

Abstract
We previously showed that loss of the high mobility group A1 (HMGA1) protein expression, induced in mice by disrupting the Hmga1 gene, considerably decreased insulin receptor expression in the major target tissues of insulin action, causing a type 2-like diabetic phenotype, in which, however, glucose intolerance was paradoxically associated with increased peripheral insulin sensitivity. Insulin hypersensitivity despite impairment of insulin action supports the existence of molecular adaptation mechanisms promoting glucose disposal via insulin-independent processes. Herein, we provide support for these compensatory pathways/circuits of glucose uptake in vivo, the activation of which under certain adverse metabolic conditions may protect against hyperglycemia. Using chromatin immunoprecipitation combined with protein-protein interaction studies of nuclear proteins in vivo, and transient transcription assays in living cells, we show that HMGA1 is required for gene activation of the IGF-binding proteins 1 (IGFBP1) and 3 (IGFBP3), two major members of the IGF-binding protein superfamily. Furthermore, by using positron emission tomography with 18F-labeled 2-fluoro-2-deoxy-d-glucose, in combination with the euglycemic clamp with IGF-I, we demonstrated that IGF-I's bioactivity was increased in Hmga1-knockout mice, in which both skeletal muscle Glut4 protein expression and glucose uptake were enhanced compared with wild-type littermates. We propose that, by affecting the expression of both IGFBP protein species, HMGA1 can serve as a modulator of IGF-I activity, thus representing an important novel mediator of glucose disposal.
We previously showed that in Hmga1-knockout mice peripheral insulin hypersensitivity paradoxically coexisted with a condition of impaired glucose tolerance and overt diabetes (1). Insulin hypersensitivity despite impairment of insulin action supports the existence of molecular adaptation mechanisms, the functional activation of which may promote tissue glucose uptake and utilization by insulin-independent mechanisms. This possibility is documented by a wide variety of observations in vivo, in animal models of diabetes (2, 3). For example, in a mouse model of insulin resistance, it has been recently shown that hepatic overexpression of XBP-1, a factor involved in the cell stress response, by inducing FoxO1 protein degradation, reduces serum glucose levels and increases glucose tolerance without improving insulin signaling (4). Also, in mice overexpressing the insulin-regulatable glucose transporter Glut4, the amount of Glut4 in the plasma membrane often increases without the need for insulin signaling, and fasting hypoglycemia can occur in these animals, even in the absence of insulin (5). However, the precise mechanism(s) by which these compensatory circuits of glucose uptake are activated and provide signals for the translocation of the intracellular Glut4-containing vesicles to the cytoplasmic membrane remain to be fully characterized and elucidated. In an attempt to clarify these mechanisms, we focus here on the role of the IGF-I/IGF binding protein (IGFBP) system in the recruitment of Glut4 to the plasma membrane. IGF-I–mediated recruitment of Glut4 to the cell surface has been observed previously in L6 muscle cells and 3T3-L1 adipocytes (6, 7). On the other hand, IGFBP1 and IGFBP3, two major members of the superfamily of IGFBP, have a high affinity for IGF-I and appear to be important regulators of IGF-I signaling and action (8, 9). IGFBP generally inhibit IGF-I action by binding competitively to it, thereby reducing its bioavailability. Cleavage of IGFBP by their specific proteases also influences IGF-I bioavailability by reducing the amount of bioavailable IGFBP (8–10). Therefore, because of their critical role in regulating the potential IGF-I's bioactivity, IGFBP1 and IGFBP3 can modulate the insulin-like metabolic effects of endogenous IGF-I on glucose metabolism.
The IGFBP1 and the IGFBP3 genes are contiguously arranged in a tail-to-tail fashion within chromosome 7. It has been suggested that the juxtaposition of the genes and their high expression within the liver could theoretically allow cellular factors to regulate the genes similarly in a given physiological condition (11). HMGA1 is a small basic protein that binds to AT-rich regions of DNA and functions mainly as a specific cofactor for gene activation (12). HMGA1 can transactivate promoters through mechanisms that facilitate the assembly and stability of stereospecific DNA-protein complexes that drive gene transcription (12). An involvement of HMGA1 in the regulation of the IGFBP1 gene has been postulated previously on the basis of in vitro evidence indicating that HMGA1 binds the insulin response element of the IGFBP1 gene promoter interfering with the inhibitory effect of insulin on IGFBP1 gene transcription (13). Consistent with this observation supporting a positive role of HMGA1 in the transcriptional activation of the IGFBP1 gene, we previously reported that IGFBP1 protein expression was reduced in Hmga1-deficient mice (1).
First, this study aimed to clarify the molecular mechanisms underlying the activation of IGFBP1 and IGFBP3 gene expression in mammals. A second aim was to provide evidence for the existence of molecular adaptation mechanisms in vivo, the functional activation of which, linked to the HMGA1-IGF-I/IGFBP system, plays a role in the recruitment of Glut4 to muscle plasma membrane and glucose uptake under adverse circumstances in which insulin action is precluded.
Materials and Methods
Cells, protein extracts, and EMSA
HepG2 human hepatoma cells, NIH-3T3 mouse fibroblasts, and MCF-7 human breast cancer cells (American Type Culture Collection, LGC Promochem; Teddington, UK) were maintained in DMEM (supplemented with 2 mmol/liter l-glutamine, 50 IU/ml penicillin and 50 μg/ml streptomycin, and 10% fetal bovine serum), and nuclear extracts from these cell lines were prepared as described elsewhere(14). Equal numbers of nuclei were homogenized, the final protein concentrations in the extracts were determined by the modified Bradford method (Bio-Rad Laboratories, Hercules, CA), and DNA-nuclear protein binding reactions were carried out by EMSA according to previous published protocols (see Supplemental Data published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org).
Chromatin immunoprecipitation (ChIP), quantitative RT-PCR, immunoprecipitation, and Western blot
These methods are described in the Supplemental Data.
Plasmid construction and transfection
Promoter elements of both human and mouse IGFBP1 and mouse IGFBP3 were prepared from genomic DNA, using sequence specific primers (see Supplemental data), and cloned into pGL3-basic reporter vector (Promega Corp., Madison, WI). Human IGFBP3 gene promoter was cloned into the pGL3-basic reporter plasmid, starting from the pGL2-IGFBP3 reporter vector (provided by Dr. Tadashi Hanafusa, Okayama University, Okayama, Japan). Recombinant Luc reporter constructs in the presence or absence of effector vectors for HMGA1 (HMGA1a isoform protein), CCAAT-enhancer binding protein β (C/EBPβ), Sp1 (15), or the hepatocyte nuclear factor-1 (HNF-1) (provided by Dr. Nobuhiko Takamatsu, Kitasato University, Kanagawa, Japan), were transiently transfected into HepG2, NIH-3T3, or MCF-7 cells using LipofectAMINE 2000 reagent (Invitrogen, Carlsbad, CA), and luciferase (Luc) activity was assayed 48 h later in a luminometer (Turner Biosystems, Mountain View, CA), using the dual-luciferase reporter assay system (Promega). Small interfering RNA (siRNA) targeted to human HMGA1 and Sp1 (16), and to C/EBPβ and HNF-1α (SMARTpool), plus nonspecific siRNA controls with a similar GC content were obtained from Dharmacon (Lafayette, CO); siRNA for mouse Hmga1 was from Santa Cruz Biotechnology (Santa Cruz, CA). 100–200 pmol siRNA duplex were transfected into cells at 50% to 60% confluency, and cells were analyzed 48–96 h later.
