

Abstract
Rationale
The contractile dysfunction that underlies heart failure involves perturbations in multiple biological processes ranging from metabolism to electrophysiology. Yet the epigenetic mechanisms that are altered in this disease state have not been elucidated. SWI/SNF chromatin-remodeling complexes are plausible candidates based on mouse knockout studies demonstrating a combined requirement for the BRG1 and BRM catalytic subunits in adult cardiomyocytes. Brg1/Brm double mutants exhibit metabolic and mitochondrial defects and are not viable although their cause of death has not been ascertained.
Objective
To determine the cause of death of Brg1/Brm double-mutant mice, to test the hypothesis that BRG1 and BRM are required for cardiac contractility, and to identify relevant downstream target genes.
Methods and results
A tamoxifen-inducible gene-targeting strategy utilizing αMHC-Cre-ERT was implemented to delete both SWI/SNF catalytic subunits in adult cardiomyocytes. Brg1/Brm double-mutant mice were monitored by echocardiography and electrocardiography, and they underwent rapidly progressive ventricular dysfunction including conduction defects and arrhythmias that culminated in heart failure and death within 3 weeks. Mechanistically, BRG1/BRM repressed c-Myc expression, and enforced expression of a DOX- inducible c-MYC trangene in mouse cardiomyocytes phenocopied the ventricular conduction defects observed in Brg1/Brm double mutants. BRG1/BRM and c-MYC had opposite effects on the expression of cardiac conduction genes, and the directionality was consistent with their respective loss- and gain-of-function phenotypes. To support the clinical relevance of this mechanism, BRG1/BRM occupancy was diminished at the same target genes in human heart failure cases compared to controls, and this correlated with increased c-MYC expression and decreased CX43 and SCN5A expression.
Conclusion
BRG1/BRM and c-MYC have an antagonistic relationship regulating the expression of cardiac conduction genes that maintain contractility, which is reminiscent of their antagonistic roles as a tumor suppressor and oncogene in cancer.
Keywords: BRG1, BRM, SWI/SNF, c-MYC, cardiac connexins, cardiomyocyte conduction, bradycardia, arrhythmias, heart failure
1. Introduction
Heart failure is a major cause of morbidity and mortality worldwide. Its complexity arises from abnormalities in many aspects of cardiac function, from metabolism to electrophysiology, resulting in impaired contraction and sudden cardiac death.1 Electrophysiological remodeling is among the most common and important alterations in human heart failure. Heart failure patients suffer from both bradyarrhythmias and tachyarrhythmias, and sudden cardiac death is 6–9-fold more common in heart failure patients than the general population [1,2]. While some genes associated with the proarrhythmic state in heart failure are known, the epigenetic mechanisms regulating their expression are unknown, as are the functional connections between metabolic and arrhythmogenic conduction that occurs in human heart failure [1]. Epigenetic mechanisms are capable of long-term regulation of gene expression, which is highly relevant to cardiomyocytes where the expression of some genes must be maintained in the same cell over an individual’s lifetime.
SWI/SNF chromatin-remodeling complexes are recruited by transcription factors to the enhancers and promoters of target genes where they reposition nucleosomes in an ATP-dependent manner to epigenetically regulate transcription [3–6]. Target genes are either activated or repressed in a context-dependent manner. Although SWI/SNF complexes are heterogeneous, BRG1 (also known as SMARCA4) and BRM (also known as SMARCA2) are the only catalytic subunits with ATPase activity. SWI/SNF complexes physically interact with cardiogenic transcription factors such as NKX2.5, TBX5, and GATA4, which make them a plausible candidate for regulating cardiomyocyte development in the embryo and contractile function in adults. Mouse knockout studies have demonstrated that BRG1 is required for cardiomyocyte development but is dispensable in adult cardiomyocytes [7–9], whereas BRM is completely dispensable [10]. To test the hypothesis that BRG1 and BRM functionally compensate in the adult cardiomyocyte, we generated Brg1fl/fl mice carrying an inducible, cardiomyocyte-specific αMHC-Cre-ERT2 transgene that were also Brm−/−. Indeed, these Brg1/Brm double mutants died within 3 weeks following the loss of Brg1, and they exhibited metabolic perturbations and mitochondrial defects in the heart [11,12]. However, their cause of death is unclear, and it is not known whether SWI/SNF is required for cardiac conduction or if this model is relevant to the study of heart failure. We therefore focused our efforts in the current study to address these issues and to identify downstream target genes that provide mechanistic insight.
2. Materials and methods
2.1. Mice
The αMHC-Cre-ERT mice [also known as B6.Cg-Tg(Myh6-cre/Esr1)1JmkJ or αMHC-MerCreMer] were obtained from The Jackson Laboratory (#005657, Bar Harbor, ME) and genotyped as previously described [13]. The Brg1 conditional mutant mouse line and Brm constitutive mutant mouse line have been described previously [10,14,15]. Genotyping of the Brg1 floxed and Δfloxed alleles and the Brm mutation were performed by PCR as previously described [10,14,15]. To induce the Brg1 conditional mutation in adult cardiomyocytes, 3–6 month old male and female mice were provided rodent chow containing tamoxifen (Sigma-Aldrich #T5648, St. Louis, MO) over a 7-day period. 500 mg of tamoxifen was mixed with 1 kg of ground-up rodent chow and then mixed with water, kneaded into pellets, and dried in a hood. Provided to mice ad libitum, the dose was estimated to be 80 mg/kg/day. After the 7-day treatment period, the tamoxifen-fortified chow was removed and replaced with the same chow lacking tamoxifen. A similar number of male and female mice were used in the study, and no phenotypic differences were observed between the two sexes as indicated in Supplemental Fig. 2.
The bi- transgenic mouse line that inducibly overexpresses the human c-MYC cDNA in cardiomyocytes under the control of the αMHC promoter has been previously described [16]. Mice were raised in the absence of doxycycline (DOX) to prevent developmental consequences from c-MYC overexpression. c-MYC was induced by feeding mice Dox-containing rodent chow (200 mg/kg, Bio-Serve, Frenchtown, NJ) ad libitum. Dox had no effect on single transgenic littermates, which were used as controls in analyses performed in this study.
All mouse experiments were approved by the Institutional Animal Care and Use Committees (IACUC) review boards at the University of North Carolina at Chapel Hill and Case Western Reserve University and were performed in accordance with federal guidelines.
2.2. Echocardiography
Conscious cardiac transthoracic echocardiography was performed on mice at the indicated time points using a VisualSonics Vevo 2100 ultrasound biomicroscopy system (VisualSonics, Inc., Toronto, Ontario, Canada) as previously described [17,18]. Two-dimensional M-mode echocardiography was performed in the parasternal long-axis view at the level of the papillary muscle on loosely restrained mice. Anterior and posterior wall thickness was measured as distance from epicardial to endocardial leading edges. Left ventricular internal diameters were also measured. Left ventricular systolic function was assessed by ejection fraction (LV EF% = [(LV Vol; d-LV Vol; s/LV Vol; d) x 100] and fractional shortening (%FS = [(LVEDD − LVESD)/LVEDD] x 100). Investigators were blinded to mouse genotype from collection through waveform measurements. Each measurement represents the average of three cardiac cycles from each mouse.
