

ABSTRACT
p53 was reported to be an attractive immunotherapy target because it is mutated in approximately half of human cancers, resulting in its inactivation and often accumulation in tumor cells. Peptides derived from p53 are presented by class I MHC molecules and may act as tumor-associated epitopes which could be targeted by p53-specific T cells. Interestingly, it was recently shown that there is a lack of significant correlation between p53 expression levels in tumors and their recognition by p53-TCR transduced T cells. To better understand the influence of the mutational status of p53 on its presentation by the MHC system and on T cell antitumor reactivity, we generated several mutant p53 constructs and expressed them in HLA-A2+/p53− cells. Upon co-culture with p53-specific T cells, we measured the specific recognition of p53-expressing target cells by means of cytokine secretion, marker upregulation and cytotoxicity, and in parallel determined p53 expression levels by intracellular staining. We also examined the relevance of antigen presentation components on p53 recognition and the impact of mutant p53 expression on cell-cycle dynamics. Our results show that selected p53 mutations altering protein stability can modulate p53 presentation to T cells, leading to a differential immune reactivity inversely correlated with measured p53 protein levels. Thus, p53 may behave differently than other classical tumor antigens and its mutational status should therefore be taken into account when elaborating immunotherapy treatments of cancer patients targeting p53.
KEYWORDS: Immunotherapy, p53, T cells, TCR, tumor immunology
Introduction
p53 is known as a stress-induced transcription factor that plays a central role in tumor suppression.1 Diverse types of cellular stress can activate p53 leading to multiple effects that include cell-cycle arrest, senescence or induction of apoptosis. Thus, it is not surprising that p53 is mutated in approximately half of human cancers.2 86% of p53 mutations are found in p53 DNA-binding domain (DBD) located between aa 102–292, most of them being missense mutations encoding a stable full-length protein.3,4 Some of these mutations harbor a dominant-negative effect, leading to a loss of transactivation activity of p535,6 or may even lead to a “gain-of-function.”7 Moreover, many p53 mutants display a reduction in the thermodynamic stability of the protein, resulting in decreased folding levels and defective functional properties.8-10 Interestingly, it was demonstrated that induced “stabilization” of mutant p53 could reactivate pro-apoptotic signaling pathways in tumor cells.9 Several second-site suppressor mutations (e.g., N239Y and N268D) that could, when combined in several p53 mutants, rescue part or all of p53 original function were identified.11-13
Because most mutations in p53 are associated with accumulation (perhaps wrongly termed as “overexpression”) of mutant p53 in cancer cells, it has been postulated that p53 may represent an attractive tumor antigen target,14,15 especially as antibodies and T cells directed against this protein were detected in cancer patients.16,17 Additionally, p53 was shown to be presented by MHC class I molecules.18 Thus, efforts have been made to redirect the immune response against p53 either by TCR-gene transfer, activated T-cells or vaccination (e.g., clinicaltrials.gov NCT00844506, NCT02275039, NCT02577588) as it was suggested that the accumulated protein should be more readily available for antigen processing and presentation than are the low levels of wild-type (wt) p53 expressed in normal cells.19-21 Importantly, due to the high frequency and the extreme diversity of the mutated state of p53 in human cancers, strategies were designed at targeting wt p53 epitopes rather than mutated ones, taking advantage of the potential over representation of these peptides following p53 accumulation in the cell.18,22
In this regard, we previously cloned from a p53-vaccinated HLA-A2 transgenic mouse a TCR specific for the wt human p53 epitope 264–272, bypassing tolerance in human for this antigen.21 We and others showed that PBLs transduced with this murine TCR isolated from T cell clones specific for the human p53264–272 epitope, could recognize p53-expressing tumor cell lines, while p53-negative lines and normal cells were mostly ignored.21,23 However, further investigation using a broad sample of tumor lines revealed that p53-specific cytotoxic T cells can eliminate tumors expressing a wide range of p53 levels,24,25 and we demonstrated a lack of correlation between p53 expression in tumors and target recognition by anti-p53 TCR-transduced T cells.25 Moreover, this finding might explain the poor objective response observed in a recent clinical trial based on the adoptive transfer of p53-TCR engineered T cells for advanced cancer treatment, in which patients were mainly selected on the basis of high p53 protein levels in their tumors.26
Therefore, to better understand the influence of p53 mutations on its recognition by T cells, we generated several mutant p53 constructs and expressed them in multiple HLA-A2+/p53− cells. Following co-culture with p53-specific T cells, we measured specific recognition of the p53-expressing target cells by means of cytokine secretion, activation marker upregulation and cytotoxicity, along with p53 expression levels. In addition, we studied the impact of p53 mutant expression on the cell-cycle dynamics and the expression of apoptotic proteins. Our results show that some p53 mutants are recognized more efficiently than others by T cells demonstrating an inverse correlation between level of p53 protein and T cell recognition.
Results
Generation of an experimental detection system
Our previous study demonstrated that there was a lack of significant correlation between the p53 protein levels and recognition.25 Antigen processing and presentation to T cells is dependent on several factors that include antigen expression levels and stability.27 Since p53 mutations can influence the latter,8,28,29 we surmised this may influence the recognition of p53-epitope by T cells. To examine this, we selected six p53 mutants based on their thermodynamic stability values (the ΔΔG presented were obtained from the IARC TP53 database).3 All these mutants result from missense mutations that are located in the DBD (Fig. 1A). More specifically, we generated constructs encoding mutant p53 proteins of three types; highly destabilizing (R175H, Y220C; ΔΔG > 2.5 kcal/mol), destabilizing (G245S, G248Q; 2.5 > ΔΔG > 0.5 kcal/mol) and stabilizing (N239Y, N268D; −0.5 > ΔΔG kcal/mol). To negate a possible role for endogenous p53 presentation by p53 already present in tumor cells, which might result in an increased background, we made use of three p53−/− tumor cell lines Saos-2, H1299-A2 and HL-60-A2 as target cell line, each of which expressed defined levels of HLA-A*0201 as exemplified in Fig. 1B. These cells were transfected with mRNA encoding the wt and the aforementioned p53 mutants. Comparable transfection efficiency was assessed by qPCR (Fig. S1). In parallel, primary human T cells were transduced to express the murine T cell receptor (TCR) specific for the HLA-A*0201/p53264–272 antigen (Fig. 1C). This TCR was shown to recognize human cancer cell lines,2021 and was recently used in a clinical trial.26
Figure 1.