Animals, IGF-I serum levels, IGF-I tolerance tests, and purification of human IGFBP1 (hIGFBP1)
Male Hmga1-deficient and wild-type mice aged 6–9 months were studied. The generation of Hmga1-mutants and many of the physiological characteristics of the mice have been described in detail (1). All animal work was carried out at the Animal Facility at the Istituto dei Tumori di Napoli, using approved animal protocols and in accordance with institutional guidelines. Blood was collected from the retro-orbital sinus, and total and free serum IGF-I concentrations were measured in mice (see Supplemental Data). IGF-I tolerance was assessed by measuring blood glucose levels in 12-h fasted conscious mice injected ip with recombinant human IGF-I (rhIGF-I) (Tercica, Inc., Brisbane, CA), 1 mg/kg body weight, either in the absence or after injection of hIGFBP1. Glycemia was measured before and 15, 30, 45, and 60 min after IGF-I injection, using the Glucocard G meter (Menarini Diagnostics s.r.l., Firenze, Italy). hIGFBP1 was isolated from midgestational amniotic fluid obtained for diagnostic purposes with approval of the local ethics committee and purified by liquid chromatography as reported elsewhere (17).
Positron emission tomography (PET)
Whole-body PET-2-fluoro-2-deoxy-d-glucose (FDG) was performed using a dedicated PET scanner [YAP(S)PET–ISE, Pisa, Italy, FWHM = 1.8 mm]. PET-FDG studies were performed in fasting anesthetized Hmga1-null (n = 4) and wild-type (n = 3) mice, after administration of 12 MBq (100–120 μl) radiolabeled FDG into the tail vein, followed by ip administration of rhIGF-I. The PET protocol included a localizer scan and a two-step whole-body PET acquisitions starting 30 min after FDG administration, with 128 views over 180 degrees (events/view: 157,500). Transverse and dorsal slices were obtained for quantitative assessment of FDG distribution, using small regions of interest positioned on brain, heart, abdomen, and the neck/forelimb region for assessment of muscle uptake. For each structure, measurements were obtained from all corresponding slices and summed. Regional tracer uptake ratios vs. whole-body counts were obtained, and FDG uptake ratios for heart, abdomen, and neck/forelimb using brain uptake as a reference value were calculated.
Euglycemic clamp with IGF-I and IGF-I/insulin signaling proteins
Euglycemic clamps with IGF-I were performed in conscious and freely moving animals, following previously published technical descriptions of the method (1). On the morning of the experiment, after a 12-h fast, mice were subjected to 120 min euglycemic clamp, using a prime dose of 33 nmol rhIGF-I/kg followed by a constant infusion of rhIGF-I (0.65 nmol/kg body weight/min). Blood samples (20 μl) were collected initially at 5-min intervals for the immediate measurement of plasma glucose concentration, and 20% glucose was infused at variable rates to maintain plasma glucose at approximately 6.0 mmol/liter during IGF-I infusion. Once the GIR had reached a plateau, the sampling interval for blood glucose was increased to 10 min. At the end of the clamp, animals were killed, and the tissue samples were removed, snap-frozen in liquid nitrogen, and stored at −80 C until processed. For IGF-I/insulin signaling protein studies, see Supplemental Data.
Statistical analysis
Statistical analyses were performed with the ANOVA test. P values < 0.05 were considered statistically significant.
Results
Role of HMGA1 in nuclear protein-DNA interactions within the IGFBP1 and IGFBP3 gene promoters
Figure 1A shows a schematic representation of the promoter region of human and mouse IGFBP1 and IGFBP3 genes. Consensus transcription factor-binding sites within these regions were explored by using the TRANSFAC database (containing a large library of predefined matrix descriptions for known transcription factor recognition sequences) searched with MatInspector professional software (18). Putative binding sites for C/EBPβ and HNF-1, both flanked by AT-rich HMGA1 binding sites, were identified in the promoter region of the human and mouse IGFBP1 genes, and initially characterized by EMSA, using sequence-specific radiolabeled fragments (Fig. 1B). Multiple DNA binding sites for HMGA1, together with putative binding motifs for C/EBPβ and HNF-1, were identified also in the promoter region of the human and mouse IGFBP3 genes, in which two additional consensus binding sites for the ubiquitously expressed transcription factor Sp1 (CG-rich region) were also found just upstream of the transcriptional start site and protein-DNA-binding activity characterized by EMSA (Fig. 1, A and B).
Fig. 1.

Analysis of DNA binding sites within the IGFBP1 and IGFBP3 gene promoters as revealed by preparative EMSA. A, Schematic representation of the regulatory regions of human (h) and mouse (m) IGFBP1 and IGFBP3 genes with HMGA1, HNF-1α, C/EBPβ, and Sp1 binding sites. IRE, insulin response element. B, Representative EMSA of radiolabeled fragments of human IGFBP1 and IGFBP3 (0.25 ng each) gene promoters with 2.5 ng of either HMGA1 (lanes 1–3 and 10–12) or pure Sp1 (lanes 19–21), in the presence of 2.0 μg of BSA, and 0.5 μg of HepG2 (lanes 4–6 and 13–15) or MCF-7 (lanes 7–9 and 16–18) nuclear extract (NE), in the presence of 0.2 μg of polydeoxyinosinic deoxycytidylic acid as the competitor DNA. In supershift assays the protein was preincubated with 1 μg of the polyclonal antibody to HMGA1 (lanes 2 and 11), HNF-1α (lanes 5 and 14), C/EBPβ (lanes 8 and 17), or Sp1 (lane 20), before addition of the probe. Control (unrelated rabbit serum IgG; data not shown) antibody did not alter the mobility of the complex. In supershift assays nuclear protein was preincubated with 1 μg polyclonal specific antibody before addition of the probe.
