

ABSTRACT
Immunotherapy, via intra-vesical instillations of BCG, is the therapy of choice for patients with high-risk non-muscle invasive bladder cancer. The subsequent recruitment of lymphocytes and myeloid cells, as well as the release of cytokines and chemokines, is believed to induce a local immune response that eliminates these tumors, but the detailed mechanisms of action of this therapy are not well understood. Here, we have studied the phenotype and function of the responding lymphocyte populations as well as the spectrum of cytokines and chemokines produced in an in vitro model of human peripheral blood mononuclear cells (PBMCs) co-cultured with BCG. Natural killer (NK) cell activation was a prominent feature of this immune response and we have studied the expansion of this lymphocyte population in detail. We show that, after BCG stimulation, CD56dim NK cells proliferate, upregulate CD56, but maintain the expression of CD16 and the ability to mediate ADCC. CD56bright NK cells also contribute to this expansion by increasing CD16 and KIR expression. These unconventional CD56bright cells efficiently degranulated against bladder cancer cells and the expansion of this population required the release of soluble factors by other immune cells in the context of BCG. Consistent with these in vitro data, a small, but significant increase in the intensity of CD16 expression was noted in peripheral blood CD56bright cells from bladder cancer patients undergoing BCG therapy, that was not observed in patients treated with mitomycin-C instillations. These observations suggest that activation of NK cells may be an important component of the anti-tumoral immune response triggered by BCG therapy in bladder cancer.
KEYWORDS: BCG, CD56bright, chemokines, cytokines, NK cell differentiation, NK receptor
Introduction
Bacille Calmette-Guérin (BCG), an attenuated strain of Mycobacterium bovis used as the vaccine for tuberculosis, is well known to be a potent enhancer of the immune response. For example, mycobacteria are a key component of Freund's adjuvant widely used in immunization.1 The immuno-stimulatory properties of mycobacteria have also been exploited as an effective treatment of bladder cancer for several decades [reviewed in Ref.2]. In fact, BCG instillation is considered the “gold-standard” treatment of non-muscle invasive bladder cancer (NMIBC) and has been demonstrated to be more effective than chemotherapy in these patients, showing statistically significant reduced recurrence, progression and mortality at 10 y. That 70% of patients respond to BCG suggests that the study of the mechanisms underlying the elimination of the tumors during BCG treatment could give more insight into how the immune system recognizes tumors. Moreover, a better understanding of how this therapy works may aid in the identification of responder and non-responder patients at an early stage of therapy, when the optimal treatment strategy for each patient needs to be decided.
Although several immune effectors, including cytotoxic T lymphocytes (CTLs), natural killer (NK) cells, monocytes and neutrophils have been suggested to be involved in the response generated after BCG instillation,3,4 data from in vitro experiments and from murine models suggest that NK cells and Natural Killer T (NKT) cells might play key roles in the immune response against bladder cancer cells.5-11 NK cells are known to be crucial players in host pathogen interactions,12-15 however, it is also now appreciated that they comprise a heterogeneous population of effector cells whose response to a large variety of stimuli (viral infection, bacterial compounds, tumor transformation, etc.) depends on a complex array of receptor–ligand interactions and signaling events. Thus, to understand the NK cell response against tumors stimulated by BCG, it is necessary to dissect the contribution of distinct populations of these innate effector cells. CD56, an isoform of the human neural cell adhesion molecule (NCAM1), that is used as general marker for human NK cells,16 divides these lymphocytes into two populations. The majority of circulating NK cells (95%) have low expression of CD56 and are considered the mature cytotoxic NK cell subset. These CD56dim cells also express high levels of the low-affinity FcγRIIIA receptor (CD16A) that mediates antibody-dependent cell-mediated cytotoxicity (ADCC) upon recognition of target cells opsonized with IgG. In contrast, the minority population of circulating CD56bright NK cells (5%) are generally considered more immature; they express little or no CD16 and respond better to soluble factors. These two subpopulations of NK cells, CD56bright and CD56dim, can be further distinguished by the differential expression of other NK receptors, such as killer immunoglobulin-like receptors (KIRs), natural cytotoxicity receptors (NCRs) and CD94/NKG2A so that, in general, the phenotype of the so called immature NK cells is CD56bright CD16lo/− CD94/NKG2Ahi KIR−, while mature cytotoxic NK cells would be CD56dim CD16+ CD94/NKG2A+/− KIRhi.17,18
Initially CD56bright NK cells were mainly thought of as cytokine-producing regulatory cells while the CD56dim subset were specialized for cytotoxicity. However, both NK cell subsets can produce large amounts of IFNγ, with the difference between them residing in the stimuli required to elicit their response. CD56bright NK cells proliferate and produce IFNγ in response to dendritic cell (DC)-derived cytokines, such as IL2, IL12, IL15 and IL18 19, while CD56dim NK cells secrete IFNγ after recognition of activating ligands on target cells.20 CD56bright and CD56dim NK cells also differ in their proliferative response to IL2, intrinsic cytotoxic capacity, NK receptor repertoire and adhesion molecule expression. We have previously reported that the recognition of bladder cancer cells by purified, IL2-activated NK cells is not affected by exposure to BCG but that co-culture with BCG and other lymphocytes did affect the NK response.11 We now report that one of the most prominent responses elicited by exposure of peripheral blood mononuclear cells (PBMCs) to BCG is the proliferation and activation of NK cells that become CD56bright while retaining many of the phenotypic and functional characteristics of mature CD56dim NK cells including a high capacity to degranulate against bladder cancer cells and to mediate ADCC. This process depends on the production of mainly innate cytokines in these cultures. Analysis of the NK cell compartment in the peripheral blood of a small cohort of bladder cancer patients revealed that peaks of CD56bright NK cells expressing significantly increased amounts of CD16 at the cell surface can be observed in patients treated with BCG, but not with mitomycin C. These observations support the idea that NK cell activation triggered by exposure of infiltrating leukocytes to BCG is an important factor in the generation of an immune response against the tumor cells.
Results
PBMCs exposed to BCG expand a cytotoxic subpopulation of CD56bright NK cells
PBMCs freshly isolated from healthy donors were incubated for a week in the presence or absence of BCG and then analyzed by flow cytometry. The most striking change noted in these cultures was a dramatic increase in the mean fluorescence intensity (MFI) of CD56 in the NK cell population accompanied by an increase in the proportion of the CD3−CD56bright population (Fig. 1A) that was consistently observed in PBMCs from multiple donors (Fig. 1B). Next, the ability of the BCG-activated NK cells to respond against target cells and, in particular, against bladder cancer cell lines was evaluated in experiments where surface expression of LAMP-1 was assayed as a marker of degranulation. Both CD56bright and CD56dim NK cells responded to a classical NK target, the erythroleukaemia cell line K562, and against a range of bladder cancer cell lines with variable intensity (Fig. 1C). Notably, BCG-stimulated CD56bright NK cells showed an increased ability to recognize bladder cancer cells and this degranulation capacity was higher than that of CD56dim NK cells exposed, or not, to BCG. The activating receptor NKG2D was important for the recognition of bladder cancer cells (Fig. S1), however, the contribution of this receptor to cytotoxicity was less pronounced for BCG-activated NK cells than for IL2-activated NK cells.11 Since CD56bright NK cells are a minority in peripheral blood, and often thought of as an immature subpopulation, this result raised several questions about the origin and functionality of these cells.
Figure 1.
