

Abstract
A limited number of studies have indicated an association of the mitotic kinase polo-like kinase 1 (PLK1) and cellular metabolism. Here, employing an inducible RNA interference approach in A375 melanoma cells coupled with a PCR array and multiple validation approaches, we demonstrated that PLK1 alters a number of genes associated with cellular metabolism. PLK1 knockdown resulted in a significant downregulation of IDH1, PDP2 and PCK1 and upregulation of FBP1. Ingenuity Pathway Analysis (IPA) identified that 1) glycolysis and the pentose phosphate pathway are major canonical pathways associated with PLK1, and 2) PLK1 inhibition-modulated genes were largely associated with cellular proliferation, with FBP1 being the key modulator. Further, BI 6727-mediated inhibition of PLK1 caused a decrease in PCK1 and increase in FBP1 in A375 melanoma cell implanted xenografts in vivo. Furthermore, an inverse correlation between PLK1 and FBP1 was found in melanoma cells, with FBP1 expression significantly downregulated in a panel of melanoma cells. In addition, BI 6727 treatment resulted in an upregulation in FBP1 in A375, Hs294T and G361 melanoma cells. Overall, our study suggest that PLK1 may be an important regulator of metabolism maintenance in melanoma cells.
Keywords: Polo like kinase, Melanoma, Metabolism
Graphical abstract
PLK1 Knockdown Affects Modulation of Metabolism-Related Genes
1. Introduction
Mammalian polo-like kinases (PLKs) are a family of highly conserved serine/threonine kinases, comprised of five known members (PLKs 1–5), which play important roles throughout the cell cycle. PLKs are known to possess a conserved kinase domain at the amino-terminus and one or more polo-box domains at the carboxy-terminus [1]. PLK1 is the most widely studied member of this kinase family that has been shown to regulate a number of important cellular processes, including regulation of spindle assembly, chromosome segregation, mitotic exit and cytokinesis, during cell cycle progression [2–4]. Dysregulation of PLK1 has been shown to disrupt many of these events [5]. Further, PLK1 overexpression has been reported in a variety of cancers including melanoma [6–8], and higher PLK1 expression has been correlated with poor prognosis of cancer [9]. Furthermore, increased PLK1 expression has been found to be involved in chemotherapy drug resistance [reviewed in [10]].
In earlier studies we have shown that PLK1 is significantly overexpressed in human melanoma and its targeted inhibition significantly inhibits melanoma cell growth, both in vitro in cell culture as well as in vivo in melanoma xenografts [6, 11]. We have also demonstrated a novel association between PLK1 and NUMB during progression of mitosis in melanoma cells [12]. Due to the critical roles of PLK1 in cancer progression and its association with drug resistance, it is important to further characterize the mechanisms of PLK1 regulation. Identifying downstream targets of PLK1 could unravel important information leading to improved cancer treatment strategies. Working on this theme, in a recent study we employed a global proteomics approach to identify the downstream targets of PLK1 in melanoma. We found that PLK1 inhibition using the small-molecule ATP-competitive PLK1 inhibitor BI 6727 (Volasertib) resulted in a decrease in multiple metabolic proteins [13]. In another study, Li and colleagues demonstrated that PLK1 phosphorylation of phosphatase and tensin homolog (PTEN) causes a tumor-promoting metabolic state, promoting the Warburg effect in prostate cancer [14]. The Warburg effect (aerobic glycolysis) is used by tumor cells to increase the rate of glucose uptake and lactate production, stimulating cellular proliferation [15]. Our current study builds on these findings in an attempt to discern the interactions of PLK1 and metabolic genes in cancer. Our findings suggest that PLK1 may be involved in the regulation of metabolism maintenance of melanoma cell growth.
2. Materials and Methods
2.1. Cell Culture
Human embryonic kidney cell line HEK293T (ATCC, CRL-3216), human melanoma cell lines A375 and Hs294T (ATCC, #s CRL-1619, HTB-140, respectively), and immortalized melanocytes (Mel-ST, a kind gift from Dr. Robert Weinberg) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% Fetal Bovine Serum (FBS). G361 (ATCC, CRL-1424) human melanoma cell line was cultured in McCoy’s 5A Medium with 10% FBS. SK-MEL-2, SK-MEL-28 and WM35 (ATCC, #s HTB-68, HTB-72, CRL-2807, respectively) melanoma cell lines were cultured in Eagle’s Minimum Essential Medium with 10% FBS. Normal human epidermal melanocytes [neonatal (HEMn, ATCC, PCS-200-012) and adult (HEMa, ATCC, PCS-200-013)] were grown in Dermal Cell Basal Medium with the appropriate melanocyte growth kit. Transfected A375 shNS and shPLK1 cell lines were cultured in DMEM with 10% FBS and 1 μg/mL puromycin. All cell lines were maintained in a humidified incubator at 37°C with 5% CO2.
2.2. Creation of Doxycycline-Inducible PLK1 Knockdown Cell Line
For virus creation, HEK293T cells (7.5×104) were plated in a 6-well plate and incubated at 37°C for 24 h. Cells were transfected as described previously [12] using 2 μg shRNA plasmid DNA [TRIPZ Inducible Lentiviral shNS Control (GE Dharmacon #RHS4743) or TRIPZ Lentiviral Human shPLK1 (GE Dharmacon, #RHS4696-99638417, -99707812, -101347498, -101349871, -101352079 or -101353597). For target transduction, A375 cells were plated in 6-well plates at a density of 7.5×104 cells per well, and transduction was done as previously described [12]. Clones were selected by adding puromycin-containing media 24 h after final transduction was removed, and maintained until discrete colonies were discernible, when 6 colonies/shRNA clone were picked. For knockdown testing, individual clones were plated at a density of 1.0×10×5 in 6-well plates and treated with 0 or 2 μg/mL doxycycline for 48–96 h to induce shPLK1 expression. Every 24 h, 1 μg/mL doxycycline was added to the media to make up for depleted doxycycline in order to ensure continued shRNA expression. Cells were then collected and tested for PLK1 knockdown by immunoblotting as described below. Clones shNS 1 and shPLK1 5d (from shRNA #RHS4696-101352079) were chosen for use in further experiments.
