

Abstract
Benign hereditary chorea is a rare autosomal-dominant disorder that is characterized by childhood-onset nonprogressive chorea and normal cognitive function. Defects in NKX2-1 on chromosome 14q13, which encodes thyroid transcription factor 1, produce a concurrent clinical manifestation of chorea, respiratory distress, and hypothyroidism known as “brain–lung–thyroid syndrome.” Here, the authors describe a video report of benign hereditary chorea in a Japanese female with a novel frameshift mutation of NKX2-1 (c.915_916insC) (p.Ala303ArgfsX132) that was initially misdiagnosed as ataxic cerebral palsy. In early infancy, especially before the appearance of chorea, benign hereditary chorea can be misdiagnosed as ataxic and dyskinetic cerebral palsy due to shared clinical features including motor delay, hypotonia, ataxic gait, and dystonia.
Keywords: brain–lung–thyroid syndrome, NKX2-1, ataxic cerebral palsy
Benign hereditary chorea is a rare autosomal-dominant disorder that is characterized by childhood-onset nonprogressive chorea. The complete phenotype of benign hereditary chorea includes a combination of chorea, hypothyroidism, and respiratory distress syndrome and is therefore termed as brain–thyroid–lung syndrome.1 Mutations of NKX2-1 on chromosome 14q13 have been identified in patients with benign hereditary chorea.2 NKX2-1 is a transcription factor essential for organogenesis of the lung, thyroid, and parts of the brain including the basal ganglia. Therefore, it is reasonable to suspect mutations of NKX2-1 and associated benign hereditary chorea in cases of neonatal respiratory distress, hypothyroidism, and chorea. However, in early childhood and especially prior to the onset of chorea, benign hereditary chorea is often misdiagnosed as ataxic cerebral palsy due to shared clinical features including hypotonia, ataxia, and motor developmental delay. Ataxic cerebral palsy is the least common type of cerebral palsy and most frequently misdiagnosed as ataxic disorders.3 Here, the authors present a video report of brain–thyroid–lung syndrome in a Japanese female with a novel frameshift mutation of NKX2-1 that was initially misdiagnosed as ataxic cerebral palsy. The authors then provide a discussion of the clinical characteristics and phenomenology of benign hereditary chorea and ataxic cerebral palsy.
Case Report
The patient was a 5-year-old Japanese female who was the second child of healthy parents. The patient had no familial history of movement disorder, but the patient’s mother experienced transient hyperthyroidism during pregnancy, the patient’s aunt had hyperthyroidism, and the patient’s grandmother had hypothyroidism.
The patient was born at term by cesarean section and was admitted to a neonatal intensive care unit at 3 days of age for severe respiratory distress syndrome that required mechanical ventilation therapy. Congenital hypothyroidism was diagnosed at 6 days of age (thyroid-stimulating hormone 100.0 μU/mL, FT4 1.0 ng/dL), and treatment with levothyroxine was initiated. The size of the thyroid was normal on thyroid echography, and thyroid hormone levels were successfully restored with levothyroxine therapy. Lung computed tomography showed cystic fibrosis and ground-glass opacities characteristic of interstitial lung disease (Figure 1), and serum KL-6 was elevated; however, bronchoalveolar lavage was not performed and SP-C (the causative gene for idiopathic interstitial pneumonitis) sequencing did not reveal a relevant alteration. The patient was accordingly treated with mechanical ventilation and hydrocortisone and prednisolone for 1 month and did not receive phototherapy at any point during hospitalization.
Figure 1.
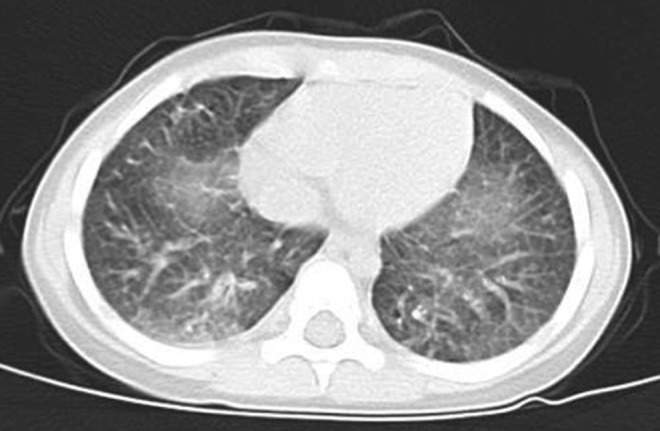
Bilateral diffuse ground-glass opacities in the lower lobe of the lung observed on computed tomography at 4 years of age.