Previously, we demonstrated that HMGA1 is required for full transactivation of the insulin receptor (INSR) gene by Sp1 and C/EBPβ (15). To see whether biochemical mechanisms of gene induction similar to those that operate during INSR gene activation were involved also in IGFBP1 and IGFBP3 gene transcription, we first analyzed the binding of HMGA1, C/EBPβ, HNF-1, and Sp1 to IGFBP1 and IGFBP3 gene promoters in vivo by DNA ChIP analysis. As shown in Fig. 2A, binding of the endogenous factors HMGA1, C/EBPβ, and HNF-1 to the native promoters of IGFBP1 and IGFBP3 genes occurred in HepG2 cells, in which these proteins are variably coexpressed under normal culture conditions (15). Perturbation of HMGA1 protein expression in HepG2 cells transfected with siRNA against HMGA1 significantly reduced the binding of HMGA1 to DNA and adversely affected protein-DNA interactions of C/EBPβ, HNF-1, and Sp1 as the amount of promoter sequence in chromatin precipitated by C/EBPβ-, HNF-1- or Sp1-specific antibody was substantially decreased in siRNA-treated cells (Fig. 2A). Occupancy on the endogenous IGFBP1/IGFBP3 locus by either C/EBPβ, HNF-1, or Sp1 was substantially unaffected in HMGA1-overexpressing HepG2 cells (data not shown), indicating that binding of HMGA1 to these promoters is required for the enhancement of C/EBPβ, HNF-1, and Sp1 binding, suggesting a molecular mechanism for HMGA1 stimulation of these IGFBP genes. Analogous results were obtained in NIH-3T3 mouse fibroblasts, a cell line naturally expressing HMGA1, in which, however, binding of endogenous C/EBPβ, HNF-1, and Sp1 to IGFBP1 (data not shown) and IGFBP3 promoters became clearly detectable after transient transfection of cells with C/EBPβ-, HNF-1-, or Sp1-specific expression vector (Fig. 2A). The decrease in DNA-protein binding induced by anti-HMGA1 siRNA in HepG2 cells paralleled the decrease in IGFBP1 and IGFBP3 mRNA and protein expression as revealed by quantitative and conventional RT-PCR and immunoprecipitation/Western blot analyses, respectively (Fig. 2B).
Fig. 2.

ChIP, IGFBP1, and IGFBP3 mRNA and protein expression. A, ChIP of human IGFBP1 and IGFBP3 gene promoters in HepG2 cells untreated or treated with siRNA targeted to HMGA1, and mouse IGFBP3 promoter in NIH-3T3 cells untransfected or transfected with either HMGA1-, C/EBPβ-, HNF-1α-, or Sp1-specific expression vector (5 μg). ChIP was done using antibodies (Ab) against HMGA1, C/EBPβ, HNF-1α, or Sp1. B, HepG2 cells were transfected with anti-siRNA (100 and 200 pmol) or a nontargeting control siRNA, and IGFBP1 and IGFBP3 mRNA levels were measured by quantitative RT-PCR (qRT-PCR), as well as conventional RT-PCR. mRNA abundance was normalized to glyceraldehydes-3-phosphate dehydrogenase (GAPDH) mRNA levels. *, P < 0.05 vs. untreated (Control) cells. IGFBP1 and IGFBP3 protein expression levels in siRNA-treated cells were determined by immunoprecipitation/Western blot (IP/WB). β-Actin, control of total cellular protein loading. Each analysis is representative of at least three separate experiments. Each analysis (ChIP, mRNA and protein expression) is representative of at least three independent experiments.
The role of HMGA1 in the recruitment and binding of C/EBPβ, HNF-1, and Sp1 to IGFBP1 and IGFBP3 gene promoters was substantiated by coimmunoprecipitation analysis of protein-protein interactions showing that physical contacts among these transcription factors occur in vivo, in the context of the intact cell. Coimmunoprecipitation studies were initially performed using an antibody against HMGA1 immobilized on protein A beads. As shown in Fig. 3A, immunoprecipitation of HMGA1 from HepG2 nuclear extracts, followed by Western blot analysis for C/EBPβ, revealed a unique specific band, which migrated in a position corresponding to the size of C/EBPβ, the faint intensity of which was compatible with the low expression levels of this nuclear protein in hepatoma cell lines (19). When the same transfer was reprobed with an anti-HMGA1 antibody, a major specific band, which migrated in a position corresponding to the size of HMGA1 was detected, indicating that these two factors physically interact in vivo, in living HepG2 cells. Moreover, when immunoprecipitation of HMGA1 from HepG2 nuclear extracts was followed by immunoblot analysis for Sp1 or HNF-1 (Fig. 3, B and C, respectively), it was found that these two proteins could be detected as well. Further evidence of the interaction between these transcription factors was obtained by performing coimmunoprecipitation experiments with HepG2 nuclear extracts and an antibody against C/EBPβ immobilized on protein A beads. As expected, immunoprecipitation of C/EBPβ, followed by immunoblot analyses with protein-specific antibodies, allowed the identification of four proteins including, in addition to C/EBPβ, HMGA1, Sp1, and HNF-1 (Fig. 3D). Similar results (data not shown) were obtained by performing reciprocal coimmunoprecipitation experiments with nuclear extracts from HepG2 cells and an antibody against HNF-1 or Sp1 immobilized on protein A beads. Together, these findings unequivocally demonstrate that physical interactions among HMGA1, C/EBPβ, HNF-1, and Sp1 occur in living cells, and that binding of these proteins to the endogenous DNA is strongly potentiated by HMGA1, which orchestrates the assembly of these transcription factors at the promoter region of human and mouse IGFBP1 and IGFBP3 genes in a way analogous to the role played by HMGA1 in the context of the INSR gene (15).
Fig. 3.

HMGA1 physically interacts with C/EBPβ, HNF-1α, and Sp1 in vivo. A–C, Immunoprecipitation (IP) of C/EBPβ, HMGA1, Sp1, and HNF-1α, by using the anti-HMGA1 antibody followed by immunoblotting with the anti-C/EBPβ antibody (lanes 1–5), the anti-Sp1 antibody (lanes 11–15), or the anti-HNF-1 antibody (lanes 21–25). Immunoblottings with the anti-HMGA1 antibody (lanes 6–10, 16–20, and 26–30) were obtained after reprobing the same transfer in each condition. D, IP of HMGA1, C/EBPβ, Sp1, and HNF-1α, by using the anti-C/EBPβ antibody, followed by immunoblotting with HMGA1 (lanes 31–33), C/EBPβ (lanes 34–36), Sp1 (lanes 37–39), or HNF-1α (lanes 40–42) specific antibody. Lanes 1, 11, 21, and 31, 5 ng of pure HMGA1; lanes 2 and 34, 10 ng of C/EBPβ (Δ198) antigen; lanes 3, 13, and 23, HepG2 nuclear extracts (NE) (30 μg); lanes 4, 14, 24, 32, 35, 38, and 41, HepG2 NE (500 μg); lanes 12 and 37, 8 ng of pure Sp1. In lanes 3, 13, and 23 the protein was directly applied to the gel without binding to and elution from protein A beads. To prove the specificity of immunoprecipitations, antibody-coupled protein A beads were tested with either Sp1 (lanes 5, 15, and 25, control) or pure HMGA1 (lanes 33, 36, 39, and 42, control) protein alone. NIH-3T3 NE (lanes 22 and 40) were used as a negative control for the presence of HNF-1α protein. Specific bands in the blots are indicated by an arrowhead.