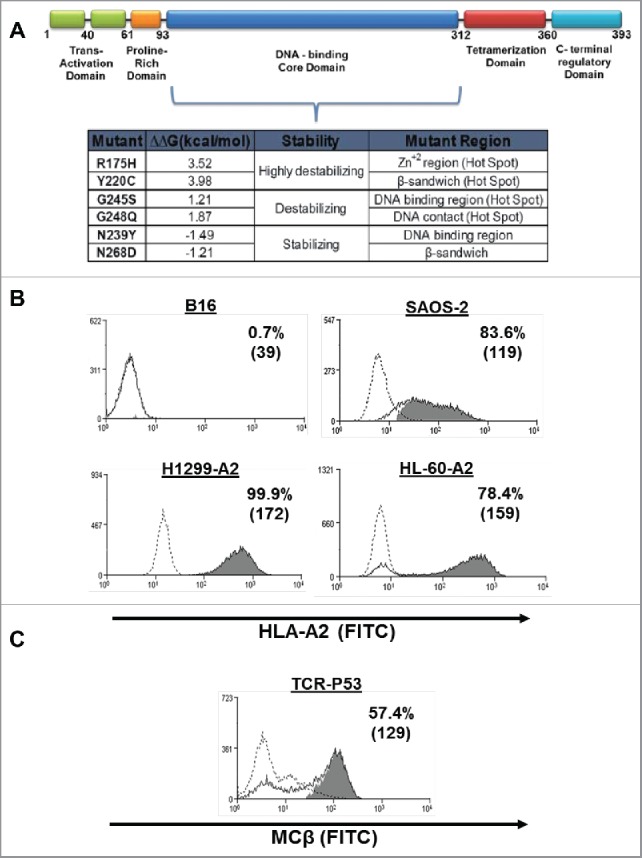
Generation of expression and recognition system. (A) Domain structure of human p53 (top) and the different p53 mutations studied in this work according to p53 core domain thermodynamic stability. ΔΔG values represent the change in free energy of urea-induced unfolding caused by mutations in Tp53 (IARC TP53 database3). (B) HLA-A2 surface expression by different cell lines measured by flow cytometry. B16 is a HLA-A2 negative cell line (control). (C) Human PBLs were transduced with a retroviral vector encoding the p53 (264–272)-specific TCR. Expression was assessed by flow cytometry 72 h after transduction by staining of mouse-constant β (MCβ) TCR. The dotted line represents the staining of the mock-transduced control. The percentage of positive cells and the MFI (in brackets) are shown. These results are representative of 10 independent experiments with at least six different donors. The difference between the p53-TCR stained population and the control population (isotype stained; dotted line) was found statistically significant (p < 0.05; Student paired t test).
Next, we set up co-cultures of the p53-transfected target cells and p53-specific T-cells and measured cytokine secretion and activation marker upregulation as a surrogate for T cell target recognition.30 As shown in Fig. 2, p53 mutants can be divided into several groups based on their recognition by T cells, by means of cytokine secretion and activation marker level; mutants R175H, Y220C triggered higher IFNγ, TNF-α and CD69 levels than wt p53 or G245S (up to 2.2-fold more than wt p53). In contrast, mutants R248Q, N239Y and N268D caused T cells to express lower IFNγ/TNF-α/CD69 levels compared with wt p53 (between 0.5 and 1-fold change).
Figure 2.
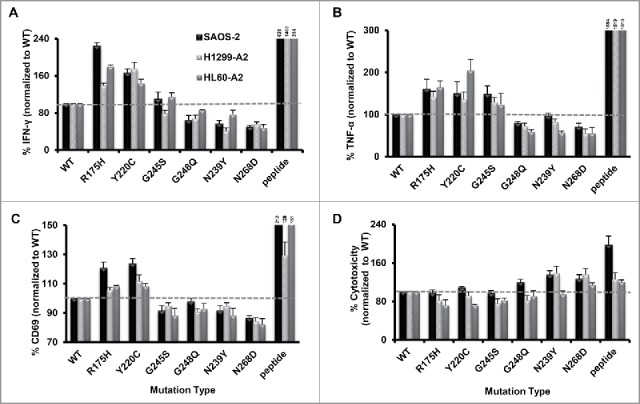
Recognition of different p53 mutants expressed in HLA-A2+/p53− tumor cell lines by the p53-TCR-transduced lymphocytes. p53-TCR transduced lymphocytes were co-cultured with tumor HLA-A2+/p53− cell lines which were electroporated with RNAs encoding different p53 mutants. 18 h after, IFNγ (A) TNF-α (B) secretion & CD69 surface expression levels (C) were assessed by ELISA or by flow cytometry, respectively. Target cells pulsed with the p53264–272 epitope was used as positive control. Data are shown as a percentage of IFNγ/TNF-α/CD69 expression levels, normalized to the results obtained with wt p53 (as mean ± SEM; n ≥ 3; average absolute values in the p53 wt co-cultures were 842 pg/mL of IFNγ, 153 pg/mL of TNFα and 51% of CD69-positivity). (D) Cell cytotoxicity levels measured in co-cultures with cells expressing p53 mutants. CFSE-labeled HLA-A2+/p53− cells were electroporated with RNAs encoding to different p53 mutations. Following a 6 h co-culture with p53-TCR-transduced lymphocytes, cytotoxicity was assessed based on the PI/CFSE double positive population to which we subtracted cytotoxicity levels from untreated p53 mutant-transfected cells. Data are shown as a percentage of PI expression levels (%killing), normalized to wt (as mean ± SEM; n = 4; average absolute values for wt were 34%).
We also examined in similar settings T cell-mediated cytotoxicity when targeting the different p53 mutants. CFSE-labeled target cells expressing the different p53 proteins were co-cultured with p53-TCR-transduced lymphocytes. Cytotoxicity was assessed based on the double positive PI/CFSE population. Though results obtained cytokine secretion are often expected to reflect T cell cytotoxic activity,31 we observed a somewhat different reactivity pattern in T cell-mediated cytotoxic assays as showed in Fig. 2D: target cells expressing mutants R175H, Y220C and G245S showed relatively similar or lower levels of cytotoxicity than wt p53 (down to 0.72-fold change) and those transfected with N239Y and N268D demonstrated in most cases higher PI levels (up to 1.9-fold more than wt p53; p < 0.05).
p53 mutant protein expression and their synthesis rate in HLA-A2+/p53− cells
To try and establish a correlation between p53 antigen expression levels and immune recognition, we quantified the expression levels of p53 protein by flow cytometry in cells electroporated with mRNA encoding the studied p53 mutants. As seen in Fig. 3A, we noticed various levels of protein expression for most of p53 mutants compared with wt p53, a pattern that was generally conserved for each mutant, independently of the host cell line tested. Mutants R175H and Y220C showed relatively low protein expression levels (down to 0.5-fold change compared with wt p53, p < 0.016), whereas mutants G248Q, N239Y and N268D showed higher expression levels of p53 protein compared with wt (up to 2.3-fold more, p < 0.04). We did not observe a significant difference in the levels of expression of mutant G245S compared with its wt version (p = 0.137 in H1299-A2 cells). As expected, measured protein expression levels reflected the expected thermodynamic stability of the different mutants. However, these results seem to negatively correlate with the recognition by anti-p53 T cells (by means of cytokine secretion and CD69 levels; coefficient of determination R2 = 0.8959) (Fig. 3B).
