

Abstract
The clinical impact of the fibrate and thiazolidinedione drugs on dyslipidemia and diabetes is driven mainly through activation of two transcription factors, peroxisome proliferator-activated receptors (PPAR)-α and PPAR-γ. However, substantial differences exist in the therapeutic and side-effect profiles of specific drugs. This has been attributed primarily to the complexity of drug-target complexes that involve many coregulatory proteins in the context of specific target gene promoters. Recent data have revealed that some PPAR ligands interact with other non-PPAR targets. Here we review concepts used to develop new agents that preferentially modulate transcriptional complex assembly, target more than one PPAR receptor simultaneously, or act as partial agonists. We highlight newly described on-target mechanisms of PPAR regulation including phosphorylation and nongenomic regulation. We briefly describe the recently discovered non-PPAR protein targets of thiazolidinediones, mitoNEET, and mTOT. Finally, we summarize the contributions of on- and off-target actions to select therapeutic and side effects of PPAR ligands including insulin sensitivity, cardiovascular actions, inflammation, and carcinogenicity.
Type 2 diabetes mellitus (T2DM) is a complex disease characterized by insulin resistance, pancreatic islet/β-cell dysfunction, hyperglycemia, dyslipidemia, and inflammation. Patients with T2DM are at high risk of cardiovascular (CV) events and microvascular complications (1, 2). The presence of multiple metabolic abnormalities underscores the need for comprehensive disease management strategies (3–5), including novel therapies.
The fibrate and thiazolidinedione (TZD) classes of drugs bind and activate two cellular transcription factors, the peroxisome proliferator-activated receptors (PPAR)-α and -γ. The impact of fibrates and TZDs on dyslipidemia and diabetes is linked primarily to their activation of these receptors (6, 7). However, substantial clinical and preclinical experience has shown that individual drugs differ from one another in therapeutic and side effect profiles (8, 9). Furthermore, the expression of PPARs in multiple tissues raises the possible value of PPAR agents in therapeutic indications as diverse as cancers (10), inflammatory-related diseases (11–13), and Alzheimer's disease (14).
The clinical use of the PPAR-γ agents pioglitazone and rosiglitazone revealed a number of common adverse effects, including weight gain, fluid retention, congestive heart failure, and bone fractures (15, 16), suggesting that these effects are likely PPAR-γ dependent (17). In contrast to these apparent class effects, substantial preclinical and clinical evidence suggests significant differences between these agents. Notably, pioglitazone and rosiglitazone have markedly different effects on the plasma lipoprotein profile in diabetic patients (8). A large meta-analysis of clinical studies suggested that pioglitazone reduced the risk of myocardial infarction (MI) and stroke (18), whereas several reports suggested rosiglitazone has no effect on CV events (19) or may increase the risk of CV events (20). A small but significantly increased incidence of bladder cancer has been observed with pioglitazone but not rosiglitazone (21). Troglitazone, another PPAR-γ agent, was withdrawn in 2000 because of hepatotoxicity, an effect that has not been a concern with either rosiglitazone or pioglitazone (22). Moreover, during development of several dual-PPAR-α/γ agonists, compound-specific safety concerns emerged, exemplified by tesaglitazar (nonreversible increases in serum creatinine), muraglitazar (increase in CV events), and ragaglitazar (bladder tumors in rodents) (23).
These data highlight an empirical view in pharmaceutical drug discovery that most drug effects are consequences of mechanisms regulated by the primary drug target, but others may result from off-target activity. In practice, a priori screening of off-target effects is generally limited to panels of known receptors/channels with arbitrary cutoff values for binding affinity. Thus, attributing a biological effect to a specific off-target protein is often challenging, especially in the in vivo setting because off-target actions are superimposed on target-mediated effects and influenced by potency, distribution, and metabolism, which are often specific to the compound and sometimes the animal model. For this reason, in vivo observations must be combined with findings from cellular models and cell-free systems. Such studies consistently suggest that some therapeutic effects or side effects of TZDs may be linked to interactions with other targets. In support of this notion, the TZD PPAR-γ agonists have been reported to interact with targets as diverse as other nuclear receptors [eg, glucocorticoid receptors, (24)], G protein-coupled receptors [eg, G protein-coupled receptor 40) (25)], protein kinases [eg, AKT and c-Jun N-terminal kinase (JNK) (26)], ion channels [eg, Ca2+ (14), K+ channels (27), and mitochondrial targets (28–31)].
Here we review the on- and off-target effects of existing PPAR agonists and where possible contextualize the contribution of these mechanisms to the overall effects of individual agents. With few data available regarding agonists of PPAR-α, our knowledge is largely limited to PPAR-γ agonists and in particular the TZDs. Hence, the major focus of this review will be on these compounds, although, where relevant, dual activators of PPAR-α/γ will also be mentioned.
Regulation of gene expression by PPAR agonists
The PPAR subfamily of nuclear hormone receptors consists of three highly homologous members [PPAR-α, PPAR-δ (also known as PPAR-β), and PPAR-γ] encoded by separate genes. The primary effect of PPAR-α activation is to improve plasma lipids [reducing levels of very low density lipoprotein triglycerides, increasing levels of high-density lipoprotein cholesterol and apolipoprotein A1, and increasing fatty acid oxidation (6, 32)]. There are two major isoforms of PPAR-γ. PPAR-γ1 is expressed in a broad range of tissues, whereas PPAR-γ2 is restricted to adipose tissue (33). PPAR-γ activation improves insulin sensitivity and glucose control and lowers the circulating levels of fatty acids and the proinflammatory markers of CV disease and atherosclerosis (Figure 1) (6, 9). Labeling studies suggest that the PPARs are primarily localized in the nucleus of cells, although nongenomic actions associated with the cytoplasmic PPARs have also been described (34).
Figure 1. Tissue-specific actions of PPARs.

Apo, apolipoprotein; HDL, high-density lipoprotein; VLDL, very low-density lipoprotein.
The mechanism of PPAR activation has been most intensively studied for PPAR-γ. PPAR activation involves the binding of ligand to a hydrophobic pocket, which causes conformational changes in the PPAR structure. This results in dissociation of cofactor proteins such as nuclear receptor corepressor (NCoR) or steroid receptor coactivator-1 (SRC-1) that inhibit or enhance gene transcription, respectively (Figure 2A) (35). The release of corepressor proteins coincides with heterodimerization of the PPAR with the retinoid X receptor (RXR). The heterodimer of PPAR-RXR then acts as a scaffold for the recruitment of coactivator proteins [eg, PPAR-γ coactivator-1 (PGC-1), p160/SRC-1, and vitamin D3 receptor interacting protein (DRIP) or thyroid hormone receptor associated protein (TRAP)-220 complexes]. This scaffold in turn recruits histone acetyl transferases and the gene transcription machinery (RNA polymerase complex), which together initiate chromatin relaxation to permit transcription of target genes (Figure 2B) (35).
Figure 2. A, Role of cofactors in PPAR function. In the unliganded state, corepressor proteins such as NCoR/silencing mediator of retinoid and thyroid receptors (SMRT) inhibit transcription by interacting with histone deacetylases. B, Upon binding of a ligand to the PPAR, the receptor undergoes a conformational change, NCoR/SMRT and histone deacetylases (HDACs) dissociate, and transcriptional coactivators such as PGC1-a or SRC-1 are recruited along with histone acetyltransferases (HAT), which open chromatin to allow interaction with RNA polymerase II. C, Each ligand may induce slightly different conformations of the receptor that influence the recruitment of coactivators and may lead to overlapping but unique transcriptional profiles in different cells and tissues. ♣, Unique genes regulated by each ligand; ♠, common target genes.

Role of differential cofactor recruitment
Ligand-induced coactivator preferences and their contribution to transcription potency and efficacy have been reported for both PPAR-α (36) and PPAR-γ (36, 37). The specific pattern of cofactors recruited to the PPAR-RXR complexes in response to different ligands leads to major differences in the transactivation of target genes and thus the biological profile of ligands (Figure 2C) (38–40). Key evidence for target gene selectivity of cofactors is exemplified by the ability of PGC-1 to potently activate the uncoupling protein (UCP)-1 gene but not the aP2 gene (41).