2.3. Trypan Blue Exclusion Assay
To determine cell growth and viability, 1.0×104 A375 shNS or shPLK1 cells were plated in 6-well plates and treated with doxycycline for 96 h as described above. Cells were then trypsinized and collected, and a 10 μL aliquot was mixed with 10 μL trypan blue and loaded into one side of a counting chamber. Counting was done using the Countess FL Automated Cell Counter (Life Technologies) in quadruplicate per sample and averaged.
2.4. MTT Assay
A375 shNS and shPLK1 cells were plated at a density of 2.0×103 in 96-well plates. 24 h after plating, cells were treated with 2 μg/mL doxycycline for 48 h to induce shPLK1 expression. After 48 h, media was removed and MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma-Aldrich) reagent was added and the plate was incubated for 2 h at 37°C. After 2 h, the crystals were dissolved by mixing with dimethyl sulfoxide (DMSO) and the plate was read at 540 nm on a BioTek Synergy H1 plate reader.
2.5. BI 6727 Treatment
Human melanoma cells were plated at a density of 7.0×104 in 6-well plates. 24 h after plating, cells were treated with vehicle control (DMSO) or BI 6727 (50–100 nM; Selleck Chemicals, #S2235). 48 h after treatment, cells were trypsinized and collected for further analyses.
2.6. Protein Isolation and Immunoblot Analysis
Protein isolation, quantification, and immunoblotting were done as reported previously [16]. Protein from mouse tissues (obtained from our previous study [11]) was extracted in supplemented RIPA buffer using ceramic bead beating tubes (MO BIO, #13113–50). For immunoblotting, 30 μg of each protein was loaded per well and subjected to SDS-PAGE before transferring onto a nitrocellulose membrane. Blots were blocked with 5% nonfat dry milk in tris-buffered saline with 0.1% Tween-20 for 1 h and then incubated with primary antibodies: PLK1, AKT, β-actin (Cell Signaling, #s 208G4, 9272, 4970, respectively), CD3 (Abcam, #Ab5690), FBP1, and PCK1 (Sigma Aldrich, #s HPA005857 and HPA006507) overnight at 4°C with gentle shaking before washing and incubating with the appropriate HRP-conjugated secondary antibody (Cell Signaling, #s 7074, 7076). Specific proteins were detected using chemiluminescent detection (ThermoFisher Scientific) on an ImageQuant LAS 4000 (GE Healthcare Bio-Sciences) imager.
2.7. RNA Isolation and Quantitative Real-Time PCR
A375 shNS and shPLK1 cells (5.0×104) and A375, Hs294T and G361 (7.0×104) were plated in 6-well plates and treated as described above. Untreated lines were maintained in T75 flasks (TPP, #90076) and pelleted as needed. For collection, cells were trypsinized and washed with PBS and pelleted by centrifugation. RNA was isolated using an RNeasy Plus Mini Kit (QIAGEN, #74134) and cDNA was synthesized using M-MLV reverse transcriptase (Promega, #M1705). The Genomic DNA Contamination Control Assay (Bio-Rad, #10025352) was used to ensure no genomic DNA contamination was present. Quantitative Real-Time PCR (qPCR) was performed in triplicate using SYBR Premix Ex Taq II (TaKaRa, #RR820) per manufacturer’s protocol using the StepOnePlus Real-Time PCR System (ThermoFisher Scientific) and the following cycling conditions: 95°C for 3 min followed by 40 cycles of 95°C for 10 sec then 55°C for 30 sec. Primer sequences for PLK1, ENO2, FBP1, IDH1, PDP2, PCK1 (PrimerBank [17]) and ACTB (Origene, #HP204660) are provided in Table S1. Relative mRNA of each gene of interest was calculated using the ΔΔCT method, with β-actin as a reference gene. Melt curve analysis was implemented to confirm the specificity of target amplicon.
2.8. Qiagen Human Glucose Metabolism RT2 Profiler PCR Array
The RT2 Profile PCR Array-Human Glucose Metabolism (Qiagen, #PAHS-006Z) was performed as above using first strand cDNA synthesized as above, with qPCR cycling conditions as follows: 95°C for 30 sec followed by 40 cycles of 95°C for 5 sec then 60°C for 30 sec. Resulting CT values were uploaded onto the data analysis portal provided by QIAGEN (http://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/). CT values were normalized based on an automatic selection from HKG panel of reference genes, with RPLP0 and ACTB used to normalize the data, as the levels were stable between treatments chosen. Three biological replicates were used to ensure reproducibility.
2.9. Pathway Analysis
To understand pathways controlled by PLK1, a list of differentially expressed genes (>2-fold) were compiled and analyzed using QIAGEN’s Ingenuity Pathway Analysis (IPA). The predicted gene gene interaction networks and canonical pathways were generated using inputs of gene identifiers and fold-changes between shNS and shPLK1 group comparisons. Analysis was conducted between June and December 2016 via the IPA web portal (www.ingenuity.com).
2.10. Statistical Analysis
Data for PCR array were analyzed using the Qiagen RT2 Profiler PCR Data Analysis software and were considered significant at >2-fold change and P < 0.05. The RT-qPCR data was analyzed using StepOne Software v2.2 RQ Study (Applied Biosystems/Life Technologies Corp.) and exported as RQmax and RQmin (2−ΔΔCt +/−ΔΔCt SD). GraphPad Prism 5 Software (GraphPad Software Inc.) was used to perform statistical analyses on RT-qPCR replicates and all other data using two-tailed unpaired t-test or one-way analysis of variance (ANOVA) followed by Sidak multiple comparison tests.