Figure 3.
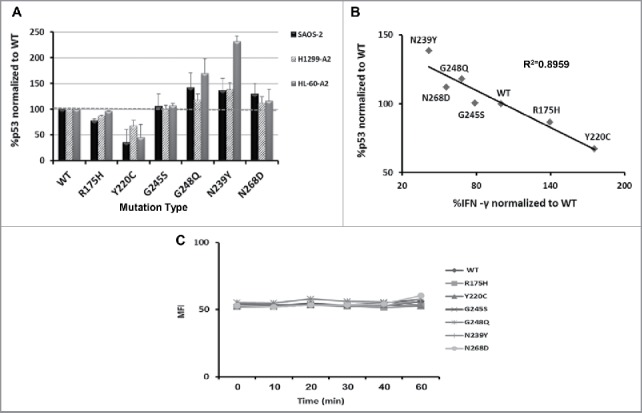
p53 mutants expression and synthesis rate (A) Cells were electroporated with mRNAs encoding different p53 mutants. Intracellular levels of p53 were determined 24 h later by flow cytometry using α-p53 antibody (DO.1) (n = 4). (B) Correlation between p53 expression levels and recognition by T cells (by means of IFN-γ secretion), relative to wt p53 (in the H1299-A2 cell line). As shown, a statistically significant inverse correlation was observed (coefficient of determination R2 = 0.8959). (C) Comparison of synthesis rate of the different p53 proteins. H1299-A2 cell were electroporated with mRNAs encoding different p53 mutants and subjected to cycloheximide. 2 h later, the cells were washed and subjected to the proteasomal inhibitor MG132 to prevent degradation of the nascent proteins. The level of p53 expression was determined at short intervals (every 10 min, up to 60 min) by flow cytometry using α-p53 antibody (DO.1). No statistically significant differences were found in the synthesis rate of the various proteins.
To further determine if the intracellular protein expression levels were due to a different protein synthesis rate rather than thermodynamic stability, cells were electroporated with the different RNAs and then exposed to protein biosynthesis inhibitor (Cycloheximide). 2 h later, cells were washed and the levels of p53 expression were determined every 10 min, in the presence of proteasome inhibitor (MG132). As shown in Fig. 3C, there was no significant difference in the protein synthesis rate of the different p53 mutants (using ANOVA; p > 0.05). These results contribute to dismiss the hypothesis that N239Y and N268D are less efficiently recognized by T cells, as a result of a possible low synthesis rate.
Influence and dependence of p53 mutants on antigen presentation components
Another possible explanation for the differential recognition of p53 mutants by T cells might be related to a change in MHC expression induced by the mutants themselves. First, we sought to determine if the studied p53 epitope 264–272 is presented via the classical MHC/TAP pathway. Indeed, MHC class I epitopes are usually derived from the degradation products of cytosolic proteins or defective ribosomal translation products (DRiPs) by the proteasome complex.27 Over 90% of these peptides are actively transported from the cytosol into the ER organelle by the TAP (peptide transporter associated with antigen processing) and loaded onto MHC class I molecules.32 We confirmed that p53 mutants are presented in a TAP-dependent mechanism since TAP-deficient T2 cells electroporated with different p53 mutants were not recognized by p53-TCR expressing T cells (Fig. 4A).
Figure 4.
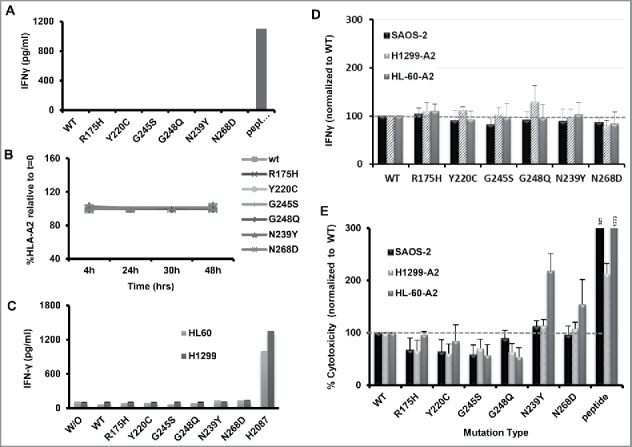
Presentation of p53 and recognition of control epitope in p53-expressing cells. (A) Dependence on TAP of p53 presentation. p53-TCR-transduced lymphocytes were co-cultured with TAP-deficient T2 cell line (HLA-A2+/TAP−) which were electroporated with RNAs encoding to different p53 mutations. 18 h after, IFNγ secretion levels were assessed by ELISA. p53 peptide-pulsed T2 cells were used as positive control. As shown, p53 recognition is dependent on TAP expression. These results are representative of three independent experiments. (B) p53 proteins do not influence HLA-A2 expression. Cells were electroporated with RNAs encoding different p53 mutants and the levels of HLA-A2 were determined after 4, 24, 30 and 48 h by flow cytometry. As shown, the expression of various p53 mutants had no significant influence of HLA-*A0201 levels. (C) Dependence on HLA-A2 presentation. p53-TCR-transduced lymphocytes were co-cultured with the parental HL-60/H1299 cell lines (HLA-A2 neg.) which were electroporated with RNAs encoding to different p53 mutants. 18 h afterward, IFNγ secretion levels were assessed by ELISA. p53+/HLA-A2+ H2087 cells were used as positive control for target. As shown, p53 mutant recognition is dependent on HLA-A2 expression. These results are representative of four independent experiments. (D–E) Influence of p53 mutations on the recognition and killing of MART-1-pulsed tumor cells by anti-MART (F4) T cells. (D) F4-TCR-transduced lymphocytes were co-cultured with tumor cell line (HLA-A2+/p53−) which were first pulsed with a MART-1 epitope and then electroporated with RNAs encoding different p53 mutations. 18 h after the beginning of the co-culture, IFNγ secretion levels were assessed by ELISA. No statistically significant differences were observed. (E) MART-1-pulsed p53-expressing tumor cells were labeled with CFSE and co-cultured with MART-1-specific T-cells. 6 h after the beginning of the co-culture, PI was added and cytotoxicity was measured by evaluating the PI/CFSE-positive population (normalized to WT; mean ± SEM; n = 4).