2.3. Electrocardiography
Continuous electrocardiographies (ECGs) were monitored by surgically implanting a TA10ETA radiotelemetry device (Data Sciences International (DSI), St. Paul, MN) into the abdomen of mice anesthetized with isoflurane and transmitting the information to APR-1 receivers under the cages that were coupled to the Ponemah v.5.0 Physiology Platform for data analysis (DSI).
2.4. RNA isolation
Cardiac tissues were homogenized using a TissueLyser LT (Qiagen N.V. #69980, Venlo, The Netherlands) according to the manufacturer’s protocols. Approximately 20–40 mg of apical ventricle was homogenized in 1 mL of Trizol (Life Technologies #15596-026, Carlsbad, CA) using a 5- mm stainless steel bead (Qiagen N.V. #69989). Chloroform (200 μL) was added, centrifuged at 12,000g (15 min at 4°C), isopropanol (0.5 mL) was then added to the aqueous phase, centrifuged at 12,000g (10 min at 4°C) and the resulting RNA pellet was washed with 1mL of 75% ethanol, centrifuged at 7500g (5 min at 4°C). The resulting pellet was dried and resuspended in RNase- free water. RNA concentrations were then determined by UV spectroscopy (absorbance of 260–280 nm).
2.5. Transcriptome profiling and RT-qPCR
RNAs were reverse-transcribed using iScript reverse transcription supermix (Bio-Rad Laboratories #170-8841, Hercules, CA). Transcriptome profiling was performed at the UNC Lineberger Comprehensive Cancer Center (LCCC) Genomic Core Facility using the Agilent Once Color 80k60k Sure Print G3 Mouse Gene Expression Array (G4858A-028005). TaqMan gene expression assays (Life Technologies) were performed using universal TaqMan master mix (Life Technologies #4304437). The method for RNA isolation and Real-time RT-PCR was described previously [19]. Primer pairs and probes for RT-qPCR assays are listed below:
F; 5′-Fluorescein (FAM)
Q; Quencher (TAMRA)
Mouse
cMyc
Forward CCA GCC CTG AGC CCC TAG T
Reverse TGC TCT TCT TCA GAG TCG CT
Probe FTG CAT GAG GAG ACA CCG CCC ACC AQ
Tbx5
Forward CCA CTG TAC CAA GAG GAA AG
Reverse TGT CTC CAT GTA CGG CTT CT
Probe FAA TGT TCC AGC ACG GAG CAC CCC TAQ
Cx40
Forward ACC ATC ATG GGC ATG ATC TG
Reverse ATA GGT GAC CCT GCC AAG AC
Probe FTG ATC GTG GAG GTC TTG CTG AGG ATG Q
Cx43
Forward CCT CTT CAA GTC TGT CTT CGA
Reverse TAG ACC GCA CTC AGG CTG AA
Probe FTG GCC TTC CTG CTG ATC CAG TGG TAQ
Trpm 7
Forward ATT CCC TTC GTT CCT GTA CC
Reverse ACT GGG AGA ACT CTC CTC CA
Probe FAC GAG GCG AGC CTG TCA CAG TGT ACQ
Scn5a
Common Forward TCA CCA ACA GCT GGA ACA TC
Full Reverse GGA GAF GAC AGT GCC AAC G D
Variant Reverse AGA GCA ACG TGC GAA CAA CG C
Variant Reverse CAA CAC CTG ACA TGT ACG CAT
Common Probe FCG ATT TCG TGG TTG TCA TCC TCT CCQ
Gapdh
Forward AGG TCG GTG TGA CCG GAT TT
Reverse GGC AAC AAT CTC CAC TTT GC
Probe FTG CAA ATG GCA GCC CTG GTG ACC AQ
Human
BRG1
Forward CGA AAG GAG CTG CCC GAG T
Reverse TGG TTG CGA ATG CGC TCC T
Probe FAC GAG CTC ATC CGC AAG CCC GTG Q
BRM
Forward GCC GTG ACG TGG ACT ACA GT
Reverse AAA TTG CCG TCT TCG ATG GC
Probe FAC GCC CTC AGG GAG AAG CAG TGG Q
cMYC
Forward TTC GGG TAG TGG AAA ACC AG
Reverse GGT CAT AGT TCC TGT TGG TG
Probe FTC CCG CGA CGA TGC CCC TCA ACG Q
SCN5A
Forward CCA ACA GCT GGA ATA TCT TCG A
Reverse TTC TGG ATG ATG TCC GAG AG
Probe FTC GTG GTT GTC ATC CTC TCC ATC GTG Q
CX40
Forward; TCT TTA TGC TGG CTG TGG CT
Reverse; GAT CTT CTT CCA GCC CAG GT
Probe; FAC TGT CCC TCC TCC TTA GCC TGG CQ
CX43
Forward AAG CAA AAG AGT GGT GCC CA
Reverse CAG CAG TTG AGT AGG CTT GA
Probe FTG TCA AGG AGT TTG CCT AAG GCG CTC Q
GAPDH
Forward ACC TCA ACT ACA TGG TTT AC
Reverse GAA GAT GGT GAT GGG ATT TC
Probe FCA AGC TTC CCG TTC TCA GCC Q
2.6. IHC and western blots
IHC was performed with a BRG1 antibody (Millipore #07-478) as described previously [12], and western blots were performed with c-MYC (Abcam ab#32072) and GAPDH (Sigma G8795) antibodies following standard procedures.
2.7. ChIP assays
ChIP assays were performed as previously described [20]. Briefly, 10–30 mg of cardiac tissues were pulverized in liquid nitrogen using a mortar and pestle and then crosslinked in 1% formaldehyde at room temperature for 10 minutes. After the crosslinking reaction was stopped with 0.125M glycine, the tissues were lysed, and the chromatin was sonicated into 200–500-bp fragments. 5% of the sonicated chromatin was removed as input, and the remainder of each sample was immunoprecipitated overnight at 4 C using a BRG1 antibody (J1, a gift from Dr. Gerald Crabtree’s lab) that cross-reacts with BRM [21]. Duplicate samples were immunoprecipitated with rabbit IgG as a negative control. Immunoprecipitants were pulled down using protein A/G beads (Santa Cruz Biotechnology), washed following standard procedures, and eluted in 10–25 μL of ddH2O.
qPCR was performed using Power SYBR Green Master Mix (Life Technologies) using the following primer pairs. Mouse: Cx40: Forward, CTTTCTCGACTGGTGAGGAA; Reverse, GAGCCTGTTAGTTGCTCCCG (450 nM final concentration of each). Cx43: Forward, CCCTTCTCGTCAGCACATTG; Reverse, AGCCACTGACTCAACTGGAA (300 nM final concentration of each). Scn5a: Forward, GTCAGAGTGGTGGGCTG; Reverse, GATCCCCACATCCCACGG (250 nM final concentration of each). Dissociation curves and agarose gels demonstrated a single PCR product in each case without primer dimers. Relative enrichment was determined by comparison to serial dilutions of input samples.
2.8. Statistics
SigmaPlot (Systat Software, Inc., San Jose, CA) was used to determine significant statistical difference by One-way ANOVA followed by post-hoc analysis using the Holm–Sidak method or a Student’s t-test. A p value < 0.05 was considered significant.