3 Results
3.1. PLK1 inhibition exerts anti-proliferative effects against A375 human melanoma cells
To confirm the anti-proliferative response of PLK1 inhibition, we first determined the effect of doxycycline-inducible lentiviral shRNA-mediated PLK1 knockdown on viability and growth of A375 melanoma cells. The knockdown was successful, as PLK1 mRNA and protein levels were significantly reduced in the selected clone (shPLK), compared to the nonsense control (shNS, Figs. 1A and 1B). As measured by trypan blue exclusion assay, cell growth was significantly decreased (23.95%) at 96 h in the shPLK1 cell line as compared to shNS (Fig. 1C). A reduction in cell viability (5.78%) was also seen in shPLK1 cells (Fig. 1D). Further, PLK1 knockdown also resulted in a decreased cellular proliferation (15.56%) as measured by MTT assay (Fig. 1E).
Figure 1. PLK1 inhibition exerts anti-proliferative effects in human melanoma cells.
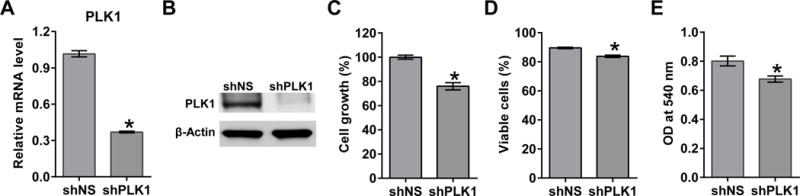
Following treatment with 2 μg/mL doxycycline for 48-96 h, PLK1 levels were analyzed as detailed in ‘Materials and Methods’. RT-qPCR was used to assess mRNA levels (A) and immunoblot analysis was used to evaluate protein levels of PLK1 (B). Cell growth was assessed by trypan blue exclusion assay and is shown normalized to the shNS control (C). Cell viability was also assessed by trypan blue exclusion assay and presented as a percentage (D). Cell proliferation was assessed using MTT assay and quantified using a BioTek plate reader (E). The data represents mean ± standard error of three experiments (*p-value <0.05).
3.2. PLK1 inhibition alters expression of multiple metabolic genes
In an effort to determine downstream metabolic targets of PLK1, we used a commercially available Glucose Metabolism PCR array to assess metabolic gene changes following PLK1 knockdown. The array contains 84 key genes involved in the regulation and enzymatic pathways of glucose and glycogen metabolism as well as appropriate PCR control primers. Detailed gene information and fold change following PLK1 knockdown are provided in Table S2. From the data, a heat map was generated which shows a general downregulation of genes involved in glucose and glycogen metabolism (Fig. 2A). Figure S1 breaks down the data as (A) a scatter plot comparing the normalized expression of every gene on the array, and (B) a heatmap clustergram to show the hierarchical clustering and genetic regulation of the entire dataset. Our results demonstrated that 29 of the 84 genes (34.5%) tested were downregulated by two-fold or more (Fig. 2B). Of these 29 genes, three were downregulated by three-fold or more: IDH1, PCK1 and PDP2, with PCK1 showing the largest fold change (-8.31 fold). Conversely, only three of the genes in the array (3.5%) were found to be >2 fold upregulated: FBP1, ENO2 and PGAM2 (Fig. 2B), where FBP1 showed the largest increase (7.96 fold).
Figure 2. PLK1 knockdown alters cellular metabolism in melanoma cells.
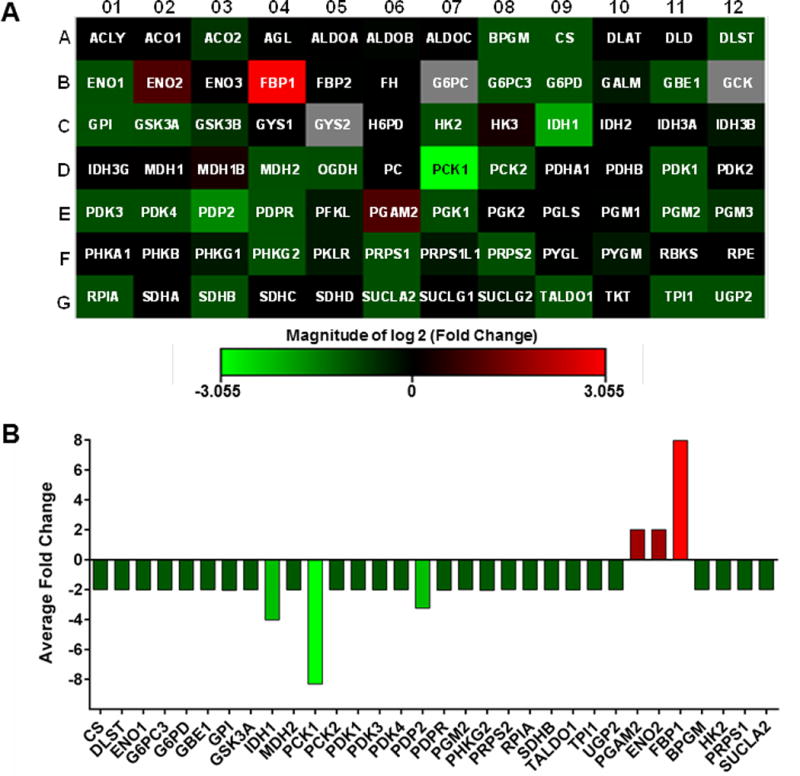
(A) A heat map was generated from the data to display gene fold changes. Upregulated genes are displayed in red and downregulated genes are shown in green, black boxes indicate no/negligible fold change in those particular genes. (B) Graphical representation of differentially expressed genes from the glucose metabolism PCR Array. Genes showing >2 fold changes are included. The data shown are representative of three biological replicates.