The patient was discharged from the neonatal intensive care unit at 2 months of age with home oxygen therapy to treat chronic lung disease. The family relocated near the hospital and introduced the patient to us for developmental follow-up. The patient’s height, weight, and general examination results were all within the normal range. No dysmorphic features were observed. Deep tendon reflex and Babinski reflex were normal, but the patient presented with hypotonia and ataxia. At 12 months of age, she was diagnosed with ataxic cerebral palsy, due to the presentation of remarkable degree of incoordination and a high frequency of falls; thus, physical therapy was initiated. Motor developmental delays were observed: roll over was observed at 9 months, sitting was observed at 15 months, walking was observed at 24 months, and speech (her first word) was noted at 30 months. As the patient matured, she exhibited choreic movement of the limbs, face, and tongue at rest and dystonic posture of the upper limbs while walking. Choreic movement and small jerking movements of the trunk, limbs, and face were apparent at rest; these movements were not exaggerated by finger-to-nose test (Supplementary Videos 1-3). To exclude other chorea-related diseases, the authors examined metabolic parameters in blood (ammonia, amino acid, lactate, pyruvate, copper, ceruloplasmin, and α-fetoprotein) and urine (organic acid). Brain magnetic resonance imaging showed perivascular space in the lower part of the right basal ganglia, but no other abnormalities related to clinical symptoms were observed (Figure 2A, B).
Figure 2.
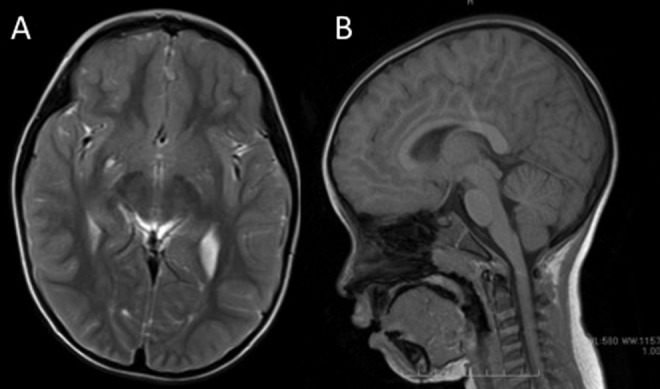
A, Axial T2-weighted and (B) sagittal T1-weighted brain magnetic resonance imaging of the patient at 4 years of age. Imaging did not reveal any abnormalities except for perivascular space in the lower part of the right basal ganglia. Cerebellar anomaly and atrophy were not observed.
A symptom triad of congenital hypothyroidism, respiratory distress, and chorea was strongly suggestive of NKX2-1 mutation. Genomic microarray analysis did not reveal any pathological copy number aberrations. Sanger sequencing of the NKX2-1 gene led to the detection of a novel heterozygous insertion (c.915_916insC) in exon 3 that resulted in a frameshift starting at amino acid position 303 and a premature stop codon (p.Ala303ArgfsX132) that rendered the protein nonfunctional. This mutation was not detected in leukocyte DNA from the patient’s mother, who had a familial history of thyroid dysfunction. The patient’s father, who had no clinical manifestation of respiratory problems, thyroid dysfunction, or chorea, did not consent to DNA analysis.
At the time of this report, the patient’s chorea is nonprogressive and she continues to demonstrate normal intelligence; the Kyoto Scale of Psychological Development 2001 at 5 years and 11 months indicated a development age of 4 years and 10 months (overall developmental quotient 81; postural–motor developmental quotient 51; cognitive–adaptive developmental quotient 76; language–social developmental quotient 88). However, the patient does have subtle difficulties with handwriting and speech due to choreic movement and accordingly requires educational support and rehabilitation. Furthermore, the patient exhibited attention-deficit hyperactivity disorder (ADHD).