2.9. Study approval for human clinical samples
Human clinical samples were from subjects consented and collected for future research by the Duke Human Heart Respository (Pro00005621). A Request for Waiver or Alteration of Consent and HIPAA Authorization” was submitted to the Duke Institutional Review Board (IRB) for the present study (Pro00060625) and approved (19 February 2015). In parallel, a request for “Exemption from IRB Review” was submitted to the University of North Carolina IRB for the present studies to be performed at UNC (14-3334) and approved (10 March 2015). Samples were de-identified without PHI and collected from heart transplant donors.
3. Results
3.1. Combined requirement for BRG1 and BRM in adult cardiomyocytes to prevent cardiomyopathy and electrophysiology defects
To investigate the combined role of BRG1 and BRM in adult cardiac function, we analyzed Brg1fl/fl mice carrying an inducible, cardiomyocyte-specific αMHC-Cre-ERT2 transgene that were also Brm−/−. We previously documented conditional loss of Brg1 in cardiomyocytes within 7 days of tamoxifen treatment in this model by PCR and IHC (Supplemental Fig. 1A–C) [11,12]. These mice (herein referred to as Brg1/Brm double mutants), which are null for BRG1 and BRM in cardiomyocytes, die at 6–22 days (mean of 11.6 ± 1.5 days) relative to the first day of tamoxifen treatment (Supplemental Fig. 1D) [12]. We have demonstrated that Brg1 conditional mutants on a wild-type background are viable, as are Brm−/− mice, indicating that the two catalytic subunits are functionally redundant in adult cardiomyocytes (Supplemental Fig. 1D) [12].
We monitored 27 Brg1/Brm double mutants and 28 controls by conscious echocardiography on a daily basis until every double mutant died. Baseline measurements prior to tamoxifen treatment demonstrated that every Brg1/Brm double mutant (Group 4) was indistinguishable from controls (Groups 1–3 and 5) with normal ejection fraction and other metrics (Fig. 1A–B). This result was expected because Brg1 had not yet been mutated and Brm is dispensable. In contrast, following tamoxifen treatment, every Brg1/Brm double mutant experienced rapid and progressive declines in cardiac function that preceded their early-onset death (Fig. 1A–B, Supplemental Fig. 2). Double mutants developed severe left ventricular (LV) systolic dysfunction as evidenced by decreased ejection fraction (EF) percentage and decreased fractional shortening percentage as well as LV dilation based on a widening LV that contracted less (Fig. 1A–B, Supplemental Table 1). A characteristic bradycardia was identified in the 24 hours before each double-mutant mouse died (herein referred to as 1-day pre- mortem) (Figure 1B, see box in last panel labeled heart rate, Supplemental Table 2). The cardiac phenotype at 1-day pre- mortem was characterized by two distinct cardiac phenotypes that are not obvious unless grouped separately: 1) a dilated cardiomyopathy with severe dysfunction and significantly thinner walls (EF%<50%, mean 26.4 ± 3.1%); and 2) a hypertrophic cardiomyopathy with less severe systolic dysfunction (EF%>50%, mean 69.1%) (Fig. 2A–B, Supplemental Table 2). However, both phenotypes had significantly decreased heart rates (422 ± 28 and 532 ± 40, respectively) by conscious echocardiography (Fig. 2B).
Figure 1.

Brg1/Brm double mutants undergo arrhythmias and heart failure. (A) Echocardiogram-based measurements of ejection fraction % (black histograms) and fractional shortening % (gray histograms) in Brg1/Brm double mutants (Group 4) and control groups at baseline (prior to loss of Brg1 via tamoxifen) and at 1-day pre-mortem (histograms enclosed by gray box at right). See key below for description of each numbered control group. (B) Six panels show left-ventricle morphometrics and heart rate, as indicated, with first 5 histograms representing baseline measurements and last 2 histograms enclosed by gray box representing 1-day pre-mortem measurements. Data represent means ± SEM with the number of mice per group indicated in each histogram. One-Way Analysis of Variance was performed followed by an all pair-wise multiple comparison procedure (Holm-Sidak method) with significant differences indicated (§, p<0.001 vs. all other groups).
Figure 2.

Two subphenotypes in Brg1/Brm double mutants. (A) The ejection fraction % and fractional shortening % data at 1-day pre-mortem from Fig. 1A are reproduced at the left. The double-mutant values are enclosed by a gray box. These data are juxtaposed with 1-day pre-mortem data from the double mutants separated out into two subsets (highlighted by arrows and histograms labeled Subset 1 and Subset 2) where the phenotypes differed with respect to wall thickening, LV dilation, and systolic dysfunction, but not heart rate (HR). (B) Six panels show left-ventricle morphometrics and heart rate that are the same as Fig. 1B except only 1-day pre-mortem data are shown and the double mutant data are shown combined and separated out into the two subgroups as indicated. Data represent means ± SEM with the number of mice per group indicated in each histogram. One-Way Analysis of Variance was performed followed by an all pair-wise multiple comparison procedure (Holm-Sidak method) with significant differences indicated (*p<0.001 vs. all other groups; **p<0.05 vs. Column 1; †p<0.01 vs. Column 4).
To demonstrate the specificity of the cardiac phenotypes identified in the Brg1/Brm double-mutant mice, we performed conscious echocardiography on 28 controls corresponding to 4 different control groups (Groups 1–3 and 5 as defined in Fig. 1A). The only abnormality that was observed among controls was a mild transient effect in Group 5 associated with tamoxifen treatment of αMHC-Cre-ERT transgenic mice in the absence of a floxed allele, which has been described previously (Supplemental Fig. 2) [22]. However, it did not result in any mortality, and baseline function was restored immediately after tamoxifen withdrawal (Supplemental Fig. 2).
To probe cardiac electrophysiological function, we performed continuous ECG telemetry on double mutants and controls. As expected, the Brg1/Brm double- mutant mice had baseline ECG measurements, prior to loss of Brg1, that were indistinguishable from controls (Baseline in Fig. 3). These measurements included a normal heart rate, PR interval, QRS duration, and corrected QT intervals at all of the time points collected. In contrast, significant repolarization abnormalities were observed in Brg1/Brm double-mutants by 13 days after the loss of Brg1 (Fig. 3). For example, one Brg1/Brm double mutant had a QT interval that was clearly >50% of the R-R interval at day 13, which is consistent with marked QT prolongation (Mouse 1 in Fig. 3). Abnormal ST segment morphology, characterized by downsloping ST depression and T-wave inversion, was also evident. Bradycardia and widening of the QRS complex was also present at day 13. These abnormalities persisted until day 17 at which time the mouse developed apparent sinus arrest with a slow ventricular escape rhythm (Fig. 2). This terminal rhythm progressed to complete cessation of electrical activity within hours. Another Brg1/Brm double mutant demonstrated abnormalities in both atrioventricular (AV) conduction and ventricular repolarization by ECG analysis at day 13 (Mouse 2 in Fig. 3). The AV conduction abnormalities initially manifested as prolongation of the PR interval (1st degree AV block) (Fig. 3) and progressed rapidly to complete heart block (3rd degree AV block) with a slow junctional escape (Fig. 3). This terminal rhythm progressed to complete cessation of electrical activity within minutes on day 13. This time point was characterized by repolarization abnormalities, initially manifest as markedly peaked (“hyperacute”) T waves, followed by ST segment depression and T wave inversion (Fig. 3). The QRS interval also became mildly prolonged. Taken together, the observations that Brg1/Brm double- mutant mice exhibit cardiomyopathy, abnormal ECG profiles, and arrhythmias that result in heart failure and death suggest that BRG1 and BRM are required for cardiac conduction within the myocardium.