In addition to differential ligand-dependent cofactor engagement, cell-based studies have shown that PPAR-γ activators can act as partial agonists in some cell types and full agonists in others, possibly due to variations in cofactor profile/amount (42). Even in the same cell type, ligands may act as a full agonist in engaging/disengaging of one cofactor (NCoR) while acting as a partial effector on others (PGC1-α and DRIP/TRAP220) (43). Several PPAR coactivators (eg, DRIP205/TRAP220 and NCoR) have been associated with adverse effects such as adipogenesis/insulin resistance, whereas others (eg, SRC-1 and PGC1-α) drive insulin-sensitizing effects (44). These findings may explain how PPAR agonist-associated side effects can relate to on-target actions of ligands but also raise the possibility that subtle differences between ligands, which impact the recruitment of specific cofactors, may result in better separation of efficacy from side effects. A structure-activity relationship of rosiglitazone and related compounds suggested the possibility to retain glycemic efficacy while reducing hemodilution (45) and was a selection criterion to progress rosiglitazone into clinical development, although the interplay of cofactors mediating this separation of effects has not been carefully evaluated.
Partial agonists and selective modulators
Further evidence for the possibility to separate efficacy from side effects comes from studies of partial PPAR-γ agonists. The intensive search for safer PPAR agonists led to the development of selective partial PPAR modulators, such as the selective partial PPAR-γ agonist INT131. INT131 recruits DRIP205 and promotes its binding to a level of approximately 30% of that conferred by the full PPAR-γ agonist rosiglitazone (46). In animal models of diabetes, INT131 caused less weight gain compared with pioglitazone or rosiglitazone while retaining efficacy to reduce plasma glucose (46, 47). Importantly, toxicology studies of INT131 (at ≥ 70 times higher than the expected human exposure at the expected therapeutic dose of 3 mg) in cynomolgus monkeys and rats was not associated with fluid retention, changes in hematocrit, or weight gain over 6 months (45, 47). However, in a phase II study, INT131 was associated with an increase in the incidence of edema, gain in body weight, and decreased hematocrit at the 10-mg dose vs placebo, highlighting the difficulty in translating promising differentiated preclinical profiles into patients (48).
Collectively these studies support the hypothesis that each ligand-PPAR receptor complex adopts a different three-dimensional structure that drives a distinct cofactor recruitment pattern and gene signature profile (49, 50). The data also rationalize many studies documenting differential but overlapping patterns of gene expression in various tissues for different PPAR-γ ligands (51–53) or dual-PPAR-α/γ agonists (53), even for highly similar compounds (51–53).
However, despite significant progress in our knowledge of the role of individual cofactors in regulating distinct pathways (35, 54), early expectations that specific cofactors may be clear drivers of certain side effects appears an oversimplification, and little progress has been made to date on translating such approaches into clinically safer molecules.
Role of balanced PPAR activation
The significant structural similarity of PPAR-α, -δ and -γ, particularly in their ligand-binding domains, has allowed the identification of several synthetic dual- or pan-PPAR agonists (Table 1). Compounds with a balanced activation of PPAR-α and -γ, such as the dual-PPAR-α/γ agonist aleglitazar, have been hypothesized to provide a better balance between efficacy and side effects vs single agonists or dual agonists with imbalanced potency (eg, tesaglitazar and muraglitazar). The 16-week phase II A Study of Aleglitazar in Patients with Type 2 Diabetes (SYNCHRONY) study in patients with T2DM showed that a 150-μg daily dose of aleglitazar significantly improved the glycemic and lipid control and was associated with edema rates similar to those of placebo (55). The 52-week phase IIB Study of Alegitazar in Type 2 Diabetes Patients with Moderate Renal Impairment (AleNephro) study confirmed that the 150-μg daily dose of aleglitazar significantly improved the glycemic and lipid profile (similar to pioglitazone 45 mg), but it exhibited a lower rate of edema (12.1% vs 19.7%) (56).
Table 1.
Transcriptional Activation Potency of Different PPAR Ligands and Their Development Status
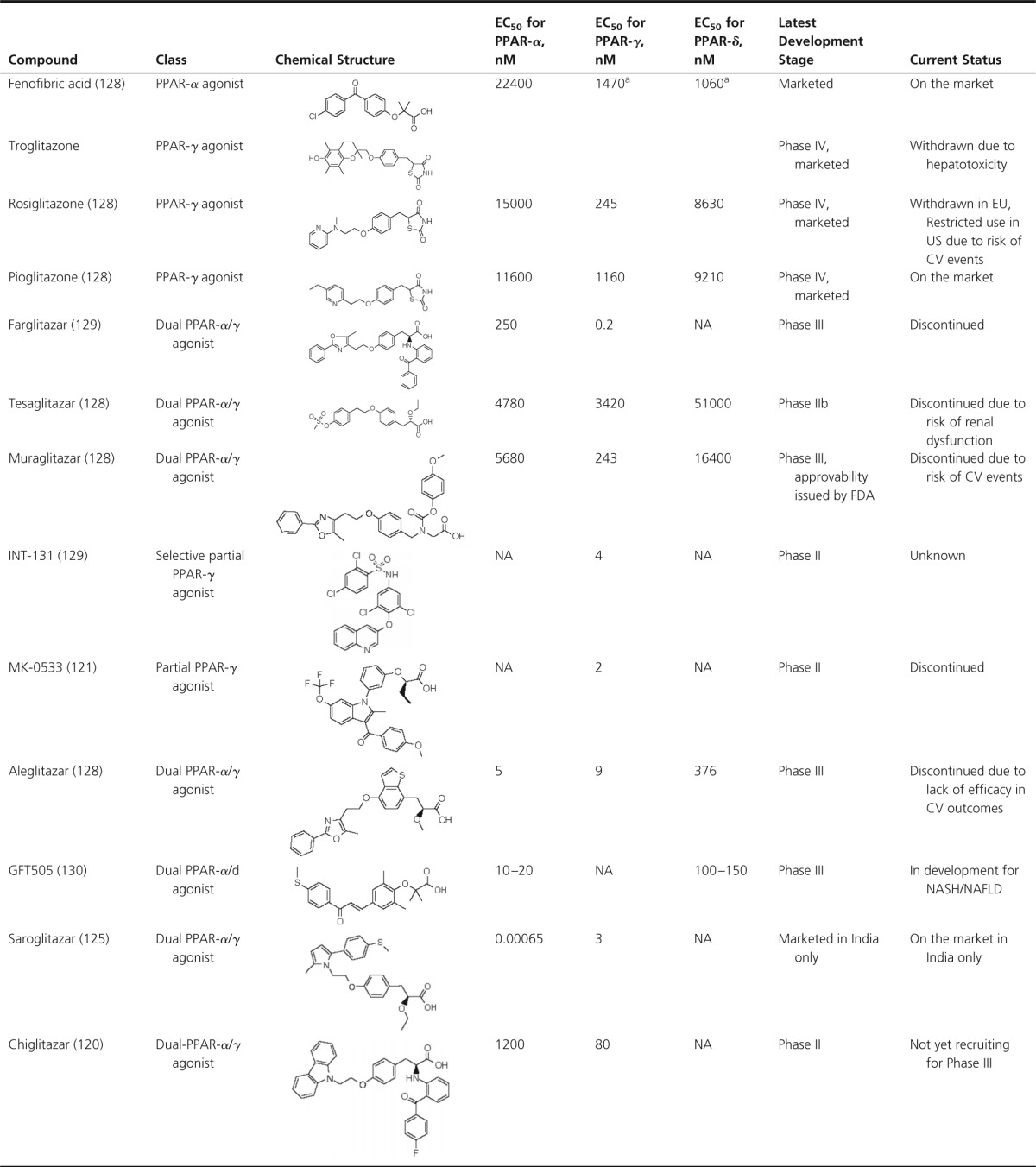
Abbreviations: EU, European Union; NA, not available; NAFLD, nonalcoholic fatty liver disease.
Value is inaccurate due to low maximal activation.