3.3. PLK1 knockdown significantly alters metabolic pathways and gene-interactions
We uploaded the 2-fold or more modulated genes in our array (a total of 32) into IPA, and identified canonical pathways associated with the modulated genes (shown in Fig. S2). Glycolysis-and Gluconeogenesis 1-pathways were the most affected pathways following PLK1 inhibition, according to p-values. The TCA Cycle II-and Pentose Phosphate-pathways were also affected by PLK1 knockdown. Using the genes listed in Table 1 to further understand the affected pathways, IPA was able to generate two pathway networks of interacting genes, one with 19 focus molecules (score=49) and one with 13 (score= 30). Focus molecules indicate the number of genes in a network that have been found in the uploaded dataset, and the score indicates the likelihood that the molecules in the network were randomly associated (higher number = less random). The network with the highest score was used for further analysis (Fig. 3), which includes the top 3 modulated genes FBP1, PCK1, and IDH1. Interestingly, many of the identified network genes have links to cancer (shown with a pink boundary in Fig. 3). In addition, ENO2, CD3 and AKT have direct links to melanoma (shown with blue letters). Importantly, CD3 and AKT were not part of the PCR array analysis, but were found only with the network analysis (Fig. 3). Using IPA, we also found several other genes outside of the array (shown as uncolored nodes) that may be potentially associated with the network of metabolic targets modulated by PLK1.
Table 1.
Key altered genes in PLK1 knockdown samples
| Gene Symbol | Entrez Gene Name | Fold Change | Location | Type(s) |
|---|---|---|---|---|
| FBP1 | fructose-bisphosphatase 1 | 7.96 | Cytoplasm | phosphatase |
| ENO2 | enolase 2 | 2.01 | Cytoplasm | enzyme |
| PGAM2 | phosphoglycerate mutase 2 | 2.00 | Cytoplasm | phosphatase |
| BPGM | bisphosphoglycerate mutase | −2.00 | Extracellular | phosphatase |
| CS | citrate synthase | −2.00 | Cytoplasm | enzyme |
| HK2 | hexokinase 2 | −2.00 | Cytoplasm | kinase |
| PGM2 | phosphoglucomutase 2 | −2.00 | Cytoplasm | enzyme |
| PRPS1 | phosphoribosyl pyrophosphate synthetase 1 | −2.00 | Cytoplasm | kinase |
| PRPS2 | phosphoribosyl pyrophosphate synthetase 2 | −2.00 | Cytoplasm | kinase |
| RPIA | ribose 5-phosphate isomerase A | −2.00 | Cytoplasm | enzyme |
| SDHB | succinate dehydrogenase complex iron sulfur subunit B | −2.00 | Cytoplasm | enzyme |
| SUCLA2 | succinate-CoA ligase ADP-forming beta subunit | −2.00 | Cytoplasm | enzyme |
| DLST | dihydrolipoamide S-succinyltransferase | −2.01 | Cytoplasm | enzyme |
| GBE1 | glucan (1,4-alpha-), branching enzyme 1 | −2.01 | Cytoplasm | enzyme |
| GSK3A | glycogen synthase kinase 3 alpha | −2.01 | Nucleus | kinase |
| PCK2 | phosphoenolpyruvate carboxykinase 2, mitochondrial | −2.01 | Cytoplasm | kinase |
| PDK1 | pyruvate dehydrogenase kinase 1 | −2.01 | Cytoplasm | kinase |
| PDK4 | pyruvate dehydrogenase kinase 4 | −2.01 | Cytoplasm | kinase |
| TPI1 | triosephosphate isomerase 1 | −2.01 | Cytoplasm | enzyme |
| UGP2 | UDP-glucose pyrophosphorylase 2 | −2.01 | Cytoplasm | enzyme |
| ENO1 | enolase 1 | −2.02 | Cytoplasm | enzyme |
| G6PC3 | glucose 6 phosphatase catalytic subunit 3 | −2.02 | Cytoplasm | phosphatase |
| G6PD | glucose-6-phosphate dehydrogenase | −2.02 | Cytoplasm | enzyme |
| MDH2 | malate dehydrogenase 2 | −2.02 | Cytoplasm | enzyme |
| PDK3 | pyruvate dehydrogenase kinase 3 | −2.02 | Cytoplasm | kinase |
| TALDO1 | transaldolase 1 | −2.02 | Cytoplasm | enzyme |
| PDPR | pyruvate dehydrogenase phosphatase regulatory subunit | −2.04 | Cytoplasm | enzyme |
| GPI | glucose-6-phosphate isomerase | −2.05 | Extracellular | enzyme |
| PHKG2 | phosphorylase kinase gamma subunit 2 | −2.05 | Cytoplasm | kinase |
| PDP2 | pyruvate dehyrogenase phosphatase catalytic subunit 2 | −3.25 | Cytoplasm | phosphatase |
| IDH1 | isocitrate dehydrogenase (NADP(+)) 1, cytosolic | −4.02 | Cytoplasm | enzyme |
| PCK1 | phosphoenolpyruvate carboxykinase 1 | −8.31 | Cytoplasm | kinase |
Figure 3. Gene network analysis showing regulatory relationships between the genes modulated upon PLK1 knockdown in melanoma cells.