Figure 3.

Brg1/Brm double mutants have conduction defects and die because of atrioventricular blockage. ECG data from double mutant and control mice at baseline and at 13 and 17 days following tamoxifen (TAM)-induced loss of Brg1 that includes measurements in the hours preceding death.
3.2. A functional link between BRG1/BRM and c-Myc
Considering the severity of the cardiac conduction phenotype and the rapid demise of Brg1/Brm double-mutant mice, the histopathology was surprisingly mild with no signs of cell death or structural damage to the tissues. Double- mutant hearts appeared normal with no significant changes in cellular composition or fibrosis [12]. These observations suggest that any alterations in cardiac gene expression in double mutants are not simply a secondary consequence of morphological defects.
To further delineate the mechanisms underlying the pathogenesis of the heart disease caused by ablating Brg1 and Brm in cardiomyocytes, we performed transcriptome-profiling experiments on hearts from Brg1/Brm double- mutant and control mice. 4 biological replicates were analyzed from each group of mice, and principal component analysis showed a marked difference between the two groups as expected (Fig. 4A). 2,291 genes were expressed at significantly different levels with a false discovery rate (FDR) of ≤0.05 (Supplemental Table 3). Unexpectedly, the gene list was highly skewed with genes upregulated in double mutants compared to controls (n=2,118) far outnumbering downregulated genes (n=173). Biological pathway analysis revealed a number of enriched networks that primarily involved cell proliferation/cell cycle, immune/cytokine/chemokine signaling, integrin/extracellular matrix (ECM) function, and lipid metabolism (Fig. 4B). Supplemental Fig. 3 shows the cell-cycle network in detail. Other potentially relevant target genes were identified that include cardiogenic transcription factors (e.g., Nkx2-5, Mef2c, Tbx18, Tbx20), sodium and calcium channels (e.g., Scn1b, Scn5a, Cacnaa1s, Atp2b1, Atp2b2), myosins (e.g., Myo1h, Myo3b, Myo1g, Myo5a), and gap junction/connexins (Supplemental Table 3).
Figure 4.

BRG1/BRM transcriptional targets including c-Myc. (A) Principal component (PC) analysis of controls and Brg1/Brm double mutants based on their transcriptome profiles. (B) Significantly enriched pathways among the genes differentially expressed between Brg1/Brm double mutants and controls. All of the listed pathways have an FDR < 0.05, and the gene set statistic on the x-axis represents the z score transformation of the mean of all genes in a set. (C) RT-qPCR analysis of c-Myc mRNA levels normalized to Gapdh in control and double-mutant hearts. Data are presented as means ± SEM based on 5 independent experiments with significant differences indicated (*p<0.05). (D) Western blot analysis of heart protein lysates from controls and double mutants probed with antibodies specific for c-MYC and GAPDH as a loading control. (E) Quantitative ChIP assays demonstrating BRG1/BRM occupancy at the c-Myc promoter in wild-type mouse heart. Histograms show the relative enrichment by comparing each ChIP sample to input by qPCR (means ± SEM for three independent samples, (*p<0.05).
We evaluated c-Myc in more detail because it was upregulated in double mutants and was represented in multiple enriched pathways. In addition to being an important cell proliferation/cell-cycle gene, c-Myc regulates immune, extracellular matrix, and metabolic function (i.e., most of the enriched pathways). Furthermore, an antagonistic relationship exists between BRG1/BRM tumor suppression and c-MYC oncogenesis, and BRG1 binds to the c-MYC promoter in lung cancer cells and represses its expression [23]. Consequently, we hypothesized that BRG1/BRM binds to the c-Myc promoter in cardiomyocytes and represses its expression. First, we confirmed that c-Myc mRNA and c-MYC protein levels are upregulated in double mutants by RT-qPCR and western blot analysis, respectively (Fig. 4C–D). Next, we performed quantitative ChIP assays and demonstrated that BRG1/BRM occupies the c-Myc promoter in cardiac tissue (Fig. 4E).
To independently evaluate the functional importance of c-Myc overexpression, which has been implicated in cardiac hypertrophy [24–27], we analyzed a transgenic mouse model that inducibly overexpresses human c-MYC in adult cardiomyocytes [16]. We confirmed that c-MYC expression was induced by DOX within 24 hours of treatment (Fig. 5A). This c-MYC induction significantly decreased the expression of the cardiac connexin Cx43 at the mRNA (Fig. 5B) and protein (Fig. 5C) levels. Importantly, MYC-ON transgenic mice exhibited rapid cardiac dysfunction compatible with loss-of-function of both CX43 and BRG1/BRM. Continuous ECG telemetry revealed a significantly decreased heart rate and significant perturbations in PR interval, QRS duration, and QTc that culminated in a complete heart block by day 6 (Fig. 5D–E). These findings indicate that c-MYC downregulation is crucial for cardiac conduction and provide support for a mechanism where BRG1/BRM represses c-Myc to maintain conduction and contractility.
Figure 5.

c-MYC gain-of-function in cardiomyocytes results in cardiac conduction defects that phenocopy BRG1/BRM loss-of-function. (A) RT-qPCR analysis of c-MYC mRNA levels in the heart of an inducible transgenic mouse line prior to induction (MYC-OFF) and 24–48 hours after DOX-mediated induction to overexpress c-Myc (MYC-ON, Day 1 and Day 2). Data are normalized to Gapdh and presented as means ± SEM based on 5 independent experiments with significant differences indicated (*p<0.05). (B) RT-qPCR analysis of Cx43 mRNA levels in heart of the same transgenic mouse line. Data are normalized to Gapdh and presented as means ± SEM based on 5 independent experiments with significant differences indicated (*p<0.05). (C) Representative western blot of CX43 protein levels in heart of same transgenic mouse line prior to induction (MYC-OFF) and after DOX-mediated induction (MYC-ON, Day 3). Actin serves as a loading control. 3 independent samples for MYC-OFF and MYC-ON are shown. (D) ECG sample trace readings from 3 MYC-OFF controls and 4 MYC-ON mice showing Wenckebach second-degree heart block by 3 days of DOX-induced c-MYC induction and a complete heart block by day 6. (E) Four panels showing ECG-based measurements from 3 MYC-OFF controls and 4 MYC-ON mice at three time points relative to DOX-mediated induction. The plots show significant differences (*p<0.05) in heart rate, PR interval, QRS duration, and QTc.
3.3. BRG1/BRM directly and indirectly regulate cardiac conduction genes
One plausible explanation for the Brg1/Brm double- mutant phenotype is that BRG1/BRM regulate genes encoding gap-junction proteins and ion channels that play crucial roles in cardiac conduction and positively regulate their expression. Although electrical impulses originate from specialized cells within the sinoatrial node, their propagation throughout the myocardium is dependent on specific gap junctions and ion channels. To follow-up on the transcriptome-profiling data, we performed RT-qPCR and analyzed the expression of Cx40 and Cx43, which encode gap-junction connexins, and Scn5a because it encodes the sodium channel Nav1.5. Indeed, Brg1/Brm double- mutant hearts had significantly decreased mRNA levels for each of these conduction genes (Fig. 6A). The expression of another ion channel gene (Trpm7) was not significantly different (Fig. 6A), which indicates there is specificity.