Furthermore, to study the influence of p53 mutants on MHC expression, the levels of HLA-A*0201 (the MHC allele presenting the p53 epitope 264–27221) were determined by flow cytometry 4, 24, 30 and 48 h following electroporation with p53 mutants. As shown in Fig. 4B, there was no significant difference in HLA-*A0201 expression (p > 0.2). Additionally, we also tested if the differential recognition of p53 mutants might be due to an epiphenomenon linked to p53 expression in the target cells and as such, unrelated to antigen presentation. To verify this point, we performed an additional series of experiments in which we used the parental HL60 and H1299 cell lines which are HLA-A0201-negative. As seen in the Fig. 4C, there was no significant interferon gamma secretion mediated by HL-60 or H1299 cells transfected with mRNA encoding any of the different p53 mutants. We thus conclude that T cell differential recognition of p53 targets is not due to a variance in MHC expression or to a cellular epiphenomenon mediated by the expression of p53 mutants.
p53 mutants do not impact on the recognition of MART-1 antigen
We observed in Fig. 3B an inverse correlation between the expression levels of p53 and T cell recognition. To test whether mutations in p53 would similarly influence the recognition of epitopes from another (control) tumor antigen, we pulsed p53-mutants expressing cells with an epitope derived from the melanoma antigen MART1 and co-cultured these with F4-TCR-expressing T-cells (the F4-TCR recognizes the MART-1 epitope 27–35, was extensively characterized in recent pre-clinical studies and shown to mediate tumor regression in melanoma patients33). As seen in Fig. 4D, we did not observe a statistically significant difference in the IFNγ secretion levels by anti-MART-1 T-cells co-cultured with target cells expressing different p53 mutants, confirming that the expression of p53 mutants did not impact the recognition of a control antigen. Additionally, cytotoxicity results (Fig. 4E) indicate that most of the mutants, namely R175H, Y220C, G245S and R248Q, exhibited similar levels of cytotoxicity, significantly lower than the wt (around 0.7-fold in average; p = 4 × 10−6). Compared to these, N239Y shows relatively higher PI levels than wt p53 (up to 2.2-fold more than wt p53, p = 0.04), mostly recapitulating the results observed using the p53 recognition system and thus, suggesting a possible influence of p53 mutants on the sensitivity to T cell cytotoxic activity regardless of the antigen targeted. Thus, while cytotoxicity sensitivity may be affected by p53 expression for certain mutants, cytokine secretion results as a surrogate for epitope recognition, obtained in control system, support the notion that the differential recognition of p53 mutants is linked to p53 itself rather than to a general influence on antigen recognition.
The mutational status of p53 can facilitate cytotoxicity
The tumor suppressor p53 is a stress responder protein, that plays a central role in anti-tumorigenesis34 facilitating cell-cycle arrest, senescence or induction of apoptosis. To further delineate possible mechanisms responsible for cytotoxic sensitization of p53 mutant-expressing cells observed when exposed to p53 or MART-1-specific T cells, we first evaluated mutant p53 transcriptional activity using a reporter system, which contains the luciferase gene under the genetic control of a synthetic promoter that includes direct repeats of the transcription recognition sequences for p53. H1299-A2 cells transfected with p53-Luc reporter plasmid were electroporated with the different p53 mutants RNAs. Luciferase activity was then assessed and as shown in Fig. 5A, only wt p53 and mutants N239Y and N268D displayed high levels of relative light units (RLU of 2,666 and 27,337, respectively), confirming a certain level of p53 activity.
Figure 5.
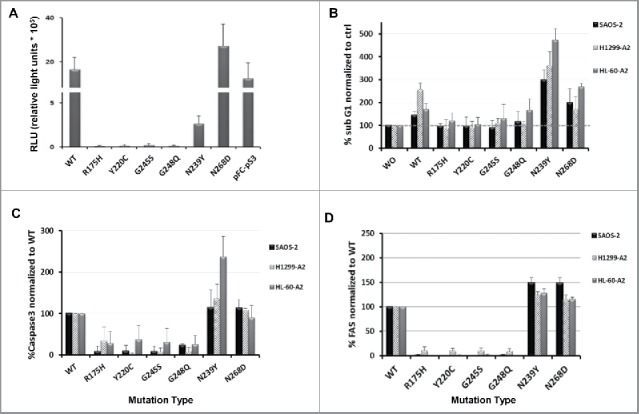
Activity in tumor cells of wild-type and mutant p53. (A) p53 activity levels in cancer cells were measured using a cis-reporter system. H1299-A2 cells were transfected with p53-Luc plasmid and electroporated 4 h later with the different p53 mutants RNAs. 48 h later, luciferase activity was measured using a luminometer and is presented herein as relative light units (RLU). pFC-p53 used as positive control plasmid for p53 expression. (B–D) Influence of various p53 proteins on the levels of subG1 population, Caspase 3 and FAS expression. Cells were electroporated with mRNAs encoding different p53 mutants. 48 h later, cell cycle (B), expression levels of the pro-apoptotic protein caspase-3 (C) and FAS receptor levels (D) were analyzed by flow cytometry. Data shown as a percentage of SubG1/Caspase-3/FAS levels, normalized to control (as mean ± SEM; n = 4).
We then examined the effect of p53 protein expression (mutant and wt) on cell cycle, focusing on changes in the proportion of the subG1 cell population. In parallel, we measured the expression levels of the pro-apoptotic protein Caspase-3 and FAS receptor that were previously shown to increase after induction of p53.35 As seen in Fig. 5(B–D) in all the three cell lines tested, expression of wt p53 and the stabilized mutants N239Y and N268D caused a shift to the subG1 phase and facilitated the expression of relatively high levels of Caspase-3 and FAS (up to 4.71-, 2.37- and 1.48-fold change compared with wt, respectively). On the other hand, mutants R175H, Y220C, G245S and R248Q display similar subG1 levels to the control group (cells w/o RNA; Fig. 5B) and relatively low/no levels of Caspase-3 and FAS, compared with wt (Figs. 5C and D, respectively). These results suggest that stabilized mutants and wt p53 (to a certain extent) may favor target sensitivity to cell death, which is accentuated when exposed to antigen-specific T cells.