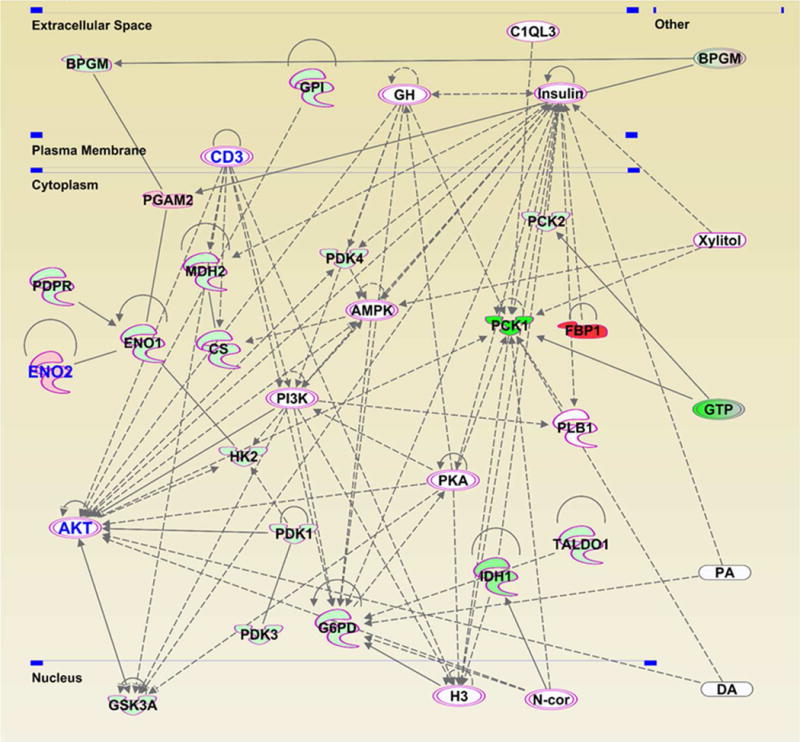
A network pathway was generated after uploading the PCR array data to the IPA software. Highlighted molecules with pink boundaries are those involved in cancer, and the ones with blue text color (CD3, ENO2 and AKT) are those specifically involved in melanoma. The gene gene interactions are indicated by arrows. The solid lines denote a robust correlation with partner genes, and dashed lines indicate statistically significant but less frequent correlations.
3.4. Validation of key altered genes in PLK1 knockdown samples
The top four genes that showed maximum change in the PCR array after PLK1 knockdown were selected for validation. These were FBP1 (8-fold), IDH1 (4-fold), PDP2 (3.3-fold) and PCK1 (8.3-fold). The rest of the genes, although they were found to be significantly modulated (2.0–2.1 fold change), were not as strongly modulated and therefore were not chosen for validation purposes (Table 1). In addition, ENO2 was also selected as it showed a direct link to melanoma in network analysis. Further, CD3 and AKT were selected due to a direct link with melanoma, as predicted by the IPA. RT-qPCR data demonstrated that the observed changes in FBP1, IDH1, PDP2, PCK1, and ENO2 were comparable to the glucose metabolism array results (Fig. 4A–E), validating the array results. We were interested to see if the observed changes at gene-level were also shown at the protein level. PCK1 expression was significantly decreased by PLK1 inhibition (Fig. 5B). However, there was no significant change in FBP1 protein expression as assessed by immunoblot analysis (Fig. 5C). No changes were found in CD3 and AKT protein expression levels (data not shown).
Figure 4. Validation of key altered genes in PLK1 knockdown samples.
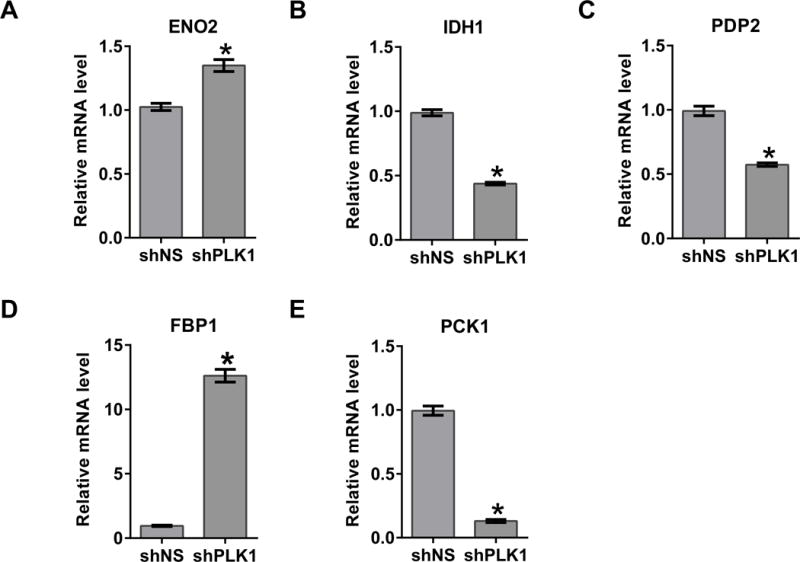
RT-qPCR analysis was performed to validate the key altered genes, ENO2 (A), IDH1 (B), PDP2 (C), FBP1 (D) and PCK1 (E) at mRNA levels in PLK1 knockdown A375 cells as detailed in ‘Materials and Methods’. Data are represented as mean value ± standard errors of minimum three biological replicates (*p-value <0.05).
Figure 5. Analysis of functional annotation of FBP1 and PCK1 in relation to antitumor efficacy of PLK1 inhibition.

(A) Using IPA, the regulatory network was generated for the functions of gene expression in PLK1-modulated genes showing inhibition of proliferation of cells via FBP1 and inhibition of quantity of monosaccharide with the involvement of PCK1. The functional annotation leading inhibition are denoted with blue dotted lines. Findings inconsistent with the state of downstream molecules are represented with the dark yellow dotted lines. The gray dotted line shows that the effect is not predicted. Immunoblot analyses of shNS and shPLK1 cell lines were performed using PCK1 (B) and FBP1 (C) antibodies. Blots were re-probed with β-actin for loading control. Further, in vivo validation was done using archived samples from a recent study published earlier [11]. Briefly, immunoblot analyses were performed in A375 xenografted melanoma tissues treated with BI 6727 (10 and 25 mg/kg body weight) or vehicle alone to determine the effect of PLK1 knockdown using (D) PCK1 and (E) FBP1 antibodies. The blots were re-probed with β-actin for loading control. Data is representative of three individual experiments.