Figure 6.

BRG1/BRM regulation of conduction genes. (A) RT-qPCR analysis of mRNA levels of cardiac connexins (Cx40, Cx43) and ion channels (Scn5a, Trpm7) normalized to Gapdh in control and double-mutant hearts. Data are presented as means ± SEM based on 5 independent experiments with significant differences indicated (*p<0.05). (B, C,) Quantitative ChIP assays demonstrating BRG1/BRM occupancy at the Cx40, Cx43, and Scn5a promoters in wild-type mouse heart tissues (B) and at the α-globin locus in wild-type heart and fetal liver (FL) tissues (C). IgG immunoprecipitations serve as a negative control. Histograms show the relative enrichment by comparing each ChIP sample to input by qPCR (means ± SEM for three independent samples, (*p<0.05). (D) Working model. The BRG1 and BRM catalytic subunits of SWI/SNF complexes directly and indirectly activate the expression of Cx40, Cx43, and Scn5a to facilitate conduction in cardiomyocytes. The direct regulation is based on ChIP assays demonstrating occupancy at each promoter. The indirect regulation is mediated by inhibition of an inhibitor (c-Myc) and activation of an activator (cardiogenic transcription factors Tbx, Nkx2-5, Mef2c).
To test the hypothesis that BRG1/BRM directly regulate conduction genes, we performed quantitative ChIP assays and observed BRG1/BRM occupancy at the promoter of Cx40, Cx43, and Scn5a in cardiac tissue (Fig. 6B). As a ChIP negative control, we evaluated BRG1/BRM occupancy at the α-globin locus in cardiac tissue because it was not on our gene list and is not expressed in cardiomyocytes. Occupancy was not observed above background levels as expected (Fig. 6C). To demonstrate that the α-globin ChIP assay was optimized and capable of detecting significant binding events, we demonstrated BRG1/BRM occupancy in fetal liver cells (Fig. 6C), which are of erythroid origin and known to bind BRG1/BRM and express α-globin [28]. These results indicate that BRG1/BRM maintain the conduction system by directly binding to the promoters of connexin and ion channel targets as well as indirectly by repressing the expression of c-Myc (an inhibitor) and by activating the expression of several cardiogenic transcription factors (that are activators) (see model in Fig. 6D).
3.4. Diminished BRG1/BRM occupancy and expression of cardiac conduction genes in human heart failure cases
To provide evidence that the BRG1/BRM and c-MYC regulatory mechanism is clinically relevant, we analyzed cardiac tissue from human heart failure cases and controls. We selected cases diagnosed with bundle branch blocks and other related arrhythmias. Controls were obtained from heart transplant donors whose hearts could not be used due to the unavailability of a suitable recipient within the required timeframe. BRG1 and BRM expression levels were not significantly different between the cases and controls (Fig. 7A). c-MYC expression levels were increased in cases (Fig. 7B), while CX43 and SCN5A expression levels were decreased (Fig. 7C). The difference in c-MYC approached statistical significance (p=0.07) despite the heterogeneity that is inherent in human clinical samples (Fig. 7B). Furthermore, when the cases were stratified into two groups, a subset corresponding to 7/10 of the cases had significantly higher c-MYC expression levels compared to controls (Fig. 7B). The decrease in CX43 and SCN5A expression levels trended toward significance (p=0.06) or were significant, respectively, in all of the cases without stratification (Fig. 7C). When the 7/10 subset of cases with elevated c-MYC were analyzed separately, CX43 was significantly lower than controls (Fig. 7C), indicating that there is a significant correlation between c-MYC high cases and significantly lower CX43 expression.
Figure 7.
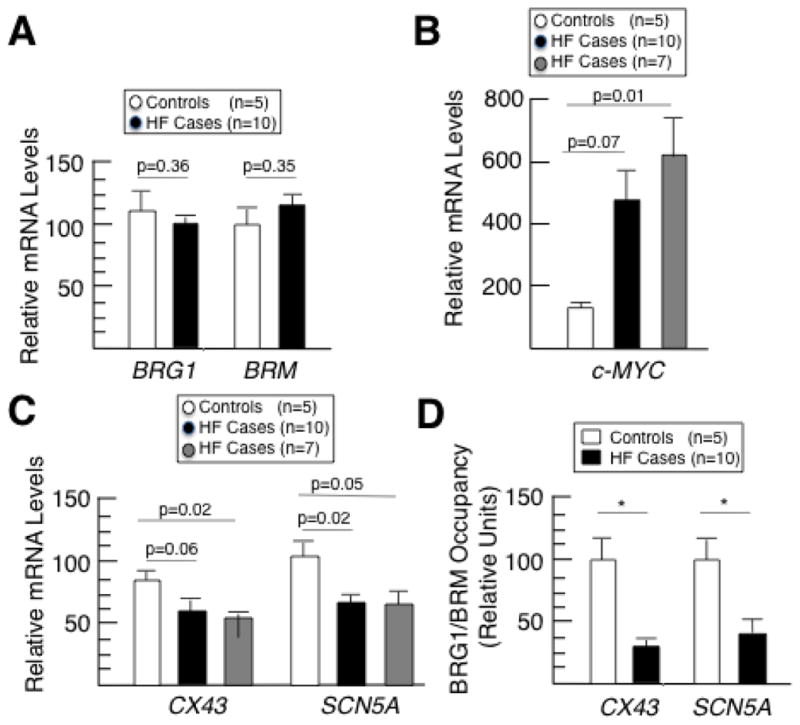
BRG1/BRM occupancy and expression of conduction genes is attenuated in human heart failure cases, while c-MYC is overexpressed. (A–C) RT-qPCR analysis of mRNA levels for human BRG1 and BRM (A), c-MYC (B), and CX43 and SCN5A (C) normalized to GAPDH mRNA levels. Data are presented as means ± SEM based on 5 controls (white), 10 heart failure cases (black), and a subset of heart failure cases with elevated c-MYC mRNA levels (gray, n=7) (as opposed to the other 3 heart failure cases that did not have elevated c-MYC) with p-values indicated. (D) Quantitative ChIP assays measuring BRG1/BRM occupancy at the human CX43 and SCN5A promoters in cardiac tissue from 5 controls and 10 heart failure cases. Each ChIP qPCR was normalized to input, and the relative enrichments are shown as means ± SEM with significant differences indicated (*p<0.05).
