MSH, NPY, or T3 sensitivity and production of hypothalamic hormones such as TRH.
Figure 1. mHypoA-2/10 and -2/12 cells resemble MSH/NPY-sensitive hypothalamic neurons.

A and B, cAMP accumulation was assessed after labeling of mHypoA-2/10 and -2/12 cells with [3H]adenine followed by the purification of [3H]cAMP and [3H]ATP by sequential chromatography. Cells were stimulated with 1 μM MSH (A) and with 1 μM FSK (B) alone or along with 100 nM NPY for 30 minutes at 37°C. Data from 5 independent experiments performed in quadruplicate are presented as means ± SEM. Hash signs indicate a significant difference from MSH-treated (A) or FSK-treated (B) cells. C and D, mHypoA-2/10 (C) and in mHypoA-2/12 (D) cells were serum starved for 16 hours and then stimulated with MSH (1 μM) for the indicated period of time. Cell lysates were subjected to Western blot analysis using a specific antiserum against murine TRH. The same blot was also analyzed with an antiserum against the total ERK-2 protein (t-ERK-2) to control for the total protein amount. One of 3 representative experiments is shown. E, mHypoA-2/10 cells were transfected with a human TRH reporter gene construct, serum starved for 16 hours, and stimulated with MSH (1 μM), NPY (100 nM), or T3 (10 nM) for various periods of time. Data from at least 4 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0).
Activation of a STAT-3–dependent reporter gene by non-CRs
To investigate STAT-3 activation by non-CRs in hypothalamic cells, we transfected a STAT-3–dependent reporter gene construct into mHypoA-2/10 and -2/12 cells and challenged these cells with saturating concentrations of either IFN-γ, EGF, BK, MSH, or NPY. IFN-γ activates hypothalamic CRs and served as a positive control. The EGFR belongs to the family of RTKs that are expressed in hypothalamic neurons and exert anorexigenic effects (50). Expression of the Gq/11-coupled bradykinin B2 receptor (B2R) has also been shown in hypothalamic cells. However, a functional role of BK in appetite or energy regulation has not been defined yet (51–53). As shown in Figure 2, MSH and NPY did not activate STAT-3 reporter activity, whereas IFN-γ, EGF, or BK profoundly induced STAT-3–dependent reporter activation in both mHypoA-2/10 and -2/12 cell lines (Figure 2), demonstrating that activation of hypothalamic STAT-3 is not restricted to CRs.
Figure 2. STAT-3 activation in hypothalamic cells.
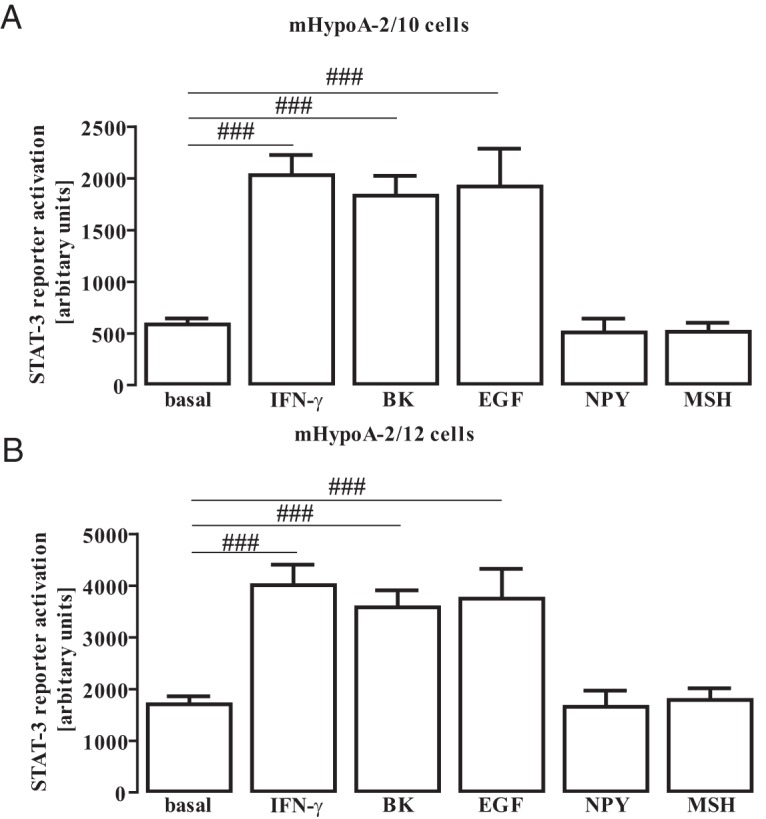
A and B, mHypoA-2/10 (A) and mHypoA-2/12 (B) cells were transfected with a reporter gene construct containing 3 times the STAT-3 binding site (5′-TTCCCGTCAA-3′), serum starved for 16 hours, and then stimulated with IFN-γ (10 ng/mL), BK (1 μM), EGF (10 ng/mL), NPY (100 nM), or MSH (1 μM) for 6 hours. Data from 10 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Hash signs indicate a significant difference from basal.
EGF and BK exclusively induce Ser-727 and IFN-γ Tyr-705 phosphorylation of STAT-3
To define the role of Tyr-705 and Ser-727 phosphorylation in the activation of hypothalamic STAT-3, we took advantage of phospho-specific antibodies against positions 705 and 727 of the murine STAT-3 protein and analyzed lysates of mHypoA-2/10 and -2/12 cells stimulated with IFN-γ, EGF, or BK for various time periods. As expected, IFN-γ induced strong STAT-3 phosphorylation at Tyr-705 after 5 to 10 minutes of stimulation followed by a further increase thereafter (Figure 3). IFN-γ did not affect Ser-727, suggesting that IFN-γ–induced STAT-3 activation is solely mediated by the established modification of Tyr-705. In sharp contrast, EGF and BK did not induce Tyr-705 phosphorylation but promoted phosphorylation of Ser-727 instead with a peak at 5 to 10 minutes (Figure 3 and Table 1). Thus, it appears that although IFN-γ, EGF, and BK induce similar STAT-3 reporter activation in hypothalamic cells, they employ distinct mechanisms.
Figure 3. Ligand-induced STAT-3 phosphorylation.

mHypoA-2/10 and -2/12 cells were serum starved for 16 hours and then stimulated with IFN-γ (10 ng/mL), BK (1 μM), or EGF (10 ng/mL) for the indicated time period. Cell lysates were subjected to Western analysis using a phospho-specific antiserum against either Tyr-705 or Ser-727. In each case the same blot was also analyzed with an antiserum against the total ERK-2 protein (t-ERK-2) to control for the total protein amount. One representative blot is shown. Data from 5 independent experiments are given in Table 1.
Table 1.
Ligand-Induced Ser-727 Phosphorylation of Hypothalamic STAT-3
| Treatment | Period of Stimulation | mHypoA-2/10 Cells |
mHypoA-2/12 Cells |
||||
|---|---|---|---|---|---|---|---|
| IFN-γ | EGF | BK | IFN-γ | EGF | BK | ||
| Basal | 2.5 min | 1.0 ± 0.2 | 1.0 ± 0.3 | 1.0 ± 0.3 | 1.0 ± 0.1 | 1.5 ± 0.3 | 1.0 ± 0.2 |
| 5.0 min | 1.0 ± 0.2 | 1.1 ± 0.2 | 1.0 ± 0.3 | 1.0 ± 0.2 | 2.1 ± 0.2* | 1.0 ± 0.9 | |
| 10 min | 1.1 ± 0.3 | 3.2 ± 0.4** | 2.5 ± 0.4** | 1.0 ± 0.2 | 3.9 ± 0.3** | 2.1 ± 0.3** | |
| 20 min | 1.1 ± 0.4 | 2.9 ± 0.3** | 2.2 ± 0.2** | 1.1 ± 0.2 | 3.2 ± 0.4** | 2.2 ± 0.2** | |
| 30 min | 1.0 ± 0.6 | 1.2 ± 0.3 | 1.3 ± 0.1 | 1.0 ± 0.4 | 1.1 ± 0.2 | 1.1 ± 0.3 | |
| DMSO | 10 min | ND | 4.3 ± 0.5** | 4.1 ± 0.3** | ND | 4.6 ± 0.4** | 4.3 ± 0.4** |
| BIM-S | 10 min | ND | ND | 4.5 ± 0.3** | ND | ND | 4.1 ± 0.3** |
| PD-184352 | 10 min | ND | 1.1 ± 0.2 | 1.1 ± 0.2 | ND | 1.1 ± 0.4 | 1.2 ± 0.2 |
| AG-1478 | 10 min | ND | ND | 1.1 ± 0.7 | ND | ND | 1.1 ± 0.3 |
Abbreviation: ND, not determined. Western blot data using a phospho-specific antiserum against Ser-727 of the murine STAT-3 protein shown in Figures 3, 4A, and 5B were compiled (n = 5) after quantification by densitometry and normalization to the time point zero (fold of basal) and are expressed as means ± SEM. Asterisks indicate a significant difference to time point zero (1.0).
