

Abstract
The osteoprotective action of estrogen in women has drawn considerable attention because estrogen deficiency-induced osteoporosis became one of the most widely spread diseases in developed countries. In men, the significance of estrogen action for bone health maintenance is also apparent from the osteoporotic phenotype seen in male patients with genetically impaired estrogen signaling. Severe bone loss and high bone turnover, including typical osteofeatures seen in postmenopausal women, can also be recapitulated in rodents after ovariectomy. However, the expected osteoporotic phenotype is not observed in female mice deficient in estrogen receptor (ER)-α or -β or both, even though the degenerative defects are clearly seen in other estrogen target tissues together with up-regulated levels of circulating testosterone. It has also been reported that estrogens may attenuate bone remodeling by cell autonomous suppressive effects on osteoblastogenesis and osteoclastogenesis. Hence, the effects of estrogens in bone appear to be complex, and the molecular role of bone estrogen receptors in osteoprotective estrogen action remains unclear. Instead, it has been proposed that estrogens indirectly control bone remodeling. For example, the enhanced production of cytokines under estrogen deficiency induces bone resorption through stimulation of osteoclastogenesis. However, the osteoporotic phenotype without systemic defects has been recapitulated in female (but not in male) mice by osteoclast-specific ablation of the ERα, proving that bone cells represent direct targets for estrogen action. An aberrant accumulation of mature osteoclasts in these female mutants indicates that in females, the inhibitory action of estrogens on bone resorption is mediated by the osteoclastic ERα through the shortened lifespan of osteoclasts.
Estrogen is osteoprotective by attenuating bone resorption in females and males due to hormonally induced death of mature osteoclasts.
Estrogen is a prime female steroid hormone as well as a pivotal regulator in many biological processes beyond development and maintenance of female reproductive organs. Among the estrogen target organs, bone has recently drawn increasing attention because postmenopausal osteoporosis induced by estrogen deficiency has emerged as the most widely spread bone/joint disease in developed countries. Osteoporosis in women and men is currently considered a serious disorder of middle-aged and elderly people because of increased risk of bone fracture, often leading to long-term incapacitation and high mortality (1, 2).
Pronounced bone mass decrease due to enhanced or imbalanced bone resorption vs. bone formation (high bone turnover) is a typical osteoporotic feature in women with estrogen deficiency or impaired estrogen signaling (Fig. 1). The osteoporotic bone phenotype can be experimentally recapitulated in female rodents by ovariectomy (OVX) and consequent estrogen depletion (3, 4). Bioavailable estrogens and selective estrogen response modulators are shown to be effective at attenuating high bone turnover and prevent bone loss in both osteoporotic patients and OVX rodents (3, 4, 5, 6). Accumulating clinical observations and genetic studies show that male patients defective in either estrogen biosynthesis or function of estrogen receptor α (ERα) display typical pathological conditions of osteoporosis (7, 8). Thus, it is evident that estrogens exert osteoprotective actions and play a significant role in skeletal maintenance in both sexes.
Fig. 1.
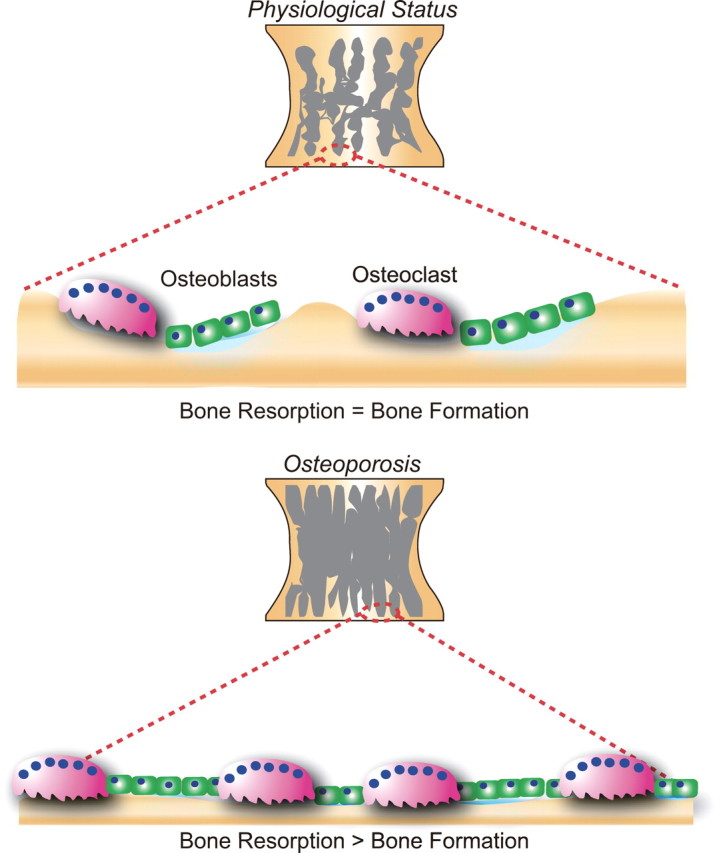
The role of bone cells in bone remodeling. Bone tissue is continuously remodeled by osteoblastic bone formation and osteoclastic bone resorption. The balance between bone formation and resorption is maintained under normal physiological conditions (upper panel). However, when this balance is altered, pathological conditions such as osteoporosis may develop in which increased osteoclastic bone resorption exceeds osteoblastic bone formation with a resultant decrease in bone mass (lower panel).