The existence of long noncoding RNAs (lncRNAs) that influence the occupancy of epigenetic enzymes at genomic loci [29] raises the possibility that BRG1/BRM occupancy of cardiac conduction genes might be diminished in heart failure cases even if global BRG1/BRM expression levels are not significantly affected. To test this hypothesis, we performed quantitative ChIP assays on cardiac tissues from the same human clinical samples. BRG1/BRM occupied the promoter of the CX43 and SCN5A genes, and occupancy was significantly diminished in the heart failure cases compared to controls (Figure 7D). Next, we analyzed the expression of MHRT because it is a lncRNA that binds to and sequesters BRG1 in cardiomyocytes [30]. Overexpression of MHRT in heart failure cases would provide an explanation for diminished BRG1/BRM occupancy at target genes without decreased global levels of BRG1/BRM protein. MRHT mRNA levels were not significantly different when all of the samples were analyzed (p=0.38), but they were significantly higher in a subset corresponding to 5/10 of the heart failure cases compared to controls (p=0.01) (Supplemental Fig. 4). Taken together, these results suggest that BRG1/BRM and c-MYC regulate the expression of cardiac conduction genes in humans and that perturbation of this mechanism contributes to heart failure and sudden death.
4. Discussion
The data presented here provide the framework for an epigenetic mechanism that regulates the coordinated contraction of cardiomyocytes in the heart. It is noteworthy that the BRG1 and BRM catalytic subunits of SWI/SNF complexes are functionally redundant in the adult heart because this is not the case during cardiomyocyte development in utero where BRG1 is essential but BRM is dispensable [7–10]. This differential requirement could be due to their relative expression levels as BRG1 is expressed in more broadly and at higher levels than BRM during embryogenesis. In fact, BRG1 is the catalytic subunit of most SWI/SNF complexes in embryonic hearts, outnumbering BRM-catalyzed complexes by a 3:1 margin [31], whereas BRG1 and BRM are expressed at similar levels in many adult tissues including the heart. A precedent for this type of differential requirement of SWI/SNF catalytic subunits during the development of a cell lineage is in vascular endothelial cells, where only BRG1 is required in the embryo but BRG1 and BRM are functionally redundant in the juvenile and adult heart [32–34]. BRG1-BRM redundancy in the adult heart may have important translational implications for anticancer therapy. SWI/SNF complexes have been investigated much more for their role in cancer than cardiovascular disease, and several groups recently demonstrated that depletion of BRM in BRG1-deficient cancer cells results in synthetic lethality [35–37]. This strategy is currently being considered as an anticancer strategy in the clinic, but care will need to be taken to avoid double depletion or perturbation of BRG1 and BRM in cardiomyocytes since this would likely result in conduction defects and sudden death.
The working model is that BRG1- and BRM-catalyzed SWI/SNF complexes directly and indirectly activate the expression of Cx40, Cx43, and Scn5a to maintain conduction. BRG1/BRM directly regulates gene expression by binding each promoter to open chromatin structure and facilitate transcription. The indirect regulation is mediated by activation of activators (cardiogenic transcription factors) and inhibition of an inhibitor (c-Myc) of the same genes. This model is compatible with the Brg1/Brm double- mutant phenotype, which is manifest as an initial prolongation of the PR interval (1st degree AV block) that progresses to lethal arrhythmias resulting from AV conduction abnormalities. The Brg1/Brm double mutants also have defects in mitochondrial dynamics (fusion/fission) and mitophagy in the myocardium as well as altered metabolism [11,12]. In this regard, it is reasonable and perhaps expected, that the contractile cardiomyocyte program is linked with mitochondrial/energy homeostasis and metabolism. The complicated nature of this phenotype suggests multiple pathways are perturbed including some that have not yet been elucidated. For example, the role of cytokine/chemokine signaling and integrins/extracellular matrix, as identified in our pathway analysis of the transcriptome data, remains to be determined. Nevertheless, the current study adds to a growing list of epigenetic factors that regulate heart physiology such as the PRC2 polycomb complex and the NuRD chromatin-remodeling complex [38–41].
The communication between cardiomyocytes necessary for the heart’s synchronous contraction occurs through ion channels and gap junctions. The role that potassium and sodium ion channels play in this process is evident based on disease-causing mutations in genes such as SCN5A that result in lethal arrhythmias due to alterations in the ability of cells to repolarize (accounting for the prolonged QT interval by ECG) [42]. Maintenance of the gap junctions between cardiomyocytes is also crucial for maintaining conduction in the heart, with the distribution and function of the gap-junction protein connexin 43 (CX43) and sodium channel Nav1.5 (encoded by SCN5A) critical to conduction. Inducible knockout of Cx43 in mice resulted in a heterogeneity of outcomes over a 2-week period, with those experiencing arrhythmias having decreased Cx43 expression (and reduced Na current) with global heterogeneity, in contrast to those without arrhythmias [43].
Several cardiogenic transcription factors including NKX2.5 have been implicated in regulating Nav1.5 and as well as CX40 and CX43 [44–47]. However, transcription factors are necessary, but not sufficient, for transcriptional regulation. To regulate transcription, transcription factors recruit different types of chromatin- modifying factors to the promoters of downstream targets. Yet a gap in our knowledge exists in terms of which chromatin- modifying factors are recruited by transcription factors to critical ion-channel and gap-junction genes in the heart to facilitate conduction. The work presented here clearly identifies SWI/SNF chromatin-remodeling complexes as a key component of the contractile cardiomyocyte program. A recent report demonstrates that BAF250A, which is an ARID-domain subunit of SWI/SNF complexes, is integral to cardiac conduction and supports our findings. Conditional deletion of cardiac Baf250a exhibited sinus bradycardia as we identified, although the conduction phenotype was not lethal as we found in the Brg1/Brm double mutants [48]. One possibility for this difference in phenotype severity is that Baf250b or Baf200, which encode alternative ARID-domain subunits of SWI/SNF complexes, are able to functionally compensate. Another possib ility is that the Baf250a deletion was restricted to the sino-atrial node, whereas Brg1/Brm were deleted throughout the myocardium. The sino-atrial node corresponds to a specialized group of cells in the right atrium and functions as the pacemaker where electrical impulses are generated to initiate heartbeat. These electrical impulses are subsequently propagated throughout the myocardium to complete heartbeat. To distinguish between these possibilities and determine the relative importance of SWI/SNF complexes in the initiation versus propagation of heartbeat, it will be necessary to utilize Hcn4-CreERT2 to create Brg1/Brm double mutants where Brg1 is deleted exclusively in the sino-atrial node.
Although c-MYC is best known as a proto-oncogene that is frequently overexpressed in cancer, overexpression in quiescent cardiomyocytes is associated with cardiac hypertrophy [24–27]. In addition to re-entering the cell cycle [49], our current work shows that c-MYC-overexpressing cardiomyocytes downregulate CX43 expression, resulting in arrhythmias. SWI/SNF and c-MYC have been reported to function cooperatively or antagonistically depending on context [23,50]. Our findings that BRG1/BRM bind to the c-Myc promoter, repress its expression, and antagonize c-MYC in terms of Cx43 expression is reminiscent of their opposing functions in cancer where BRG1 and BRM function as tumor suppressors and c-MYC functions as a proto-oncogene. In fact, BRG1 has been reported to directly repress c-Myc expression in primary tumor cells [23]. When another SWI/SNF subunit (BAF155) is methylated by CARM1, it is localized to c-MYC target genes and regulates the expression of genes in the c-MYC pathway [51]. The SWI/SNF-c-MYC antagonistic relationship is also compatible with similar metabolic changes occurring in Brg1/Brm cardiac knockout mice and transgenic mice that overexpress c-MYC in the heart [11,26,27].