To investigate signaling cascades leading to EGF- or BK-mediated activation of hypothalamic STAT-3, we used chemical kinase inhibitors. The ERK-1/2 inhibitor PD-184352 (10 μM) completely blocked BK-mediated phosphorylation of STAT-3, whereas BIM-X (1 μM) targeting protein kinase C had no effect (Figure 4A and Table 1). PD-184352 also blunted BK-induced activation of the STAT-3 reporter (Figure 4B, right panel), implying that ERK-1/2 are responsible for the effects of BK on STAT-3. EGF-induced STAT-3 phosphorylation (Figure 4A and Table 1) and reporter activation (Figure 4B, left panel) was also sensitive to PD-184352, indicating that ERK-1/2 plays a major role in the modulation of STAT-3 activity by RTKs and G protein–coupled receptor–mediated phosphorylation at Ser-727. In accord with the prominent role of ERK-1/2 for EGF- and BK-, but not for IFN-γ–induced STAT-3 activation, ERK-1/2 phosphorylation in mHypoA-2/10 and -2/12 cell lines was observed in response to EGF and BK but not to IFN-γ (Figure 4C).
Figure 4. ERK-1/2 activity is required for BK- or EGF-induced STAT-3 activation.

A, mHypoA-2/10 or -2/12 cells were serum starved for 16 hours, preincubated for 30 minutes with BIM-X (1 μM), PD-184352 (10 μM), or the carrier DMSO (0.1%) and were then stimulated for 10 minutes with either BK (1 μM) or EGF (10 ng/mL). Cell lysates were subjected to Western blot analysis using either a phospho-specific antiserum against Ser-727 or against the total ERK-2 protein (t-ERK-2) to control for the total protein amount. One representative blot is shown. Data from 7 independent experiments are given in Table 1. B, mHypoA-2/10 or -2/12 cells were transfected with a STAT-3–dependent reporter gene construct, serum starved for 16 hours, preincubated for 30 minutes with PD-184352 (10 μM) or the carrier DMSO (0.1%), and then stimulated for 6 hours with either 10 ng/mL EGF (left panel) or 1 μM BK (right panel). The effects of PD-184352 on STAT-3 activation in 5 independent experiments performed in quadruplicate were compiled by defining ligand-induced reporter activation in the presence of DMSO as 100%. Ligand-induced STAT-3 activation is expressed as the mean ± SEM, and asterisks indicate a significant difference from 100%. C, mHypoA-2/10 (left panel) or mHypoA-2/12 (right panel) cells were serum starved for 16 hours and then were stimulated with IFN-γ (10 ng/mL), BK (1 μM), or EGF (10 ng/mL) for the indicated period of time. ERK-1/2 phosphorylation was detected by performance of a whole-cell ELISA with a p-ERK-1/2–specific antiserum. Ligand-induced ERK-1/2 phosphorylation measured in 5 independent experiments performed in quadruplicate was normalized to basal and is expressed as the mean ± SEM.
BK-induced ERK-1/2 and STAT-3 activation requires EGFR transactivation
Multiple signaling pathways link BK with ERK-1/2 in nonhypothalamic cells (54–56). To identify pertinent signaling cascades in mHypoA-2/12 cells, we established a whole-cell ELISA based on an ERK-1/2 phospho-specific antibody. As shown in Figure 5A (left panel), we detected robust BK-dependent signals and were able to screen for signaling components using a host of chemical inhibitors. As indicated in Figure 5A (right panel), BK-induced ERK-1/2 phosphorylation was sensitive to inhibitors of src kinases (PP-2), tyrosine kinases (genistein), Gβγ-subunits (gallein), phospholipase C (U-73122), metalloproteases (GM-6001), and EGFR (AG-1478). This pattern of inhibitors points to a signaling pathway recently established in other cell systems (57), encompassing Ca2+- and Gβγ-dependent src kinase activation, resulting in metalloprotease-mediated shedding of membrane-bound EGF that in turn activates ERK-1/2 via the EGFR (see Figure 10). Accordingly, BK-induced STAT-3 phosphorylation and activation were strongly inhibited after preincubation of mHypoA-2/10 and -2/12 cells with the EGFR kinase inhibitor AG-1478 (Figure 5, B and C, and Table 1).
Figure 5. BK-induced ERK-1/2 and STAT-3 activation activity required EGFR activity.

A (left panel), mHypoA-2/12 cells were serum starved for 16 hours, preincubated for 30 minutes with PD-184352 (10 μM) or the carrier DMSO (0.1%), and then stimulated for 10 minutes with BK (1 μM). ERK-1/2 phosphorylation was detected by performance of a whole-cell ELISA with a p-ERK-1/2–specific antiserum. Data from 1 representative experiment performed in quadruplicate are given. Hash signs indicate significant differences from basal (−). A (right panel), mHypoA-2/12 cells were serum starved for 16 hours, preincubated for 16 hours with PTX (100 ng/mL), or for 30 minutes with BIM-X (1 μM), AG-1478 (100 nM), PP-2 (10 μM), genistein (50 μM), gallein (10 μM), Ly-294002 (20 μM), U-73122 (10 μM), GM-6001 (1 μM), AG-1296 (100 nM), or the carrier DMSO (0.1 and 0.2%), and then stimulated for 10 minutes with BK (1 μM). Inhibitor concentrations were chosen based on data from the literature, and inhibitors with no effects were tested in different assays. For example, PTX blocked the effects of NPY on cAMP and BIM-X blocked the effects of phorbol ester on ERK-1/2. ERK-1/2 phosphorylation was detected by performing a whole-cell ELISA with a p-ERK-1/2–specific antiserum. Data from 5 independent experiments performed in quadruplicate were compiled by calculating the inhibition as a percentage. Asterisks indicate a significant difference from 0%. B, mHypoA-2/10 or -2/12 cells were serum starved for 16 hours, preincubated for 30 minutes with AG-1478 (10 μM), and then stimulated for 10 minutes with BK (1 μM). Cell lysates were subjected to Western analysis using either a phospho-specific antiserum against Ser-727 of STAT-3 or against the total ERK-2 protein (t-ERK) to control for the total protein amount. One representative blot is shown. Data from 5 independent experiments are given in Table 1. C, mHypoA-2/10 or -2/12 cells were transfected with a STAT-3–dependent reporter gene construct, serum starved for 16 hours, preincubated for 30 minutes with AG-1478 (100 nM), and stimulated for 6 hours with 1 μM BK. The effects of AG-1478 on STAT-3 activation in 5 independent experiments performed in quadruplicate were compiled by defining ligand-induced reporter activation under basal conditions as 100%. Ligand-induced STAT-3 activation is expressed as the mean ± SEM, and asterisks indicate a significant difference from 100%.