HMGA1 is required for transactivation of IGFBP1 and IGFBP3 gene transcription
We next performed experiments to see whether HMGA1, C/EBPβ, HNF-1, and Sp1 cooperate to activate IGFBP1 and IGFBP3 gene promoters at the transcriptional level. To test this possibility, HepG2 and NIH-3T3 cells were cotransfected transiently with Luc reporter plasmids containing human or mouse IGFBP1 or IGFBP3 gene promoter and various effector vectors, in the absence or presence of siRNA against HMGA1. As shown in Fig. 4A, simultaneous overexpression of C/EBPβ and HNF-1 in HepG2 and NIH-3T3 cells led to a significant increment in the Luc activity of IGFBP1 gene promoter that exceeded that seen with either factor alone. Induction of Luc activity by C/EBPβ and HNF-1 together was virtually abolished in both cell lines in the presence of siRNA targeting HMGA1 and was further enhanced by coexpression of increasing amounts of HMGA1, indicating that transactivation of this gene promoter by C/EBPβ and HNF-1 requires HMGA1. Overexpression of Sp1 in these cells had no effect on IGFBP1 promoter activity, and this is consistent with the observation that no putative binding sites for Sp1 were identified in the regulatory region of both human and mouse IGFBP1 genes. Vice versa, overexpression of Sp1 in HepG2 and NIH-3T3 cells significantly increased Luc activity of human and mouse IGFBP3 promoters (Fig. 4B), in which consensus sequences for Sp1 have been instead identified. At these promoter levels, a functional cooperativity between C/EBPβ and Sp1 occurred, as demonstrated by transfecting IGFBP3 reporter constructs in HepG2 and NIH-3T3 cells, in which forced expression of both C/EBPβ and Sp1 was induced simultaneously. As shown in Fig. 4B, Sp1 and C/EBPβ together activated this promoter to levels of approximately 9-fold above that observed with human or mouse pIGFBP3Luc alone. This increase was higher than the sum of the levels observed for transactivations with the two proteins individually, implying that Sp1 and C/EBPβ act synergistically to drive IGFBP3 gene transcription. Again, induction of promoter activity by Sp1 and C/EBPβ together was virtually abolished in the presence of siRNA directed against endogenous HMGA1 and was increased further by cotransfecting the cells with HMGA1 expression vector, confirming the central importance of this nuclear protein in this scenario. The induction of promoter function by HNF-1 seemed specific for the IGFBP1 gene because Luc activity was significantly reduced in HepG2 and NIH-3T3 cells cotransfected along with HNF-1 and pIGFBP3Luc reporter constructs (Fig. 4B). The fact that HNF-1 exhibited opposite transcriptional properties in the context of IGFBP1 and IGFBP3 genes underlies the high specificity of this factor for these promoters and is consistent with the notion that the rate of gene transcription is primarily regulated through sequence-specific as well as sequence-independent interactions between trans-acting protein factors and cis-acting DNA. The interplay among HMGA1, C/EBPβ, HNF-1, and Sp1 at these promoter sites had functional sequelae also in MCF-7 cells, a cell line ideally suited for studying the effects of these proteins on transcription because it does not express HNF-1 (20), whereas HMGA1 and Sp1 are barely detectable and endogenous C/EBPβ is relatively abundant (15). As shown in Fig. 4C, coexpression of HMGA1 and HNF-1 or HMGA1 and Sp1 in MCF-7 cells led to a significant increment in IGFBP1 and IGFBP3 promoter activity, respectively, that exceeded that seen with either factor alone and that, in both cases, was, at least in part, dependent on endogenous C/EBPβ, as revealed by C/EBPβ siRNA knockdown experiments. The positive role of HNF-1 and Sp1 in these promoter functions was substantiated by demonstrating that transactivation of reporter constructs was significantly blunted in MCF-7 cells pretreated with siRNA directed toward HNF-1 or Sp1. As in HepG2 and NIH-3T3 cells, transactivation of both IGFBP reporter constructs was virtually abolished in MCF-7 cells in the presence of siRNA directed against HMGA1, suggesting that these factors interact directly in delivering an activation signal to the basic transcription machinery. As measured by Western blot analysis, functional activity of IGFBP1 and IGFBP3 promoter gene constructs correlated strictly with nuclear protein expression levels of HMGA1, C/EBPβ, HNF-1, and/or Sp1 in all cell lines investigated (Fig. 4, A–C).
Fig. 4.
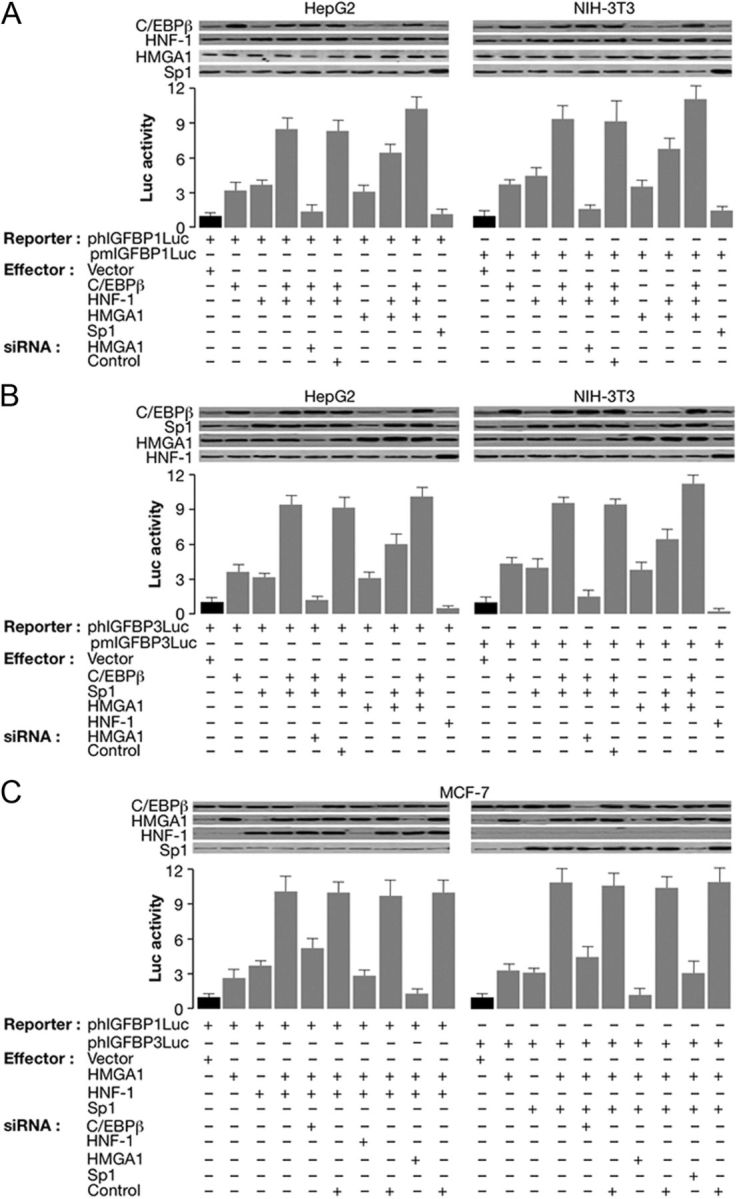
In vivo functional analysis of IGFBP1 and IGFBP3 gene promoters. A and B, HepG2 and NIH-3T3 cells were transfected with human (phIGFBP1Luc or phIGFBP3Luc) or mouse (pmIGFBP1Luc or pmIGFBP3Luc) Luc reporter plasmids (2 μg), respectively, in the absence or presence of the indicated effector vectors (5 μg), either alone or in combination. For specific knockdown of HMGA1, the cells were treated with anti-HMGA1 siRNA (100 pmol) or a nontargeting control siRNA 6 h after transfection, and Luc activity was measured 48–96 h later. C, MCF-7 cells were transfected as in panels A and B, along with HMGA1 and/or HNF-1, and HMGA1 and/or Sp1 expression plasmids, alone or in combination, as indicated, in the absence of C/EBPβ expression vector. Specific knockdown of endogenous C/EBPβ was achieved by transfection of an anti-C/EBPβ siRNA (100 pmol), and Luc activity was determined 48 h later. siRNA for HNF-1, HMGA1, or Sp1, plus nonspecific siRNA controls were transfected into MCF-7 cells together with expression vectors for the corresponding proteins, and Luc activity was measured 48–96 h later. Data represent the means ± se for a minimum of three separate experiments for each condition; values are expressed as factors by which Luc activity increased or decreased as compared with the level of Luc activity obtained in transfections with reporter vector alone (black columns), which is assigned an arbitrary value of 1. Immunoblots for the expression of specific nuclear binding proteins in each condition are shown in the autoradiograms.