Genetic Analysis
Genomic DNA from the patient and the patient’s mother was extracted from leukocytes using a DNA isolation kit (Wako, Japan). Primers were designed corresponding to the intronic sequences flanking the 3 exons of the NKX2-1 gene using Primer3 software. Polymerase chain reaction (PCR) was performed using GoTaq (Promega, Madison, Wisconsin) under standard conditions, PCR products were purified using ExoSAP (USB, Cleveland, Ohio), and products were sequenced for both forward and reverse strands using the BigDye Terminator Chemistry Kit (version 3; Applied Biosystems, Foster City, California) according to the standard protocol. Sequences were obtained with the ABI Genetic Analyzer 3100 (Applied Biosystems) and the sequence analysis software program GENETYX (version 9; Genetyx, Japan). Genomic copy numbers were analyzed using the Human Genome comparative genomic hybridization Microarray 105K (Agilent Technologies, Santa Clara, California) as described previously.4
Discussion
In general, the phenotype of benign hereditary chorea is consistent with mutation of NKX2-1. Large NKX2-1 deletions have been reported in infants with respiratory distress, congenital hypothyroidism, delayed motor milestones, and ataxia, whereas missense mutations and late protein truncations of NKX2-1 have been associated with milder but similar phenotypes.2 In this case report, the authors anticipated the observation of a large NKX2-1 deletion given severe respiratory distress and congenital hypothyroidism in the neonatal period and the later development of chorea. However, the comparative genomic hybridization array showed no abnormalities in chromosome 14 and the authors instead identified a novel heterozygous loss-of-function mutation, c.915_916insC (p.Ala303ArgfsX132), by Sanger sequencing analysis. The authors suspect that this mutation rendered the protein dysfunctional: Ala303 is in close proximity to the NK2-specific domain, which is thought to function as an accessory DNA-binding domain or as an interface for protein–protein interaction.5 A lack of functional NKX2-1 protein in neurons is known to impair developmental differentiation and organization of basal ganglia and basal forebrain and causes aberrant trajectory of the dopaminergic pathway in the developing hypothalamus of mice.6 This can explain the symptomatic manifestations of NKX2-1 mutation in this patient.
Table 1 shows NKX2-1 mutations that produced severe neonatal respiratory distress requiring mechanical ventilation, congenital hypothyroidism, and chorea similar to this case.1,7-15 In 11 severe neonatal respiratory distress cases, there were 4 deletion mutations (2 macrodeletions and 2 microdeletions), 4 point mutations (2 splice site mutations and 2 nonsense mutations), and 3 insertion mutations. The heterogeneity of mutation type and location in these cases of brain–lung–thyroid syndrome with severe neonatal respiratory distress suggests that there is no correlation between the severity of respiratory problems in benign hereditary chorea and specific mutations of NKX2-1. It remains unclear why some cases present with irreversible lung failure from birth, whereas others only demonstrate transient and mild respiratory complications. Additionally, some cases of NKX2-1 mutation that present with mild chorea improve with age, whereas other cases have severe chorea and myoclonus into adulthood. Other factors, such as modifying genes, hormonal factors, and environmental factors, can underlie phenotypic heterogeneity in benign hereditary chorea.2,10
Table 1.