Based on the early promise of the SYNCHRONY trial, Roche Pharmaceuticals initiated a phase III trial [A Study With Alegitazar in Patients With a Recent Acute Coronary Syndrome and Type 2 Diabetes Mellitus (AleCardio); clinical trials registration NCT01042769] to determine the impact of aleglitazar on CV events in patients with T2DM and a recent acute coronary syndrome event. The Safety and Efficacy Study to Evaluate the Potential of Aleglitazar to Reduce Cardiovascular Risk in Coronary Heart Disease (CHD) Patients With a Recent Acute Coronary Syndrome (ACS) Event and Type 2 Diabetes Mellitus (T2D) trial was recently terminated due to the lack of efficacy in CV outcomes and the presence of PPAR-related class side effects (57). Moreover, an increase in gastrointestinal bleeding was reported in patients randomized to aleglitazar. This was an unexpected finding not observed in prior studies of aleglitazar, although patients with a recent acute coronary syndrome heavily treated with multiple antiplatelet and anticoagulant therapies had not been enrolled in these studies. The mechanism of this adverse event is unknown because previous safety and clinical studies showed no gastrointestinal toxicity or hemostatic abnormalities with aleglitazar (57). The termination of this program further underscores the challenges in developing dual-PPAR therapies for cardiometabolic diseases.
Emerging mechanisms of PPAR regulation
Phosphorylation and posttranslational regulation
Several compelling new mechanisms of posttranslational control of PPAR action have recently been described, including phosphorylation, sumoylation (involving the covalent attachment of small ubiquitin-like modifier peptides to lysine residues), ubiquitination (leading to increased degradation of PPAR-γ) and nitration (eg, of protein tyrosine residues in response to increased concentrations of nitric oxide radicals) (58).
The phosphorylation of PPAR-γ occurs at several sites via different kinases including MAPKs. An N-terminal serine phosphorylation (Ser-82 in PPAR-γ1 and Ser-112 in PPAR-γ2), mediated by MAPKs, reduces the transcriptional activity of PPAR-γ1 [in cells activated by epidermal growth factor or platelet derived growth factor (59)] and PPAR-γ2 (in cells activated by serum) (60).
Recently a new role for phosphorylation as a mechanism of action for PPAR-γ agonists, along with an intriguing new molecular link between obesity, adipose inflammation, and insulin resistance, was described (61). In addition to enhancing the transcriptional activity of PPAR-γ, rosiglitazone, was found to inhibit the phosphorylation of PPAR-γ at Ser273 by cyclin-dependent kinase 5 (CDK5) in adipose tissue, preserving the transcription of insulin-response genes (such as adiponectin and adipsin) and correlating with antidiabetic activity. A second PPAR-γ agent, MRL24, was as effective as rosiglitazone at blocking phosphorylation and improving diabetes in animal models, despite being only a partial PPAR-γ agonist. Taken together, these results suggest that the insulin-sensitizing benefits of PPAR-γ agonists are due in part to their ability to block phosphorylation and not solely to their agonist activity. Because CDK5 was activated by inflammatory mediators, the inflammation/insulin resistance component commonly found in obesity may be explained partly by CDK5/PPAR-γ regulation (61). Ember Therapeutics aims to exploit this novel finding to develop inhibitors of CDK5 phosphorylation of PPAR-γ devoid of classical PPAR-γ agonism, with the aim to achieve antidiabetic effects with reduced side effects compared with TZDs (62).
Nongenomic regulation
Recent evidence also suggests the potential role of nongenomic regulation of PPAR-γ and PPAR-α (58), mediated by interaction with cytosolic second messengers, including kinases and phosphatases (58). The MAP/ERK kinase, MAPK kinase (MEK)-1, was reported to bind directly to the AF2 domain of PPAR-γ in response to mitogenic stimulation, leading to the sequestration of PPAR-γ in the cytoplasm (63). In gastric cancer cells, PPAR-γ nuclear-cytoplasmic shuttling has been proposed to serve as a mechanism for spatial regulation of PPAR-γ signaling. In this model, MEK-1 or cavelin-1 binds to surfaces on the PPAR-γ ligand-binding domain, altering its subcellular localization and activity (64). Selective inhibition of MEK-1/PPAR-γ interactions has recently been proposed as a concept for treatment of cancer, inflammation, and metabolic disorders but has yet to gain significant acceptance (63).
Understanding on-target vs off-target actions of PPAR ligands
Insulin-sensitizing and metabolic effects: identification of non-PPAR-γ targets of TZDs
The divergent rank order potency for PPAR-γ activity of TZDs vs their antiinflammatory and antiproliferative effects has long suggested additional pharmacological activity that may contribute to their activity (65, 66). Several non-PPAR-γ proteins have been identified as high-affinity binders of TZDs. Colca et al (29) used tritiated [3H]pioglitazone and a structurally related iodinated photoaffinity probe to identify binding sites in mitochondria. A saturable and specific binding site for [3H]pioglitazone to a 17-kDa protein was detected. The protein was isolated and identified by mass spectrometry analysis and amino-terminal sequencing as the mitochondrial protein mitoNEET (29).
mitoNEET is an iron sulfur-cluster transfer protein that has been shown to inhibit mitochondrial iron transport, which may in turn result in decreased respiratory activity and enhanced fatty acid uptake (Figure 3) (67, 68). mitoNEET is broadly expressed in insulin-sensitive tissues including liver, muscle, adipose, and heart (69) and appears to be involved in regulating mitochondrial oxidative capacity (69) and redox-sensitive signaling (28). mitoNEET protein exists in low levels in preadipocytes, and its expression increases substantially upon differentiation to mature adipocytes. Transgenic mice overexpressing mitoNEET in adipocytes become extremely obese when challenged with a high-fat diet but maintain normal insulin sensitivity, low levels of ectopic fat deposition, and low levels of reactive oxygen species (68). In contrast, mitoNEET knockdown mice, despite gaining less weight on a high-fat diet, have increased reactive oxygen species-induced protein damage and are less glucose tolerant (68). Future studies on mitoNEET should address its role in different cell types and molecular pharmacology as a drug target.
Figure 3. Mitochondrial targets of thiazolidinediones.

A, MitoNeet. MitoNEET is an integral iron-sulfur (FeS)-containing protein in the outer mitochondrial membrane and regulates mitochondrial respiratory rates [adapted from Kusminski et al (68)]. B, MPC/mTOT. After glycolysis in the cytosol, pyruvate diffuses through the outer mitochondrial membrane and is then transported by the mitochondrial pyruvate carrier, MPC/mTOT, across the inner mitochondrial membrane. In the matrix, pyruvate is oxidized to produce ATP, supporting the processes of gluconeogenesis and lipogenesis. Both proteins have been shown to be non-PPAR targets of the thiazolidinedione class of diabetes agents. Acetyl CoA, acetyl–coenzyme A.
A second mitochondrial target of the TZDs (mTOT) has recently been described. The mitochondrial pyruvate transporter was recently reported to be composed of the mitochondrial pyruvate carrier (Mpc-1 and Mpc-2) proteins (70–72). Almost coincidentally, another group identified Mpc-2 (mTOT) to be a second direct mitochondrial target of pioglitazone, by photoaffinity and mass spectrometry-based proteomics approaches (Figure 3) (31). Two mTOT-binding TZDs with little effect on PPAR-γ (MSDC-0160 and MSDC-0602) were shown to enhance brown adipose tissue formation and improve insulin sensitivity in mice, whereas the deletion of the Mpc-2/mTOT gene resulted in a loss of brown adipose tissue formation (31). A phase IIb study in patients with diabetes suggested that MSDC-0160 may have similar glucose-lowering efficacy to pioglitazone, with preliminary hints of fewer side effects (30). MSDC-0160 was associated with a lower level of fluid retention (as evidenced by reduction in hematocrit, red blood cells, and total hemoglobin) compared with pioglitazone (30). These data suggest that specifically targeting Mpc-2/mTOT may have potential as a therapy for diabetes. Although highly novel and interesting, MSDC-0160 is structurally related to pioglitazone, differing only in a carbonyl group. Similar results with non-TZD drugs optimized against the pyruvate transporter directly would add greater confidence in this potential target.