Thus, taken together, these results provide new insights into the molecular mechanisms regulating IGFBP1 and IGFBP3 gene expression and consistently support the role of HMGA1 as an architectural element tightly linked to the formation of highly specific and transcriptionally active multiprotein-DNA complexes necessary for the up-regulation of target genes involved in glucose metabolism, such as the INSR gene (1), the leptin gene (21), and for the note, the IGFBP1 and IGFBP3 genes.
IGFBP1 and IGFBP3 expression and IGF-I tolerance tests in Hmga1-knockout mice
Under physiological circumstances (e.g., in response to food and exercise), both IGFBP1 and IGFBP3 seem to regulate bioavailable IGF-I (8, 9, 22). As a step toward understanding the molecular adaptation mechanisms promoting glucose disposal via insulin-independent processes, we first measured IGF-I, IGFBP1, and IGFBP3 serum levels, together with IGFBP1 and IGFBP3 mRNA abundance in liver from age- and body weight-matched Hmga1-mutant animals and littermate controls. Whereas IGF-I serum levels did not differ among mice with diverse genotypes at 6 and 9 months of age, the free IGF-I concentrations were higher in Hmga1-deficient animals (Fig. 5A), in which plasma levels of IGFBP1 and IGFBP3 were lower compared with controls (Fig. 5B). Free IGF-I serum levels were measured using a method originally described for the assay of free IGF-I in humans, which may affect the precision of free IGF-I measurements in rodents (see Supplemental Data). Consistent with our observations above, implying a positive role of HMGA1 in IGFBP1 and IGFBP3 gene expression, IGFBP1 and IGFBP3 mRNA abundance was reduced in liver from Hmga1-deficient mice compared with controls (Fig. 5C). No significant changes were detected in expression of either IGFBP2 or IGFBP5 mRNA (data not shown), although a compensatory increase in IGFBP2 or IGFBP5 protein levels in serum cannot be excluded. These findings, in animals with a disadvantageous metabolic risk profile like that affecting Hmga1-mutant mice, may reflect an adaptive mechanism to increase IGF-I's bioactivity, leading to the recruitment of Glut4 to muscle plasma membrane and improvement of glucose tolerance. In agreement with this interpretation, the glucose-lowering effect of exogenous IGF-I was more pronounced in Hmga1-deficient mice with decreased levels of IGFBP1 and IGFBP3 than in control animals having normal expression of both IGFBP. As shown in Fig. 5D, whereas blood glucose in fasted wild-type mice subjected to ip injection of rhIGF-I decreased approximately 2-fold within 30 min of injection, glycemia decreased 3.5- and 5-fold in heterozygous and homozygous mutants, respectively, revealing a dosage-dependent effect of IGFBP1 and IGFBP3 expression on glucose uptake in mice that have a genetic ablation of Hmga1. These findings are consistent with previous observations indicating that the glucose-lowering effect of exogenous insulin was enhanced in Hmga1-deficient mice during insulintolerance test, and the glucose infusion rate necessary to maintain euglycemia was higher in mutant mice during hyperinsulinemic-euglycemic clamp (1). Similar results were obtained after daily injections of IGF-I for 7 d (data not shown), indicating that IGF-I has both acute and chronic effects to improve glucose uptake in Hmga1-deficient mice. To demonstrate that the increased response to IGF-I injection in mutant mice was in part dependent on HMGA1 regulation of IGFBP1, the effect of IGFBP1 administration on the glucose fall induced by rhIGF-I was determined in these genotypes. As shown in Fig. 5D, whereas injection of hIGFBP1 in heterozygous and homozygous mutants increased basal plasma glucose levels, it considerably decreased the hypoglycemic response to the ip IGF-I. By itself, hIGFBP1 had no effect on basal blood glucose but completely inhibited the hypoglycemic response to rhIGF-I in control mice (Fig. 5D).
Fig. 5.

IGF-I, IGFBP1, and IGFBP3 levels, and plasma glucose concentration in mice with different genotypes. A and B, IGF-I (total and free), IGFBP1, and IGFBP3 serum levels from fasted mice at 6 and 9 months of age. Inset shows a representative serum immunoprecipitation/Western blot (IP/WB) of IGFBP1 and IGFBP3. Each bar represents the mean ± sem (n = 6–15); black bars, Hmga1+/+; gray bars, Hmga1+/−; white bars, Hmga1−/−. *, P < 0.05 vs. Hmga1+/+. C, Liver IGFBP1 and IGFBP3 mRNA as measured by quantitative RT-PCR (qRT-PCR) and conventional RT-PCR. mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels. Hmga1+/+, n = 12; Hmga1+/−, n = 6; Hmga1−/−, n = 10. *, P < 0.05 vs. Hmga1+/+. D, Blood glucose levels before (−) and after (+) administration of rhIGF-I in mice at 9 months of age. Conscious mice fasted for 12 h (at least 12 animals of each genotype) were injected ip with rhIGF-I (1 mg/kg body weight) (37), and plasma glucose levels were measured 30 min after injection. Where indicated, the effect of rhIGF-I was evaluated in Hmga1-deficient animals (four to six mice per group) as above, 10 min after iv injection of hIGFBP-1 (100 μg). Each bar represents the mean ± sem. Black bars, Hmga1+/+; gray bars, Hmga1+/−; white bars, Hmga1−/−. *, P < 0.05 vs. untreated (−) mice for each genotype group; **, P < 0.05 vs. untreated Hmga1+/+.