Figure 10. STAT-3 activity regulating the signaling network induced by BK or EGF in hypothalamic cells.

EGF-induced EGFR activation enhances ERK-1/2 activity, which leads to Ser-727 phosphorylation of STAT-3 and c-fos induction. Ser-727 phosphorylated STAT-3 together with AP-1 orchestrates transient SOCS-3 and putatively TRH expression via coactivation of SP-1, as indicated by the dashed arrow and previously proposed by others (69) TRH expression. BK-induced EGFR transactivation via src kinase and metalloproteases (MP) mimics the effects of EGF on ERK-1/2 and STAT-3. HB-EGF, heparin-binding EGF; PLC, phospholipase B.
STAT-3 phosphorylation at Tyr-705 or Ser-727 differently affects expression of SOCS-3
Several studies have shown that activation of hypothalamic STAT-3 leads to the expression of SOCS-3 proteins (58). Thus, to analyze the effects of Tyr-705 and Ser-727 on STAT-3–dependent gene expression, we stimulated mHypoA-2/12 cells with IFN-γ, EGF, or BK for 30 or 120 minutes and detected mRNA levels of SOCS-3 by RT-PCR. As illustrated in Figure 6, all stimuli induced SOCS-3 mRNA expression in hypothalamic cells. Of note, EGF and BK induced transient mRNA expression of SOCS-3 after 30 minutes, whereas IFN-γ elevated SOCS-3 mRNA levels over a longer time period (120 minutes), suggesting that Tyr-705- and Ser-727-mediated STAT-3 activation targets the same gene with distinct kinetics.
Figure 6. EGF, BK, and IFN-γ induce SOCS-3 expression.
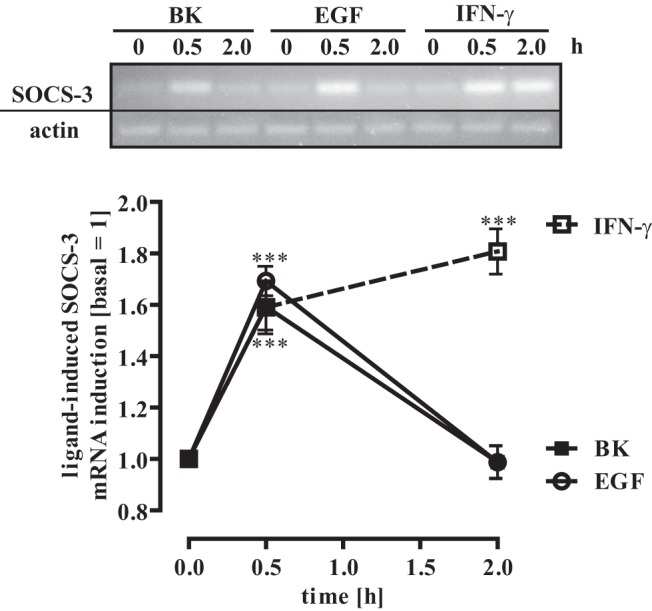
mHypoA-2/12 cells were serum starved for 16 hours and then stimulated for 30 or 120 minutes with EGF (10 ng/mL), BK (1 μM), or IFN-γ (10 ng/mL). SOCS-3 (40 cycles) or β-actin (30 cycles) mRNA expression was determined by RT-PCR. One representative blot is shown. Blots were quantified by densitometry, SOCS-3 expression was normalized to β-actin expression, and the time point zero is set to 1.0. Data are given as means ± SEM from 5 independent experiments, and asterisks indicate a significant difference from time point zero (1.0).
The minimal SOCS-3 promoter contains a proximal and a distal STAT-3 binding site as well as an activating protein-1 (AP-1) responsive element near the distal STAT-3 site (36). We studied the effects of IFN-γ, EGF, or BK on the activity of this minimal SOCS-3 promoter and found that all stimuli similarly activated the SOCS-3 reporter construct in mHypoA-2/12 cells (Figure 7A). Of note, the BK-induced effects on SOCS-3 were inhibited by AG-1478, indicating that EGFR transactivation is involved. The STAT-3 inhibitor STATTIC (59) blocked IFN-γ-, EGF-, and BK-induced SOCS-3 promoter activation, indicating that all ligands use the STAT-3 pathway to induce SOCS-3. However, SR-11302, an inhibitor of AP-1–dependent gene expression (60), decreased EGF- and BK- but not IFN-γ–induced SOCS-3 promoter activation, suggesting that promoter activation by EGF or BK requires STAT-3 and AP-1, whereas IFN-γ–promoted STAT-3 activation alone is sufficient to activate the SOCS-3 promoter.
Figure 7. Regulation of the SOCS-3 promoter by distinct STAT-3 activating stimuli.

A, mHypoA-2/12 cells were transfected with a SOCS-3 reporter gene construct containing AP-1, the distal dis-STAT-3 (5′-TTCCAGGA-3′) and the proximal pro-STAT-3 site (5′-TTACAAGAA-3′), serum starved for 16 hours, preincubated for 1 hour with the STAT-3 inhibitor STATTIC (10 μM) or the AP-1 inhibitor SR-11 302 (1 μM), and then stimulated with IFN-γ (10 ng/mL), EGF (10 ng/mL), or BK (1 μM) for 6 hours. Data from 5 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0) and hash signs from untreated cells. B, mHypoA-2/12 cells were transfected with a SOCS-3 reporter gene mutant lacking the AP-1 and the distal dis-STAT-3 site, serum starved for 16 hours, and then stimulated with IFN-γ (10 ng/mL), EGF (10 ng/mL), or BK (1 μM) for 6 hours. Data from 5 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0). C, mHypoA-2/12 cells were transfected with a SOCS-3 reporter gene mutant containing the AP-1 and the proximal pro-STAT-3 site, serum starved for 16 hours, and then stimulated with IFN-γ (10 ng/mL), EGF (10 ng/mL), or BK (1 μM) for 6 hours. Data from 5 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0). D, mHypoA-2/12 cells were transfected with an SRF/TCF-dependent reporter gene construct, serum starved for 16 hours, preincubated for 30 minutes with PD-184253 (10 μM), and then stimulated with IFN-γ (10 ng/mL), EGF (10 ng/mL), or BK (1 μM) for 6 hours. Data from 5 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0) and hash signs from untreated cells. E, mHypoA-2/12 cells were serum starved for 16 hours, preincubated for 30 minutes with PD-184253 (10 μM), and then stimulated for 30 or 120 minutes with EGF (10 ng/mL), BK (1 μM), or IFN-γ (10 ng/mL). c-fos (40 cycles) or β-actin (30 cycles) mRNA expression was determined by RT-PCR. Blots were quantified by densitometry, c-fos expression was normalized to β-actin expression, and time point zero was set to 1.0. Data are given as means ± SEM from 5 independent experiments, and asterisks indicate a significant difference from time point zero.
To gain deeper insight into STAT-3–mediated SOCS-3 expression, we took advantage of previously established SOCS-3 promoter mutants (36). The first mutant lacked the AP-1 and the distal but contained the proximal STAT-3 site. In contrast to EGF and BK, IFN-γ still activated the truncated SOCS-3 promoter (Figure 7B), indicating that STAT-3 proteins that are activated by IFN-γ utilize the proximal binding site and those activated by EGF or BK the distal binding site. To test this hypothesis we also used a SOCS-3 promoter mutant that lacked the distal but still contained the AP-1 and the proximal STAT-3 sites. Depletion of the distal site completely abolished EGF- and BK-induced SOCS-3 promoter activation but did not affect activation by IFN-γ (Figure 7C). Hence, it appears that although all 3 ligands induce SOCS-3 expression via STAT-3, depending on the stimulus, STAT-3 either activates the SOCS-3 promoter alone (in the case of IFN-γ) by binding to the proximal STAT-3 site or does so in concert with AP-1 (in the case of EGF or BK) by binding to the distal STAT-3 site.