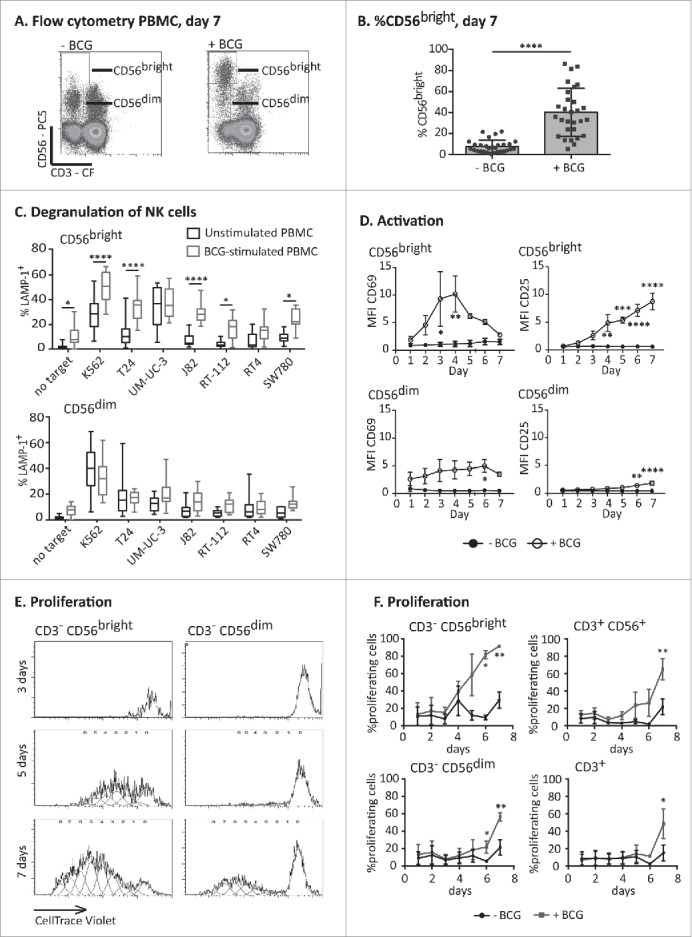
Expansion and activation of an anti-tumoral CD56bright NK cell population. PBMCs from healthy donors were incubated with or without BCG at a 1:50 ratio (viable bacteria to PBMC). At day 7, cells in suspension were recovered from the co-culture, centrifuged and analyzed. (A) Flow cytometry of PBMCs at day 7 showing the percentage of NK cells as CD56bright and CD56dim populations. A representative experiment is shown. (B) Statistical analysis. The plot represents the comparison using a Student t-test of the percentage of CD56bright within total NK cells in PBMCs from 29 different donors stimulated or not with BCG (**** means significant, p <0.0001). (C) Degranulation of NK cells against bladder cancer cells (T24, UM-UC-3, RT-112, RT4, J82, SW780) and positive control (K562 cells) was measured by analyzing surface LAMP-1 (CD107a) within the CD3−CD56+ regions; CD56bright and CD56dim were analyzed separately. The distribution and average of degranulation percentages obtained from, at least, nine assays are represented. Statistical analysis was performed using one-way ANOVA (*p <0.05; ****p <0.0001). (D) Activation markers. Surface expression of CD69 (left) and CD25 (right) on NK cells was analyzed by flow cytometry each day of the week of treatment with BCG in three independent experiments. In the CD56bright, the MFI represented for CD25 corresponds to nearly 100% of the population, while the MFI for CD69 corresponds to 72 ± 14% (see text). Statistical analysis was performed using Two-way ANOVA (*p <0.05; ***p <0.001). (E and F) Proliferation assays. PBMCs from healthy donors were labeled with CellTrace™ Violet before the incubation with or without BCG. Proliferation was analyzed evaluating the amount of dye per cell by flow cytometry in the CD3−CD56bright and CD3−CD56dim regions (E) and the percentage of cells that had proliferated within CD3−CD56bright, CD3−CD56dim, CD3+ and CD3+CD56+ regions (F) in four independent experiments. Statistical analysis was performed using two-way ANOVA (*p <0.05; **p <0.01).
Analysis of activation markers on both NK cell subpopulations revealed that CD56bright NK cells were activated in response to mycobacteria. A peak of high expression of the activation marker CD69 was observed after 3–4 d of co-culture between PBMCs and BCG (72 ± 14% of the CD56bright cells were positive for CD69). The expression of CD25 (IL2Rα) increased starting day 4 until day 7 in nearly 100% of the population (95 ± 2%) (Fig. 1D). In contrast, the expression of activation markers by CD56dim NK cells in the culture was always lower than that observed for the CD56bright subset after BCG stimulation and only in a low percentage of cells (7% for CD69 at day 4; 27% for CD25, at day 7).
CD56bright NK cells are, in general, a cytokine-secreting population,19 with only a limited capacity to mediate natural cytotoxicity.21 Thus, to better understand the origin and the basis of these antitumor CD56bright NK cells, the expansion of these populations of NK cells in the presence of BCG was further characterized. Proliferation experiments, using PBMCs labeled with CellTrace™ Violet stain revealed that CD56bright NK cells started proliferating on day 4 of the co-culture of PBMCs with BCG, while no proliferating CD56dim NK cells were observed during the first week of co-culture in the presence of BCG (Fig. 1E). In fact, the fraction of CD56dim cells decreased over this time and death of CD56dim NK cells could be observed from day 3 of the co-culture with BCG (data not shown and Fig. S2). Proliferation of CD3+ T cells and CD3+CD56+ was detected only after 6–7 d of co-culture (Fig. 1F) suggesting that proliferation of a subpopulation of CD56bright NK cells with an increased tumor recognition capacity is a dominant feature of the early lymphocyte response to BCG.
BCG-stimulated CD56bright NK cells have an intermediate phenotype between mature and immature NK cells
The expression of other NK receptors on the BCG-activated CD56bright NK cell population was analyzed by flow cytometry. Peripheral blood CD56bright cells usually express CD94, in association with NKG2A, while they are negative for CD16 and KIR (Fig. S3).16,17 BCG-activated CD56bright NK cells expressed CD94 but, interestingly, the fluorescence intensity for this receptor was higher in the BCG-activated cells than in untreated CD56bright cells, and this was true at both days 3 (data not shown) and 7 (Fig. 2). In contrast to the typical phenotype of CD56bright cells in peripheral blood, which do not express KIR or CD16, ∼25 and ∼50% of the BCG-activated CD56bright population stained for KIR and for CD16, respectively, and the fluorescence intensity of CD16 was higher than in the CD56dim cells. CD57, a marker associated with terminal stages of NK cell differentiation, was also expressed by the BCG-activated population of CD56bright CD16+ NK cells but not by CD56bright from untreated cultures (Fig. 2 and Figs. S4A–E). Although there is no relationship between any particular KIR and the expansion of the activated population, the percentage of KIR+ cells was increased among BCG-stimulated CD56bright cells that also expressed CD16 and CD57, compared with the KIR+ expression on the whole population of activated CD56bright (Figs. 4D and E). It is also interesting to note that the percentage of CD57+ cells was increased in CD16+ vs CD16− CD56bright cells. Thus, these detailed studies of the BCG-activated CD56bright NK cells, led to the conclusion that they were not “classical” CD56bright NK cells, but rather activated NK cells. Thus, for the rest of this paper, although we will continue to use the nomenclature CD56bright and dim, to compare NK cells expressing high vs. low amounts of CD56, we need to clarify that “BCG-stimulated CD56bright NK cells” are not the usual CD56 bright subset, usually found in a low percentage peripheral blood, but instead represent a newly activated NK cell population. Indeed, the observed phenotype, especially their newly acquired degranulation capacity, implied that BCG exposure generated an anti-tumoral subset of CD56bright NK cells, many of which express high levels of CD16 and can co-express CD57 and KIR.
Figure 2.
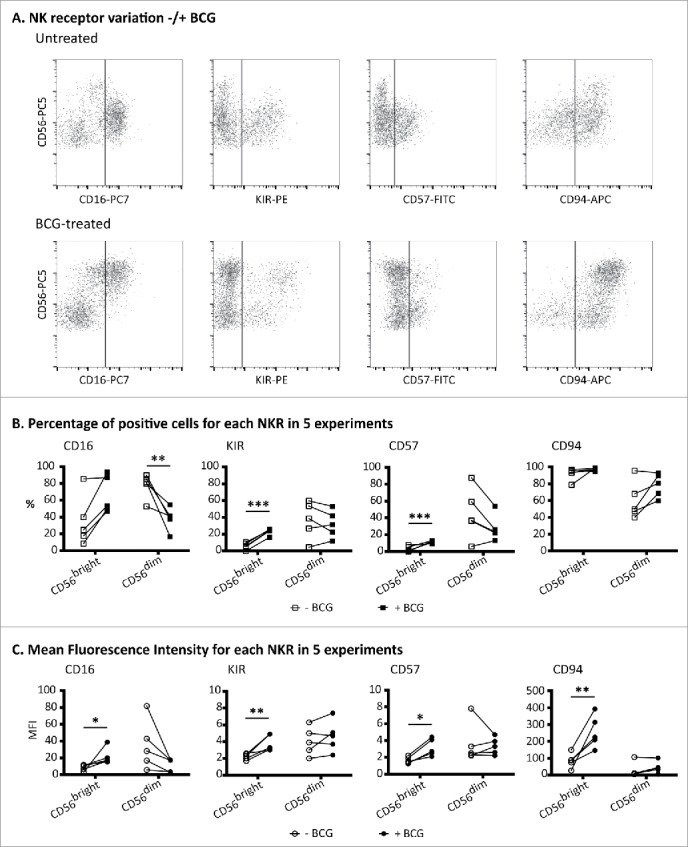
BCG-stimulated CD56bright NK cells express CD16, KIRhi, CD57 and CD94hi. PBMCs from healthy donors were incubated for 1 week with or without BCG at a 1:50 ratio. Samples were analyzed by flow cytometry at day 7. (A) Expansion of the CD56bright subset and NK receptor phenotyping, gating inside the CD3−CD56+ region, corresponding to a representative donor. (B) Percentages of NK receptors in the CD56bright and CD56dim populations. (C) Mean fluorescence intensity of NK receptors in the CD56bright and CD56dim populations. (B) and (C) show the statistical analysis of the phenotypes obtained in five independent experiments using a Student's t-test (* means significant, p <0.05; **p <0.01; ***p <0.001). For comparison, an example of normal peripheral blood NK receptor values is shown in Fig. 3.