Reported Severe Neonatal Respiratory Distress Requiring Mechanical Ventilation Produced by NKX2-1 Mutations.
| Patient No. | NKX2-1 Mutations | Chorea | Hypothyroidism | References |
|---|---|---|---|---|
| Age at Diagnosis | Age of Onset | |||
| 1 | c.786_787del | 3 years | Congenital | Gras, 2012 |
| 2 | 14q13-21 Macrodeletion | 2 years | Congenital | Devriendt, 1998 |
| 3 | C1302A (nonsense) | Childhood | Congenital (agenesis) | Krude, 2002 |
| 4 | (14)(11.2;13.3) deletion | Infancy | Congenital (hypoplasia) | Krude, 2002 |
| 5 | Intron 2-2A>G | Infancy | Congenital | Doyle, 2004 |
| 6 | 376-2A>G (intron 2) | 5 years | Congenital | Carre, 2009 |
| 7 | c.278_308del | Died at 10 months | Congenital | Kleinlein, 2011 |
| 8 | C609A | 4 years | Congenital | Salvatore, 2010 |
| 9 | 86insG | 4.5 years | 2.5 years | Pohlenz, 2002 |
| 10 | 859_860insC | Childhood | 16 months | Willemsen, 2005 |
| 11 | c.915_916insC | 2 years | Congenital | This case |
The major characteristic of benign hereditary chorea is childhood-onset nonprogressive chorea. Other accompanying features include gait disturbance, dystonia, ataxia, myoclonus, and dysarthria.13 Prior to the onset of chorea, motor symptoms such as motor delay, ataxia, and gait disturbance can lead to the misdiagnosis of benign hereditary chorea as ataxic cerebral palsy. McMichael and colleagues16 reported the misdiagnosis of a father and his 2 children with ataxic dyskinetic cerebral palsy; in fact, a 7-base pair deletion within exon 1 of NKX2-1. In this study, motor delay and ataxia were observed in early infancy, whereas choreiform movements and unsteady gait appeared in a later stage of development. Doyle and colleagues11 reported a case study of siblings with an NKX2-1 splice mutation that first exhibited congenital hypothyroidism, motor delay, and ataxia and later presented chorea and dysarthria. The mother of the siblings had been diagnosed with cerebral palsy. In the present case, a wide-based gait was initially diagnosed as ataxic gait; however, the authors hypothesize that it can have been a compensatory movement for the truncal jerking rather than cerebellar ataxia. Consistent with this hypothesis, no cerebellar abnormalities were observed in brain images (Figure 2B). These studies underscore the need for careful evaluation of cases of ataxia and motor delay in young children who meet the criteria for ataxic cerebral palsy.
After appearance of dyskinetic movements, such as chorea, myoclonus, and dystonia, it can be difficult to distinguish benign hereditary chorea from myoclonus–dystonia syndrome resultant from epsilon sarcoglycan (SGCE) gene mutations.17 Both diseases share a similar age of onset, dominant inheritance, minimal progression, and additional dystonia with no other neurological abnormalities. Asmus and colleagues18 reported that patients with myoclonus–dystonia showed “lightning-like” myoclonic jerks during the finger-to-nose test, whereas patients with benign hereditary chorea showed small jerking movements but no exacerbation of choreic movement. Salvatore and colleagues15 also reported the case of 3 patients from an Italian family with the S145X NKX2-1 mutation and indicated that while dyskinetic movements, chorea, myoclonus, and jerky dystonia were apparent walking and sitting, dyskinetic movements seemed to improve during the finger-to-nose test. In the present study, the patient exhibited small jerks of the arm during the finger-to-nose test, but no exacerbation of choreic movement; rather, chorea was less noticeable during intentional tasks than at rest (Supplementary Videos 1 and 3). Myoclonus dystonia presents with action myoclonus when the arms are held outstretched and during the finger-to-nose test; alternatively, benign hereditary chorea presents with small jerking movements that are spontaneous, unrelated to intentional movement, and slower than myoclonus. These clinical features of benign hereditary chorea are notable and can thus be used to distinguish benign hereditary chorea from myoclonus dystonia syndrome in patients.