Discussion
Herein, we analyzed the influence of different mutations in p53 on the recognition of this protein by T cells. Interestingly, our results support an inverse correlation between the levels of p53 protein levels measured in cells and their susceptibility to stimulate p53-TCR-expressing T lymphocytes. This finding was not necessarily anticipated as one might expect that the higher an antigen is detected in a target cell, the more it can be recognized.27,36 This assumption was considered to be valid also for mutated p53 as many assume that it can be “overexpressed” in cancer cells leading to its recognition by T cells.18,29,37
In contrast, our previous study failed to demonstrate a defined relationship between p53 protein levels and recognition by the anti-p53 TCR-transduced T cells using an extensive panel of human tumors25—only a subtle associative link was observed between MHC expression levels on these different cells and T cell reactivity. Additionally, it was also shown that p53-specific cytotoxic T cells can eliminate tumors that do not express detectable levels of p53, like in the case of human papilloma virus (HPV16) infection which could lead to enhanced proteasomal degradation of p53.24,38 These observations, strengthening our present results, implied that p53 protein levels per se might not necessarily determine whether or not a given cell is sensitive to recognition by p53-specific T cells. Rather, this suggested that the frequency of p53 epitopes presented by the class I MHC molecules on these cells could be related to the rate by which this protein could be degraded.
To eliminate any bias due to different levels of MHC expression or other target cell properties that may impact on T cell recognition, we made use in the present study of a system in which the only variable was the mutation in p53. We have shown herein that p53-bearing destabilizing mutations are recognized more effectively by p53-specific T cells than stabilized p53 mutants. One of the mechanisms facilitating the rapid presentation by class I MHC of self-epitopes is linked to the processing of defective ribosomal products (DRiPs) which are unstable/mis-folded proteins being degraded during or shortly after translation.27 This supports our results as illustrated by two of the p53 mutants tested, R175H and Y220C, which are known to display considerable thermodynamic instability; in turn, the latter may result in a decrease of properly folded proteins and defective functional properties of the p53 protein8-10 (corroborated herein by relatively low p53 protein levels measured without any detectable p53 activity), which would be more efficiently processed by the proteasome and presented to T cells. Importantly, this differential T cell recognition pattern of p53 proteins measured by IFNγ secretion seems to be confined to p53 as an antigen as expression of different p53 mutants neither altered HLA-A*0201 surface expression nor impacted on the recognition of another (control) antigen (MART-1).
p53 is considered by many a valid immune target in cancer mainly due to its ubiquitous nature and “overexpression” or accumulation in tumors relying on a differential antigenic display between tumor versus normal cells.18,39 Moreover, the presence of antibodies16,40 and T cell responses19,41,42 against p53 in cancer patients demonstrate that immune tolerance for this self-antigen is not absolute. Interestingly, we did show herein that T cell recognition of wt p53 is still possible, though at lower levels than several mutants. The in vivo biologic significance of this observation remains to be determined but this warrants caution when elaborating T cell immunotherapies targeting p53 as off-target effects might be plausible. Interestingly, wt p53 recognition could also lend support to previous observations of a fratricide effect in HLA-A2+ T cells expressing a p53-specific TCR25 or to the stimulatory potential of dendritic cells transfected with wt p53 in breast cancer patients.43
Additionally, the development of immunotherapy strategies to target p53 in the past decades include recombinant viruses-based vaccines, p53 peptides44,45 or dendritic cells expressing p53 epitope.46-48 Although, some of these approaches could lead to the induction of reasonably strong p53-specific immune response, their clinical efficacy and their tumor regression-mediating potential are still limited. This may be the result of peripheral tolerance enforcement, in situ immunosuppression that might be overcome using immune-modulating agents49 and/or as emphasized by our results, deficient presentation/recognition of p53 epitopes due to protein stability. We therefore suggest that one of the biomarkers to be considered when administering an immunotherapy targeting p53 should be the type of mutation expressed rather than p53 protein levels only, even when targeting a wt p53 epitope. Thus, our results may have significant implications for the efficient design of successful p53-based immunotherapy of cancer.
Materials and methods
Patient PBMCs and cell lines
PBLs used in this study were from normal donors from the Israeli Blood Bank (Tel-Hashomer, Israel). p53-/HLA-A2+ cell lines were Saos-2,21 H1299-A2 and HL-60-A2 (H1299-A2 and HL-60-A2 cells were transduced to express HLA-A*0201). Packaging line 293GP (expressing GAG and POL) was described.50 Tumor cells were cultured in RPMI (Invitrogen, Carlsbad, CA) and PG13 cells in DMEM (Invitrogen, Carlsbad, CA), both supplemented with 10% FBS (Biological Industries, Beth Haemek, Israel). Lymphocytes were cultured in BioTarget medium (Biological Industries, Beth Haemek, Israel), 10% FBS and 300 IU/mL IL-2. Cells were maintained at 37°C and 5% CO2.
DNA constructs
p53 cDNA was cloned into the pGEM-4Z/64A vector as described.51 The p53 mutants were created by site-directed mutagenesis using overlapping PCR. The retroviral vector backbone pMSGV1, is a derivative of the MSCV-based splice-gag vector and has been described.33 The isolation and cloning of the HLA-A*0201/p53264–272-specific TCR in MSGV1 and the generation of PG13/p53-TCR retrovirus packaging cell clones was described previously21
Retroviral transduction of PBLs and HLA- tumor cells
PBLs were collected by leukopheresis, and lymphocytes were separated by centrifugation on a Ficoll/Hypaque cushion were stimulated with 300 IU/mL IL-2 and 50ng/mL OKT3 (eBioscience, San Diego, CA). Retroviral supernatant produced from PG13/p53TCR was collected and stimulated PBLs were transduced with retroviral particles as described previously51
Electroporation of tumor cells
This technique has been described in our previous reports.30,52 Briefly, in vitro-transcribed mRNA for p53 (mutant or wild-type) was generated using AmpliCap-Max T7 high-yield message maker kit (Cellscript, Madison, WI) and purified using RNAcleanup kit (Norgen Biotek, Canada). Electroporation was performed at 400V/500us using an ElectroSquare Porator ECM 830 (BTX, San Diego, CA). The amount of in vitro-transcribed mRNA was 2 ug per 0.5 × 106 tumor cells.