Figure 4.
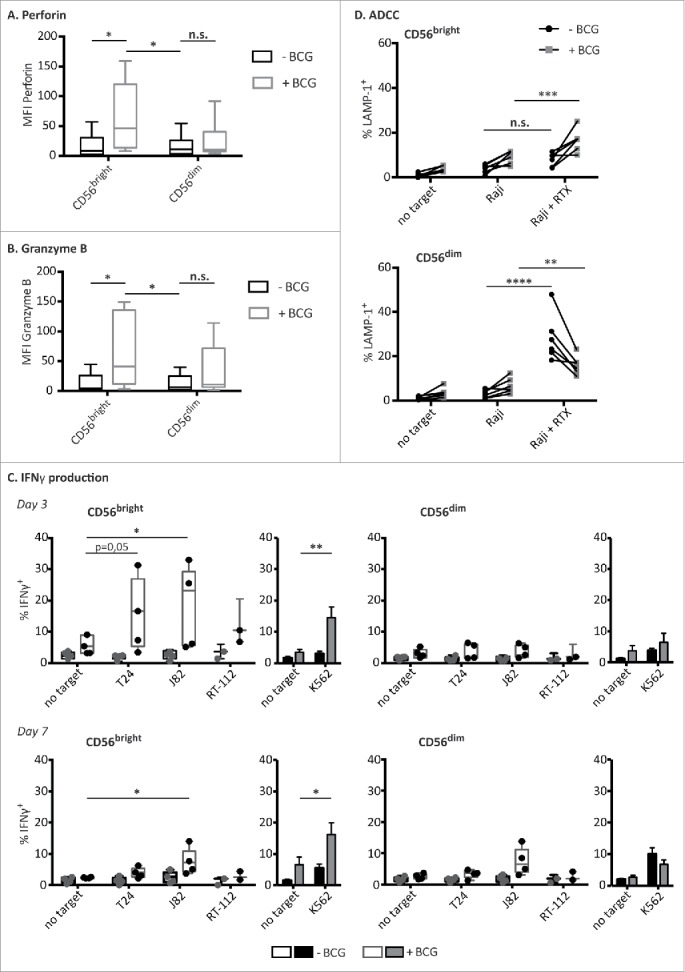
BCG-activated CD56bright NK cells capacities. PBMCs from healthy donors were incubated with or without BCG at a 1:50 ratio. Perforin (A) and granzyme B (B) content of the different NK cell subpopulations were analyzed by flow cytometry at day 7. The figures show the average and distribution of the mean fluorescence intensity obtained in each CD3−CD56+ subpopulation as indicated. The MFIs represented corresponds to nearly 100 % of the population. Comparison of the results obtained from untreated vs. BCG-treated cultures, from a total of eight independent experiments, was performed using a Student's t-test (* means significant, p <0.05; n.s., non-significant). (C) IFNγ release upon target recognition. NK cells (CD3−CD56+), CD56bright and CD56dim subpopulations, were analyzed for their ability to release IFNγ after being co-cultured with BCG for 3 (upper panels) or 7 d (bottom panels) and further incubated with the indicated target cells. Statistical analysis of 3–5 independent experiments were done using a Student's t-test (*p <0.05; **p <0.01). (D) ADCC. After one week in culture with BCG, CD56bright and CD56dim NK cell subpopulations were analyzed for their capacity to perform ADCC against Raji cells pre-incubated with rituximab. Figure shows the results obtained from six independent experiments. Statistical analysis was performed using a two-way ANOVA (*p <0.05, **p <0.01, p <0.05; ***p <0.001, ****p <0.0001).
A majority of BCG-stimulated CD56bright NK cells derive mainly from CD56dim NK cells
To define more precisely the origin of the cytotoxic CD56bright NK cell population that expanded after BCG exposure, experiments were performed using PBMC preparations from which one of the CD56-expressing NK subpopulations had been depleted. PBMCs from healthy donors were labeled with CD3 and CD56 antibodies and PBMCs lacking only the CD56bright or the CD56dim subpopulation were obtained by cell sorting. Then, co-cultures with BCG were set up, either using unfractionated PBMCs or the sorted PBMCs lacking one CD56 subpopulation (Fig. 3A). After one week in culture, these PBMC populations were analyzed by flow cytometry (Fig. 3B). As expected, exposure of whole PBMCs to BCG, led to a significant expansion of CD56bright NK cells. Strikingly, a very similar subpopulation was also observed in the co-culture initiated with PBMCs containing only CD56dim NK cells, suggesting that many of the BCG-stimulated cytotoxic CD56bright population actually derived from CD56dim cells. This CD56bright population also showed an increased expression of CD16, KIR, CD57 and CD94, demonstrating that the CD56dim population of NK cells can upregulate the expression of CD56 at the cell surface while maintaining mature-phenotype receptor expression and functional capacities. Interestingly, CD56bright NK cells expressing CD16 and increased levels of CD94 could also be generated from CD56dim-depleted PBMCs exposed to BCG, suggesting that differentiation of the CD56bright NK subset after exposure to BCG also contributed to the observed expansion of phenotypically mature and cytotoxic CD56bright NK cells.
Figure 3.
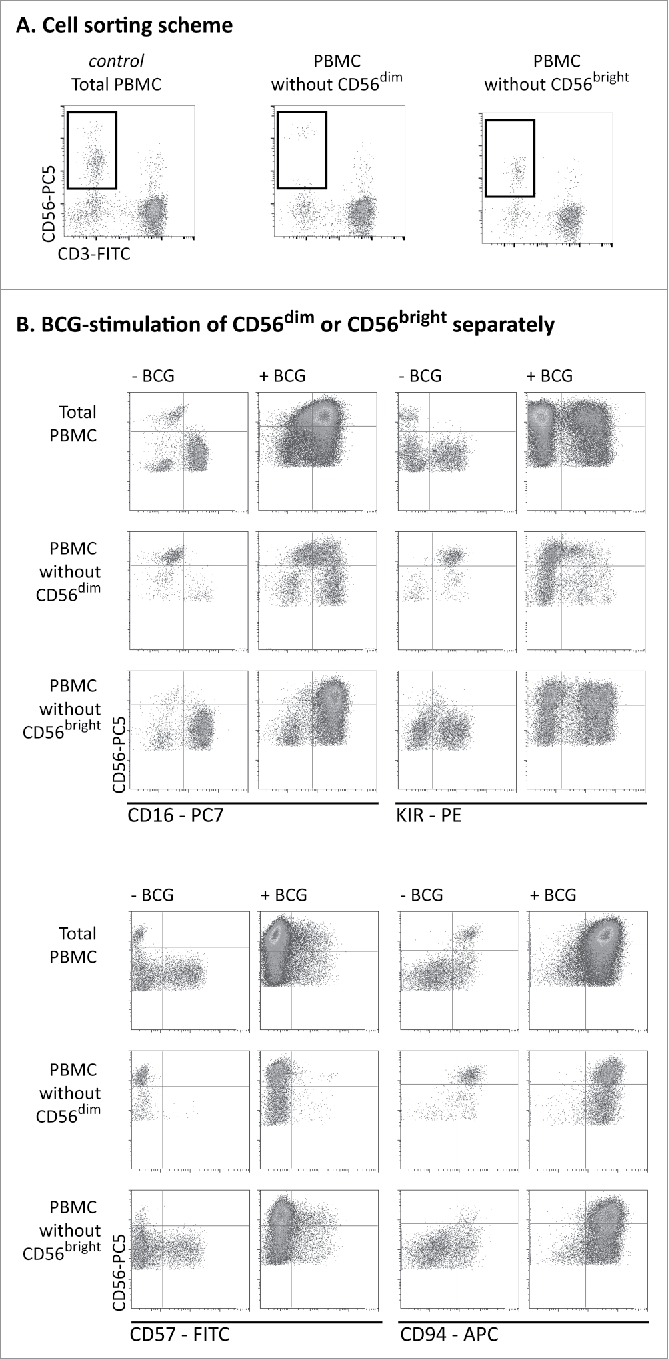
BCG-stimulated CD56bright NK cells originate mainly from CD56dim cells. (A) PBMCs from healthy donors were labeled with directly conjugated antibodies. CD3− CD56bright and CD3−CD56dim populations were eliminated by cell sorting and the resulting PBMC population was analyzed by flow cytometry. (B) The cells obtained in (A) were incubated for 1 week with or without BCG at a 1:50 ratio. Samples were analyzed by flow cytometry at day 7. Figure shows the expansion of the CD56bright subset and NK receptor phenotyping in a representative donor. This experiment was repeated three times.