Cardiovascular effects of glitazones: involvement of protein kinase pathways and ion channels
Although PPAR-γ is predominantly expressed in adipose tissue, it is also produced in the myocardium (73) in which evidence suggests that it has metabolic and antiinflammatory effects (74–77). PPAR-γ is also expressed in vascular endothelial cells and is implicated in endothelial progenitor cell function and vascular repair (78, 79). Aortic expression of PPAR-γ is subject to circadian variation and has been proposed to participate in regulation of CV rhythm such as circadian variations in blood pressure and heart rate (80).
Early clinical studies with troglitazone, prior to its market withdrawal due to hepatotoxicity, demonstrated an increased cardiac output in patients with T2DM indicative of a role for PPAR-γ in cardiac function (81). In obese Zucker fatty rats, treatment with rosiglitazone for 14 days normalized glucose uptake and protected the heart from ischemic injury (75). These cardioprotective effects were attributed to PPAR-γ activation because they were observed for a number of different drugs (troglitazone, pioglitazone, and rosiglitazone), and pretreatment protocols were thought to allow sufficient time to lead to gene expression changes after transcriptional activation of PPAR-γ. However, in an experimental model of myocardial infarction in male Lewis rats, rosiglitazone improved myocardial function and reduced infarct size acutely after a single iv dose prior to the induction of ischemia. The rapidity of the onset of protection suggested the effects may be independent of PPAR-γ activation (82).
Additional studies support this hypothesis. In isolated hearts (74) or in vivo (26), rosiglitazone suppressed JNK kinase and activated the AMP-activated protein kinase and AKT pathways, effects that could not be fully blocked by a PPAR-γ antagonist (26). Similarly, studies in the pig demonstrated rapid effects of troglitazone in the recovery of left ventricular function after ischemia/reperfusion injury (83, 84). The effect of troglitazone in this model was found to be imparted not by the TZD moiety but also by its tocopherol moiety, which does not activate PPAR-γ (84). Together these data highlight the existence of non-PPAR-γ off-target CV effects of some TZDs.
In addition to cardioprotective effects seen in preclinical studies, the proarrhythmic effects of TZDs have also been reported. Evaluation of direct (acute) cardiac effects of pioglitazone, rosiglitazone, and troglitazone indicated proarrhythmic potential, which may increase the propensity for ventricular fibrillation under ischemic conditions (85). These effects were observed rapidly after TZD administration, suggesting that they may occur independently of PPAR-γ transcriptional activation. Intravenous infusion of troglitazone, rosiglitazone, and pioglitazone in the pig, at clinically relevant doses, led to the attenuation of action potential shortening in response to experimental myocardial ischemia, consistent with inhibition of cardiac sarcolemmal ATP-regulated potassium (KATP) channels (85). Because the opening of KATP channels during ischemia is generally believed to be an adaptive/protective energy-sparing response, the effect of glitazones on KATP channels could theoretically impair contractile recovery and/or result in ventricular fibrillation (85).
Rosiglitazone has been shown to suppress L-type calcium currents and outward potassium currents (86) in patch clamp studies of dispersed ventricular cells from dog hearts. This appears to be due to direct inhibition of Kv4.3 channel activity as demonstrated in a recombinant system (27). Although highly interesting, the contribution of PPAR-γ-independent effects of TZDs on calcium or potassium channel activity to their CV effects remains to be clarified.
Anti-inflammatory effects of PPAR-γ agonists: receptor-dependent and -independent effects
PPAR-γ activators have broad antiinflammatory effects based on consistent reductions in serum markers of inflammation such as C-reactive protein, IL-6, and TNF-α, suggesting a potential impact on atherogenesis (87), renal dysfunction (88), and autoimmune diseases. The mechanisms for the antiinflammatory effects of TZDs have been studied primarily in monocytic and dendritic cells, in which PPAR-γ activation attenuates expression and secretion of proinflammatory cytokines (including IL-1β, IL-6, and TNF-α) associated with M1 macrophages (89) and reduces macrophage activity via the transrepression of nuclear factor-κB (90). PPAR-γ is also expressed in B cells (91), T cells (92), and natural killer cells (93).
The antiinflammatory activity of PPAR-γ activation may reduce atherosclerosis (34) and provide renoprotection (94). PPAR-γ agonists have also shown efficacy in animal models of inflammatory bowel disease (11), autoimmune myocarditis (12), and autoimmune encephalomyelitis (95). In a mouse model of inflammatory bowel disease, troglitazone and rosiglitazone reduced colonic inflammation (11) consistent with data in colonic epithelial cells showing that PPAR-γ ligands attenuate cytokine gene expression through the inhibition of nuclear factor-κB activation.
The PPAR-γ ligands 15-δ-prostaglandin (PG)-J2 and pioglitazone markedly reduced the severity of myocarditis (12), accompanied by a reduced expression of myocardial inflammatory cytokines. In addition, the proliferative response and interferon-γ production of T cell-enriched splenocytes was suppressed in animals treated with PPAR-γ activators (12).
Experimental allergic encephalomyelitis (EAE) is a Th1 cell-mediated inflammatory demyelinating autoimmune disease model of multiple sclerosis. Supporting a critical physiological role for PPAR-γ as a regulator of neuronal inflammation and demyelination, PPAR-γ-deficient heterozygous mice develop exacerbated EAE with exaggerated clinical symptoms, associated with an increased expansion of CD4 and CD8 T cells and expression of CD40 and major histocompatibility complex class II molecules (13). Consistent with the potential of the PPAR-γ agonists in suppressing central nervous system inflammation, 15-δ-PGJ2 and ciglitazone were able to inhibit EAE by blocking IL-12 production and signaling and suppressing neural antigen-induced Th1 differentiation in vivo (95).
Several pharmacological and genetic studies suggest that some of the antiinflammatory effects of TZDs (and 15-δ-PGJ2) may be independent of PPAR-γ. For example, the rank order potency of drugs to activate PPAR-γ is often inconsistent with the rank order of antiinflammatory efficacy, and expression of PPAR-γ does not always correlate with the observed antiinflammatory effects. Most convincingly, some antiinflammatory effects of 15-δ-PGJ2 and TZDs were shown to be unaffected in PPAR-γ knockout models; secretion of both IL-6 and TNF-α is inhibited equally in wild-type and PPAR-γ-deficient macrophages (96). Similarly, interferon-γ induction of inducible nitric oxide synthase and cyclooxygenase-2 is unaffected by PPAR-γ deficiency (96).
In summary, it appears that some antiinflammatory effects of glitazones are PPAR-γ independent, but the mechanisms are poorly understood. Conceptually, these effects may derive from the engagement of cyclooxygenase-2 or JNK/ERK protein kinase pathways.
Tumorigenicity of PPAR agonists: receptor-dependent and -independent effects
The potential of PPAR-γ or dual PPAR-α/γ agonists to induce tumors in rodents has been disclosed in regulatory submissions to the Food and Drug Administration (FDA). Common findings in rodent carcinogenicity studies include hemangioma/hemangiosarcoma, urinary bladder/renal pelvic tumors, lipoma/liposarcoma, skin fibrosarcoma, and, less frequently, mammary adenocarcinoma, gall bladder adenoma, and hepatic tumors (97). Not all molecules exhibit carcinogenic effects, and in many cases effects were observed with significant safety margins vs therapeutic exposure. To date, the only clinical finding of concern is a small increased risk of bladder cancer in patients on long-term therapy with pioglitazone but not rosiglitazone (21). Several dual-PPAR-α/γ agonists, muraglitazar (98), naveglitazar (99), and ragaglitazar (100), but not aleglitazar (101), induce bladder tumor formation in rodents. In 2-year rat and mouse carcinogenicity studies of aleglitazar, no urinary bladder hyperplasia or tumors were reported despite exposure multiples (26-fold in rat and 123-fold in mice) greater than exposure at the 150-μg dose tested in human phase III studies (101).