Finally, the current work extends our understanding of BRG1 and BRM in human cardiac disease. Heterozygous mutations of several SWI/SNF subunits including BRG1 and BRM cause Coffin-Siris syndrome and Nicolaides-Baraitser syndrome, which include congenital heart defects such as patent ductus arteriosus and ventricular septal defects [3,52]. Additionally, BRG1 is upregulated in a subset of human cardiac hypertrophy cases, and BRG1 upregulation in response to cardiac stress is required for cardiac hypertrophy in a mouse model [7]. In the present study, BRG1/BRM occupancy of the CX43 and SCN5A promoters is downregulated in human heart failure cases associated with arrhythmias and bundle branch blocks. It is not yet clear how BRG1/BRM occupancy is diminished at these sites, but long noncoding RNAs (lncRNAs) are strong candidates. MHRT and SChLAP1 are recently identified lncRNAs that bind to and sequester BRG1 and BAF47/SNF5, respectively [30,53]. In so doing, these lncRNAs could directly (MHRT) or indirectly (SChLAP1) decrease BRG1/BRM occupancy at specific downstream target genes without altering global BRG1/BRM expression levels. Considering that other chromatin- modifying factors, such as the EZH2 catalytic subunit of PRC2, are bound by many different lncRNAs [29], it is plausible that MHRT, SChLAP1, or another lncRNA that sequesters BRG1 or other SWI/SNF subunits is upregulated in heart failure cases. To lend credence to this mechanism, MHRT was upregulated in a subset of heart failure cases analyzed in this study. Regardless of the precise details, the current findings combined with previous work reveal that BRG1 is dosage sensitive in the heart with upregulation [7] and downregulation (this study) each being deleterious and associated with cardiac disease.
Supplementary Material
Highlights.
Mouse BRG1 and BRM functionally compensate in adult cardiomyocytes as opposed to fetal cardiomyocytes, where BRG1 is essential but BRM is dispensable.
BRG1 and BRM regulate the expression of cardiac conduction genes to prevent arrhythmias, heart failure, and death.
BRG1 and BRM regulate the expression of downstream target genes both directly and indirectly via the activation of cardiogenic transcription factors and inhibition of c-Myc.
The phenotype of the BRG1/BRM double mutant mouse model is phenocopied by inducible overexpression of c-Myc.
Human heart failure cases exhibit increased expression of c-Myc, diminished BRG1/BRM occupancy of conduction genes, and decreased conduction gene expression.
Acknowledgments
We would like to thank Dr. Mark Zylka’s laboratory for lending their implantable telemetry devices for us to use in this study. We would like to acknowledge Dr. Dawn Bowles and Michael Watson at the Duke Human Heart Repository for providing the human heart tissues. Finally, we would like to thank Drs. Gerald Crabtree, Weidong Wang, and Keji Zhao for the J1 antibody and Dr. Frank Conlon for constructive comments on the manuscript. This work was supported by funding from the National Institutes of Health (RO1HL104129 to M.W. and CA125237 to S.B.), a Jefferson-Pilot Corporation (fellowship to M.W.), and the Leducq Foundation (to M.W.).
Footnotes
Disclosures of conflict of interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gloschat C, Koppel A, Aras K, Brennan J, Holzem K, Efimov I. Arrhythmogenic and metabolic remodelling of failing human heart. J Physiol. 2016;594:3963–3980. doi: 10.1113/JP271992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomaselli G, Zipes D. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 3.Bevilacqua A, Willis MW, Bultman SJ. SWI/SNF chromatin-remodeling complexes in cardiovascular development and disease. Cardiovasc Pathol. 2014;23:85–91. doi: 10.1016/j.carpath.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang CP, Bruneau BG. Epigenetics and cardiovascular development. Annu Rev Physiol. 2012;74:41–68. doi: 10.1146/annurev-physiol-020911-153242. [DOI] [PubMed] [Google Scholar]
- 5.Han P, Hang CT, Yang J, Chang CP. Chromatin remodeling in cardiovascular development and physiology. Circ Res. 2011;108:378–396. doi: 10.1161/CIRCRESAHA.110.224287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruneau BG. Chromatin remodeling in heart development. Curr Opin Genet Dev. 2010;20:505–511. doi: 10.1016/j.gde.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, Zhou B, Chang CP. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stankunas K, Hang CT, Tsun ZY, Chen ZYH, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado-Olguin P, Holloway AK, Mori AD, Wylie JN, Zhu Y, Zhou YQ, Yeh RF, Henkelman RM, Harvey RP, Metzger D, Chambon P, Stainier DY, Pollard KS, Scott IC, Bruneau BG. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat Commun. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv Y. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) EMBO J. 1998;17:6979–6991. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee R, Bultman SJ, Holley D, Hillhouse C, Bain JR, Newgard CB, Muehlbauer MJ, Willis MS. Non-targeted metabolomics of Brg1/Brm double-mutant cardiomyocytes reveals a novel role for SWI/SNF complexes in metabolic homeostasis. Metabolomics. 2015;11:1287–1301. doi: 10.1007/s11306-015-0786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bultman SJ, Holley DW, de Ridder GG, Pizzo SV, Sidorova TN, Murray KT, Jensen BC, Wang Z, Bevilacqua A, Chen X, Quintana MT, Tannu M, Rosson GB, Pandya K, Willis MS. BRG1 and BRM ATPases redundantly maintain cardiomyocyte homeostasis by regulating carddiomyocyte mitophagy and mitochondrial dynamics in vivo. Cardiovasc Pathol. 2016;25:258–269. doi: 10.1016/j.carpath.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen- inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 14.Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- 15.Sumi-Ichinose C, Ichinose H, Metzger D, Chambon P. SNF2beta-BRG1 is essential for the viability of F9 murine embryonal carcinoma cells. Mol Cell Biol. 1997;17:5976–5986. doi: 10.1128/mcb.17.10.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HG, Chen Q, Wolfram JA, Richardon SL, Liner A, Siedlak SL, Zhu X, Ziats NP, Fujioka H, Felsher DW, Castellani RJ, Valencik ML, McDonald JA, Hoit BD, Lesnefsky EJ, Smith MA. PLoS One. 2009;4:e7172. doi: 10.1371/journal.pone.0007172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willis MS, Rojas M, Li L, Selzman CH, Tang RH, Stansfield WE, Rodriguez JE, Glass DJ, Patterson C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Physiol. 2009;296:H997–H1006. doi: 10.1152/ajpheart.00660.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willis MS, Schisler JC, Li L, Rodriguez JE, Hilliard EG, Charles PC, Patterson C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res. 2009;105:80–88. doi: 10.1161/CIRCRESAHA.109.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HS, Lee G, John SMW, Maeda N, Smithies O. Molecular phenotyping for analyzing subtle genetic effects in mice: Application to an angiotensinogen gene titration. Proc Nat Acad Sci USA. 2002;99:4602–4607. doi: 10.1073/pnas.072083799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pandya K, Pulli B, Bultman S, Smithies O. Reversible epigenetic modifications of the two cardiac myosin heavy chain genes during changes in expression. Gene Expr. 2010;15:51–59. doi: 10.3727/105221611x12973615737505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith-Roe SL, Bultman SJ. Combined gene dosage requirement for SWI/SNF catalytic subunits during early mammalian development. Mamm Genome. 