In general, the first-line therapy for chorea is levodopa, but few cases of benign hereditary chorea report significant benefit.20,21 In contrast, Chen and colleagues22 reported the improvement of symptoms with a low dose of tetrabenazine, an agent used to treat Huntington’s disease. Gras and colleagues13 similarly reported a moderate-to-marked beneficial effect of low-dose tetrabenazine on chorea and motor function in children and adults with benign hereditary chorea. Tetrabenazine acts in the central nervous system to deplete monoamines such as serotonin from nerve terminals by inhibiting their incorporation into presynaptic vesicles. Other agents such as trihexyphenidyl, corticosteroids, sodium valproate, and propranolol have also been used for the treatment of chorea and involuntary movement in benign hereditary chorea, but there is no consensus regarding a first-line therapy.23
Cognitive impairments have not been historically documented in benign hereditary chorea. However, prior to the discovery of NKX2-1, several reports observed reduced intelligent quotient in individuals with clinical benign hereditary chorea syndrome. Gras and colleagues13 reported NKX2-1 mutations in 28 patients with benign hereditary chorea from 13 families; of these patients, 71% experienced learning difficulties. A quantitative evaluation of cognitive performance in 14 children from this study identified mental retardation in 2 children and borderline mental retardation in 2 additional children.13 Our patient’s postural–motor developmental quotient was low because of choreic movement, but no apparent cognitive impairment has been observed. The patient did, however, exhibit dysgraphia and learning disability due to choreic movement and thus continues to require educational support. Congenital hypothyroidism for itself could be a cause of cognitive impairment and learning disability if not diagnosed and treated. But this patient had been diagnosed with early neonatal period and treated with levothyroxine. Furthermore, this patient exhibited ADHD. If benign hereditary chorea is pathologically related to dysfunction of the motor and associative striatal networks, chorea and ADHD are logical symptomatic manifestations. However, it remains unclear whether functional deficits reflect an integral part of the benign hereditary chorea phenotype or a secondary consequence of social embarrassment and isolation.
If an attending physician is aware of brain–lung–thyroid syndrome and its characteristic clinical features (respiratory distress despite birth at term, congenital hypothyroidism, and chorea), it can be diagnosed without difficulty in most cases. However, in early childhood, benign hereditary chorea can be difficult to identify prior to the appearance of chorea and can be mistaken for ataxic or dyskinetic cerebral palsy.19 Ataxic cerebral palsy is one of the least common types of cerebral palsy and is frequently associated with hypotonia and cerebellar features including tremor, gait disturbance, and poor coordination.16 Accordingly, benign hereditary chorea can be misdiagnosed as ataxic cerebral palsy in early childhood based on the observable symptoms. Phenotyping should therefore be performed with care in order to avoid the confusion of chorea and myoclonus with ataxia. It should be noted that, in this patient, despite severe respiratory distress in the neonatal period that may have caused hypoxic damage, a diagnosis of ataxic and/or dyskinetic cerebral palsy in the absence of abnormal imaging findings should be regarded as putative and thus continuously reevaluated according to the development of the clinical course.
Supplementary Material
Footnotes
Author Contributions: TT wrote the first draft of the manuscript. SK and TK performed Sanger sequencing analysis of NKX2-1. TY performed CGH array analysis. KY gave valuable suggestions for patient diagnosis. AN, TC, MM, and HH reviewed and approved the final manuscript.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval: A written and informed consent was taken from the parents for publication of the case report.
Supplemental Material: The online supplementary videos are available at http://cno.sagepub.com/supplemental.