The AP-1 transcription factor consists of the c-fos/c-jun heterodimer activated by ERK-1/2–dependent induction of either protein. Because EGF and BK activated ERK-1/2 in mHypoA-2/12 cells, it is tempting to assume that ERK-1/2 mediates not only EGF- and BK-induced STAT-3 activation but also AP-1 induction. Thus, we analyzed whether or not EGF, BK, or IFN-γ would activate ERK-1/2–dependent gene expression by using a serum response factor/ternary complex factor (SRF/TCF)–dependent reporter construct. Because of its inability to activate ERK-1/2, IFN-γ failed to induce SRF/TCF-dependent reporter activation (Figure 7D). In contrast, EGF or BK induced SRF/TCF-dependent reporter activation in an ERK-1/2 dependent manner. Furthermore, EGF or BK but not IFN-γ induced mRNA expression of c-fos via ERK-1/2 (Figure 7E), suggesting that ERK-1/2 orchestrates EGF-induced SOCS-3 expression via Ser-727 phosphorylation of STAT-3 and c-fos induction via SRF/TCF.
EGF but not IFN-γ induces TRH expression
The TRH promoter contains binding sites for the cAMP response element binding protein (CREB) that allows MSH to induce TRH expression via the Gs pathway as shown in Figure 1. However, the TRH promoter also contains a STAT-3 binding site. Thus, we tested the effects of IFN-γ-– and EGF-induced STAT-3 activation on TRH expression in mHypoA-2/10 cells, by stimulating the cells with either hormone for up to 6 hours (Figure 8). EGF induced significant TRH expression comparable to MSH, whereas IFN-γ was without effect, suggesting that STAT-3 activation alone is not sufficient to promote TRH expression. Alternatively, STAT-3 may target distinct genes depending on the phosphorylation site. To exclude a putative role of the CREB/CRE pathway for EGF-promoted TRH induction, we stimulated mHypoA-2/10 cells stably expressing a CREB-dependent reporter gene construct with MSH or EGF for 4 hours (Figure 9A). As expected, MSH significantly activated the CREB-dependent reporter, whereas EGF failed to do, indicating that activation of the CREB pathway does not account for EGF-induced TRH expression. To dissect the molecular events leading to EGF-promoted TRH expression, we used a reporter gene construct coding for Renilla luciferase under the control of the human TRH promoter. In accord with our data observed on the protein level in Figure 8, MSH and EGF promoted TRH promoter activity in mHypoA-2/10 cells, whereas IFN-γ was without effect (Figure 9B). To investigate a role of STAT-3 in EGF-induced and basal TRH expression, we used the structurally unrelated STAT-3 inhibitors STATTIC and S3I-201 (59, 61). First, we observed that both inhibitors significantly inhibited basal TRH promoter activity (Figure 9C), indicating that STAT-3 is involved in the regulation of basal TRH expression in mHypoA-2/10 cells. Second, we found that both STAT-3 inhibitors abolished EGF- but not MSH-induced TRH promoter activation (Figure 9C), indicating that EGF-induced STAT-3 activation via Ser-727 phosphorylation is involved in EGF-promoted TRH expression.
Figure 8. EGF but not IFN-γ induces TRH expression.
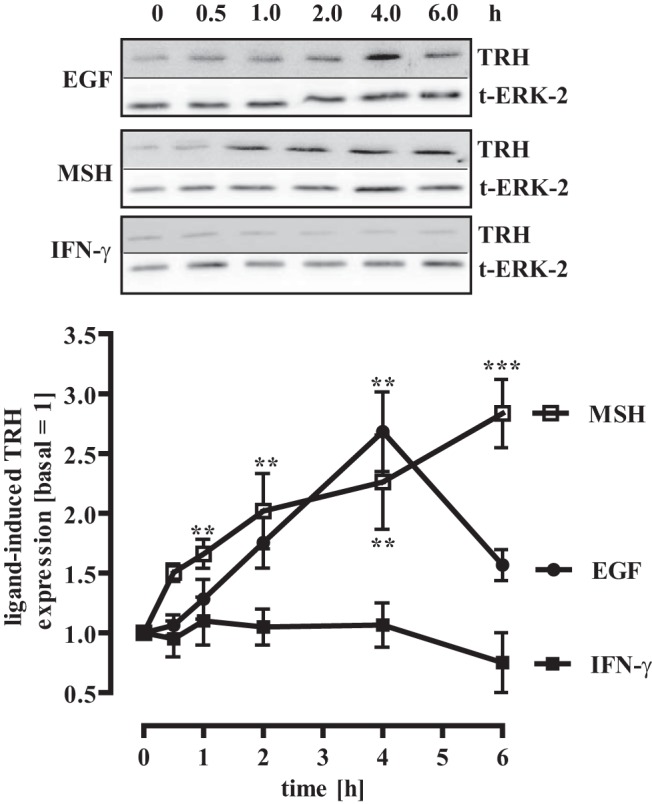
mHypoA-2/10 cells were serum starved for 16 hours and then stimulated with IFN-γ (10 ng/mL), MSH (1 μM), or EGF (10 ng/mL) for the indicated period of time. Cell lysates were subjected to Western blot analysis using an antiserum against murine TRH. The same blot was also analyzed with an antiserum against the total ERK-2 protein (t-ERK-2) to control for the total protein amount. One representative blot is shown. Data from 4 independent experiments were quantified by densitometry and are given as means ± SEM. Asterisks indicate a significant difference from time point zero (1.0).
Figure 9. Regulation of the human TRH promoter by MSH, EGF, or IFN-γ.
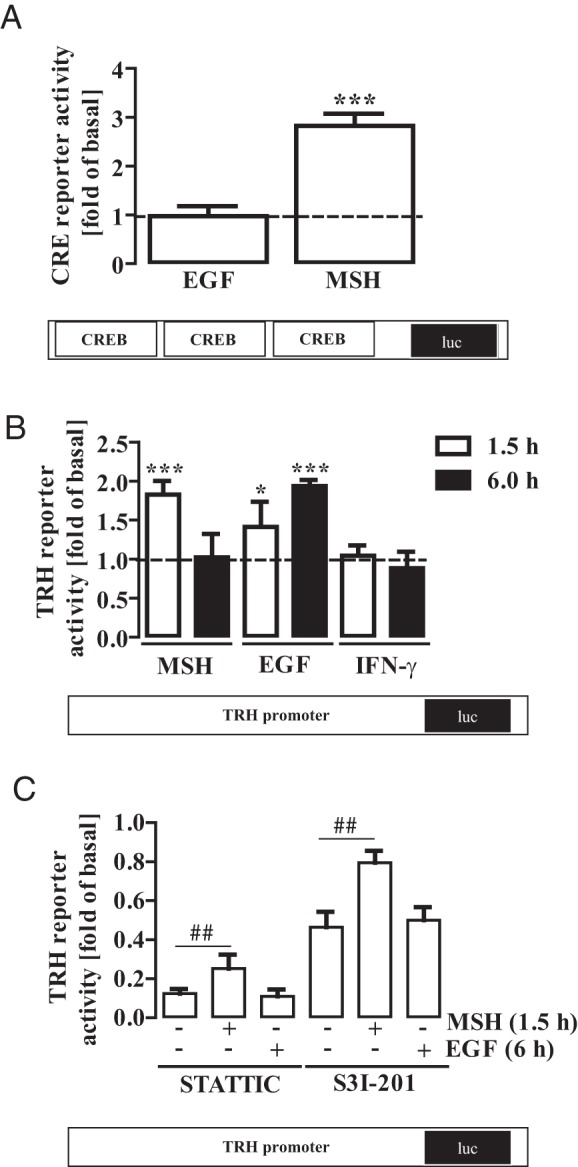
A, mHypoA-2/10 cells stably expressing a CREB-dependent reporter gene construct were serum starved for 16 hours and then stimulated with MSH (1 μM) or EGF (10 ng/mL) for 4 hours. Data from 5 independent experiments performed in quadruplicate were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0). B, mHypoA-2/10 cells were transfected with a human TRH reporter gene construct containing the STAT-3 site 5′-TTTCCGGAGGAG-3′ at the same position as the murine promoter contains the 5′-TTTCCGGAAAG-3′ site. Afterward cells were serum starved for 16 hours and stimulated with MSH (1 μM), EGF (10 ng/mL), or IFN-γ (10 ng/mL) for 1.5 and 6 hours. Data from 5 independent experiments performed in quadruplicates were compiled and are expressed as means ± SEM. Asterisks indicate a significant difference from basal (1.0). Data from 3 independent experiments performed in quadruplicates were compiled and normalized to basal and are expressed as means ± SEM. Asterisks indicate a significant difference from 100%. C, mHypoA-2/10 cells were transfected with the TRH reporter gene construct, serum starved for 16 hours, preincubated for 1 hour with the STAT-3 inhibitor STATTIC (10 μM) or STA-21 (5 μM), and then stimulated with 1 μM MSH for 1.5 hours and with 10 ng/mL EGF for 6 hours. Data from 4 independent experiments performed in quadruplicates were compiled and normalized to basal and are expressed as means ± SEM. Hash signs indicate a significant difference for cells treated solely with STATTIC or S3I-201.