Glucose uptake in Hmga1-knockout and control mice
PET-FDG has proven valuable for assessing tissue-specific glucose uptake and glucose metabolism in vivo. By using PET-FDG, we measured the relevance of IGF-I on organ glucose uptake after administration of rhIGF-I in wild-type and mutant mice expressing variable amounts of IGFBP1 and IGFBP3. In agreement with the increased IGF-I's bioactivity observed in mice with lower levels of IGFBP1 and IGFBP3, IGF-I-stimulated skeletal muscle uptake of the positron-emitting glucose analog FDG was significantly higher in mutant mice compared with controls, whereas tracer uptake ratios did not differ among these genotypes at either the brain, heart, and abdomen level (Fig. 6A). Consistent with these observations, skeletal muscle glucose uptake was higher in mutant mice under euglycemic clamp with IGF-I, whereas no significant differences with controls were found under basal conditions, although an increased tendency in glucose uptake was noted in mutant mice (Fig. 6B). Accordingly, under clamp conditions, Hmga1-deficient mice showed an increase in the whole-body glucose uptake compared with littermate controls (Fig. 6C), whereas the rates of hepatic glucose production were suppressed by 50–60% in normal mice injected with rhIGF-I (Fig. 6D). Thus, in contrast to insulin, which failed to suppress hepatic glucose production in Hmga1-deficient mice under hyperinsulinemic conditions (1), infusion of IGF-I markedly decreased the rates of hepatic glucose production also in mutant animals (Fig. 6D), indicating the existence of a distinct IGF-I signaling pathway through which IGF-I's biological activity is manifest during IGF-I clamp. A role for IGFBP1 and IGFBP3 in these metabolic events was supported further by the demonstration that, in control mice, the IGF-I infusion was less effective in suppressing hepatic glucose production than in Hmga1-deficient mice (Fig. 6D), in which the reduced expression of both IGFBP per se may lead to higher IGF-I bioavailability and ultimately to increased IGF-I's bioactivity. Thus, the increased responsiveness to IGF-I represents the primary mechanism for promoting glucose disposal in Hmga1-deficient mice with defective INSR and insulin action.
Fig. 6.

PET and glucose turnover studies in wild-type and mutant mice. A, Whole-body reconstructions of PET studies. Shown are coronal PET scan images (top panels) of a representative control mouse (left) and a representative Hmga1-knockout mouse (right). Regional ratios of tracer uptake vs. whole-body counts are shown in the bar graphs at the bottom, together with FDG uptake ratios for heart, abdomen, and neck/forelimb using brain uptake as reference (light blue bars, Hmga1+/+; dark blue bars, Hmga1−/−). Data represent mean ± sem for at least three mice of each genotype. B, Bladder; H, heart; M, muscle (neck/forelimb). B–D, In vivo glucose transport in soleus skeletal muscle tissue, and whole-body glucose uptake and hepatic glucose production in conscious freely moving animals under basal and euglycemic IGF-I clamp conditions. Results are the mean values ± sem for eight animals per genotype. *, P < 0.01 vs. littermate control mice.
IGF-I receptor signaling in Hmga1-knockout mice
We next carried out studies to determine the primary site of action of IGF-I in mice with diverse genotypes. As measured by immunoprecipitation and Western blot analyses, total expression of IGF-I receptor (IGF-IR) was similar in hindlimb muscle and liver extracts from wild-type and mutant mice (Fig. 7A). This in vivo observation contradicts a previous in vitro report (23) that HMGA1 is a positive regulator of the IGF-IR gene. The reason for the different outcomes is not clear; however, in agreement with our previous observations (1), whereas IGF-IR protein expression was similar in wild-type and Hmga1-mutant mice, tissue expression of the closely related INSR was severely attenuated in mutant animals, and no differences were found among these genotypes in the expression of the INSR substrate 1 (IRS1) and the other components of the insulin/IGF-I signaling pathway, such as the regulatory p85 subunit of phosphatidylinositol (PI) 3-kinase and the protein kinase Akt (Fig. 7A). In these animals, autophosphorylation of IGF-IR was measured in hindlimb muscle (and liver) extracts, together with the capacity of tyrosine-phosphorylated IGF-IR to catalyze the association of the p85 subunit of PI 3-kinase with IRS1. As shown in Fig. 7A, IGF-I-induced tyrosine phosphorylation of the IGF-IR β-subunit (IGF-IRβ) was considerably less effective in wild-type controls than in Hmga1-deficient mice, in which increased IGF-IRβ autophosphorylation paralleled the increase in IRS1 and Akt phosphorylation in skeletal muscle. The association of tyrosine-phosphorylated IRS1 with the p85 subunit of PI 3-kinase is required to elicit both insulin and IGF-I effects on glucose transporter translocation (24). Thus, the relevance of IGF-I on glucose uptake in Hmga1-deficient mice was substantiated in antiphosphotyrosine immunoprecipitation-ELISA assays, showing increased PI 3-kinase activity in muscle from fasted mutant animals acutely injected with rhIGF-I. As shown in Fig. 7B, whereas IGF-I caused a 2.4-fold increase in PI 3-kinase activity in skeletal muscle of control mice, acute treatment with IGF-I led to a 3- to 5-fold increase in PI 3-kinase activity in Hmga1-deficient mice. No additive effect on PI 3-kinase activity was found when the two peptides, IGF-I and insulin, were coinfused (data not shown), thus seemingly excluding the existence of independent pathways modulating distinct pools of PI 3-kinase/Akt linked to insulin and IGF-I receptors in these animals. After IGF-I administration, we also evaluated the activation state of the protein kinase Akt, an important downstream target of PI 3-kinase regulating insulin/IGF-I serum effects on Glut4 translocation and carbohydrate metabolism (25). To this end, Western blot analysis was performed using an antibody against the phosphorylated active form of Akt on muscle lysates from IGF-I treated animals. As shown in Fig. 7C, phospho-Akt immunoreactivity was higher in skeletal muscle from Hmga1-deficient mice compared with wild-type controls, and this increase paralleled closely the increase of Glut4 protein in skeletal muscle plasma membranes from heterozygous and homozygous mutants, in which a direct effect of HMGA1 on Glut-4 mRNA expression had been excluded.
Fig. 7.

Analysis of insulin/IGF-I signaling proteins in mice with different genotypes. A, Immunoblots of total and phosphorylated (pIGF-IRβ, pIRS1, pAkt) signaling proteins in skeletal muscle of mice after a 120-min euglycemic clamp period. Total IGF-IR and pIGF-IRβ are also shown in liver. Representative autoradiograms of eight to 10 mice per group are shown. B, PI 3-kinase activity. A representative autoradiogram from skeletal muscle of mice before (−) and after (+) rhIGF-I injection is shown. The position of PI 3-phosphate (PI3P) is indicated. Results are presented as increase in PI 3-kinase activity over basal after rhIGF-I stimulation. Values are the mean ± sem for six to eight mice for each group. Black bars, Hmga1+/+; gray bars, Hmga1+/−; white bars, Hmga1−/−. *, P < 0.01 vs. control mice. C, Skeletal muscle protein abundance of pAkt (top) parallels the expression of Glut4 protein (bottom) in muscle plasma membranes from wild-type and mutant mice. Densitometric analyses of eight to 10 independent Glut4 immunoblots are shown in bar graphs. Black bars, Hmga1+/+, n = 9; gray bars, Hmga1+/−, n = 8; white bars, Hmga1−/−, n = 10. *, P < 0.01 vs. Hmga1+/+.
All together, these findings highlight the importance of the IGF-I/IGFBP system in maintaining glucose homeostasis in vivo, and more definitively demonstrate that, under conditions of reduced insulin action, alternative molecular mechanisms can be envisaged for the translocation of Glut4 to the plasma membrane, ensuring peripheral glucose uptake.