3.5. Modulations in FBP1 and PCK1 are associated with antitumor response of PLK1 inhibition
Further IPA exploration of the metabolism array-identified gene changes following PLK1 inhibition found two significant functional annotations. The first is in proliferation of cells (activation z-score-2.043), where FBP1 appears as a key regulatory player showing interaction with several genes in the network. The second is in quantity of monosaccharide (activation z-score −2.000), which includes PCK1, a highly downregulated gene that interacts with fewer genes (Fig. 5A). The activation z-score measures the match between expected relationship direction and observed gene expression (≥2 is considered significant). IPA predicted the activation state for these two functional annotations as decreased, as most of the genes are related to inhibition (denoted with blue dotted lines). The findings of downregulated ENO1 and GP1, and upregulated PGAM2 were inconsistent with the predicted state of downstream molecules (represented with the yellow dotted lines). The functional annotation of DLST (denoted with a gray dotted line) was unpredictable.
We were also interested in validating our findings in vivo. For this, we employed archived samples from a recent study in which we demonstrated antitumor effects of the second-generation small molecule PLK1 inhibitor BI 6727 on A375 melanoma cell implanted xenografts in immunocompromised mice [11]. Immunoblot analyses were performed on A375 xenografted melanoma tissues treated with BI 6727 (10 and 25 mg/kg body weight) or vehicle alone. As shown in Fig. 5D, PCK1 was found to be significantly decreased in BI 6727 treatment groups, at both the doses. Interestingly, FBP1 protein levels were found to be increased in BI 6727 treated tissues (Fig. 5E). These results show a similar trend to the PCR array results.
3.6. Tumor suppressor FBP1 is downregulated in human melanoma and enhanced by PLK1 knockdown
Since studies have shown that FBP1 1) is an important regulator of glycolysis, 2) has been shown to be a tumor suppressor, and 3) appears to be an important downstream target of PLK1, we determined its expression profile in human melanocytes and melanoma cells. As shown in Figs. 6A and 6B, FBP1 mRNA and protein levels were significantly downregulated in a panel of melanoma cells (A375, Hs294T, G361, SK-MEL-2, SK-MEL-28, WM35) compared to normal melanocytes (HEMa and HEMn). Interestingly, we observed decreased expression of FBP1 in the immortalized melanocyte cell line (Mel-ST), which despite being normal (non-tumorigenic), have a high proliferative index.
Figure 6. Analysis of inverse association of FBP1 and PLK1 in melanoma.

(A) RT-qPCR and (B) immunoblot analyses were performed to assess FBP1 mRNA and protein levels in normal melanocytes and in a panel of melanoma cell lines. (C) FBP1 mRNA and (D) protein levels were also analyzed in BI 6727 treated A375, Hs294T, G361 and SK-MEL-2 human melanoma cells. For comparative analysis (E) PLK1 mRNA levels were also analyzed in BI 6727 treated A375, Hs294T, G361 cells. The blots were re-probed with β-actin for loading control. Data is representative of three individual experiments. RT-qPCR data are represented as mean value ± standard errors of three biological replicates (*p-value <0.05).
We also determined the effect of BI 6727 (50 nM and 100 nM) treatment on FBP1 in melanoma cells. BI 6727 treatment resulted in a significant increase in FBP1 mRNA levels in A375 and Hs294T melanoma cells at both concentrations, as well in G361 cells at 100 nM (Fig. 6C). FBP1 protein expression was elevated in BI 6727 treated Hs294T cells, but not in A375 or SK-MEL-2 (Fig. 6D). Interestingly, PLK1 mRNA was significantly decreased with BI 6727 treatment, showing a strong inverse correlation of FBP1 with PLK1 inhibition (Fig. 6E).
4. Discussion
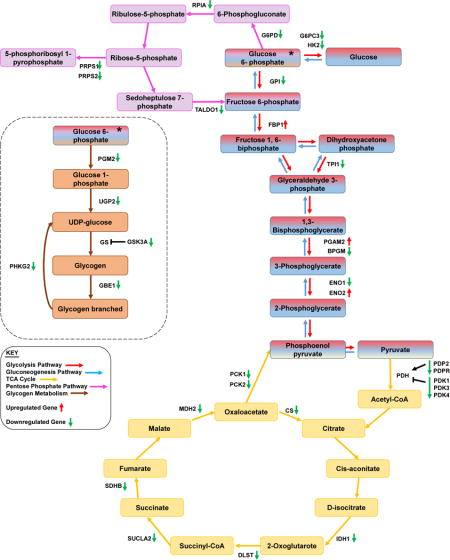
PLK1 has emerged as an attractive ‘druggable’ target for the management of cancer and PLK1 inhibitors are being tested in preclinical and clinical trials for their potential clinical use. However, so far the pre-clinical success of PLK1 inhibitors has not translated into clinical settings. Therefore, it is important to intensify our efforts to better-understand the upstream and downstream mechanisms of PLK1. Earlier work has suggested that PLK1 may modulate cellular metabolism [13, 14]. However, additional studies are needed to further discern the mechanism of PLK1 ’s association with downstream metabolic targets and their interactions. Our current study is an effort in this direction. To attempt to uncover which genes or pathways associated with cellular metabolism may be modulated by PLK1, we used a doxycycline-inducible PLK1 knockdown melanoma cell line in conjunction with a PCR array focused on glucose metabolism. As discussed above, we found that a number of genes known to be involved in glucose-and glycogen-pathways were altered following PLK1 knockdown, suggesting a potential modulation of multiple metabolic processes by PLK1 (also outlined in Fig. S3). The five genes that were validated are discussed here in relevance to melanoma development and progression.