Discussion
The hypothalamic-pituitary-thyroid (HPT) axis is of prime importance in the central regulation of appetite and energy metabolism (62). The hypothalamic nucleus arcuatus receives peripheral satiety signals such as leptin or insulin and reacts either by releasing NPY or MSH. NPY and MSH act on neurons of the PVN or DMH and thereby control the release of TRH, resulting in TSH secretion from the pituitary gland. TSH affects the metabolic rate by promoting the secretion of T3 and T4. Regulation of transcriptional activity is a major control mechanism within the HPT axis, and STAT-3 has been shown to act as a major regulator of hypothalamic transcription (6, 7). So far, activation of the hypothalamic STAT-3 pathway via Tyr-705 phosphorylation has been exclusively attributed to leptin-induced signaling in neurons of the nucleus arcuatus (14, 63). Here, we analyzed the regulation of STAT-3 activity in mHypoA-2/10 and -2/12 cells that resemble PVN neurons as deduced from the inhibitory effects of the thyroid hormone T3 on TRH expression (Figure 1E). TRH gene expression has been shown to be regulated by T3 only in the PVN, even though TRH is widely expressed in many other areas of the hypothalamus (64). The results shown in here (Figure 1E) suggest that at least a significant proportion of mHypoA-2/10 and -2/12 cells behave as TRH-positive neurons of the PVN and may serve as valuable cellular model systems in future investigations.
Besides Tyr-705, mammalian STAT-3 proteins contain a second phosphorylation site at Ser-727. Several endogenous stimuli have been shown to induce STAT-3 phosphorylation at Ser-727 by employing various serine/threonine kinases in nonhypothalamic cell models or tissues. Two studies reported that Ser-727 becomes phosphorylated when cells are stimulated either with cytokines or growth factors, such as EGF, and that Ser-727 phosphorylation is required for full STAT-3 activation (30, 31). However, shortly thereafter, other researchers proposed that Ser-727 affects neither STAT-3 dimerization nor DNA binding (22). Furthermore, EGF-induced Ser-727 phosphorylation via ERK-1/2 diminished Tyr-705 phosphorylation and STAT-3 activity in Swiss 3T3 or NIH 3T3 fibroblasts (23, 25). In line with these data, ionomycin, granulocyte/macrophage colony-stimulating factor or phorbol esters blocked IL-6–induced STAT-3 activation in various cell types via ERK-1/2–mediated STAT-3 phosphorylation at Ser-727 (24, 65). Similar effects were observed after overexpression of ERK-2 or its upstream kinase MEK-2 (66). Inhibition of STAT-3 activity due to Ser-727 phosphorylation was also observed after UV- or anisomycin-induced c-Jun NH2-terminal kinase or EGF-induced phosphatidylinositol 3-kinase–AKT activation (27, 28). In stark contrast, glial cell line–derived neurotrophic factor activated STAT-3 via ERK-1/2–mediated Ser-727 phosphorylation in NIH 3T3 cells as well as IL-6 in Schwann cells, neurturin in Neuro2A cells, and phorbol esters in melanocytes (29, 32, 33, 67). Furthermore, neonatal diethylnitrosamine treatment of mice contributed to hepatocarcinogenesis via Ser-727 phosphorylation of STAT-3, and transgenic mice carrying a STAT-3 Ser-727-Ala-allele showed decreased embryonic and prenatal growth, most likely due to altered IGF serum levels (34, 35). Even though a salient role of Ser-727 in the regulation of STAT-3 is undisputed, the cellular context determining enhancement or silencing of STAT-3 activity due to Ser-727 phosphorylation still remains ill defined. Strikingly, no data considering the impact of Ser-727 on the regulation of hypothalamic STAT-3 are available to date.
Here, we show that the activity of hypothalamic STAT-3 in MSH/NPY-sensitive cells is not only enhanced by CR/JAK-mediated Tyr-705 phosphorylation but also by EGF-promoted ERK-1/2 activation, leading to subsequent STAT-3 phosphorylation at Ser-727. In a first set of experiments we used a STAT-3–dependent reporter gene construct and stimulated the cells with IFN-γ, EGF, BK, MSH, or NPY. We found that IFN-γ, EGF, or BK induced comparable reporter gene activation. When analyzing effects of these hormones on Tyr-705 phosphorylation, we observed Tyr-705 phosphorylation induced by IFN-γ but no effects of EGF and BK, indicating that EGF- and BK-mediated STAT-3 activation occurred independently from Tyr-705. Corresponding results were observed when a Ser-727–specific antiserum was used, suggesting that BK and EGF mediate their effects on STAT-3 activity via Ser-727 phosphorylation. The MEK-1/2 inhibitor PD-184253 blocked EGF-promoted phosphorylation at Ser-727 as well as STAT-3–dependent reporter activation, highlighting the importance of the MEK/ERK-1/2 pathway for the functional interactions between EGF and STAT-3. Of note, 2 independent studies reported that Ser-727 might actually be a direct target of MEK-1/2 and not of ERK-1/2 (66, 68). Therefore, additional studies are required to define the exact role of MEK-1/2 for the regulation of hypothalamic STAT-3. The sensitivity of the Ser-727–specific antiserum appeared to be lower than that of the Tyr-705 antiserum. Therefore, we cannot entirely rule out the possibility that IFN-γ might also affect Ser-727 phosphorylation and, thus, mediate its effects on STAT-3 activity via Tyr-705 and Ser-727 modification. However, given that IFN-γ did not activate ERK-1/2 (Figure 4C) or ERK-1/2–dependent reporter activity (Figure 9C), we consider this possibility as less likely.
Growth factors such as EGF have been reported to release TRH from hypothalamic neurons and to induce central anorexigenic effects (50, 69). Deletion experiments revealed that the −130/−84 region of the TRH promoter is responsible for EGF-induced promoter activation (69). Because this region contains a binding site for the SP-1 transcription factor, a role for SP-1 in EGF-induced TRH expression has been proposed (69). Interestingly, the −130/−84 region also contains the STAT-3 binding site of the TRH promoter, which is used by leptin to induce TRH expression (44). Thus, STAT-3 may also play a role in EGF-promoted TRH expression. In line with this assumption, we show herein that 2 structurally unrelated small molecule STAT-3 inhibitors blocked EGF-induced activation of the TRH reporter construct. Thus, our data highlight a new mechanism by which EGF affects the HPT axis via ERK-1/2–promoted STAT-3 activation in MSH/NPY-sensitive hypothalamic neurons. Although the physiological role of EGF-induced TRH expression is unclear at this point, it may shed new light on pathophysiological conditions involving EGF. Cancer-induced cachexia is a life-threatening condition frequently occurring in types of cancer with increased EGF serum levels and enhanced thyroid function (70–75). Thus, EGF-induced STAT-3 activation via Ser-727 may contribute to cancer-induced cachexia and thus could be of significant therapeutic interest. However, additional in vivo studies are required to test this intriguing hypothesis.