RNA extraction and cDNA synthesis
Real-time qPCR reactions were performed to quantitate p53 gene expression and to ensure comparable mRNA transfection levels. Briefly, total RNA was isolated from the cultured cells following RNA electroporation using Tri reagent (Ambion® Life technologies Co. GI, NY, USA) according to manufacturer's instructions. Up to 1 μg of RNA was used for cDNA synthesis using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Life technologies Co.GI, NY, USA) according to the manufacturer protocol. Each PCR reaction contains 2 μL of serially diluted cDNA samples, 10 µmoles of each forward and reverse primer, complementary to the tested gene. As a loading control, and 10 μL KAPA Syber FAST ABI Prism qPCR Kit (KapaBiosystems Inc. Woburn, MA, USA). Reactions were run on 7900HT Real-time PCR (Applied Biosystems, Life technologies Co.GI, NY, USA) instrument in FAST mode with standard curve program keeping the manufacturer defaults. The primers sequences were as follows: p53: For 5′-CCCCTCCTGGCCCCTGTCATCTT-3′ Rev 5′-GCCTCACAACCTCCGTCATGTGC-3′. βactin: For 5′-AGCGAGCATCCCCCAAAGTT-3′ Rev 5′-GGGCACGAAGGCTCATCATT-3′.
FACS analysis and antibodies
Fluorophore-labeled anti-human p53 (DO-1), CD69, active-caspase-3, CD95 (FAS) and anti-murine Cβ were purchased from BioLegend (San Diego, CA). Immunofluorescence, analyzed as the relative log fluorescence of live cells, was measured using a CyAn-ADP flow cytometer (Beckman Coulter, Brea). Approximately 1 × 104 cells were analyzed. Cells were stained in a FACS buffer made of PBS, 0.5% BSA, and 0.02% sodium azide.
Cytokine release assays
PBL cultures were tested for reactivity in cytokine release assays using commercially available ELISA kits for IFNγ and TNFα (R&D Systems, Minneapolis, MN). For these assays, 1 × 105 responder cells (PBL) and 1 × 105 stimulator cells (tumor cells) were incubated in a 0.2-mL culture volume in individual wells of 96-well plates for 18 h. Cytokine secretion was measured in culture supernatants diluted to be in the linear range of the assay. As a positive control for T cell activity, we used target cells loaded with p53264–272 epitope.
Synthesis protein rate determination
Tumor cells were electroporated with the different mRNAs and then exposed to a protein biosynthesis inhibitor—cycloheximide (200 µg/mL(. 2 h later, the cells were washed and the p53 levels of expression were determined by flow cytometry every 10 min, in the presence of the proteasome inhibitor MG132 (10 µM).
Cell-mediated cytotoxicity assay
Target cells were labeled with 2 µM CFSE (eBioscience, San Diego, CA) for 6 min and then co-cultured with transduced lymphocytes at 37°C for 6 h, at E:T ratio of 5:1. After the co-culture, propidium iodide (PI) 1 µM (Sigma-Aldrich, Israel) was added for assigning the ratio of cell death. Samples were analyzed by flow cytometry.
Cell-cycle analysis
To test the survival of tumor cells that express p53 mutants, tumor cells were electroporated with different mRNAs and incubated at 37°C. 48 h later, cells were harvested and washed twice in cold PBS. Then, cells were fixed for 24 h at 4 °C, using 4 mL cold 70% ethanol. For PI staining and flow cytometric analysis, fixed cells were washed and centrifuge in 500 g, for 5 min. Cell pellet then was resuspended in 400 µL PBS supplement with 8 µL RNAse (1 µg/mL) and 4 µL PI (2 µg/mL). Samples were incubated for 10 min in the dark, before analyzed by flow cytometry.
p53 activity assay
p53 activity levels were determined by using signal cis- reporter system (Agilent Technologies), which contains the luciferase gene under the genetic control of a synthetic promoter that includes direct repeats of the transcription recognition sequences for the protein p53. H1299-A2 cells were transfected with p53-Luc plasmid and electroporated with the different p53 mutants RNAs, 4 h later. Luciferase activity assay was preformed 48 h later and displayed as RLU, using the luminometer. pFC-p53 was used as positive control plasmid.
Supplementary Material
Disclosure of potential conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Prof. Varda Rotter for helpful discussions, Dr Rachel Levy-Drummer for help with statistical analysis and Dr Alona Zilberberg for help with quantitative assays and data analysis.
Funding
This work was supported by the Israel Cancer Association and the Israel Science Foundation (1422/15).
References
- 1.Levyne AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749-58; PMID:19776744; http://dx.doi.org/ 10.1038/nrc2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levyne AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323-31; PMID:9039259; http://dx.doi.org/ 10.1016/S0092-8674(00)81871-1 [DOI] [PubMed] [Google Scholar]
- 3.Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Adv Cancer Res 2011; 110:107-39; PMID:21704230; http://dx.doi.org/ 10.1016/B978-0-12-386469-7.00005-0 [DOI] [PubMed] [Google Scholar]
- 4.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2:a001008; PMID:20182602; http://dx.doi.org/ 10.1101/cshperspect.a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MK, Sabapathy K. The R246S hot-spot p53 mutant exerts dominant-negative effects in embryonic stem cells in vitro and in vivo. J Cell Sci 2008; 121:1899-906; PMID:18477611; http://dx.doi.org/ 10.1242/jcs.022822 [DOI] [PubMed] [Google Scholar]
- 6.de VA, Flores ER, Miranda B, Hsieh HM, van Oostrom CT, Sage J, Jacks T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci U S A 2002; 99:2948-53; PMID:11867759; http://dx.doi.org/ 10.1073/pnas.052713099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2010; 2:a001107; PMID:20182618; http://dx.doi.org/ 10.1101/cshperspect.a001107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bullock AN, Henckel J, DeDecker BS, Johnson CM, Nikolova PV, Proctor MR, Lane DP, Fersht AR. Thermodynamic stability of wild-type and mutant p53 core domain. Proc Natl Acad Sci U S A 1997; 94:14338-42; PMID:9405613; http://dx.doi.org/ 10.1073/pnas.94.26.14338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 2000; 19:1245-56; PMID:10713666; http://dx.doi.org/ 10.1038/sj.onc.1203434 [DOI] [PubMed] [Google Scholar]