BCG-stimulated CD56bright NK cells contain cytotoxic granules and mediate ADCC
The function of the CD56bright NK cell population that expanded after co-culture with BCG was studied next. CD56dim NK cells are usually preloaded with granzyme B and perforin-containing granules. When PBMCs were cultured in medium alone, perforin and granzyme expression decreased (data not shown). In contrast, after co-culture with BCG, the CD56bright NK cells contained significantly more perforin and granzyme B, consistent with their cytotoxic capacities (Figs. 4A and B). There was no statistically significant increase in perforin and granzyme B-containing granules of CD56dim NK cells. Since CD56bright NK cells are usually efficient cytokine producers,13 the ability of BCG-stimulated NK cells to secrete IFNγ after target-cell recognition was also analyzed (Fig. 4C). IFNγ production by CD56bright NK cells was increased at day 3 of BCG co-culture, when compared with the unstimulated CD56bright NK cells, however, at day 7, no IFNγ production was triggered by bladder cancer cells under any condition. This result indicates that, before the burst of NK cell proliferation and the increase in surface CD56 expression, each subpopulation still displayed their main functional capacity: i.e., CD56bright being secretory and CD56dim cytotoxic. CD56 upregulation was evident from days 4–5 of the co-culture (data not shown). By day 7, when the CD56bright NK cell subpopulation was mainly composed of CD56dim cells with increased levels of cell surface CD56, the main functional characteristic of this population was cytotoxicity.
Consistent with the expression of CD16 on the BCG-stimulated CD56bright cells (Fig. 2), these effector cells could also mediate ADCC against Raji cells that had been previously sensitized with Rituximab (Fig. 4D). Also, the loss of CD16 in BCG-treated CD56dim NK cells resulted in a decrease of ADCC activity.
All these data are consistent with the idea that the majority of these CD56bright cells originate from CD56dim NK cells which have upregulated cell surface expression of the CD56 molecule and undergone intense cell proliferation. A transformation that likely reflects alterations in the maturation and activation status of these NK cells.
Expansion and priming of BCG-stimulated CD56bright NK cells depend on soluble factors
The expansion of BCG-stimulated CD56bright NK cells primed to degranulate in response to cancer cells could depend on direct interactions with BCG, cross-talk with other cellular populations from peripheral blood or soluble factors secreted within the co-culture. Our previous experiments argued against the first possibility,11 thus whether the priming and/or the expansion of CD56bright population depended on the presence of soluble factors was tested. Supernatants obtained after one week of PBMC and BCG co-culture were prepared and used to stimulate a new culture of PBMCs without BCG addition. In this culture, a marked expansion of the CD56bright NK cells (Fig. 5A), as well as the potentiation of the degranulation capacity against bladder tumor cells (Fig. 5B) were also observed. However, only modest increases in the degranulation capacity of CD56dim NK cells in these cultures were observed. These data indicate that soluble factors are involved in the expansion of BCG-activated CD56bright NK cells. To confirm this hypothesis and check whether direct contact with other immune populations was needed to generate the population of activated CD56bright, experiments where purified CD56bright or CD56dim NK cells were separated by a transwell from a PBMC/BCG co-culture were performed. Under these conditions, CD56dim cells acquired the CD56bright phenotype (Fig. 5C), accompanied by expression of CD16 (Fig. 5D) and other markers usually present in the CD56dim population, such as KIR and CD57 (data not shown), even though they only had contact with the supernatants generated in PBMCs co-cultured with BCG. Interestingly, the degree of CD56 acquisition varied slightly between different donors, probably reflecting variation in the time required by NK cells from each donor to achieve full activation.
Figure 5.
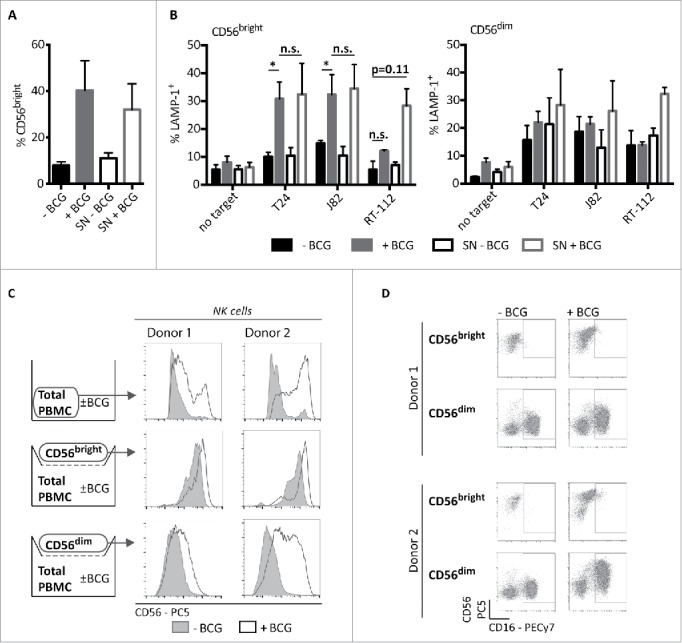
Antitumor CD56bright induction by BCG requires soluble factors. CD56bright expansion (A) and degranulation (B) in experiments using BCG-conditioned media. PBMCs from healthy donors were supplemented as indicated with supernatant (SN) from a previous 7-d co-culture of PBMCs with BCG (BCG-conditioned SN) or from a 7-d culture of PBMCs alone (untreated SN). For reference, untreated and BCG-treated PBMCs were used. At day 7, cells were recovered and the percentage of CD56bright NK cells was analyzed by flow cytometry (A) or degranulation assays against bladder cancer cells were performed (B). As positive control for the assay, NK cell activity against K562 cells was analyzed (not shown). Statistical analysis of four independent experiments was performed using Student t-test (* means significant, p <0.05; n.s., not significant). CD56bright expansion (C) and phenotype (D), after NK cell subpopulation sorting and incubation using Transwell. PBMCs from three healthy donors (two donors shown) were labeled with directly conjugated antibodies. CD3− CD56bright and CD3−CD56dim populations were separated by cell sorting and set in the upper chamber of a transwell. In the lower chamber, total PBMCs were incubated for 7 d in the presence or absence of BCG. After this time, NK cells subpopulations from the upper chamber were analyzed by flow cytometry.
Next, the amounts of an extensive panel of cytokines and chemokines were measured at different time points in supernatants from in vitro co-cultures of BCG and PBMCs of three healthy donors (Fig. 6). In parallel, the expansion and degranulation capacity of CD56bright NK cells after BCG exposure were also evaluated (Fig. S5). These three donors showed similar patterns and levels of production of soluble factors, suggesting only limited variation in the response to BCG between donors. IL2 was found at a similar concentration in both untreated and BCG-treated co-cultures, and may be responsible for the survival of T and NK cells in the culture. IL4, IL8, IL9, IL22, IL23 and MIG were also found in both conditions, although the amounts of these cytokines were higher in the BCG-treated cultures. The levels of IL9, IL22 and IL23 (from day 1) and MIG (from day 3) increased during the week in co-culture with BCG, although some of these cytokines could also be detected at low concentrations in PBMC cultures not exposed to BCG.
Figure 6.