We found that PLK1 knockdown resulted in a significant decrease in PDP2 and IDH1 in melanoma cells. PDP2 is an enzyme that activates the pyruvate dehydrogenase complex (PDH), which converts pyruvate into acetyl-CoA to be used in cellular respiration [18]. Thus, our data suggests that PLK1 may potentially be involved in pyruvate to acetyl-CoA conversion in the TCA cycle. The other significantly downregulated gene, IDH1, is a cytosolic enzyme involved in catalyzing the conversion of isocitrate to 2-oxoglutarate [19]. IDH1 is one of the most frequently mutated metabolic genes in a variety of cancer types [20, 21]. Seltzer et al. have shown that inhibition of glutaminase decreases the growth of glioma cells with mutations in IDH1 [22]. Interestingly, Shibata and colleagues identified IDH1 mutations in melanoma that were shown to confer a tumor growth advantage [23]. Thus it is possible that the observed downregulation of IDH1 following PLK1 inhibition may help limit melanoma cell growth.
ENO2 was one of the genes found to be upregulated following PLK1 inhibition in the PCR array. ENO2 is a key component of the glycolytic pathway and is involved in converting 2-phosphoglycerate into phosphoenolpyruvate. The role of ENO2 in cancer is not clearly understood. In renal cell carcinoma, ENO2 was not found to affect tumor growth [24]. However, another recent study has shown a correlation in glioblastomas between increased ENO2 expression and tumor progression in vivo [25]. Further studies will be required to determine the role of ENO2 in melanoma. It is also possible that the increased expression of ENO2 following PLK1 knockdown may be to compensate for the downregulation of its counterpart ENO1. Interestingly, ENO1 knockdown has previously been shown to result in suppression of glioma cell growth [26].
Another key component of the gluconeogenesis pathway, PCK1, was found to be dramatically downregulated following PLK1 knockdown. The PCK1 gene encodes a cytosolic isozyme that uses GTP to catalyze the conversion of oxaloacetate to phosphoenolpyruvate [27]. An important paralog of this gene, PCK2, was also found to be downregulated upon PLK1 inhibition, though not as much as PCK1. The expression of PCK1 is regulated by several other molecules, as appeared in our gene network (Fig. 3). However, these connections are less frequently correlated (as shown by dashed lines), but are still statistically significant. Overexpression of PCK1 has been implicated in type 2 diabetes due to its role in excessive glucose production [28]. Previously, Li and colleagues discovered that tumor-repopulating cells (TRCs) upregulate PCK1 while downregulation of PCK1 results in slower growth of TRCs in vitro [29]. Furthermore, they noted that knockdown of PCK1 reduced lactate release, slowing metabolism. In an in vivo study, mice injected with DU145 prostate cancer cells plus prostate-derived stromal cells had higher expression of PCK1 and more aggressive tumor growth than mice injected with DU145 cells alone [30]. Thus, our finding of PCK1 decrease following PLK1 knockdown samples is consistent with reported studies and suggests that an overexpression of PLK1 might play a role in upregulating PCK1, which can increase glucose metabolism and tumor growth potential.
Of the three genes that were found to be upregulated upon PLK1 knockdown, fructose-bisphosphatase 1 (FBP1) was found to be the most significantly altered. The protein encoded by this gene has been traditionally known as the rate-limiting enzyme in gluconeogenesis. In fact, FBP1 is an enzyme that resists glycolysis and supports gluconeogenesis through making of fructose 6-phosphate from fructose 1, 6-biphosphate (Fig. S3). Interestingly, in recent studies, FBP1 appears to act as a tumor suppressor in hepatocellular, colon, renal, and melanoma cancers [31–33]. Chen et al. observed decreased FBP1 expression in human hepatocellular carcinoma and colon cancer cell lines that occurred as a result of promoter hypermethylation [31]. Wu and colleagues have found that FBP1 is often downregulated in melanoma tissues, and when FBP1 is restored, melanoma proliferation is impeded in vitro and in vivo [33]. Li et al. have shown that FBP1 reduces renal carcinoma progression and downregulates glycolysis and the pentose phosphate pathway by inhibiting hypoxia-inducible factors (HIFs). They also found that FBP1 antagonizes glycolytic flux to prevent the Warburg effect from occurring [32]. Ning et al. observed higher frequencies of low FBP1 expression in tumor tissues in comparison to healthy tissues, as well as an association between low FBP1 expression and high HIF1α and erythropoietin expression in clear cell renal cell carcinoma [34]. This suggests that the effects of FBP1 may be conserved across tumor types. Our data showing a significant increase in FBP1 expression upon PLK1 knockdown is an important finding, as PLK1 has not previously been linked with FBP1. This data was also confirmed when we found similar effects on FBP1 following a small molecule inhibition of PLK1. Importantly, FBP1 mRNA and protein expression were found to be significantly downregulated in human melanoma cells, compared to normal melanocytes. Together, these results suggest that PLK1 inhibitory approaches may be useful in reversing the FBP1 downregulation in melanoma in order to activate the tumor suppressive function of FBP1.
In this study, we have shown that PLK1 knockdown significantly alters various metabolic genes associated with a decrease in cellular metabolism. Specifically, many of the modulated genes seem to disturb the process of gluconeogenesis (Fig. S3), a pathway essential for cancer cell survival. Our study also provides evidence of a novel relationship between PLK1 and the tumor suppressor FBP1, suggesting that this axis may be an important new area of study for melanoma research.
Supplementary Material
Highlights.
-
➢
PLK1 knockdown alters expression of metabolic genes in A375 melanoma cells.
-
➢
PLK1 knockdown causes an alteration in FBP1 and PCK1.
-
➢
BI 6727-mediated inhibition of PLK1 increases FBP1 and decreases PCK1 in vivo.
-
➢
FBP1 is significantly downregulated in human melanoma cells.
Acknowledgments
This work was supported by the National Institutes of Health (grant numbers R01AR059130 and R01CA176748) and the Department of Veterans Affairs (VA Merit Review Award number 1I01BX001008). In addition, we acknowledge the core facilities supported by the Skin Diseases Research Center (SDRC) Core Grant P30AR066524 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None.