- 10.Mayer S, Rudiger S, Ang HC, Joerger AC, Fersht AR. Correlation of levels of folded recombinant p53 in escherichia coli with thermodynamic stability in vitro. J Mol Biol 2007; 372:268-76; PMID:17631895; http://dx.doi.org/ 10.1016/j.jmb.2007.06.044 [DOI] [PubMed] [Google Scholar]
- 11.Wieczorek AM, Waterman JL, Waterman MJ, Halazonetis TD. Structure-based rescue of common tumor-derived p53 mutants. Nat Med 1996; 2:1143-6; PMID:8837616; http://dx.doi.org/ 10.1038/nm1096-1143 [DOI] [PubMed] [Google Scholar]
- 12.Brachmann RK, Yu K, Eby Y, Pavletich NP, Boeke JD. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J 1998; 17:1847-59; PMID:9524109; http://dx.doi.org/ 10.1093/emboj/17.7.1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baroni TE, Wang T, Qian H, Dearth LR, Truong LN, Zeng J, Denes AE, Chen SW, Brachmann RK. A global suppressor motif for p53 cancer mutants. Proc Natl Acad Sci U S A 2004; 101:4930-5; PMID:15037740; http://dx.doi.org/ 10.1073/pnas.0401162101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lane DP, Brown CJ, Verma C, Cheok CF. New insights into p53 based therapy. Discov Med 2011; 12:107-17; PMID:21878188 [PubMed] [Google Scholar]
- 15.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253:49-53; PMID:1905840; http://dx.doi.org/ 10.1126/science.1905840 [DOI] [PubMed] [Google Scholar]
- 16.Winter SF, Minna JD, Johnson BE, Takahashi T, Gazdar AF, Carbone DP. Development of antibodies against p53 in lung cancer patients appears to be dependent on the type of p53 mutation. Cancer Res 1992; 52:4168-74; PMID:1322237 [PubMed] [Google Scholar]
- 17.Lambeck A, Leffers N, Hoogeboom BN, Sluiter W, Hamming I, Klip H, ten Hoor K, Esajas M, van Oven M, Drijfhout JW et al.. P53-specific T cell responses in patients with malignant and benign ovarian tumors: implications for p53 based immunotherapy. Int J Cancer 2007; 121:606-14; PMID:17415711; http://dx.doi.org/ 10.1002/ijc.22710 [DOI] [PubMed] [Google Scholar]
- 18.Theobald M, Biggs J, Dittmer D, Levyne AJ, Sherman LA. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci U S A 1995; 92:11993-7; PMID:8618830; http://dx.doi.org/ 10.1073/pnas.92.26.11993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albers AE, Ferris RL, Kim GG, Chikamatsu K, DeLeo AB, Whiteside TL. Immune responses to p53 in patients with cancer:enrichment in tetramer+ p53 peptide-specific T cells and regulatory T cells at tumor sites. Cancer Immunol Immunother 2005; 54(11):1072-81; PMID:15959774; http://dx.doi.org/ 10.1007/s00262-005-0670-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuball J, Schmitz FW, Voss RH, Ferreira EA, Engel R, Guillaume P, Strand S, Romero P, Huber C, Sherman LA et al.. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity 2005; 22:117-29; PMID:15664164; http://dx.doi.org/ 10.1016/j.immuni.2004.12.005 [DOI] [PubMed] [Google Scholar]
- 21.Cohen CJ, Zheng Z, Bray R, Zhao Y, Sherman LA, Rosenberg SA, Morgan RA. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol 2005; 175:5799-808; PMID:16237072; http://dx.doi.org/ 10.4049/jimmunol.175.9.5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe J, Shatz M, Resnick MA, Menendez D. Modulation of immune responses by the tumor suppressor p53. Bio Discovery 2013; 8:1-12 [DOI] [PubMed] [Google Scholar]
- 23.Voss RH, Kuball J, Engel R, Guillaume P, Romero P, Huber C, Theobald M. Redirection of T cells by delivering a transgenic mouse-derived MDM2 tumor antigen-specific TCR and its humanized derivative is governed by the CD8 coreceptor and affects natural human TCR expression. Immunol Res 2006; 34:67-87; PMID:16720899; http://dx.doi.org/ 10.1385/IR:34:1:67 [DOI] [PubMed] [Google Scholar]
- 24.Vierboom MP, Zwaveling S, Bos GMJ, Ooms M, Krietemeijer GM, Melief CJ, Offringa R. High steady-state levels of p53 are not a prerequisite for tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. Cancer Res 2000; 60:5508-13; PMID:11034095 [PubMed] [Google Scholar]
- 25.Theoret MR, Cohen CJ, Nahvi AV, Ngo LT, Suri KB, Powell DJ Jr, Dudley ME, Morgan RA, Rosenberg SA. Relationship of p53 overexpression on cancers and recognition by anti-p53 TCR Transduced T cells. Hum Gene Ther 2008; 19(11):1219-32; PMID:19848582; http://dx.doi.org/ 10.1089/hum.2008.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis JL, Theoret MR, Zheng Z, Lamers CH, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res 2010; 16:5852-61; PMID:21138872; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anton LC, Yewdell JW. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J Leukoc Biol 2014; 95:551-62; PMID:24532645; http://dx.doi.org/ 10.1189/jlb.1113599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Offringa R, Vierboom MP, van der Burg SH, Erdile L, Melief CJ. p53: a potential target antigen for immunotherapy of cancer. Ann N Y Acad Sci 2000; 910:223-33; PMID:10911916; http://dx.doi.org/ 10.1111/j.1749-6632.2000.tb06711.x [DOI] [PubMed] [Google Scholar]
- 29.Gnjatic S, Cai Z, Viguier M, Chouaib S, Guillet JG, Choppin J. Accumulation of the p53 protein allows recognition by human CTL of a wild-type p53 epitope presented by breast carcinomas and melanomas. J Immunol 1998; 160:328-33; PMID:9551988 [PubMed] [Google Scholar]
- 30.Tal Y, Yaakobi S, Horovitz-Fried M, Safyon E, Rosental B, Porgador A, Cohen CJ. An NCR1-based chimeric receptor endows T-cells with multiple anti-tumor specificities. Oncotarget 2014; 5:10949-58; PMID:25431955; http://dx.doi.org/ 10.18632/oncotarget.1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilde S, Sommermeyer D, Leisegang M, Frankenberger B, Mosetter B, Uckert W, Schendel DJ. Human antitumor CD8+ T cells producing Th1 polycytokines show superior antigen sensitivity and tumor recognition. J Immunol 2012; 189:598-605; PMID:22689880; http://dx.doi.org/ 10.4049/jimmunol.1102165 [DOI] [PubMed] [Google Scholar]