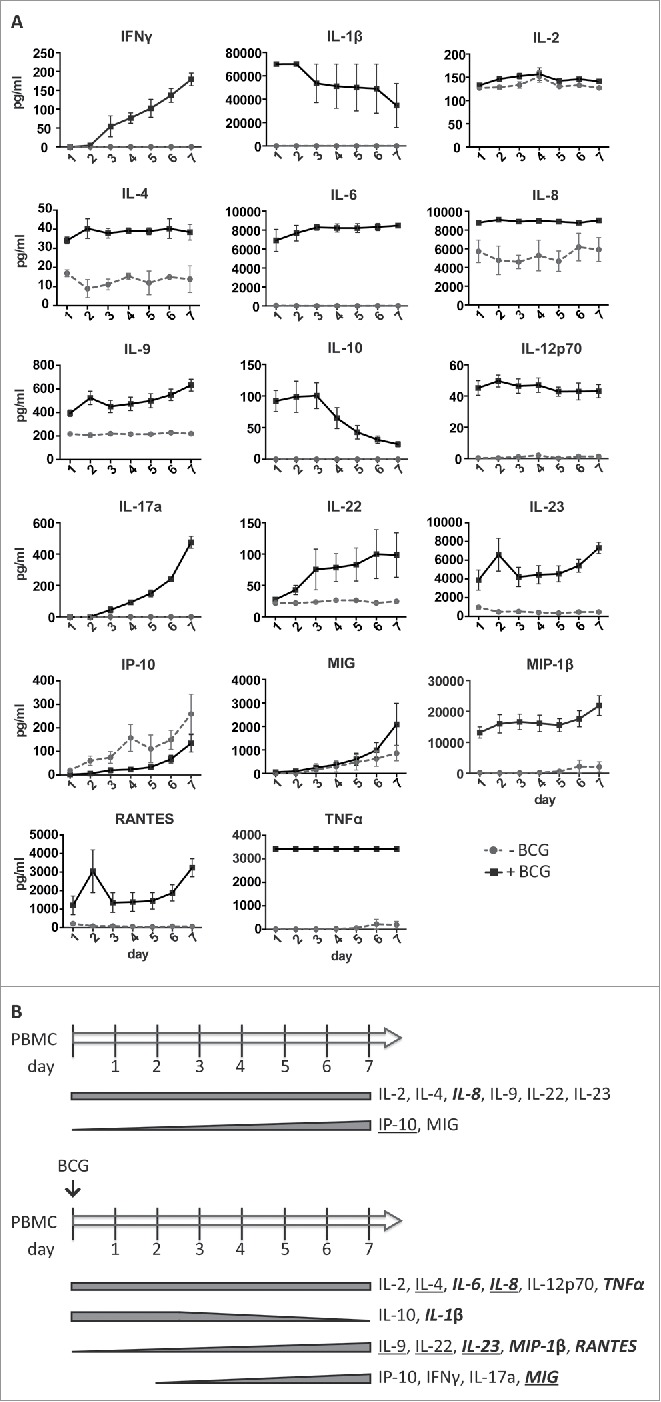
Soluble factors released by PBMCs in the presence of BCG during one week of co-incubation. PBMCs from three healthy donors were incubated with or without BCG at a 1:50 ratio. Daily, supernatants were recovered, centrifuged to eliminate cells and bacteria, and used to determine the content in soluble factors by Luminex. NK cells from the different cultures were analyzed at day 7 to control the expansion and capacity to respond by degranulation against different bladder cell lines (Fig. S4). (A) Quantitation of the indicated cytokines or chemokines contained in the supernatants during 7 d. (B) Summary of the cytokines and chemokines released by PBMCs co-cultured in vitro with or without BCG. Bold and italics: soluble factors detected at ≥ 1,000 pg/mL; Underlined: condition in which the concentration was higher, when a soluble factor was detected in cultures with or without BCG.
IL6, IL12p70, TNFα were found at constant concentrations in the supernatants of the BCG-treated PBMCs from the first day of co-culture, but were undetectable in the untreated cultures. Many of these cytokines have been found in urine collected from patients immediately after the BCG instillations.22 High amounts of IL10 and IL1β were detected on day 1 in BCG containing co-cultures (and undetectable in the absence of BCG), but then decreased over time. Increasing concentrations of MIP-1β and RANTES (from day 1) and IFNγ and IL17a (from day 3) were detected in the presence of BCG but not found in untreated cultures. Increasing concentrations of IP10 were found in both conditions, suggesting that myeloid-lineage cells may be activated in these cultures.
In general, the pattern of cytokines found in the co-cultures during the first days agrees with the initial pro-inflammatory response detected in patient samples collected immediately after the instillations where a high degree of acute inflammation was accompanied by elevated levels of TNFα, IL6, IL1β and IL8 [22–24 and references therein]. However, our new data reveal a marked increase in the release of a range of other innate and pathogen-induced soluble factors, in the days following the first 48 h after exposure to BCG. These data suggest that a second wave of factors, including IFNγ, IL17a, IL22, IL23 and MIP-1 β, might contribute to modulate the immune response and help in the elimination of the tumor.
NK cell phenotype in BCG-treated bladder cancer patients
In light of the above in vitro experiments, it was of interest to analyze whether related changes could be detected in the immune response of patients in vivo. For this purpose, immune changes in PBMCs of a cohort of 10 bladder cancer patients treated with BCG were followed for 18 mo. Samples were obtained either 7 d or 3 mo after each instillation, to focus on monitoring the long-lasting features of the response (Fig. 7A). As a control, seven bladder cancer patients receiving mitomycin C instillations, with a different schedule of treatment, were also included in the study. NK cells were studied in these blood samples (Fig. 7B). This approach was limited by the fact that NK cells that had re-circulated through the bladder would only be a fraction of peripheral blood NK cells. Nevertheless, a higher percentage of CD56bright NK cells was present on average in BCG-treated patients compared with mitomycin C-treated patients, which was closer to the proportion (5%) observed in healthy donors (Fig. 7C). NK cells from peripheral blood of the BCG-treated patients also showed an increase in the MFI of CD16 when comparing the samples obtained at time 0 with those obtained one year later (Fig. 7D). This increased CD16 expression was statistically significant for CD56bright NK cells from BCG-treated patients, but not for the CD56dim subpopulation. No statistically significant changes for either NK cell subpopulation was noted in mitomycin C-treated patients after 3 mo. This observation is consistent with the data obtained in our in vitro experiments. Altogether, these results suggest that a larger cohort including a panel of NK cell markers should be studied to establish the link between levels of CD56 and response to treatment. Furthermore, since the changes in PBMCs are likely to be mild, a detailed study of the immune cells found in urine might provide additional useful information.
Figure 7.
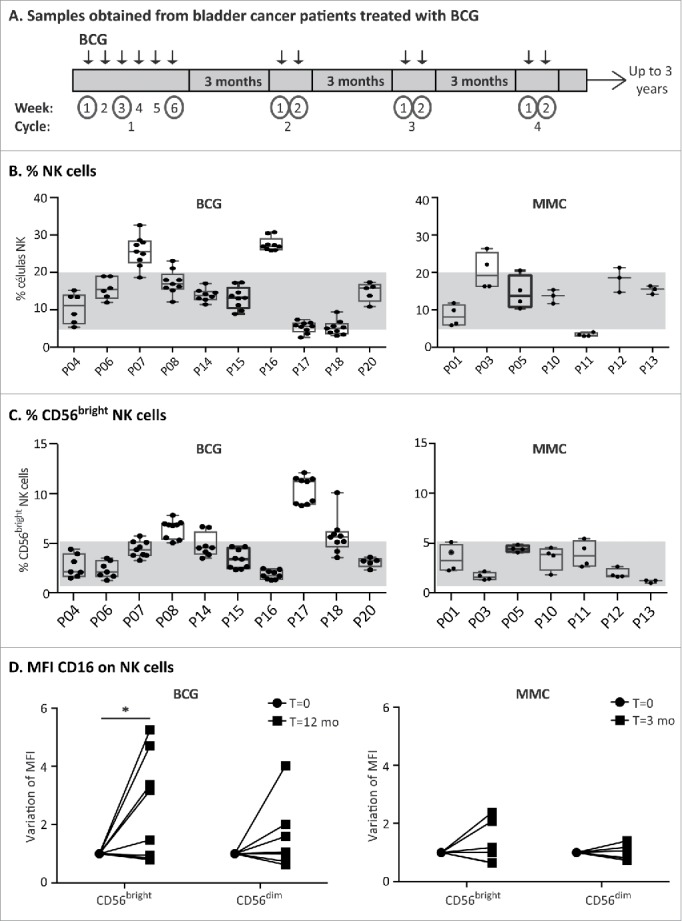
Analysis of blood samples obtained from bladder cancer patients treated with BCG. (A) Schematic representation of sample collection. A cohort of bladder cancer patients (Ta/T1G3 or CIS, mean age 72.8 y old) receiving intravesical BCG instillations was recruited. During the first cycle, patients received one weekly instillation for 6 weeks (indicated by arrows). After a 3-mo rest period, they received a second cycle of 2 weekly instillations and they continued this treatment of up to 3 y. Samples were obtained just before receiving an instillation at the time points indicated with a circle. Therefore, the sample obtained just before the first instillation of the first cycle is the basal level. Remaining samples were obtained a week after the patient received an instillation or after the 3-mo rest period. (B and C) NK cell analysis of patient PBMCs. Whole blood was analyzed by multiparametric flow cytometry. Gating was performed by selecting lymphocytes by FSC/SSC and the percentage of total NK cells (B) as well as CD56bright (C) on total NK cells was obtained. The figure represents the average and standard deviation of the CD56 percentages of all samples obtained for each patient along the treatment. (D) CD16. The MFI of CD16 was determined in the different NK cell populations of the patients at different times (12 time points for BCG-treated patients; 4 time points for mitomycin C-treated patients). CD16 MFI ranged between 0.6 and 8 in CD56bright, and 9.8 and 40.3 for CD56dim within these patients cohorts (data not shown). The values of the CD16 MFI from the initial (before starting the treatment) and final samples (12 mo for BCG, 3 mo for mitomycin C) were normalized for comparison and statistical analysis. The significance of the variation between the initial and final sample was analyzed by t-test using the Sidak–Bonferroni method (* means significant, p <0.05).
Discussion
Here, we report that the exposure of PBMCs to BCG results in the activation and proliferation of an unconventional cytotoxic subpopulation of CD56bright NK cells that maintain expression of receptors such as CD16 and KIR. The expansion of these cells depends on soluble factors, mainly innate cytokines. We show that, after one week of co-culture with BCG, these activated CD56bright NK cells have an unusual CD3− CD56bright CD94bright CD16+ KIR2D+ phenotype and have become specialized to mediate cytotoxicity. We also report that, in a small cohort of bladder cancer patients treated with BCG, a small, but significant, increase in numbers of CD56bright CD16+ NK cells circulating in peripheral blood was observed.