References
- 1.de Career G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle. 2011;10:2255–2262. doi: 10.4161/cc.10.14.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sumara I, Gimenez-Abian JF, Gerlich D, Hirota T, Kraft C, de la Torre C, Ellenberg J, Peters JM. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14:1712–1722. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 3.Shao H, Huang Y, Zhang L, Yuan K, Chu Y, Dou Z, Jin C, Garcia-Barrio M, Liu X, Yao X. Spatiotemporal dynamics of Aurora B-PLK1-MCAK signaling axis orchestrates kinetochore bi-orientation and faithful chromosome segregation. Sci Rep. 2015;5:12204. doi: 10.1038/srep12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell. 2007;12:713–725. doi: 10.1016/j.devcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 6.Schmit TL, Zhong W, Setaluri V, Spiegelman VS, Ahmad N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J Invest Dermatol. 2009;129:2843–2853. doi: 10.1038/jid.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takai N, Miyazaki T, Fujisawa K, Nasu K, Hamanaka R, Miyakawa I. Expression of polo-like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett. 2001;164:41–49. doi: 10.1016/s0304-3835(00)00703-5. [DOI] [PubMed] [Google Scholar]
- 8.Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger HM, Rubsamen-Waigmann H, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14:543–549. doi: 10.1038/sj.onc.1200862. [DOI] [PubMed] [Google Scholar]
- 9.Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int J Oncol. 1999;15:687–692. doi: 10.3892/ijo.15.4.687. [DOI] [PubMed] [Google Scholar]
- 10.Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther. 2016;15:1427–1435. doi: 10.1158/1535-7163.MCT-15-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cholewa BD, Ndiaye MA, Huang W, Liu X, Ahmad N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2016;385:179–187. doi: 10.1016/j.canlet.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmit TL, Nihal M, Ndiaye M, Setaluri V, Spiegelman VS, Ahmad N. Numb regulates stability and localization of the mitotic kinase PLK1 and is required for transit through mitosis. Cancer Res. 2012;72:3864–3872. doi: 10.1158/0008-5472.CAN-12-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cholewa BD, Pellitteri-Hahn MC, Scarlett CO, Ahmad N. Large-scale label-free comparative proteomics analysis of polo-like kinase 1 inhibition via the small-molecule inhibitor BI 6727 (Volasertib) in BRAF(V600E) mutant melanoma cells. J Proteome Res. 2014;13:5041–5050. doi: 10.1021/pr5002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Li J, Bi P, Lu Y, Burcham G, Elzey BD, Ratliff T, Konieczny SF, Ahmad N, Kuang S, Liu X. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol Cell Biol. 2014;34:3642–3661. doi: 10.1128/MCB.00814-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, Hu W, Feng Z. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cholewa BD, Ndiaye MA, Huang W, Liu X, Ahmad N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2017;385:179–187. doi: 10.1016/j.canlet.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 19.Geisbrecht BV, Gould SJ. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem. 1999;274:30527–30533. doi: 10.1074/jbc.274.43.30527. [DOI] [PubMed] [Google Scholar]
- 20.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Carli E, Wang X, Puget S. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:2248. doi: 10.1056/NEJMc090593. author reply 2249. [DOI] [PubMed] [Google Scholar]
- 22.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol. 2011;178:1395–1402. doi: 10.1016/j.ajpath.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang T, Niu X, Liao L, Cho EA, Yang H. The contributions of HIF-target genes to tumor growth in RCC. PLoS One. 2013;8:e80544. doi: 10.1371/journal.pone.0080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanzey M, Abdul Rahim SA, Oudin A, Dirkse A, Kaoma T, Vallar L, Herold-Mende C, Bjerkvig R, Golebiewska A, Niclou SP. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS One. 2015;10:e0123544. doi: 10.1371/journal.pone.0123544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Y, Luo Q, Long H, Hu Z, Que T, Zhang X, Li Z, Wang G, Yi L, Liu Z, Fang W, Qi S. Alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol Cancer. 2014;13:65. doi: 10.1186/1476-4598-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beale EG, Harvey BJ, Forest C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem Biophys. 2007;48:89–95. doi: 10.1007/s12013-007-0025-6. [DOI] [PubMed] [Google Scholar]
- 28.Beale EG, Hammer RE, Antoine B, Forest C. Disregulated glyceroneogenesis: PCK1 as a candidate diabetes and obesity gene. Trends Endocrinol Metab. 2004;15:129–135. doi: 10.1016/j.tem.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Luo S, Ma R, Liu J, Xu P, Zhang H, Tang K, Ma J, Zhang Y, Liang X, Sun Y, Ji T, Wang N, Huang B. Upregulation of cytosolic phosphoenolpyruvate carboxykinase is a critical metabolic event in melanoma cells that repopulate tumors. Cancer Res. 2015;75:1191–1196. doi: 10.1158/0008-5472.CAN-14-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng YB, Zhou J, Gao Y, Li YH, Wang H, Zhang M, Ma LM, Chen Q, Da J, Wang Z, Li R. Normal prostate-derived stromal cells stimulate prostate cancer development. Cancer Sci. 2011;102:1630–1635. doi: 10.1111/j.1349-7006.2011.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen M, Zhang J, Li N, Qian Z, Zhu M, Li Q, Zheng J, Wang X, Shi G. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS One. 2011;6:e25564. doi: 10.1371/journal.pone.0025564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TP, Keith B, Nissim I, Simon MC. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–255. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y, Ge Y, Lei T. ZFX-mediated down-regulation of FBP1 confers to growth in melanoma. Int J Clin Exp Pathol. 2016;9:1222–1230. [Google Scholar]
- 34.Ning XH, Li T, Gong YQ, He Q, Shen QI, Peng SH, Wang JY, Chen JC, Guo YL, Gong K. Association between FBP1 and hypoxia-related gene expression in clear cell renal cell carcinoma. Oncol Lett. 2016;11:4095–4098. doi: 10.3892/ol.2016.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.