Discussion
As a peculiarity of the Hmga1-knockout mouse, it was found that major defects in both peripheral insulin action and β-cell insulin secretion were coexpressed simultaneously in this animal, leading to the highest percentage of diabetes in a genetically modified mouse model (1). However, compared with control animals, the glucose-lowering effect of exogenous insulin was enhanced in Hmga1-deficient mice during insulin-tolerance test, and the glucose infusion rate necessary to maintain euglycemia was higher in mutant mice during hyperinsulinemic-euglycemic clamp, indicating hypersensitivity to insulin (1). Insulin hypersensitivity despite impairment of insulin action is consistent with the existence of compensatory signaling pathways, the functional activation of which may promote tissue glucose uptake and utilization by insulin-independent mechanisms. The role of insulin-independent mechanisms in mammalian glucose regulation is well established (26–29). However, how these mechanisms are activated and contribute to glucose regulation, under conditions of reduced insulin action, remains largely unexplained. Thus, the Hmga1-knockout mouse model appears to be useful for understanding the molecular mechanisms of tissue and cellular adaptation to conditions in which insulin action becomes compromised.
The importance of IGF-I for normal carbohydrate metabolism is witnessed by numerous results of experimental and clinical studies (30, 31). IGFBP1 and IGFBP3, by influencing both the bioavailability and distribution of IGF-I in the extracellular environment, hold a central position in IGF-I ligand-receptor interactions. Because of their critical role in modulating IGF-I's bioactivity, IGFBP1 and IGFBP3 can modulate the insulin-like metabolic effects of IGF-I, thus representing important determinants in the maintenance of glucose homeostasis. In this regard, although hypoglycemia can be detected in the presence of increasing amounts of free IGF-I in the circulation, fasting hyperglycemia with impaired glucose tolerance and insulin resistance has been demonstrated in rats after the injection of IGFBP1 and IGFBP3 (32, 33), as well as in transgenic mice overexpressing either IGFBP1 or IGFBP3 (34, 35).
In the light of our results indicating that levels of IGFBP1 and IGFBP3 are considerably decreased in Hmga1-knockout mice, it is tempting to hypothesize that, under certain adverse metabolic conditions, functional inactivation of HMGA1, by adversely affecting the expression of both IGFBP protein species, may reflect an adaptive mechanism to increase IGF-I's bioactivity, ensuring recruitment of Glut4 to muscle plasma membrane and tissue glucose disposal. To probe this hypothesis, we initially performed experiments designed to explore the biochemical mechanisms involved in HMGA1 activation of IGFBP1 and IGFBP3 gene expression in transient transfection studies using the IGFBP1 and IGFBP3 promoter-reporter constructs. In HepG2 cells, a cell line readily expressing both IGFBP protein species, we demonstrated, for the first time, that HMGA1 indeed plays a major role in the transcriptional activation of these genes by potentiating the recruitment and binding of transcriptional coactivators such as C/EBPβ, HNF-1, and Sp1 to both IGFBP1 and IGFBP3 gene promoters. Then, by employing the Hmga1-knockout mouse model, we provided compelling evidence for the hypothesis that, under adverse circumstances in which insulin action is precluded, additional molecular mechanisms do exist that may lead to translocation of Glut4 to plasma membrane and enhancement of muscle glucose uptake. Given that INSR expression in Hmga1-deficient mice is considerably decreased in the major target tissues of insulin action, our observation consistently supports this hypothesis and agrees closely with other authors' observations indicating that IGF-I can lower glycemia through its own receptor, in the absence of INSR (36). Although we cannot exclude the possibility that some minor action of IGF-I may be mediated through the residual INSR, our results lend support to the existence of compensatory mechanisms/pathways based on IGF-I receptor signaling able to bypass the INSR defect. It is tempting to hypothesize that the molecular events underlying IGF-I action on glucose uptake/metabolism in skeletal muscle from Hmga1-mutant mice are exerted by bypassing the INSR but later sharing a common intracellular postreceptor pathway with insulin.
In conclusion, our findings provide mechanistic insight into the molecular processes regulating muscle glucose uptake by insulin-independent mechanisms and emphasize the role of HMGA1 in this process in vivo, by finely modulating the expression and function of IGFBP1 and IGFBP3. Elucidating the mechanisms leading to improvement of glucose tolerance is of potential importance in the development of new therapeutic strategies for patients with metabolic disorders such as diabetes mellitus and other pathological states with impairment of insulin signaling and action.
Acknowledgments
We thank Drs. Tadashi Hanafusa and Nobuhiko Takamatsu for providing plasmids. We also thank Dr. Ross G. Clark (Tercica, Inc.) for the generous gift of rhIGF-I.
This work was supported by Telethon-Italy (Grant GGP04245) and MIUR (Italian Ministry of University and Research protocol 2006061742_002) (to A.B.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- C/EBPβ
- CCAAT-enhancer binding protein β
- ChIP
- chromatin immunoprecipitation
- FDG
- 2-fluoro-2-deoxy-d-glucose
- HMGA1
- high mobility group A1
- HNF-1
- hepatocyte nuclear factor-1
- IGFBP
- IGF binding protein
- IGF-IR
- IGF-I receptor
- INSR
- insulin receptor
- IRS1
- INSR substrate 1
- PET
- positron emission tomography
- PI
- phosphatidylinositol
- rhIGF-I
- recombinant human IGF-I
- siRNA
- small interfering RNA
References
- 1. Foti D , Chiefari E , Fedele M , Iuliano R , Brunetti L , Paonessa F , Barbetti F , Brunetti A , Croce CM , Fusco A , Brunetti A. 2005. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med 11:765–773 [DOI] [PubMed] [Google Scholar]
- 2. Kitamura T , Kahn CR , Accili D. 2003. Insulin receptor knockout mice. Annu Rev Physiol 65:313–332 [DOI] [PubMed] [Google Scholar]
- 3. Saltiel AR , Pessin JE. 2002. Insulin signaling pathways in time and space. Trends Cell Biol 12:65–71 [DOI] [PubMed] [Google Scholar]
- 4. Zhou Y , Lee J , Reno CM , Sun C , Park SW , Chung J , Lee J , Fisher SJ , White MF , Biddinger SB , Ozcan U. 2011. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med 17:356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belke DD , Larsen TS , Gibbs EM , Severson DL. 2001. Glucose metabolism in perfused mouse hearts overexpressing human GLUT-4 glucose transporter. Am J Physiol Endocrinol Metab 280:E420–E427 [DOI] [PubMed] [Google Scholar]
- 6. Bilan PJ , Ramlal T , Klip A. 1991. IGF-I mediated recruitment of glucose transporters from intracellular membranes to plasma membranes in L6 muscle cells. Adv Exp Med Biol 293:273–288 [DOI] [PubMed] [Google Scholar]