While the generation of a CD56bright NK cell subset in response to live BCG or other bacterial compounds has been described previously,5,25,26 in this paper, we report for the first time that the NK cells expanded on co-culture with BCG are mainly derived from the CD56dim subpopulation, although CD56bright cells also can increase the MFI of other NK receptors. These NK cells have upregulated CD56 to become CD56bright effectors and are activated (as shown by the expression of CD25 and CD69), proliferate vigorously and degranulate against bladder cancer cells. Moreover, they maintain many of the phenotypic and functional characteristics of a mature NK subpopulation, including the high expression of several NK receptors and the capacity to mediate ADCC. In contrast to what is known about NK cells responding to viruses, BCG-stimulated CD56bright cells do not seem to correspond to a population that can be defined by the expression of single or a limited number of cell surface receptors. For example, HCMV infection is associated with the expansion of a late differentiated CD57+ NKG2C+ population,27,28 that is usually CD16low, while a CD56dim NKG2D+ NKG2A+ CD57− KIR− early differentiated subpopulation expands after EBV infections.29,30 In contrast, BCG exposure clearly activates a wider range of NK cells including CD56dim CD94+ KIR− and CD56dim CD57+ KIR+ NK cells (Figs. S4D and E). Thus, these data on changes in NK cells induced by BCG exposure provide a clear example of NK cell plasticity. These observations of the retention of CD16 expression and ADCC function are also in marked contrast to the matrix metalloprotease mediated shedding that produces a loss of CD16, which generally accompanies CD56 upregulation after NK stimulation by target cells and cytokines.31,32
It is also interesting to note that sorted CD56bright NK cells can also contribute to the expansion of CD16+CD94+KIR+ NK cells on co-culture with BCG. Human NK cells develop from CD34+ haematopoietic stem cells (HSC) that acquire different receptors sequentially, allowing characterization of several intermediate immature stages.33,34 In a late stage, CD56bright cells (stage 4), characterized by high expression of CD94 and the absence of CD16, have been proposed to become mature CD56dim cells (stage 5), acquiring CD16 and KIR expression and losing CD94 and CD117 in this process,19,35,36 although the exact sequence of events during these later stages of NK cell differentiation is still debated.32 To date, it has proven difficult to drive the differentiation of purified CD56bright stage 4 NK cells toward later stages of NK cell development in vitro. Our data have shown that BCG treatment of PBMC cultures can also provoke CD56bright CD16− NK cells to upregulate CD16 and CD94 and transition toward a CD56bright cytotoxic population with a reduced ability to produce IFNγ that could correspond to an intermediate between the defined differentiation stages 4 and 5 of NK cell differentiation.36,37 Double-positive CD56bright CD16+ cells are found in small numbers in peripheral blood and have been observed in other situations such as aging,38,39 infection 26 and transplantation.40 CD56bright NK cells expressing CD16 have also been described in metastatic lymph nodes in melanoma 41 and breast cancer 42 and are a subject of active research. The data reported here suggest that the cytotoxic CD56bright CD16+ cells that expand after BCG stimulation could represent an alternative pathway of NK cell maturation from CD56dim precursors, perhaps related to IL12 and IL15-stimulated NK cells, which also retain the CD16+ phenotype 26 and emphasize that there may be considerably more plasticity in the pathways of NK cell maturation than currently appreciated.
In this paper, we also demonstrate that the soluble factors released after BCG stimulation of PBMCs are sufficient to drive the expansion and priming of the anti-tumoral CD56bright NK cells. Our analysis of cytokines present in the in vitro co-cultures of PBMCs and BCG revealed interesting features about the cellular populations that could be involved in the response, since several of the factors are most likely produced by myeloid cells and other innate immune cells. The co-culture of PBMCs with BCG is enough to initiate a strong cytokine response with high amounts of a range of soluble factors, including TNFα, increasing amounts of monokines, such as RANTES, MIP-1β and IP10, as well as an initial pulse of IL10 that subsequently declines and practically disappears. T cells are probably responsible for the secretion of IL2, detected both in the presence and absence of BCG and, thus, it seems reasonable to assume that both T cells and antigen-presenting cells probably participate in the secretion of cytokines that drive the NK expansion and differentiation in the culture.
Finally, we have looked in vivo for correlates of our in vitro observations. We have evaluated the presence of CD3−CD56bright CD16+ effector NK cells in blood samples from bladder cancer patients treated with either BCG instillation or mitomycin C. These analyses are difficult because detection of small populations of effector cells in peripheral blood is a very indirect manner of evaluating the local response in the bladder and is further complicated by the usually low percentage of circulating CD3−CD56bright cells. Nevertheless, we could observe a clear pattern of increased CD56 expression in 6 out of 10 BCG-treated patients that was not noted in mitomycin C-treated patients. Importantly, CD16 expression was brighter on CD56bright NK cells from bladder cancer patients that had undergone BCG treatment compared with NK cells from mitomycin C-treated patients, again confirming the in vitro data. These initial data should be further confirmed by analyzing PBMCs from a larger cohort as well as different time points. However, since our analyses of patients at least partially validate the in vitro model, it also seems plausible that analysis of the immune response activated in the bladder might provide more direct and thus stronger data than that obtained from studies on peripheral blood. For this reason, we are currently recruiting patients for a new study characterizing the immune cells shed into urine, and thus susceptible to analysis by flow cytometry, with the idea that this will allow exploration of whether the CD56bright CD16+ effector NK cell population is recruited to, or expanded in, cancerous bladder tissue.
Materials and methods
Reagents and antibodies
Directly-labeled antibodies for analysis of blood lymphocytes were from Biolegend and Immunotools (Table S1). Biotinylated anti-human granzyme B antibody was purchased from MABTECH and biotinylated anti-human perforin from Ancell; APC-Cy7-conjugated streptavidin from Biolegend. PE-conjugated anti-human IFNγ antibody was from BD PharMingen. Secondary FITC- and PE-conjugated anti-mouse Ig antibodies were from DakoCytomation. Blocking antibodies, specific for anti-human NKG2D (clone 149810) and NKp46/NCR1 (clone 195314) were from R&D.
BCG Tice strain (from Merck Canada Inc.) was used. Aliquots of reconstituted BCG were prepared in RPMI 10% DMSO and stored at −20˚C. The viability of the bacteria was not affected by this process.
Cell lines and peripheral blood mononuclear cell culture
Culture of the bladder cancer cell lines UM-UC-3, RT-112, J82, T24, SW780 and RT4 used in this study has been described previously.11
PBMCs from healthy volunteer buffy coats (Regional Transfusion Center, Madrid), were isolated by centrifugation on Ficoll-HyPaque and cultured in complete (4 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 U/mL streptomycin, 10 mM Hepes, 50 µM β-mercaptoethanol) RPMI-1640 medium (Lonza) supplemented with 5% FBS, 5% male AB human serum, (Biowest). K562 cells were used for functional assays and grown in complete RPMI-1640 medium (Lonza) supplemented with 10% FBS.
PBMCs were incubated in 24-well plates at 106 cells/mL with or without BCG at a 1:50 ratio (viable bacteria to PBMC). At the days indicated for each experiment, cells in suspension were recovered from the co-culture, centrifuged, analyzed by flow cytometry or used as effector cells in functional experiments. In experiments using BCG-conditioned medium, supernatants were recovered at day 7, centrifuged at 200 × g to eliminate cells and at 13,000 × g to eliminate bacteria. Supernatants were added to a fresh PBMC culture, diluting 1:2 with fresh complete RPMI supplemented with 5% FBS, 5% male AB human serum. Cells were incubated for a further week.
Degranulation assays and ADCC
Untreated or BCG-treated PBMCs were co-cultured with target cells, pretreated with HP1F7 antibody to block MHC-I,43 for 2 h at an E:T ratio of 5:1 [i.e., 1:2 NK:target ratio]. NK cells would represent around 10% of total PBMCs. Although some donor-to-donor variation was encountered, the difference between treated and untreated was not significant, thus not affecting the comparison. In experiments where NK cell receptors were blocked, monoclonal antibodies were included in the medium to a final concentration of 5 µg/mL for 20 min, before co-incubation with target cells. K562 cells were used as positive control targets for NK degranulation. For ADCC, Raji cells, pretreated or not with Rituximab (RTX), were used as targets in 2 h experiments at an E:T ratio of 5:1 [i.e., 1:2 NK:target ratio]. Surface expression of LAMP1 (CD107a) was analyzed by flow cytometry. Statistical analyses were performed using the Prism 6 software.