- 32.Leonhardt RM, Abrahimi P, Mitchell SM, Cresswell P. Three tapasin docking sites in TAP cooperate to facilitate transporter stabilization and heterodimerization. J Immunol 2014; 192:2480-94; PMID:24501197; http://dx.doi.org/ 10.4049/jimmunol.1302637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314:126-9; PMID:16946036; http://dx.doi.org/ 10.1126/science.1129003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogelstein B, Lane D, Levyne AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; http://dx.doi.org/ 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- 35.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci 2003; 116:4077-85; PMID:12972501; http://dx.doi.org/ 10.1242/jcs.00739 [DOI] [PubMed] [Google Scholar]
- 36.Riker AI, Kammula US, Panelli MC, Wang E, Ohnmacht GA, Steinberg SM, Rosenberg SA, Marincola FM. Threshold levels of gene expression of the melanoma antigen gp100 correlate with tumor cell recognition by cytotoxic T lymphocytes. Int J Cancer 2000; 86:818-26; PMID:10842196; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 37.Tokunaga N, Murakami T, Endo Y, Nishizaki M, Kagawa S, Tanaka N, Fujiwara T. Human monocyte-derived dendritic cells pulsed with wild-type p53 protein efficiently induce CTLs against p53 overexpressing human cancer cells. Clin Cancer Res 2005; 11:1312-8; PMID:15709203 [PubMed] [Google Scholar]
- 38.Sirianni N, Ha PK, Oelke M, Califano J, Gooding W, Westra W, Whiteside TL, Koch WM, Schneck JP, DeLeo A et al.. Effect of human papillomavirus-16 infection on CD8+ T-cell recognition of a wild-type sequence p53264-272 peptide in patients with squamous cell carcinoma of the head and neck. Clin Cancer Res 2004; 10:6929-37; PMID:15501971; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0672 [DOI] [PubMed] [Google Scholar]
- 39.Terashima T, Mizukoshi E, Arai K, Yamashita T, Yoshida M, Ota H, Onishi I, Kayahara M, Ohtsubo K, Kagaya T et al.. P53, hTERT, WT-1, and VEGFR2 are the most suitable targets for cancer vaccine therapy in HLA-A24 positive pancreatic adenocarcinoma. Cancer Immunol Immunother 2014; 63:479-89; PMID:24633336; http://dx.doi.org/ 10.1007/s00262-014-1529-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crawford LV, Pim DC, Bulbrook RD. Detection of antibodies against the cellular protein p53 in sera from patients with breast cancer. Int J Cancer 1982; 30:403-8; PMID:6292117; http://dx.doi.org/ 10.1002/ijc.2910300404 [DOI] [PubMed] [Google Scholar]
- 41.Hoffmann TK, Donnenberg AD, Finkelstein SD, Donnenberg VS, Friebe-Hoffmann U, Myers EN, Appella E, DeLeo AB, Whiteside TL. Frequencies of tetramer+ T cells specific for the wild-type sequence p53(264-272) peptide in the circulation of patients with head and neck cancer. Cancer Res 2002; 62:3521-9; PMID:12067999 [PubMed] [Google Scholar]
- 42.Black AP, Bailey A, Jones L, Turner RJ, Hollowood K, Ogg GS. p53-specific CD8+ T-cell responses in individuals with cutaneous squamous cell carcinoma. Br J Dermatol 2005; 153:987-91; PMID:16225611; http://dx.doi.org/ 10.1111/j.1365-2133.2005.06878.x [DOI] [PubMed] [Google Scholar]
- 43.Met O, Balslev E, Flyger H, Svane IM. High immunogenic potential of p53 mRNA-transfected dendritic cells in patients with primary breast cancer. Breast Cancer Res Treat 2011; 125:395-406; PMID:20336365; http://dx.doi.org/ 10.1007/s10549-010-0844-9 [DOI] [PubMed] [Google Scholar]
- 44.Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008; 8:351-60; PMID:18418403; http://dx.doi.org/ 10.1038/nrc2373 [DOI] [PubMed] [Google Scholar]
- 45.Bijker MS, Melief CJ, Offringa R, van der Burg SH. Design and development of synthetic peptide vaccines: past, present and future. Expert Rev Vaccines 2007; 6:591-603; PMID:17669012; http://dx.doi.org/ 10.1586/14760584.6.4.591 [DOI] [PubMed] [Google Scholar]
- 46.Schuler PJ, Harasymczuk M, Visus C, Deleo A, Trivedi S, Lei Y, Argiris A, Gooding W, Butterfield LH, Whiteside TL et al.. Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin Cancer Res 2014; 20:2433-44; PMID:24583792; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Svane IM, Pedersen AE, Johnsen HE, Nielsen D, Kamby C, Gaarsdal E, Nikolajsen K, Buus S, Claesson MH. Vaccination with p53-peptide-pulsed dendritic cells, of patients with advanced breast cancer: report from a phase I study. Cancer Immunol Immunother 2004; 53:633-41; PMID:14985857; http://dx.doi.org/ 10.1007/s00262-003-0493-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svane IM, Pedersen AE, Johansen JS, Johnsen HE, Nielsen D, Kamby C, Ottesen S, Balslev E, Gaarsdal E, Nikolajsen K et al.. Vaccination with p53 peptide-pulsed dendritic cells is associated with disease stabilization in patients with p53 expressing advanced breast cancer; monitoring of serum YKL-40 and IL-6 as response biomarkers. Cancer Immunol Immunother 2007; 56:1485-99; PMID:17285289; http://dx.doi.org/ 10.1007/s00262-007-0293-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardwick N, Chung V, Cristea M, Ellenhorn JD, Diamond DJ. Overcoming immunosuppression to enhance a p53MVA vaccine. Oncoimmunology 2014; 3:e958949; PMID:25941580; http://dx.doi.org/ 10.4161/21624011.2014.958949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wargo JA, Robbins PF, Li Y, Zhao Y, El-Gamil M, Caragacianu D, Zheng Z, Hong JA, Downey S, Schrump DS et al.. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol Immunother 2009; 58:383-94; PMID:18677478; http://dx.doi.org/ 10.1007/s00262-008-0562-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haga-Friedman A, Horovitz-Fried M, Cohen CJ. Incorporation of transmembrane hydrophobic mutations in the TCR enhance its surface expression and T cell functional avidity. J Immunol 2012; 188:5538-46; PMID:22544927; http://dx.doi.org/ 10.4049/jimmunol.1103020 [DOI] [PubMed] [Google Scholar]
- 52.Bialer G, Horovitz-Fried M, Ya'acobi S, Morgan RA, Cohen CJ. Selected murine residues endow human TCR with enhanced tumor recognition. J Immunol 2010; 184:6232-41; PMID:20427762; http://dx.doi.org/ 10.4049/jimmunol.0902047 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.