The mechanism(s) of tumorigenicity have not been fully elucidated for most PPAR-associated tumors, although there is evidence to suggest some tumor types are drug specific. In a comprehensive review of preclinical and clinical data, Pruimboom-Brees et al (102) recently proposed a hypothetical mechanism that may help to explain the varied properties of different PPAR agonists in inducing sc (lipo)sarcomas. The hypothesis is based on the tumor initiation/promotion concept leading to tumorigenesis: initiation, during which DNA damage occurs, and promotion, involving aberrant cell proliferation. In this model, initiation would occur independently of PPAR-γ and involve drug-specific effects leading to mitochondrial dysfunction in sc stromovascular cells, leading to oxidative stress and oxidative DNA damage (102). The subsequent promotion stage would require PPAR-γ and lead to recruitment, proliferation, and differentiation of stromovascular cells, driving thermogenesis and adipogenesis, and subsequent generation of oxidative free radicals (102). According to this model, and in agreement with available safety data, the rank order of potency of PPAR-γ agonists to increase mitochondrial oxidative stress is as follows: troglitazone (most potent) greater than ciglitazone greater than darglitazone/muraglitazar greater than rosiglitazone/pioglitazone (least potent) (102). Thus, screening to exclude compounds that induce mitochondrial dysfunction may help reduce the potential for tumor-promoting effects of new agents.
Despite these hypotheses, regulatory agencies remain cautious about PPAR agents. The FDA continues to require completion of 2-year preclinical carcinogenicity studies with an acceptable profile prior to the initiation of clinical studies longer than 6 months (97).
Potential antitumor effects
Substantial evidence for antitumor effects of synthetic and natural PPAR-γ agonists has been reported in several cancers in vitro and in vivo (10, 103–106). Early studies indicated PPAR-γ to be up-regulated in several human cancer lines compared with normal lines. Treatment with TZDs in vitro (in particular troglitazone and ciglitazone) led to cell cycle arrest and/or increased apoptosis. In a clinical trial in patients with advanced prostate cancer, troglitazone therapy resulted in the stabilization of prostate-specific antigen levels, which continued to increase in placebo-treated subjects, suggesting the stabilization of disease (107, 108). In vitro studies in primary cultures of human prostate cancer cells also suggested an antiproliferative effect of rosiglitazone, associated with increased expression of transcription repressors as well as morphological changes indicative of terminal differentiation (109). In addition, PPAR-γ agonists inhibit in vitro motility and invasiveness of glioma cells and decrease glioma progression and improve survival in rodent models (110, 111). However, in mouse and human glioma cell lines, inhibition of cell growth by troglitazone was similar to the effects observed with its PPAR-γ inactive derivative Δ2-troglitazone (112). Furthermore, PPAR-γ knockdown or blockade, by small interfering RNA or with the antagonist GW9662, failed to counteract the effect of troglitazone in a variety of cell types (113), suggesting PPAR-γ-independent effects. In contrast, other studies found that PPAR-γ overexpression in a variety of neoplastic cells inhibited cell motility and invasiveness, reinforcing a role for PPAR-γ in cellular differentiation. Similar conclusions were reached using pharmacological activation of PPAR-γ with structurally different classes of compounds. For example, PPAR-γ agonists or overexpression of PPAR-γ in non-small cell lung cancer cells inhibits transformed growth and invasiveness and promotes differentiation to an epithelial phenotype in a variety of lines (114, 115).
Overall, these data suggest that the antitumor effects of TZDs in some cell types require PPAR-γ and are mediated by PPAR-γ-dependent pathways, whereas in other cell types they occur independently. Further systematic investigations in different tumor cell types are required to elucidate the utility of TZDs in different cancers.
Conclusion and future directions
After the discovery of PPAR-α and -γ as targets of fibrate and glitazone classes of antihyperlipidemic and antidiabetic agents (116), the broader role of PPARs in vascular and metabolic health has been heavily investigated. Although PPAR-α agonists (fibrates) are generally well tolerated, PPAR-γ activation is associated with weight gain, fluid retention, and bone fractures (33, 117). Efforts to improve the efficacy and/or safety of first-generation PPAR-γ agents (the TZDs) have led to understanding of the complexity of PPAR regulation, including the importance of coactivator and corepressor proteins and, most recently, phosphorylation and posttranslational mechanisms (35–42, 118, 119). We believe that exploiting cofactor biology to derive better agents has not delivered on its initial promise. Despite the demonstration of differential cofactor recruitment and better preclinical efficacy/safety profiles vs TZDs, new molecules have not convincingly translated these profiles in the clinic (48, 122–123).
The future of PPAR-targeted agents for cardiometabolic therapy remains uncertain, particularly in light of the recent termination of the CV outcome study of the dual PPAR-α/γ agonist aleglitazar. However, several late-stage molecules may hold promise. Saroglitazar, a PPAR agonist with predominant PPAR-α and moderate PPAR-γ activity, was launched exclusively in India for the control of dyslipidemia (124, 125). However, few data are available on its molecular profile, and the treatment duration and low patient number in its phase III program make it impossible to draw conclusions regarding its CV and long-term safety profiles. Genfit is developing a dual PPAR-α/δ agonist (GFT505) for the treatment of nonalcoholic steatohepatitis (NASH), which is associated with insulin resistance and higher risk of CV events. The company recently reported that it has received the FDA's fast-track designation for development in this indication (126). Thus, new agents that see limited use or in diseases, such as NASH, with high unmet need and no approved therapies may emerge.
Future directions in PPAR research are likely to focus on optimizing the PPAR subtype interaction profile, as well as maximizing inhibition of PPAR-γ phosphorylation, and screening against off-target activity. In addition, biochemical studies have identified the mitochondrial targets mitoNEET and mTOT (29, 127) as TZD targets that may have roles in targeting insulin resistance and diabetes in their own right and will clearly be a focus of future investigation.
Acknowledgments
Current affiliation for M.T.: Pharmidex, Stevenage Science BioCatalyst Campus, SG1 2FX.
Funding for the manuscript was provided by F. Hoffman-La Roche (Basel, Switzerland).
Disclosure Summary: M.B.W., M.Bor., and M.Bop. are employees of F. Hoffmann-La Roche (Basel, Switzerland). M.T. was Scientific Advisor at MediTech Media (London, United Kingdom), which provides scientific and editorial support to Hoffmann-La Roche Ltd.
Funding Statement
Funding for the manuscript was provided by F. Hoffman-La Roche (Basel, Switzerland).
Footnotes
- AleCardio
- A Study With Alegitazar in Patients With a Recent Acute Coronary Syndrome and Type 2 Diabetes Mellitus
- AleNephro
- A Study of Alegitazar in Type 2 Diabetes Patients with Moderate Renal Impairment
- CDK5
- cyclin-dependent kinase 5
- CV
- cardiovascular
- DRIP
- vitamin D3 receptor interacting protein
- EAE
- experimental allergic encephalomyelitis
- JNK
- c-Jun N-terminal kinase
- KATP
- ATP-regulated potassium
- MEK
- MAPK kinase
- Mpc
- mitochondrial pyruvate carrier
- NASH
- nonalcoholic steatohepatitis
- NCoR
- nuclear receptor corepressor
- PG
- prostaglandin
- PGC-1
- PPAR-γ coactivator-1
- PPAR
- peroxisome proliferator-activated receptor
- RXR
- retinoid X receptor
- SRC-1
- steroid receptor coactivator-1
- SYNCHRONY
- A Study of Aleglitazar in Patients with Type 2 Diabetes
- T2DM
- type 2 diabetes mellitus
- TRAP
- thyroid hormone receptor associated protein
- TZD
- thiazolidinedione.