Flow cytometry
Cells were incubated with the appropriate primary antibodies, followed by either streptavidin-APC-Cy7, or PE- or FITC-labeled F(ab')2 fragments of goat anti-mouse Ig (Dako), or directly with the conjugated antibodies for surface markers. For intracellular staining (granzyme B, perforin, IFNγ), cells were fixed with 2% para-formaldehyde at room temperature (RT) for 10 min and permeabilized with 0.2% saponin at RT for 10 min. For identification of cell death, cells were stained, washed and incubated with Annexin V-PE (Immunostep) and 7-AAD (Sigma), following the manufacturer's instructions, and then analyzed by flow cytometry. Samples were analyzed using BD FACSCalibur (Becton Dickinson), Gallios Flow Cytometer or Cytomics FC 500 (Beckman Coulter). Analysis of the experiments was performed using Kaluza or FlowJo softwares.
Proliferation assays
PBMCs were incubated with 2 μM CellTrace™ Violet stain (Molecular Probes) for 20 min at 37˚C 5% CO2. RPMI 10% FBS was then added for 5 min and the cells were washed once with complete medium and resuspended again before plating in 24-well plates in the presence or absence of BCG. At different times (days 1–7), cells were recovered and analyzed by flow cytometry.
Measurement of IFNγ production
Untreated or BCG-treated PBMCs were co-cultured with target cells for 6 h at 37˚C 5% CO2 at a PBMC:target ratio of 5:1 [i.e., 1:2 NK:target ratio]. After 1 h of co-incubation, monensin was added to a final concentration of 2.5 μM. After 6 h, cells were recovered, fixed, permeabilized and intracellular IFNγ was analyzed by flow cytometry.
Cell sorting experiments
PBMCs were stained in sterile conditions with directly conjugated anti-CD3-FITC and anti-CD56-APC antibodies and, after washing and filtering, they were processed through a HIPERSORT MoFlow XDP sorter cytometer to eliminate either CD56bright or CD56dim NK subpopulations. The composition of the sorted populations was assessed by flow cytometry and PBMC containing only one of the NK cell subsets were analyzed in the experiments.
Cytokine measurements
Tissue culture supernatants were centrifuged at 200 × g to eliminate cells and stored at −80°C until Luminex analysis. Samples were analyzed in duplicates using human magnetic Luminex® screening assays (R&D Systems) and Luminex® 100™ or 200™ (Qiagen) Luminex analyzer instruments, according to manufacturers' instructions. Five-parameter logistic standard curves were generated and analyte concentrations within each sample were interpolated (taking into account the dilution factor) using the Bio-Plex Manager™ Software (Bio-Rad).
Patient samples
Samples were obtained with the understanding and the informed consent of each participant and approved by local and regional ethical committees (CEIC La Paz Hospital, CEI Infanta Sofía Hospital and CSIC Local Ethical Committee). Peripheral blood was obtained from a cohort of bladder cancer patients (Ta/T1 G3 or CIS, mean age 72.8) receiving BCG instillations, at different times during treatment at Hospital Infanta Sofía (Madrid, Spain). As control, bladder cancer patients (Ta/T1 G2, mean age 70.1) receiving mitomycin-C instillations were also recruited. 100 µL of whole blood were stained using directly conjugated antibodies and analyzed by flow cytometry for the expression of CD56 and CD16 using a Gallios Flow cytometer and Kaluza software. Data processing was performed using an algorithm created to compile information for further statistical analysis using the GraphPad Prism 6 package. Unless otherwise indicated, statistical significance was assessed using multiple t-tests and corrected for multiple comparisons using the Sidak–Bonferroni method.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank the collaboration of patients and nurses from Infanta Sofía Hospital, MC Moreno and S. Escudero from the Cytometry Service at the CNB, A. Valés-Gómez for informatics support and Immunotools for the gift of antibodies [IT-BOX-139 2012, IT-special-Award 2014].
Funding
This work was supported by grants from Madrid Regional Government “INMUNOTHERCAN” [S2010/BMD-2326 (LMP, MVG)]; the Spanish Ministries of Economy and Health [SAF-2012–32293, SAF2015–69169-R (MVG) and SAF2014–58752-R (HTR)]; EMGC and SLC are recipients of Fellowships from La Caixa and Spanish Ministry of Education (FPU), respectively.
Author contributions
EMGC, GE and OA optimized the assays, acquired and analyzed data and contributed to writing the manuscript. SLC, MAM and AL acquired data. MMH provided material support and supervised the study. LMP conceived and designed the patient study, provided material support and supervised the study. MVG and HTR acquired data, conceived, designed and supervised the study, provided material support and wrote the manuscript.
References
- 1.Freund J. The mode of action of immunologic adjuvants. Bibliotheca tuberculosea 1956:130-48; PMID:13341884 [PubMed] [Google Scholar]
- 2.Gandhi NM, Morales A, Lamm DL. Bacillus Calmette-Guerin immunotherapy for genitourinary cancer. BJU Int 2013; 112:288-97; PMID:23517232; http://dx.doi.org/ 10.1111/j.1464-410X.2012.11754.x [DOI] [PubMed] [Google Scholar]
- 3.Brandau S, Suttmann H. Thirty years of BCG immunotherapy for non-muscle invasive bladder cancer: a success story with room for improvement. Biomed Pharmacother 2007; 61:299-305; PMID:17604943; http://dx.doi.org/ 10.1016/j.biopha.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 4.Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer-a current perspective. Nat Rev Urol; 11:153-62; PMID:24492433; http://dx.doi.org/ 10.1038/nrurol.2014.15 [DOI] [PubMed] [Google Scholar]
- 5.Brandau S, Bohle A. Activation of natural killer cells by Bacillus Calmette-Guerin. Eur Urol 2001; 39:518-24; PMID:11464031; http://dx.doi.org/ 10.1159/000052497 [DOI] [PubMed] [Google Scholar]
- 6.Brandau S, Riemensberger J, Jacobsen M, Kemp D, Zhao W, Zhao X, Jocham D, Ratliff TL, Böhle A. NK cells are essential for effective BCG immunotherapy. Int J Cancer 2001; 92:697-702; PMID:11340575; http://dx.doi.org/ 10.1002/1097-0215(20010601)92:5%3c697::AID-IJC1245%3e3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 7.Brandau S, Suttmann H, Riemensberger J, Seitzer U, Arnold J, Durek C, Jocham D, Flad HD, Böhle A. Perforin-mediated lysis of tumor cells by Mycobacterium bovis Bacillus Calmette-Guerin-activated killer cells. Clin Cancer Res 2000; 6:3729-38; PMID:10999767 [PubMed] [Google Scholar]
- 8.Suttmann H, Jacobsen M, Reiss K, Jocham D, Bohle A, Brandau S. Mechanisms of bacillus Calmette-Guerin mediated natural killer cell activation. J Urol 2004; 172:1490-5; PMID:15371877; http://dx.doi.org/ 10.1097/01.ju.0000131944.52354.63 [DOI] [PubMed] [Google Scholar]
- 9.Higuchi T, Shimizu M, Owaki A, Takahashi M, Shinya E, Nishimura T, Takahashi H. A possible mechanism of intravesical BCG therapy for human bladder carcinoma: involvement of innate effector cells for the inhibition of tumor growth. Cancer Immunol Immunother 2009; 58:1245-55; PMID:19139883; http://dx.doi.org/ 10.1007/s00262-008-0643-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naoe M, Ogawa Y, Takeshita K, Morita J, Iwamoto S, Miyazaki A, Yoshida H. Bacillus Calmette-Guerin-pulsed dendritic cells stimulate natural killer T cells and gammadeltaT cells. Int J Urol 2007; 14:532-8; discussion 8; PMID:17593099; http://dx.doi.org/ 10.1111/j.1442-2042.2006.01697.x [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Cuesta EM, Lopez-Cobo S, Alvarez-Maestro M, Esteso G, Romera-Cardenas G, Rey M, Cassady-Cain RL, Linares A, Valés-Gómez A, Reyburn HT et al.. NKG2D is a key receptor for recognition of bladder cancer cells by IL-2-activated NK cells and BCG promotes NK cell activation. Front Immunol 2015; 6:284; PMID:26106390; http://dx.doi.org/ 10.3389/fimmu.2015.00284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lodoen MB, Lanier LL. Natural killer cells as an initial defense against pathogens. Curr Opin Immunol 2006; 18:391-8; PMID:16765573; http://dx.doi.org/ 10.1016/j.coi.2006.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 2013; 31:227-58; PMID:23516982; http://dx.doi.org/ 10.1146/annurev-immunol-020711-075005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol 2013; 132:536-44; PMID:23906377; http://dx.doi.org/ 10.1016/j.jaci.2013.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strauss-Albee DM, Blish CA. Human NK Cell Diversity in Viral Infection: Ramifications of Ramification. Front Immunol 2016; 7:66; PMID:26973646; http://dx.doi.org/ 10.3389/fimmu.2016.00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol 2001; 22:633-40; PMID:11698225; http://dx.doi.org/ 10.1016/S1471-4906(01)02060-9 [DOI] [PubMed] [Google Scholar]