References
- 1. Shimojima K, Komoike Y, Tohyama J, et al. TULIP1 (RALGAPA1) haploinsufficiency with brain development delay. Genomics. 2009;94(6):414–422. [DOI] [PubMed] [Google Scholar]
- 2. Inzelberg R, Weinberger M, Gak E. Benign hereditary chorea: an update. Parkinsonism Relat Disord. 2011;17(5):301–307. [DOI] [PubMed] [Google Scholar]
- 3. Lee RW, Poretti A, Cohen JS, et al. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med. 2014;16(4):821–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilbertz T, Maier S, Perner S. NKX2-1 (NK2 homeobox 1). Atlas Genet Cytogenet Oncol Haematol. 2011;15(1):19–28. [Google Scholar]
- 5. Watada H, Mirmira RG, Kalamaras J, German MS. Intramolecular control of transcriptional activity by the NK2-specific domain in NK-2 homeodomain proteins. Proc Natl Acad Sci U S A. 2000;97(17):9443–9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Willemsen MA, Breedveld GJ, Wouda S, et al. Brain-Thyroid-Lung syndrome: a patient with a severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur J Pediatr. 2005;164(1):28–30. [DOI] [PubMed] [Google Scholar]
- 7. Pohlenz J, Dumitrescu A, Zundel D, et al. Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest. 2002;109(4):469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schneider SA, Lang AE, Moro E, Bader B, Danek A, Bhatia KP. Characteristic head drops and axial extension in advanced chorea-acanthocytosis. Mov Disord. 2010;25(10):1487–1491. [DOI] [PubMed] [Google Scholar]
- 9. Kleinlein B, Griese M, Liebisch G, et al. Fatal neonatal respiratory failure in an infant with congenital hypothyroidism due to haploinsufficiency of the NKX2-1 gene: alteration of pulmonary surfactant homeostasis. Arch Dis Child Fetal Neonatal Ed. 2011;96(6):F453–F456. [DOI] [PubMed] [Google Scholar]
- 10. Carre A, Szinnai G, Castanet M, et al. Five new TTF1/NKX2.1 mutations in brain-lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum Mol Genet. 2009;18(12):2266–2276. [DOI] [PubMed] [Google Scholar]
- 11. Doyle DA, Gonzalez I, Thomas B, Scavina M. Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a mutation of NKX2-1. J pediat. 2004;145(2):190–193. [DOI] [PubMed] [Google Scholar]
- 12. Devriendt K, Vanhole C, Matthijs G, de Zegher F. Deletion of thyroid transcription factor-1 gene in an infant with neonatal thyroid dysfunction and respiratory failure. N Eng J Med. 1998;338(18):1317–1318. [DOI] [PubMed] [Google Scholar]
- 13. Gras D, Jonard L, Roze E, et al. Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the TITF1/NKX2-1 gene. J Neurol Neurosurg Psychiatry. 2012;83(10):956–962. [DOI] [PubMed] [Google Scholar]
- 14. Krude H, Schütz B, Biebermann H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002;109(4): 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salvatore E, Di Maio L, Filla A, et al. Benign hereditary chorea: clinical and neuroimaging features in an Italian family. Mov Disord. 2010;25(10):1491–1496. [DOI] [PubMed] [Google Scholar]
- 16. McMichael G, Haan E, Gardner A, et al. NKX2-1 mutation in a family diagnosed with ataxic dyskinetic cerebral palsy. Eur J Med Genet. 2013;56(9):506–509. [DOI] [PubMed] [Google Scholar]
- 17. Asmus F, Salih F, Hjermind LE, et al. Myoclonus-dystonia due to genomic deletions in the epsilon-sarcoglycan gene. Ann Neurol. 2005;58(5):792–797. [DOI] [PubMed] [Google Scholar]
- 18. Asmus F, Devlin A, Munz M, Zimprich A, Gasser T, Chinnery PF. Clinical differentiation of genetically proven benign hereditary chorea and myoclonus-dystonia. Mov Disord. 2007;22(14):2104–2109. [DOI] [PubMed] [Google Scholar]
- 19. Albright AL. Spasticity and movement disorders in cerebral palsy. J Child Neurol. 1996;11 suppl 1:S1–S4. [DOI] [PubMed] [Google Scholar]
- 20. Konishi T, Kono S, Fujimoto M, et al. Benign hereditary chorea: dopaminergic brain imaging in patients with a novel intronic NKX2.1 gene mutation. J Neurol. 2013;260(1):207–213. [DOI] [PubMed] [Google Scholar]
- 21. Asmus F, Horber V, Pohlenz J, et al. A novel TITF-1 mutation causes benign hereditary chorea with response to levodopa. Neurology. 2005;64(11):1952–1954. [DOI] [PubMed] [Google Scholar]
- 22. Chen JJ, Ondo WG, Dashtipour K, Swope DM. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487–1504. [DOI] [PubMed] [Google Scholar]
- 23. Peall KJ, Kurian MA. Benign hereditary chorea: an update. Tremor Other Hyperkinet Mov (N Y). 2015;5:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.