- 7. Weiland M , Bahr F , Höhne M , Schümann A , Ziehm D , Joost HG. 1991. The signaling potential of the receptors for insulin and insulin-like growth factor 1 (IGF-I) in 3T3–L1 adipocytes: comparison of glucose transport activity, induction of oncogene c-fos, glucose transporter mRNA, and DNA-synthesis. J Cell Physiol 149:428–435 [DOI] [PubMed] [Google Scholar]
- 8. Baxter RC. 2000. Insulin-like growth factor (IGF)-binding proteins: interactions with IGFs and intrinsic bioactivities. Am J Physiol Endocrinol Metab 278:E967–E976 [DOI] [PubMed] [Google Scholar]
- 9. Clemmons DR. 1997. Insulin-like growth factor binding proteins and their role in controlling IGF actions. Cytokine Growth Factor Rev 8:45–62 [DOI] [PubMed] [Google Scholar]
- 10. Sandhu MS , Dunger DB , Giovannucci EL. 2002. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J Natl Cancer Inst 94:972–980 [DOI] [PubMed] [Google Scholar]
- 11. Villafuerte BC , Zhao W , Herington AC , Saffery R , Phillips LS. 1997. Identification of an insulin-responsive element in the rat insulin-like growth factor-binding protein-3 gene. J Biol Chem 272:5024–5030 [DOI] [PubMed] [Google Scholar]
- 12. Bustin M , Reeves R. 1996. High-mobility group proteins: architectural components that facilitates chromatin function. Prog Nucleic Acid Res Mol Biol 54:35–100 [DOI] [PubMed] [Google Scholar]
- 13. Allander SV , Durham SK , Scheimann AO , Wasserman RM , Suwanichkul A , Powell DR. 1997. Hepatic nuclear factor 3 and high mobility group I/Y proteins bind the insulin response element of the insulin-like growth factor-binding protein-1 promoter. Endocrinology 138:4291–4300 [DOI] [PubMed] [Google Scholar]
- 14. Brunetti A , Manfioletti G , Chiefari E , Goldfine ID , Foti D. 2001. Transcriptional regulation of human insulin receptor gene by the high-mobility group protein HMGI(Y). FASEB J 15:492–500 [DOI] [PubMed] [Google Scholar]
- 15. Foti D , Iuliano R , Chiefari E , Brunetti A. 2003. A nucleoprotein complex containing Sp1, C/EBPβ, and HMGI-Y controls human insulin receptor gene transcription. Mol Cell Biol 23:2720–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paonessa F , Foti D , Costa V , Chiefari E , Brunetti G , Leone F , Luciano F , Wu F , Lee AS , Gulletta E , Fusco A , Brunetti A. 2006. Activator protein-2 overexpression accounts for increased insulin receptor expression in human breast cancer. Cancer Res 66:5085–5093 [DOI] [PubMed] [Google Scholar]
- 17. Cingel-Ristic V , Van Neck JW , Frystyk J , Drop SL , Flyvbjerg A. 2004. Administration of human insulin-like growth factor-binding protein-1 increases circulating levels of growth hormone in mice. Endocrinology 145:4401–4407 [DOI] [PubMed] [Google Scholar]
- 18. Cartharius K , Frech K , Grote K , Klocke B , Haltmeier M , Klingenhoff A , Frisch M , Bayerlein M , Werner T. 2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942 [DOI] [PubMed] [Google Scholar]
- 19. Friedman AD , Landschulz WH , McKnight SL. 1989. CCAAT/enhancer binding protein activates the promoter of the serum albumin gene in culture hepatoma cells. Genes Dev 3:1314–1322 [DOI] [PubMed] [Google Scholar]
- 20. Bach I , Yaniv M. 1993. More potent transcriptional activators or a transdominant inhibitor of the HNF1 homeoprotein family are generated by alternative RNA processing. EMBO J 12:4229–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Melillo RM , Pierantoni GM , Scala S , Battista B , Fedele M , Stella A , De Biasio MC , Chiappetta G , Fidanza V , Condorelli G , Santoro M , Croce CM , Viglietto G , Fusco A. 2001. Critical role of the HMGI(Y) proteins in adipocytic cell growth and differentiation. Mol Cell Biol 21:2485–2495 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Sandhu MS , Heald AH , Gibson JM , Cruickshank JK , Dunger DB , Wareham NJ. 2002. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet 359:1740–1745 [DOI] [PubMed] [Google Scholar]
- 23. Aiello A , Pandini G , Sarfstein R , Werner H , Manfioletti G , Vigneri R , Belfiore A. 2010. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur J Cancer 46:1919–1926 [DOI] [PubMed] [Google Scholar]
- 24. Myers MJ , Sun XJ , Cheatham B , Jachna BR , Glasheen EM , Backer JM , White MF. 1993. IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology 132:1421–1430 [DOI] [PubMed] [Google Scholar]
- 25. Cho H , Mu J , Kim JK , Thorvaldsen JL , Chu Q , Crenshaw EB , Kaestner KH , Bartolomei MS , Shulman GI , Birnbaum MJ. 2001. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292:1728–1731 [DOI] [PubMed] [Google Scholar]
- 26. DeFronzo R. 2009. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58:773–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rahmoune H , Thompson PW , Ward JM , Smith CD , Hong G , Brown J. 2005. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54:3427–3434 [DOI] [PubMed] [Google Scholar]
- 28. Brown GK. 2000. Glucose transporters: structure, function and consequences of deficiency. J Inherit Metab Dis 23:237–246 [DOI] [PubMed] [Google Scholar]
- 29. Gerich JE. 2000. Physiology of glucose homeostasis. Diabetes Obes Metab 2:345–350 [DOI] [PubMed] [Google Scholar]
- 30. Woods KA , Camacho-Hübner C , Bergman RN , Barter D , Clark AJ , Savage MO. 2000. Effects of insulin-like growth factor 1 (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J Clin Endocrinol Metab 85:1407–1411 [DOI] [PubMed] [Google Scholar]
- 31. Yakar S , Liu JL , Fernandez AM , Wu Y , Schally AV , Frystyk J , Chernausek SD , Mejia W , Le Roith D. 2001. Liver specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes 50:1110–1118 [DOI] [PubMed] [Google Scholar]
- 32. Lewitt MS , Denyer GS , Cooney GJ , Baxter RC. 1991. Insulin-like growth factor binding protein-1 modulates blood glucose levels. Endocrinology 129:2254–2256 [DOI] [PubMed] [Google Scholar]
- 33. Muzumdar RH , Ma X , Fishman S , Yang X , Atzmon G , Vuguin P , Einstein FH , Hwang D , Cohen P , Barzilai N. 2006. Central and opposing effects of IGF-I and IGF-binding protein-3 on systemic insulin action. Diabetes 55:2788–2796 [DOI] [PubMed] [Google Scholar]
- 34. Rajkumar K , Krsek M , Dheen ST , Murphy LJ. 1996. Impaired glucose homeostasis in insulin-like growth factor binding protein-1 transgenic mice. J Clin Invest 98:1818–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silha JV , Gui Y , Murphy LJ. 2002. Impaired glucose homeostasis in insulin-like growth factor-binding protein-3-transgenic mice. Am J Physiol Endocrinol Metab 283:E937–E945 [DOI] [PubMed] [Google Scholar]
- 36. Di Cola G , Cool MH , Accili D. 1997. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J Clin Invest 99:2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Higaki K , Matsumoto Y , Fujimoto R , Kurosaki Y , Kimura T. 1997. Pharmacokinetics of recombinant human insulin-like growth factor-I in diabetic rats. Drug Metab Dispos 25:1324–1327 [PubMed] [Google Scholar]