Interestingly, although IFN-γ activated the STAT-3 reporter and induced SOCS-3 expression via STAT-3, it failed to induce TRH expression via STAT-3. One explanation for the disparate actions of EGF and IFN-γ may be that Tyr-705–phosphorylated STAT-3 does not bind to the STAT-3 site of the TRH reporter. However, leptin-induced TRH expression appears to be mediated by Tyr-705–phosphorylated STAT-3 (13, 14). Notably, the SP-1 transcription factor proposed to be involved in EGF-induced TRH expression is activated by ERK-1/2, and the anorexigenic effects induced by leptin require STAT-3 and ERK-1/2 signaling in hypothalamic neurons (76–78). Thus, neither Ser-727– nor Tyr-705–phosphorylated STAT-3 may be sufficient to activate the TRH promoter but may require coactivation of SP-1. In such a model, leptin would induce TRH expression via JAK-induced STAT-3 and ERK-1/2–promoted SP-1 activation and EGF via ERK-1/2-mediated activation of STAT-3 and SP-1, whereas IFN-γ would fail to induce TRH expression despite its strong effects on STAT-3 because of its inability to activate SP-1 via ERK-1/2 (see Figure 10).
To further dissect the effects of Tyr-705 vs Ser-727 phosphorylation on STAT-3 activity, we studied the regulation of SOCS-3 expression by IFN-γ and EGF. STAT-3 activation mediated by Tyr-705 phosphorylation has been reported to induce expression of SOCS-3 proteins, exerting a negative feedback on hypothalamic STAT-3 signaling (5). The minimal SOCS-3 promoter contains a proximal and a distal STAT-3 site. The later site is near an AP-1 site and a recent study reported that phorbol ester–induced Ser-727 phosphorylation via ERK-1/2 enhanced SOCS-3 expression in human umbilical vein endothelial cells in cooperation with AP-1 (36). We observed that IFN-γ and EGF increased SOCS-3 mRNA levels in hypothalamic cells, indicating that both stimuli target the SOCS-3 promoter. While IFN-γ–induced sustained SOCS-3 expression, EGF elevated SOCS-3 mRNA levels in a transient fashion. Interestingly, similar distinct kinetics were also observed on the level of STAT-3 phosphorylation shown in Figure 3. Distinct effects of the kinetics of STAT-3 phosphorylation/activation by the CR/JAK or the RTK/ERK-1/2 pathway might be due to (1) differences in the pharmacodynamics of CRs and RTKs (eg, receptor desensitization), (2) distinct kinetics of JAK and ERK-1/2 activity, (3) distinct regulation of STAT-3 dephosphorylation (eg, kinetics of Ser and Tyr phosphatases might differ), or (4) distinct events on the SOCS-3 promoter due to differential STAT-3 phosphorylation (eg, difference in the transcriptional cofactors required).
To gain deeper insight into the exact molecular events leading to IFN-γ and EGF-induced SOCS-3 expression in hypothalamic cells, we used mutated SOCS-3 reporter constructs and chemical inhibitors of the STAT-3 and AP-1 pathway (36). We observed (1) that STAT-3 activity is required for IFN-γ and EGF-induced SOCS-3 expression, (2) that EGF additionally requires AP-1 activity, and (3) that IFN-γ utilizes the proximal and EGF the distal STAT-3 site to induce SOCS-3 expression (Figure 10). Thus, we propose the new concept that Ser-727 not only is a regulator of hypothalamic STAT-3 activity but also enhances cooperation or functional interactions of STAT-3 with distinct cofactors and thereby directs activated STAT-3 to different gene regulatory elements compared with Tyr-705 phosphorylation. The fact that EGF-promoted activation of the SOCS-3 promoter via Ser-727–phosphorylated STAT-3 requires coactivation of AP-1 resembles our results obtained for the effects of EGF on the TRH promoter, indicating that EGF requires coactivation of STAT-3 and SP-1. With the assumption that data obtained in hypothalamic cells also apply to other cell systems, they may offer an explanation for the contrasting reports on the functional corollaries of Ser-727 phosphorylation. Because Ser-727-phosphorylated STAT-3 requires cofactors for gene regulation, it might either increase or decrease promoter activity of different genes depending on the cellular context.
The kallikrein-kinin system (KKS) consisting of the BK precursor kininogen, the kinogenase kallikrein and the B2R is expressed in the mammalian central nervous system including hypothalamic neurons (79–85). Obesity induced by leptin deficiency correlates with decreased B2R expression and diabetes type 1 induced in obese Zucker rats or type 2 induced by streptozotocin with increased B2R expression in hypothalamic neurons (79, 80). Apparently, there is a physiological link between the brain KKS and the HPT axes. So far, BK-induced signaling in hypothalamic neurons is ill defined, and the effects of BK on hypothalamic gene expression have not been reported yet. Here we show that MSH/NPY-sensitive hypothalamic cells respond to BK, resulting in EGFR transactivation with STAT-3–dependent SOCS-3 induction. Thus, our data represent the first molecular link by which the KKS affects the HPT axis on the level of gene regulation in hypothalamic cells.
In summary, we show that ERK-1/2–promoted Ser-727 phosphorylation is sufficient to activate hypothalamic STAT-3 and to enhance TRH and SOCS-3 expression in cooperation with other transcription factors such as c-fos. Our data underline the important role of Ser-727 for STAT-3 activation in general and provide a new mechanistic link between hypothalamic STAT-3 and signaling of non-CRs, such as the EGFR.
Acknowledgments
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AP-1
- activating protein-1
- BK
- bradykinin
- B2R
- bradykinin B2 receptor
- CR
- cytokine receptor
- CRE
- cAMP response element
- CREB
- cAMP response element binding protein
- DMH
- dorsomedial hypothalamic nucleus
- DMSO
- dimethylsulfoxide
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- FSK
- forskolin
- HPT
- hypothalamic-pituitary-thyroid
- JAK
- Janus kinase
- KKS
- kallikrein-kinin system
- NPY
- neuropeptide Y
- p
- phospho
- PTX
- pertussis toxin
- PVN
- paraventricular nucleus
- RTK
- receptor tyrosine kinase
- SRF/TCF
- serum response factor/ternary complex factor
- STAT-3
- signal transducer and activator of transcription-3
- SOCS-3
- suppressor of cytokine signaling-3
- TRH
- thyroliberin-releasing hormone.