References
- 1. Centers for Disease Control and Prevention. National Diabetes Fact Sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta: US Department of Health and Human Services; 2011. [Google Scholar]
- 2. Yau JW, Rogers SL, Kawasaki R, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leiter LA, Fitchett DH, Gilbert RE, et al. Identification and management of cardiometabolic risk in Canada: a position paper by the cardiometabolic risk working group (executive summary). Can J Cardiol. 2011;27:124–131. [DOI] [PubMed] [Google Scholar]
- 4. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287:2570–2581. [DOI] [PubMed] [Google Scholar]
- 5. Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008;371:1800–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lalloyer F, Staels B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2010;30:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812:1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. [DOI] [PubMed] [Google Scholar]
- 9. Kahn SE, Haffner SM, Viberti G, et al. Rosiglitazone decreases C-reactive protein to a greater extent relative to glyburide and metformin over 4 years despite greater weight gain: observations from a Diabetes Outcome Progression Trial (ADOPT). Diabetes Care. 2010;33:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Panigrahy D, Shen L, Kieran MW, Kaipainen A. Therapeutic potential of thiazolidinediones as anticancer agents. Exp Opin Invest Drugs. 2003;12:1925–1937. [DOI] [PubMed] [Google Scholar]
- 11. Su CG, Wen X, Bailey ST, et al. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yuan Z, Liu Y, Liu Y, et al. Peroxisome proliferation-activated receptor-γ ligands ameliorate experimental autoimmune myocarditis. Cardiovasc Res. 2003;59:685–694. [DOI] [PubMed] [Google Scholar]
- 13. Bright JJ, Natarajan C, Muthian G, Barak Y, Evans RM. Peroxisome proliferator-activated receptor-γ-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J Immunol. 2003;171:5743–5750. [DOI] [PubMed] [Google Scholar]
- 14. Pancani T, Phelps JT, Searcy JL, et al. Distinct modulation of voltage-gated and ligand-gated Ca2+ currents by PPAR-γ agonists in cultured hippocampal neurons. J Neurochem. 2009;109:1800–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation. 2003;108:2941–2948. [DOI] [PubMed] [Google Scholar]
- 16. Betteridge DJ. Thiazolidinediones and fracture risk in patients with type 2 diabetes. Diabet Med. 2011;28:759–771. [DOI] [PubMed] [Google Scholar]
- 17. Food and Drug Administration. Advisory Committee Briefing Document. Preclinical pharmacology and toxicology summary. Drug: Pargluva® (muraglitazar, BMS-298,585). Bethesda, MD: Food and Drug Administration; 2005. [Google Scholar]
- 18. Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188. [DOI] [PubMed] [Google Scholar]
- 19. Mahaffey KW, Hafley G, Dickerson S, et al. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J. 2013;166:240–249. [DOI] [PubMed] [Google Scholar]
- 20. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. [DOI] [PubMed] [Google Scholar]
- 21. Friedland SN, Leong A, Filion KB, et al. The cardiovascular effects of peroxisome proliferator-activated receptor agonists. Am J Med. 2012;125:126–133. [DOI] [PubMed] [Google Scholar]
- 22. Scheen AJ. Thiazolidinediones and liver toxicity. Diabetes Metab. 2001;27:305–313. [PubMed] [Google Scholar]
- 23. Conlon D. Goodbye glitazars? Br J Diabetes Vasc Dis. 2006;6:135–137. [Google Scholar]
- 24. Matthews L, Berry A, Tersigni M, D'Acquisto F, Ianaro A, Ray D. Thiazolidinediones are partial agonists for the glucocorticoid receptor. Endocrinology. 2009;150:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mieczkowska A, Basle MF, Chappard D, Mabilleau G. Thiazolidinedione induced osteocyte apoptosis by a GPR40-dependent mechanism. J Biol Chem. 2012;287:23517–23526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morrison A, Yan X, Tong C, Li J. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt and JNK signalling in nondiabetic mice. Am J Physiol. 2011;301:H895–H902. [DOI] [PubMed] [Google Scholar]
- 27. Jeong I, Choi BH, Hahn SJ. Rosiglitazone inhibits Kv4.3 potassium channels by open-channel block and acceleration of closed-state inactivation. Br J Pharmacol. 2011;163:510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paddock ML, Wiley SE, Axelrod HL, et al. MitoNEET is a uniquely folded 2Fe-2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci USA. 2007;104:14342–14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colca JR, McDonald WG, Waldon DJ, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab. 2004;286:E252–E260. [DOI] [PubMed] [Google Scholar]
- 30. Colca JR, VanderLugt JT, Adams WJ, et al. Clinical proof-of-concept study with MSDC-0160, a prototype mTOT-modulating insulin sensitizer. Clin Pharmacol Ther. 2013;93:352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colca JR, McDonald WG, Cavey GS, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)—relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013;8:e61551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Staels B, Maes M, Zambon A. Fibrates and future PPARα agonists in the treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2008;5:542–553. [DOI] [PubMed] [Google Scholar]
- 33. Kota BP, Huang THW, Roufgalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51:85–94. [DOI] [PubMed] [Google Scholar]
- 34. Li D, Chen K, Sinha N, et al. The effects of PPAR-γ ligand pioglitazone on platelet aggregation and arterial thrombus formation. Cardiovasc Res. 2005;65:907–912. [DOI] [PubMed] [Google Scholar]
- 35. Lonard DM, O'Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8:598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mukherjee R, Sun S, Santomenna L, et al. Ligand and coactivator recruitment preferences of peroxisome proliferator activated receptor alpha. J Steroid Biochem Mol Biol. 2002;81:217–225. [DOI] [PubMed] [Google Scholar]
- 37. Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. Corepressors selectively control the transcriptional activity of PPARγ in adipocytes. Genes Dev. 2005;19:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kodera Y, Takeyama K, Murayama A, Suzawa M, Masuhiro Y, Kato S. Ligand type-specific interactions of peroxisome proliferator-activated receptor γ with transcriptional coactivators. J Biol Chem. 2000;275:33201–33204. [DOI] [PubMed] [Google Scholar]
- 39. Shao G, Heyman RA, Schulman IG. Three amino acids specify coactivator choice by retinoid X receptors. Mol Endocrinol. 2000;14:1198–1209. [DOI] [PubMed] [Google Scholar]
- 40. Rocchi S, Auwerx J. Peroxisome proliferator-activated receptor γ, the ultimate liaison between fat and transcription. Br J Nutr 2000;84(suppl 2):S223–S227. [DOI] [PubMed] [Google Scholar]
- 41. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. [DOI] [PubMed] [Google Scholar]
- 42. Camp HS, Li O, Wise SC, et al. Differential activation of peroxisome proliferator-activated receptor-γ by troglitazone and rosiglitazone. Diabetes. 2000;49:539–547. [DOI] [PubMed] [Google Scholar]
- 43. Weidner C, de Groot JC, Prasad A, et al. Amorfrutins are potent antidiabetic dietary natural products. Proc Natl Acad Sci USA. 2012;109:7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang F, Lavan BE, Gregoire FM. Selective modulators of PPAR-γ activity: molecular aspects related to obesity and side effects. PPAR Res. 2007;2007:32696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pickavance L, Tadayyon M, Widdowson PS, Buckingham RE, Wilding JPH. Therapeutic index for rosiglitazone in dietary obese rats: separation of efficacy and haemodilution. Br J Pharmacol. 1999;128:1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Motani A, Wang Z, Weiszmann J, et al. INT131: a selective modulator of PPAR γ. J Mol Biol. 2009;386:1301–1311. [DOI] [PubMed] [Google Scholar]
- 47. DePaoli A, Higgins LS. INT131, a non-TZD selective PPARγ modulator (SPPARM), does not cause toxicities typical of TZD full PPARγ agonists following 6-month high dose treatment of rats or monkeys. Diabetologia. 2008;51:S369 (Abstract). [Google Scholar]