2013;24:21–29. doi: 10.1007/s00335-012-9433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen- induced MerCreMer gene deletion models. Circ Res. 2009;105:12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romero OA, Setien F, John S, Gimenez-Xavier P, Gomez-Lopez G, Pisano D, Condom E, Villanueva A, Hager GL, Sanchez-Cespedes M. The tumour suppressor and chromatin-remodelling factor BRG1 antagonizes Myc activity and promotes cell differentiation in human cancer. EMBO Mol Med. 2012;4:603–616. doi: 10.1002/emmm.201200236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starksen NF, Simpson PC, Bishopric N, Coughlin SR, Lee WM, Escobedo JA, Williams LT. Cardiac myocyte hypertrophy is associated with c-myc protooncogene expression. Proc Natl Acad Sci USA. 1986;83:8348–8350. doi: 10.1073/pnas.83.21.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olson AK, Ledee D, Iwamoto K, Kajimoto M, O’Kelly Priddy C, Isern N, Portman MA. C-Myc induced compensated cardiac hypertrophy increases fatty acid utilization for the citric acid cycle. J Mol Cell Cardiol. 2013;55:156–164. doi: 10.1016/j.yjmcc.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, Wang Y, Jin ES, Portman M, Maclellan WR. Myc controls transciptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest. 2010;120:1494–1505. doi: 10.1172/JCI38331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson T, Allard MF, Sreenan CM, Doss LK, Bishop SP, Swain JL. The c-myc proto-oncogene regulates cardiac development in mice. Mol Cell Biol. 1990;10:3709–3716. doi: 10.1128/mcb.10.7.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim SI, Bresnick EH, Bultman SJ. BRG1 directly regulates nucleosome structure and chromatin looping of the α globin locus to activate transcription. Nucl Acids Res. 2009;37:6019–6027. doi: 10.1093/nar/gkp677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:v145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han P, Li W, Lin CH, Yang J, Nurnberg ST, Jin KK, Xu W, Lin CY, Lin CJ, Chien HC, Zhou B, Ashley E, Bernstein D, Chen PS, Chen HS, Quertermous T, Chang CP. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh AP, Archer TK. Analysis of the SWI/SNF chromatin-remodeling complex during early heart development and BAF250a repression cardiac gene transcription during P19 cell differentiation. Nucl Acids Res. 2014;42:2958–2975. doi: 10.1093/nar/gkt1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin CT, Brennan J, Magnuson T. The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development. 2008;135:493–500. doi: 10.1242/dev.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willis MS, Homeister JW, Rosson GB, Annayev Y, Holley D, Holly SP, Madden VJ, Godfrey V, Parise LV, Bultman SJ. Functional redundancy of SWI/SNF catalytic subunits in maintaining vascular endothelial cells in the adult heart. Circ Res. 2012;111:e111–122. doi: 10.1161/CIRCRESAHA.112.265587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiley MM, Muthukumar V, Griffin TM, Griffin CT. SWI/SNF chromatin-remodeling enzymes Brahma-related gene 1 (BRG1) and Brahma (BRM) are dispensable in multiple models of postnatal angiogenesis but are required for vascular integrity in infant mice. J Am Heart Assoc. 2015;4:e001972. doi: 10.1161/JAHA.115.001972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman GR, Rahal R, Buxton F, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci USA. 2014;111:3128–3133. doi: 10.1073/pnas.1316793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oike T, Ogiwara H, Tominaga Y, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73:5508–5518. doi: 10.1158/0008-5472.CAN-12-4593. [DOI] [PubMed] [Google Scholar]
- 37.Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, Hahn WC, Roberts CW. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol. 2014;34:1136–1144. doi: 10.1128/MCB.01372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delgado-Olguin P, Huang Y, Li X, Christodoulou D, Seidman C, Seidman J, Tarakhovsky A, Bruneau B. Epigenetic repression of cardiac progenior gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;22:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montgomery R, Davis C, Potthoff M, Haberland M, Fielitz J, Qi X, Hill J, Richardson J, Olson E. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waldron L, Steimle J, Greco T, Gomez N, Dorr K, Kweon J, Temple B, Yang X, Wilczewski C, Davis I, Cristea I, Moskowitz I, Conlon F. The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev Cell. 2016;8:262–275. doi: 10.1016/j.devcel.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez-Del Arco P, Perdiguero E, Yunes-Leites P, Acin-Perez R, Zeini M, Garcia-Gomez A, Lopez-Maderuelo D, Ornes B, Jimenez-Borreguero L, D’Amato G, Enshell-Seijiffers D, Morgan B, Georgopoulos K, Islam A, Braun T, de la Pompa J, Kim J, Enriquez J, Ballestar E, Munoz-Canoves P, Redondo J. Cell Metab. 2016;10:881–892. doi: 10.1016/j.cmet.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Moreno JD, Yang PC, Bankston JR, Grandi E, Bers DM, Kass RS, Clancy CE. Ranolazine for congenital and acquired late INa-linked arrhythmias: in silico pharmacological screening. Circ Res. 2013;113:e50–61. doi: 10.1161/CIRCRESAHA.113.301971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012;9:600–607. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasahara H, Ueyama T, Wakimoto H, Liu MK, Maguire CT, Converso KL, Kang PM, Manning WJ, Wawitts J, Paul DL, Berul CI, Izumo S. Nkx2.5 homeoprotein regulates expression of gap junction protein connexin 43 and sacromere organization in postnatal cardiomyocytes. J Mol Cell Cardiol. 2003;35:243–56. doi: 10.1016/s0022-2828(03)00002-6. [DOI] [PubMed] [Google Scholar]
- 45.Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012;122:2509–2518. doi: 10.1172/JCI62617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda M, Briggs LE, Wakimoto H, Marks MH, Warren SA, Lu JT, Weinberg EO, Robertson KD, Chien KR, Kasahara H. Slow progressive conduction and contraction defects in loss of Nkx2-5 mice after cardiomyocyte terminal differentiation. Lab Invest. 2009;89:989–993. doi: 10.1038/labinvest.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2013;31:1119–1126. doi: 10.3892/ijmm.2013.1316. [DOI] [PubMed] [Google Scholar]
- 48.Wu M, Peng S, Yang J, Tu Z, Cai X, Cai CL, Wang Z, Zhao Y. Baf250a orchestrates an epigenetic pathway to repress the Nkx2.5-directed contractile cardiomyocyte program in the sinoatrial node. Cell Res. 2014;24:1201–1213. doi: 10.1038/cr.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, Fishbein MC, MacLellan WR. Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res. 2001;89:1122–1129. doi: 10.1161/hh2401.100742. [DOI] [PubMed] [Google Scholar]
- 50.Shi J, Whyte WA, Zepeda-Mendoza CJ, et al. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW, Yuan M, Ahlquist P, Xu W. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell. 2014;25:21–36. doi: 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kosho T, Okamoto N, Ohashi H, et al. Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am J Med Genet A. 2013;161A:1221–1237. doi: 10.1002/ajmg.a.35933. 161. [DOI] [PubMed] [Google Scholar]
- 53.Prensner JR, Iyer MK, Sahu A, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet. 2013;45:1392–1398. doi: 10.1038/ng.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.