References
- 1. Schindler C, Fu XY, Improta T, Aebersold R, Darnell JE Jr. Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon α. Proc Natl Acad Sci USA. 1992;89:7836–7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fu XY, Schindler C, Improta T, Aebersold R, Darnell JE Jr. The proteins of ISGF-3, the interferon α-induced transcriptional activator, define a gene family involved in signal transduction. Proc Natl Acad Sci USA. 1992;89:7840–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frias MA, Montessuit C. JAKSTAT signaling and myocardial glucose metabolism. JakStat. 2013;2:e26458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li J. JAKSTAT and bone metabolism. JakStat. 2013;2:e23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wunderlich CM, Hovelmeyer N, Wunderlich FT. Mechanisms of chronic JAKSTAT3-SOCS3 signaling in obesity. JakStat. 2013;2:e23878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ladyman SR, Grattan DR. JAK-STAT and feeding. JakStat. 2013;2:e23675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cui Y, Huang L, Elefteriou F, et al. Essential role of STAT3 in body weight and glucose homeostasis. Mol Cell Biol. 2004;24:258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao Q, Wolfgang MJ, Neschen S, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci USA. 2004;101:4661–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gong L, Yao F, Hockman K, et al. Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology. 2008;149:3346–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park OK, Schaefer LK, Wang W, Schaefer TS. Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J Biol Chem. 2000;275:32244–32249. [DOI] [PubMed] [Google Scholar]
- 11. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. [DOI] [PubMed] [Google Scholar]
- 12. Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. [DOI] [PubMed] [Google Scholar]
- 13. McCowen KC, Chow JC, Smith RJ. Leptin signaling in the hypothalamus of normal rats in vivo. Endocrinology. 1998;139:4442–4447. [DOI] [PubMed] [Google Scholar]
- 14. Frontini A, Bertolotti P, Tonello C, et al. Leptin-dependent STAT3 phosphorylation in postnatal mouse hypothalamus. Brain Res. 2008;1215:105–115. [DOI] [PubMed] [Google Scholar]
- 15. Guo F, Bakal K, Minokoshi Y, Hollenberg AN. Leptin signaling targets the thyrotropin-releasing hormone gene promoter in vivo. Endocrinology. 2004;145:2221–2227. [DOI] [PubMed] [Google Scholar]
- 16. Ellacott KL, Cone RD. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Progr Horm Res. 2004;59:395–408. [DOI] [PubMed] [Google Scholar]
- 17. Rahmouni K, Haynes WG. Leptin signaling pathways in the central nervous system: interactions between neuropeptide Y and melanocortins. Bioessays. 2001;23:1095–1099. [DOI] [PubMed] [Google Scholar]
- 18. Hommel JD, Trinko R, Sears RM, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. [DOI] [PubMed] [Google Scholar]
- 19. Håkansson ML, Meister B. Transcription factor STAT3 in leptin target neurons of the rat hypothalamus. Neuroendocrinology. 1998;68:420–427. [DOI] [PubMed] [Google Scholar]
- 20. Håkansson-Ovesjo ML, Collin M, Meister B. Down-regulated STAT3 messenger ribonucleic acid and STAT3 protein in the hypothalamic arcuate nucleus of the obese leptin-deficient (ob/ob) mouse. Endocrinology. 2000;141:3946–3955. [DOI] [PubMed] [Google Scholar]
- 21. Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. [DOI] [PubMed] [Google Scholar]
- 22. Wen Z, Darnell JE Jr. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian ZJ, An W. ERK1/2 contributes negative regulation to STAT3 activity in HSS-transfected HepG2 cells. Cell Res. 2004;14:141–147. [DOI] [PubMed] [Google Scholar]
- 25. Waitkus MS, Chandrasekharan UM, Willard B, Haque SJ, DiCorleto PE. STAT3-mediated coincidence detection regulates noncanonical immediate early gene induction. J Biol Chem. 2013;288:11988–12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA. 1998;95:11107–11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem. 1999;274:31055–31061. [DOI] [PubMed] [Google Scholar]
- 28. Jain N, Zhang T, Fong SL, Lim CP, Cao X. Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene. 1998;17:3157–3167. [DOI] [PubMed] [Google Scholar]
- 29. Murase S, McKay RD. Neuronal activity-dependent STAT3 localization to nucleus is dependent on Tyr-705 and Ser-727 phosphorylation in rat hippocampal neurons. Eur J Neurosci. 2014;39:557–565. [DOI] [PubMed] [Google Scholar]
- 30. Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. [DOI] [PubMed] [Google Scholar]
- 31. Wen Z, Zhong Z, Darnell JE Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. [DOI] [PubMed] [Google Scholar]
- 32. Plaza-Menacho I, van der Sluis T, Hollema H, et al. Ras/ERK1/2-mediated STAT3 Ser727 phosphorylation by familial medullary thyroid carcinoma-associated RET mutants induces full activation of STAT3 and is required for c-fos promoter activation, cell mitogenicity, and transformation. J Biol Chem. 2007;282:6415–6424. [DOI] [PubMed] [Google Scholar]
- 33. Lee HK, Jung J, Lee SH, Seo SY, Suh DJ, Park HT. Extracellular signal-regulated kinase activation is required for serine 727 phosphorylation of STAT3 in Schwann cells in vitro and in vivo. Korean J Physiol Pharmacol. 2009;13:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, Darnell JE Jr. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol. 2004;24:407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miyakoshi M, Yamamoto M, Tanaka H, Ogawa K. Serine 727 phosphorylation of STAT3: an early change in mouse hepatocarcinogenesis induced by neonatal treatment with diethylnitrosamine. Mol Carcinog. 2014;53:67–76. [DOI] [PubMed] [Google Scholar]
- 36. Wiejak J, Dunlop J, Gao S, Borland G, Yarwood SJ. Extracellular signal-regulated kinase mitogen-activated protein kinase-dependent SOCS-3 gene induction requires c-Jun, signal transducer and activator of transcription 3, and specificity protein 3 transcription factors. Mol Pharmacol. 2012;81:657–668. [DOI] [PubMed] [Google Scholar]
- 37. Damm E, Buech TR, Gudermann T, Breit A. Melanocortin-induced PKA activation inhibits AMPK activity via ERK-1/2 and LKB-1 in hypothalamic GT1–7 cells. Mol Endocrinol. 2012;26:643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dalvi PS, Belsham DD. Glucagon-like peptide-2 directly regulates hypothalamic neurons expressing neuropeptides linked to appetite control in vivo and in vitro. Endocrinology. 2012;153:2385–2397. [DOI] [PubMed] [Google Scholar]
- 39. Dalvi PS, Erbiceanu FD, Irwin DM, Belsham DD. Direct regulation of the proglucagon gene by insulin, leptin, and cAMP in embryonic versus adult hypothalamic neurons. Mol Endocrinol. 2012;26:1339–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dhillon SS, Belsham DD. Estrogen inhibits NPY secretion through membrane-associated estrogen receptor (ER)-α in clonal, immortalized hypothalamic neurons. Int J Obes. 2011;35:198–207. [DOI] [PubMed] [Google Scholar]
- 41. Dhillon SS, Gingerich S, Virtanen C, Belsham DD. Gene array analysis of embryonic- versus adult-derived hypothalamic NPY-expressing cell lines. Mol Cell Endocrinol. 2012;358:116–126. [DOI] [PubMed] [Google Scholar]
- 42. Dhillon SS, McFadden SA, Chalmers JA, Centeno ML, Kim GL, Belsham DD. Cellular leptin resistance impairs the leptin-mediated suppression of neuropeptide Y secretion in hypothalamic neurons. Endocrinology. 2011;152:4138–4147. [DOI] [PubMed] [Google Scholar]
- 43. Belsham DD, Fick LJ, Dalvi PS, et al. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. 2009;23:4256–4265. [DOI] [PubMed] [Google Scholar]