- 48. Dunn FL, Higgins LS, Fredrickson J, DePaoli AM. Selective modulation of PPARγ activity can lower plasma glucose without typical thiazolidinedione side effects in patients with type 2 diabetes. J Diabetes Complications. 2011;25:151–158. [DOI] [PubMed] [Google Scholar]
- 49. Pickavance L, Widdowson PS, King P, Ishii S, Tanaka H, Williams G. The development of overt diabetes in young Zucker Diabetic Fatty (ZDF) rats and the effects of chronic MCC-555 treatment. Br J Pharmacol. 1998;125:767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choi J, Park Y, Lee HS, Yang Y, Yoon S. 1,3-Diphenyl-1H-pyrazole derivatives as a new series of potent PPARγ partial agonists. Bioorg Med Chem. 2010;18:8315–8323. [DOI] [PubMed] [Google Scholar]
- 51. Guo L, Zhang L, Sun Y, et al. Differences in hepatotoxicity and gene expression profiles by anti-diabetic PPAR γ agonists on rat primary hepatocytes and human HepG2 cells. Mol Divers. 2006;10:349–360. [DOI] [PubMed] [Google Scholar]
- 52. Rogue A, Lambert C, Josse R, et al. Comparative gene expression profiles induced by PPARγ and PPARα/γ agonists in human hepatocytes. PLoS One. 2011;6:e18816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deehan R, Maerz-Weiss P, Catlett NL, et al. Comparative transcriptional network modeling of three PPAR-α/γ co-agonists reveals distinct metabolic gene signatures in primary human hepatocytes. PLoS One. 2012;7:e35012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu C, Lin JD. PGC-1 coactivators in the control of energy metabolism. Acta Biochim Biophys Sin (Shanghai). 2011;43:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, Herz M. Effect of the dual peroxisome proliferator-activated receptor-α/γ agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet. 2009;374:126–135. [DOI] [PubMed] [Google Scholar]
- 56. Ruilope LM, Hanefeld M, Lincoff AM, et al. Effects of aleglitazar on renal function in patients with stage 3 chronic kidney disease and type 2 diabetes. J Am Soc Nephrol. 2012;23:9B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lincoff AM, Tardif JC, Schwartz GG, et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA. 2014;311(15):1515–1525. [DOI] [PubMed] [Google Scholar]
- 58. Luconi M, Cantini G, Serio M. Peroxisome proliferator-activated receptor γ (PPARγ): Is the genomic activity the only answer? Steroids. 2010;75:585–594. [DOI] [PubMed] [Google Scholar]
- 59. Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor γ activity by mitogen-activated protein kinase. J Biol Chem. 1997;272:10811–10816. [DOI] [PubMed] [Google Scholar]
- 60. Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science. 1996;274:2100–2103. [DOI] [PubMed] [Google Scholar]
- 61. Choi JH, Banks AS, Estall JL, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature. 2010;466:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ember Therapeutics. Our Science. Watertown, MA: Ember Therapeutics; 2013. Available at: http://www.embertx.com/approach.php. . [Google Scholar]
- 63. Burgermeister E, Seger R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARγ. Cell Cycle. 2007;6:1539–1548. [DOI] [PubMed] [Google Scholar]
- 64. Burgermeister E, Friedrich T, Hitkova I, et al. The Ras inhibitors caveolin-1 and docking protein 1 activate peroxisome proliferator-activated receptor γ through spatial relocalization at helix 7 of its ligand-binding domain. Mol Cell Biol. 2011;31:3497–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reginato MJ, Bailey ST, Krakow SL, et al. A potent antidiabetic thiazolidinedione with unique peroxisome proliferator-activated receptor-activating properties. J Biol Chem. 1998;273:32679–32684. [DOI] [PubMed] [Google Scholar]
- 66. Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors and are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. [DOI] [PubMed] [Google Scholar]
- 67. Zuris JA, Harir Y, Conlan AR, et al. Facile transfer of [2Fe-2S] clusters from the diabetes drug target mitoNEET to an apo-acceptor protein. Proc Natl Acad Sci USA. 2011;108:13047–13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kusminski CM, Holland WL, Sun K, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. 2012;18:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc Natl Acad Sci USA. 2007;104:5318–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bricker DK, Taylor EB, Schell JC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Herzig S, Raemy E, Montessuit S, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. [DOI] [PubMed] [Google Scholar]
- 72. Divakaruni AS, Murphy AN. Cell biology. A mitochondrial mystery, solved. Science. 2012;337:41–43. [DOI] [PubMed] [Google Scholar]
- 73. Gilde AJ, Van Bilsen M. Peroxisome proliferator-activated receptors (PPARS): regulators of gene expression in heart and skeletal muscle. Acta Physiol Scand. 2003;178:425–434. [DOI] [PubMed] [Google Scholar]
- 74. Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B, Bril A. Rosiglitazone, a peroxisome proliferator-activated receptor-, inhibits the Jun NH2-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes. 2002;51:1507–1514. [DOI] [PubMed] [Google Scholar]
- 75. Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the Zucker Fatty rat heart. Diabetes. 2002;51:1110–1117. [DOI] [PubMed] [Google Scholar]
- 76. Edgley AJ, Thalén PG, Dahllöf B, Lanne B, Ljung B, Oakes ND. PPARγ agonist induced cardiac enlargement is associated with reduced fatty acid and increased glucose utilization in myocardium of Wistar rats. Eur J Pharmacol. 2006;538:195–206. [DOI] [PubMed] [Google Scholar]
- 77. Okada M, Yan SF, Pinsky DJ. Peroxisome proliferator-activated receptor-γ activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB J. 2002;16:1861–1868. [DOI] [PubMed] [Google Scholar]
- 78. Magri CJ, Fava S. Albuminuria and glomerular filtration rate in type 2 diabetes mellitus. Minerva Urol Nefrol. 2011;63:273–280. [PubMed] [Google Scholar]
- 79. Biscetti F, Pola R. Endothelial progenitor cells and angiogenesis join the PPAR-γ. Circ Res. 2008;103:7–9. [DOI] [PubMed] [Google Scholar]
- 80. Wang N, Yang G, Jia Z, et al. Vascular PPAR-γ controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab. 2008;8:482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ghazzi MN, Perez JE, Antonucci TK, et al. Cardiac and glycemic benefits of troglitazone treatment in NIDDM. The Troglitazone Study Group. Diabetes. 1997;46:433–439. [DOI] [PubMed] [Google Scholar]
- 82. Yue T-L, Chen J, Bao W, et al. In vivo myocardial protection from ischemia/reperfusion injury by the PPAR-γ agonist rosiglitazone. Circulation. 2001;104:2588–2594. [DOI] [PubMed] [Google Scholar]
- 83. Zhu P, Lu L, Xu Y, Schwartz GG. Troglitazone improves recovery of left ventricular function after regional ischemia in pigs. Circulation. 2000;101:1165–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xu Y, Gen M, Lu L, et al. PPAR-γ activation fails to provide myocardial protection in ischemia and reperfusion in pigs. Am J Physiol Heart Circ Physiol. 2005;288:H1314–H1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lu L, Reiter MJ, Xu Y, Chicco A, Greyson CR, Schwartz GG. Thiazolidinedione drugs block cardiac KATP channels and may increase propensity for ischaemic ventricular fibrillation in pigs. Diabetologia. 2008;51:675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Szentandrássy N, Harmati G, Bárándi L, et al. Effects of rosiglitazone on the configuration of action potentials and ion currents in canine ventricular cells. Br J Pharmacol. 2011;163:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ohshima K, Mogi M, Horiuchi M. Role of peroxisome proliferator-activated receptor-γ in vascular inflammation. Int J Vasc Med. 2012;2012:508416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sarafidis PA, Bakris GL. Protection of the kidney by thiazolidinediones: an assessment from bench to bedside. Kidney Int. 2006;70:1223–1233. [DOI] [PubMed] [Google Scholar]
- 89. Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. [DOI] [PubMed] [Google Scholar]
- 90. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. [DOI] [PubMed] [Google Scholar]
- 91. Padilla J, Leung E, Phipps RP. Human B lymphocytes and B lymphomas express PPAR-γ and are killed by PPAR-γ agonists. Clin Immunol. 2002;103:22–33. [DOI] [PubMed] [Google Scholar]