- 17.Voss SD, Daley J, Ritz J, Robertson MJ. Participation of the CD94 receptor complex in costimulation of human natural killer cells. J Immunol 1998; 160:1618-26; PMID:9469418 [PubMed] [Google Scholar]
- 18.Andre P, Spertini O, Guia S, Rihet P, Dignat-George F, Brailly H, Sampol J, Anderson PJ, Vivier E. Modification of P-selectin glycoprotein ligand-1 with a natural killer cell-restricted sulfated lactosamine creates an alternate ligand for L-selectin. Proc Natl Acad Sci U S A 2000; 97:3400-5; PMID:10725346; http://dx.doi.org/ 10.1073/pnas.97.7.3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, Carson WE, Caligiuri MA. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001; 97:3146-51; PMID:11342442; http://dx.doi.org/ 10.1182/blood.V97.10.3146 [DOI] [PubMed] [Google Scholar]
- 20.Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010; 115:2167-76; PMID:19965656; http://dx.doi.org/ 10.1182/blood-2009-08-238469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bade B, Boettcher HE, Lohrmann J, Hink-Schauer C, Bratke K, Jenne DE, Jr Virchow JC, Luttmann W. Differential expression of the granzymes A, K and M and perforin in human peripheral blood lymphocytes. Int Immunol 2005; 17:1419-28; PMID:16186162; http://dx.doi.org/ 10.1093/intimm/dxh320 [DOI] [PubMed] [Google Scholar]
- 22.Bisiaux A, Thiounn N, Timsit MO, Eladaoui A, Chang HH, Mapes J, Mogenet A, Bresson JL, Prié D, Béchet S et al.. Molecular analyte profiling of the early events and tissue conditioning following intravesical bacillus calmette-guerin therapy in patients with superficial bladder cancer. J Urol 2009; 181:1571-80; PMID:19230924; http://dx.doi.org/ 10.1016/j.juro.2008.11.124 [DOI] [PubMed] [Google Scholar]
- 23.Zuiverloon TC, Nieuweboer AJ, Vekony H, Kirkels WJ, Bangma CH, Zwarthoff EC. Markers predicting response to bacillus Calmette-Guerin immunotherapy in high-risk bladder cancer patients: a systematic review. Eur Urol 2011; 61:128-45; PMID:22000498; http://dx.doi.org/ 10.1016/j.eururo.2011.09.026 [DOI] [PubMed] [Google Scholar]
- 24.Luo Y, Chen X, O'Donnell MA. Mycobacterium bovis bacillus Calmette-Guerin (BCG) induces human CC- and CXC-chemokines in vitro and in vivo. Clin Exp Immunol 2007; 147:370-8; PMID:17223980; http://dx.doi.org/ 10.1111/j.1365-2249.2006.03288.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batoni G, Esin S, Favilli F, Pardini M, Bottai D, Maisetta G, Florio W, Campa M. Human CD56bright and CD56dim natural killer cell subsets respond differentially to direct stimulation with Mycobacterium bovis bacillus Calmette-Guerin. Scand J Immunol 2005; 62:498-506; PMID:16316416; http://dx.doi.org/ 10.1111/j.1365-3083.2005.01692.x [DOI] [PubMed] [Google Scholar]
- 26.Takahashi E, Kuranaga N, Satoh K, Habu Y, Shinomiya N, Asano T, Seki S, Hayakawa M. Induction of CD16+ CD56bright NK cells with antitumour cytotoxicity not only from CD16- CD56bright NK Cells but also from CD16- CD56dim NK cells. Scand J Immunol 2007; 65:126-38; PMID:17257217; http://dx.doi.org/ 10.1111/j.1365-3083.2006.01883.x [DOI] [PubMed] [Google Scholar]
- 27.Guma M, Budt M, Saez A, Brckalo T, Hengel H, Angulo A, López-Botet M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006; 107:3624-31; PMID:16384928; http://dx.doi.org/ 10.1182/blood-2005-09-3682 [DOI] [PubMed] [Google Scholar]
- 28.Lima JF, Oliveira LM, Pereira NZ, Mitsunari GE, Duarte AJ, Sato MN. Distinct natural killer cells in HIV-exposed seronegative subjects with effector cytotoxic CD56(dim) and CD56(bright) cells and memory-like CD57(+)NKG2C(+)CD56(dim) cells. J Acqu Immune Defici Synd 2014; 67:463-71; PMID:25230289; http://dx.doi.org/ 10.1097/QAI.0000000000000350 [DOI] [PubMed] [Google Scholar]
- 29.Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, Staubli G, Gysin C, Berger C, Münz C et al.. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 2014; 124:2533-43; PMID:25205117; http://dx.doi.org/ 10.1182/blood-2014-01-553024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatton O, Strauss-Albee DM, Zhao NQ, Haggadone MD, Pelpola JS, Krams SM, Martinez OM, Blish CA. NKG2A-expressing natural killer cells dominate the response to autologous lymphoblastoid cells infected with Epstein-Barr virus. Front Immunol 2016; 7:607; PMID:28018364; http://dx.doi.org/ 10.3389/fimmu.2016.00607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B et al.. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013; 121:3599-608; PMID:23487023; http://dx.doi.org/ 10.1182/blood-2012-04-425397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, Zimmer J. Human CD56bright NK Cells: An Update. J Immunol 2016; 196:2923-31; PMID:26994304; http://dx.doi.org/ 10.4049/jimmunol.1502570 [DOI] [PubMed] [Google Scholar]
- 33.Freud AG, Yokohama A, Becknell B, Lee MT, Mao HC, Ferketich AK, Caligiuri MA. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med 2006; 203:1033-43; PMID:16606675; http://dx.doi.org/ 10.1084/jem.20052507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol 2013; 34:573-82; PMID:24055329; http://dx.doi.org/ 10.1016/j.it.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagler A, Lanier LL, Cwirla S, Phillips JH. Comparative studies of human FcRIII-positive and negative natural killer cells. J Immunol 1989; 143:3183-91; PMID:2530273 [PubMed] [Google Scholar]
- 36.Yu J, Mao HC, Wei M, Hughes T, Zhang J, Park IK, Liu S, McClory S, Marcucci G, Trotta R et al.. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood 2010; 115:274-81; PMID:19897577; http://dx.doi.org/ 10.1182/blood-2009-04-215491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev 2006; 214:56-72; PMID:17100876; http://dx.doi.org/ 10.1111/j.1600-065X.2006.00451.x [DOI] [PubMed] [Google Scholar]
- 38.Campos C, Pera A, Sanchez-Correa B, Alonso C, Lopez-Fernandez I, Morgado S, Tarazona R, Solana R. Effect of age and CMV on NK cell subpopulations. Experimental Gerontology 2014; 54:130-7; PMID:24440462; http://dx.doi.org/ 10.1016/j.exger.2014.01.008 [DOI] [PubMed] [Google Scholar]
- 39.Solana R, Campos C, Pera A, Tarazona R. Shaping of NK cell subsets by aging. Curr Opin Immunol 2014; 29:56-61; PMID:24792889; http://dx.doi.org/ 10.1016/j.coi.2014.04.002 [DOI] [PubMed] [Google Scholar]
- 40.Dulphy N, Haas P, Busson M, Belhadj S, Peffault de Latour R, Robin M, Carmagnat M, Loiseau P, Tamouza R, Scieux C et al.. An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J Immunol 2008; 181:2227-37; PMID:18641363; http://dx.doi.org/ 10.4049/jimmunol.181.3.2227 [DOI] [PubMed] [Google Scholar]
- 41.Messaoudene M, Fregni G, Fourmentraux-Neves E, Chanal J, Maubec E, Mazouz-Dorval S, Couturaud B, Girod A, Sastre-Garau X, Albert S et al.. Mature cytotoxic CD56(bright)/CD16(+) natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res 2014; 74:81-92; PMID:24225017; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1303 [DOI] [PubMed] [Google Scholar]
- 42.Mamessier E, Pradel LC, Thibult ML, Drevet C, Zouine A, Jacquemier J, Houvenaeghel G, Bertucci F, Birnbaum D, Olive D. Peripheral blood NK cells from breast cancer patients are tumor-induced composite subsets. J Immunol 2013; 190:2424-36; PMID:23359508; http://dx.doi.org/ 10.4049/jimmunol.1200140 [DOI] [PubMed] [Google Scholar]
- 43.Perez-Villar JJ, Melero I, Navarro F, Carretero M, Bellon T, Llano M, Colonna M, Geraghty DE, López-Botet M. The CD94/NKG2-A inhibitory receptor complex is involved in natural killer cell-mediated recognition of cells expressing HLA-G1. J Immunol 1997; 158:5736-43; PMID:9190923 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.