- 44. Harris M, Aschkenasi C, Elias CF, et al. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest. 2001;107:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cyr NE, Toorie AM, Steger JS, et al. Mechanisms by which the orexigen NPY regulates anorexigenic α-MSH and TRH. Am J Physiol Endocrinol Metab. 2013;304:E640–E650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nillni EA. Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin-releasing hormone and proopiomelanocortin. Endocrinology. 2007;148:4191–4200. [DOI] [PubMed] [Google Scholar]
- 47. Sarkar S, Légrádi G, Lechan RM. Intracerebroventricular administration of α-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res. 2002;945:50–59. [DOI] [PubMed] [Google Scholar]
- 48. Guissouma H, Ghorbel MT, Seugnet I, Ouatas T, Demeneix BA. Physiological regulation of hypothalamic TRH transcription in vivo is T3 receptor isoform specific. FASEB J. 1998;12:1755–1764. [DOI] [PubMed] [Google Scholar]
- 49. Guissouma H, Ghaddab-Zroud R, Seugnet I, Decherf S, Demeneix B, Clerget-Froidevaux MS. TRα2 exerts dominant negative effects on hypothalamic Trh transcription in vivo. PLoS One. 2014;9:e95064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Plata-Salamán CR. Food intake suppression by growth factors and platelet peptides by direct action in the central nervous system. Neurosci Lett. 1988;94:161–166. [DOI] [PubMed] [Google Scholar]
- 51. Tsuda K, Tsuda S, Nishio I, Masuyama Y, Goldstein M. Synergistic effects of Bay K 8644 and bradykinin on norepinephrine release in the hypothalamus of spontaneously hypertensive rats. Clin Exp Pharmacol Physiol Suppl. 1995;22:S54–S57. [DOI] [PubMed] [Google Scholar]
- 52. Corrêa FM, Innis RB, Uhl GR, Snyder SH. Bradykinin-like immunoreactive neuronal systems localized histochemically in rat brain. Proc Natl Acad Sci USA. 1979;76:1489–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen EY, Emerich DF, Bartus RT, Kordower JH. B2 bradykinin receptor immunoreactivity in rat brain. J Comp Neurol. 2000;427:1–18. [PubMed] [Google Scholar]
- 54. El-Dahr SS, Dipp S, Baricos WH. Bradykinin stimulates the ERK→Elk-1→Fos/AP-1 pathway in mesangial cells. Am J Physiol. 1998;275:F343–F352. [DOI] [PubMed] [Google Scholar]
- 55. Hayashi R, Yamashita N, Matsui S, et al. Bradykinin stimulates IL-6 and IL-8 production by human lung fibroblasts through ERK- and p38 MAPK-dependent mechanisms. Eur Respir J. 2000;16:452–458. [DOI] [PubMed] [Google Scholar]
- 56. Srivastava S, Sharma K, Kumar N, Roy P. Bradykinin regulates osteoblast differentiation by Akt/ERK/NFκB signaling axis. J Cell Physiol. 2014;229:2088–2105 [DOI] [PubMed] [Google Scholar]
- 57. Feng PH, Hsiung TC, Kuo HP, Huang CD. Cross-talk between bradykinin and epidermal growth factor in regulating IL-6 production in human airway smooth muscle cells. Chang Gung Med J. 2010;33:92–99. [PubMed] [Google Scholar]
- 58. Myers MG., Jr Leptin receptor signaling and the regulation of mammalian physiology. Recent Progr Horm Res. 2004;59:287–304. [DOI] [PubMed] [Google Scholar]
- 59. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. [DOI] [PubMed] [Google Scholar]
- 60. Huang C, Ma WY, Dawson MI, Rincon M, Flavell RA, Dong Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc Natl Acad Sci USA. 1997;94:5826–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pang M, Ma L, Gong R, et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010;78:257–268. [DOI] [PubMed] [Google Scholar]
- 62. Boelen A, Wiersinga WM, Fliers E. Fasting-induced changes in the hypothalamus-pituitary-thyroid axis. Thyroid. 2008;18:123–129. [DOI] [PubMed] [Google Scholar]
- 63. Rosenblum CI, Tota M, Cully D, et al. Functional STAT 1 and 3 signaling by the leptin receptor (OB-R); reduced expression of the rat fatty leptin receptor in transfected cells. Endocrinology. 1996;137:5178–5181. [DOI] [PubMed] [Google Scholar]
- 64. Segerson TP, Kauer J, Wolfe HC, et al. Thyroid hormone regulates TRH biosynthesis in the paraventricular nucleus of the rat hypothalamus. Science. 1987;238:78–80. [DOI] [PubMed] [Google Scholar]
- 65. Terstegen L, Gatsios P, Bode JG, Schaper F, Heinrich PC, Graeve L. The inhibition of interleukin-6-dependent STAT activation by mitogen-activated protein kinases depends on tyrosine 759 in the cytoplasmic tail of glycoprotein 130. J Biol Chem. 2000;275:18810–18817. [DOI] [PubMed] [Google Scholar]
- 66. Lim CP, Cao X. Regulation of Stat3 activation by MEK kinase 1. J Biol Chem. 2001;276:21004–21011. [DOI] [PubMed] [Google Scholar]
- 67. Sakaguchi M, Oka M, Iwasaki T, Fukami Y, Nishigori C. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J Invest Dermatol. 2012;132:1877–1885. [DOI] [PubMed] [Google Scholar]
- 68. Ceresa BP, Horvath CM, Pessin JE. Signal transducer and activator of transcription-3 serine phosphorylation by insulin is mediated by a Ras/Raf/MEK-dependent pathway. Endocrinology. 1997;138:4131–4137. [DOI] [PubMed] [Google Scholar]
- 69. Ren Y, Satoh T, Yamada M, et al. Stimulation of the preprothyrotropin-releasing hormone gene by epidermal growth factor. Endocrinology. 1998;139:195–203. [DOI] [PubMed] [Google Scholar]
- 70. Smyth PP. The thyroid, iodine and breast cancer. Breast Cancer Res. 2003;5:235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ditsch N, Liebhardt S, Von Koch F, et al. Thyroid function in breast cancer patients. Anticancer Res. 2010;30:1713–1717. [PubMed] [Google Scholar]
- 72. Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89:381–410. [DOI] [PubMed] [Google Scholar]
- 73. Kim BK, Lee JW, Park PJ, et al. The multiplex bead array approach to identifying serum biomarkers associated with breast cancer. Breast Cancer Res. 2009;11:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marinho LA, Rettori O, Vieira-Matos AN. Body weight loss as an indicator of breast cancer recurrence. Acta Oncol. 2001;40:832–837. [DOI] [PubMed] [Google Scholar]
- 75. Yotsumoto F, Yagi H, Suzuki SO, et al. Validation of HB-EGF and amphiregulin as targets for human cancer therapy. Biochem Biophys Res Commun. 2008;365:555–561. [DOI] [PubMed] [Google Scholar]
- 76. Chupreta S, Du M, Todisco A, Merchant JL. EGF stimulates gastrin promoter through activation of Sp1 kinase activity. Am J Physiol Cell Physiol. 2000;278:C697–C708. [DOI] [PubMed] [Google Scholar]
- 77. Rahmouni K, Sigmund CD, Haynes WG, Mark AL. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes. 2009;58:536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Toda C, Shiuchi T, Kageyama H, et al. Extracellular signal-regulated kinase in the ventromedial hypothalamus mediates leptin-induced glucose uptake in red-type skeletal muscle. Diabetes. 2013;62:2295–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Abe KC, Mori MA, Pesquero JB. Leptin deficiency leads to the regulation of kinin receptors expression in mice. Regul Pept. 2007;138:56–58. [DOI] [PubMed] [Google Scholar]
- 80. Qadri F, Stark E, Häuser W, Jöhren O, Dendorfer A, Dominiak P. Expression of kinin receptor mRNA in the HPA axis of type 1 and type 2 diabetic rats. Int Immunopharmacol. 2004;4:571–576. [DOI] [PubMed] [Google Scholar]
- 81. Qadri F, Schwartz EC, Häuser W, Jöhren O, Müller-Esterl W, Dominiak P. Kinin B2 receptor localization and expression in the hypothalamo-pituitary-adrenal axis of spontaneously hypertensive rats. Int Immunopharmacol. 2003;3:285–292. [DOI] [PubMed] [Google Scholar]
- 82. Raidoo DM, Bhoola KD. Kinin receptors on human neurones. J Neuroimmunol. 1997;77:39–44. [DOI] [PubMed] [Google Scholar]
- 83. Raidoo DM, Ramsaroop R, Naidoo S, Bhoola KD. Regional distribution of tissue kallikrein in the human brain. Immunopharmacology. 1996;32:39–47. [DOI] [PubMed] [Google Scholar]
- 84. Lewis RE, Childers SR, Phillips MI. [125I]Tyr-bradykinin binding in primary rat brain cultures. Brain Res. 1985;346:263–272. [DOI] [PubMed] [Google Scholar]
- 85. Snider RM, Richelson E. Bradykinin receptor-mediated cyclic GMP formation in a nerve cell population (murine neuroblastoma clone N1E-115). J Neurochem. 1984;43:1749–1754. [DOI] [PubMed] [Google Scholar]