- 92. Harris SG, Phipps RP. The nuclear receptor PPAR γ is expressed by mouse T lymphocytes and PPAR γ agonists induce apoptosis. Eur J Immunology. 2001;31:1098–1105. [DOI] [PubMed] [Google Scholar]
- 93. Zhang X, Rodriguez-Galán MC, Subleski JJ, et al. Peroxisome proliferator-activated receptor-γ and its ligands attenuate biologic functions of human natural killer cells. Blood. 2004;104:3276–3284. [DOI] [PubMed] [Google Scholar]
- 94. Yang J, Zhou Y, Guan Y. PPARγ as a therapeutic target in diabetic nephropathy and other renal diseases. Curr Opin Nephrol Hypertens. 2012;21:97–105. [DOI] [PubMed] [Google Scholar]
- 95. Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-γ agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. [DOI] [PubMed] [Google Scholar]
- 96. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evan RM. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. [DOI] [PubMed] [Google Scholar]
- 97. Aoki T. current status of carcinogenicity assessment of PPAR agonists by the US FDA and a mode-of-action approach to the carcinogenic potential. J Toxicol Pathol. 2007;20:197–202. [Google Scholar]
- 98. Tannehill-Gregg SH, Sanderson TP, Minnema D, et al. Rodent carcinogenicity profile of the antidiabetic dual PPAR α and γ agonist muraglitazar. Toxicol Sci. 2007;98:258–270. [DOI] [PubMed] [Google Scholar]
- 99. Long GG, Reynolds VL, Lopez-Martinez A, Ryan TE, White SL, Eldridge SR. Urothelial carcinogenesis in the urinary bladder of rats treated with naveglitazar, a γ-dominant PPAR alpha/gamma agonist: lack of evidence for urolithiasis as an inciting event. Toxicol Pathol. 2008;36:218–231. [DOI] [PubMed] [Google Scholar]
- 100. Egerod FL, Nielsen HS, Iversen L, Thorup I, Storgaard T, Oleksiewicz MB. Biomarkers for early effects of carcinogenic dual-acting PPAR agonists in rat urinary bladder urothelium in vivo. Biomarkers. 2005;10:295–309. [DOI] [PubMed] [Google Scholar]
- 101. Bortolini M, Wright MB, Bopst M, Balas B. Examining the safety of PPAR agonists—current trends and future prospects. Expert Opin Drug Saf. 2013;12:65–79. [DOI] [PubMed] [Google Scholar]
- 102. Pruimboom-Brees IM, Francone O, Pettersen JC, et al. The development of subcutaneous sarcomas in rodents exposed to peroxisome proliferators agonists: hypothetical mechanisms of action and de-risking attitude. Toxicol Pathol. 2012;40:810–818. [DOI] [PubMed] [Google Scholar]
- 103. Chaffer CL, Thomas DM, Thompson EW, Williams ED. PPAR-γ-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer. 2006;6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Panigrahy D, Huang S, Kieran MW, Kaipainen A. PPAR-γ as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol Ther. 2005;4:687–693. [DOI] [PubMed] [Google Scholar]
- 105. Elstner E, Müller C, Koshizuka K, et al. Ligands for peroxisome proliferator-activated receptor-γ and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA. 1998;95:8806–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kubota T, Koshizuka K, Williamson EA, et al. Ligand for peroxisome proliferator-activated receptor γ (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998;58:3344–3352. [PubMed] [Google Scholar]
- 107. Mueller E, Smith M, Sarraf P, et al. Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proc Natl Acad Sci USA. 2000;97:10990–10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hisatake JI, Ikezoe T, Carey M, Holden S, Tomoyasu S, Koeffler HP. Down-regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor γ in human prostate cancer. Cancer Res. 2000;60:5494–5498. [PubMed] [Google Scholar]
- 109. Xu Y, Iyengar S, Roberts RL, Shappell SB, Peehl DM. Primary culture model of peroxisome proliferator-activated receptor γ activity in prostate cancer cells. J Cell Physiol. 2003;196:131–143. [DOI] [PubMed] [Google Scholar]
- 110. Grommes C, Landreth GE, Schlegel U, Heneka MT. The nonthiazolidinedione tyrosine-based peroxisome proliferator-activated receptor ligand GW7845 induces apoptosis and limits migration and invasion of rat and human glioma cells. J Pharmacol Exp Ther. 2005;313:806–813. [DOI] [PubMed] [Google Scholar]
- 111. Grommes C, Landreth GE, Sastre M, et al. Inhibition of in vivo glioma growth and invasion by peroxisome proliferator-activated receptor agonist treatment. Mol Pharmacol. 2006;70:1524–1533. [DOI] [PubMed] [Google Scholar]
- 112. Seufert S, Coras R, Trankle C, et al. PPAR-γ activators: off-target against glioma cell migration and brain invasion. PPAR Res. 2008;2008:513943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wei S, Yang J, Lee SL, Kulp SK, Chen CS. PPARγ-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009;276:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wick M, Hurteau G, Dessev C, et al. PPAR-γ is a target of nonsteroidal anti-inflammatory drugs mediating cyclooxygenase-independent inhibition of lung cancer cell growth. Mol Pharmacol. 2002;62:1207–1214. [DOI] [PubMed] [Google Scholar]
- 115. Bren-Mattison Y, Meyer AM, Van Putten V, et al. Antitumorigenic effects of PPAR-γ in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor κ-B. Mol Pharmacol. 2008;73:709–717. [DOI] [PubMed] [Google Scholar]
- 116. Davis TM, Ting R, Best JD, et al. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia. 2010;54:280–290. [DOI] [PubMed] [Google Scholar]
- 117. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang W, Rachez C, Freedman LP. Discrete roles for peroxisome proliferator-activated receptor γ and retinoid X receptor in recruiting nuclear receptor coactivators. Mol Cell Biol. 2000;20:8008–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Picard F, Gehin M, Annicotte J, et al. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell. 2002;111:931–941. [DOI] [PubMed] [Google Scholar]
- 120. Li PP, Shan S, Chen YT, et al. The PPARα/γ dual agonist chiglitazar improves insulin resistance and dyslipidemia in MSG obese rats. Br J Pharmacol. 2006;148:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Acton JJ III, Akiyama TE, Chang CH, et al. Discovery of (2R)-2-(3-{3-[(4-methoxyphenyl)carbonyl]-2-methyl-6-(trifluoromethoxy)-1H-indol-1-yl}phenoxy)butanoic acid (MK-0533): a novel selective peroxisome proliferator-activated receptor γ modulator for the treatment of type 2 diabetes mellitus with a reduced potential to increase plasma and extracellular fluid volume. J Med Chem. 2009;52:3846–3854. [DOI] [PubMed] [Google Scholar]
- 122. Larsen PJ, Lykkegaard, Larsen LK, Fleckner J, Sauerberg P, Wassermann K, Wulff EM. Dissociation of antihyperglycemic and adverse effects of partial PPAR-γ agonist balaglitzone. Eur J Pharmacol. 2008;596:173–179. [DOI] [PubMed] [Google Scholar]
- 123. Feldman P, Lambert MH, Henke BR. PPAR modulators and PPAR pan agonists for metabolic diseases: the next generation of drugs targeting peroxisome proliferator-activated receptors? Curr Topics Med Chem. 2008;8:728–749. [DOI] [PubMed] [Google Scholar]
- 124. Agrawal R. The first approved agent in the glitazar's class: saroglitazar. Curr Drug Targets. 2014;15:151–155. [DOI] [PubMed] [Google Scholar]
- 125. Jain MR, Giri S, Sundar R, Swain P, Ranvir R. ZYH1, a novel PPAR agonist that shows lipid-lowering and insulin-sensitizing effects with good safety profile in preclinical models. Diabetes. 2012;61(1):269. [Google Scholar]
- 126. Genfit. The FDA grants fast track designation to GFT505 in NASH Reuters. Cambridge, MA: Genfit; 2014. Available at: http://www.genfit.com/wp-content/uploads/2014/03/2014.02.17_PR-GENFIT_EN.pdf. Accessed February 17, 2014. [Google Scholar]
- 127. Colca JR, Kletzien RF. What has prevented the expansion of insulin sensitisers? Exp Opin Invest Drugs. 2006;15:205–210. [DOI] [PubMed] [Google Scholar]
- 128. Dietz M, Mohr P, Kuhn B, et al. Comparative molecular profiling of the PPARα/γ activator aleglitazar: PPAR selectivity, activity and interaction with cofactors. ChemMedChem. 2012;7:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Nevin DK, Lloyd DG, Fayne D. Rational targeting of peroxisome proliferating activated receptor subtypes. Curr Med Chem. 2011;18:5598–5623. [DOI] [PubMed] [Google Scholar]
- 130. Cariou B, Zair Y, Staels B, Bruckert E. Effects of the new dual PPARα/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011;34:2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]