The first conventional gene disruption of the mouse ERα locus was achieved in the early 1990s (9). Paradoxically, however, neither males nor female ERα-deficient mice exhibited typical osteoporotic bone phenotypes (10, 11). Thereafter, the role of ERs in bone health remained obscure. Instead, indirect mechanisms via extraskeletal tissues have been postulated to account for the osteoprotective actions of estrogen (12, 13). In this review, we will describe the skeletal and extraskeletal activities of ERs in mediating osteoprotective estrogen actions.
Nuclear ERs
Both subtypes of nuclear ERs, α and β, are members of the nuclear steroid hormone receptor gene superfamily and mediate most biological effects of estrogens (9, 14, 15). Nuclear estrogen-bound ERs are responsible for the genomic actions of estrogen through estrogen response element-dependent transcriptional control of target genes (16, 17, 18, 19) (Fig. 2). Rapid estrogen responses, so-called nongenomic actions, likely require cytoplasmic ERs and/or uncharacterized atypical ERs on the cell membrane (20).
Fig. 2.

The molecular mechanism of gene regulation by sex steroid hormones. Sex steroid hormones bind to their cognate nuclear receptors, and the ligand-bound receptors act as transcription factors regulating expression of specific target genes. Testosterone (weak androgen) can be converted by 5α-reductase into an active form of androgen, dihydrotestosterone (DHT). At the same time, testosterone serves as a precursor for an active estrogen, 17β-estradiol, through conversion by aromatase.
Both ERα and ERβ recognize and specifically bind to estrogen response elements in the target gene promoters as homodimers (α-α or β-β) and/or heterodimers (α-β) (9, 14, 15, 21). No clear difference in the binding of endogenous estrogens has been observed between ERα and ERβ; however, the ERs appear to exhibit different affinities for selective estrogen response modulators (22). In comparison with ERα, ERβ apparently has a lower capacity for hormone-induced transcriptional activation (23). Therefore, ERβ can be thought of as a dominant-negative counterpart of ERα that moderates the induction of endogenous estrogen target genes as well as transcriptional responses to estrogens. The molar, or quantitative, ratio of ERβ to ERα in a given cell is thus considered to define the cell’s sensitivity to estrogens and the extent of its biological responses to the hormone (24). Expression patterns of ERα and ERβ overlap in many organs and tissues, whereas in some types of cells, only one ER subtype is detectable.
ER-Deficient Female Mice Display Expected Abnormalities in the Estrogen Target Tissues But Not in Bones
The first generated ERα knockout (ERαKO) mice exhibited a wide spectrum of phenotypic abnormalities. Consistent with previously accumulated in vitro and in vivo findings, female reproductive organs in these ERαKO mice were poorly developed, and their phenotypes closely resembled features of OVX mice (9). However, the first ERαKO reportedly expressed shortened ER transcripts, and a residual ERα activity was suspected to mediate the endothelial effects of estrogens (25). Later, complete ERαKO mice (ERα−/−) expressing no detectable ERα transcripts were generated. Phenotypes of the ERα−/− mice verified and confirmed the ERα impact on female reproductive organs reported earlier in the ERαKO mice (26). Mice with a disrupted ERβ gene have been obtained by several groups; however, the described phenotypes of independently generated ERβ-deficient mice have not been consistent in the resultant abnormalities of estrogen target tissues (11, 24). Inconsistency between these initial studies was apparently based on analyses of mice with incomplete knockout of the ERβ (ERβKO) that could express abnormal transcripts. The phenotypic difference could be also due to residual ERβ activity, but the molecular basis of the varied observations still remains unclear at this stage. Interestingly, fewer abnormalities have been observed in recently generated complete ERβKO (ERβ−/−) mice (26, 27). Because ERα−/− mice exhibit far more severe abnormalities in estrogen target tissues than ERβ−/− mice (9, 10, 11, 27, 28), it appears that ERα is the prime receptor mediating major physiological actions of estrogens (Table 1).
Table 1.
Phenotypes of ERKOs in reproductive organs
| ERαKO | ERβKO | ERαβKO | |
|---|---|---|---|
| Pituitary gland | High LH | Normal | High LH |
| Hemorrhagic cystic | |||
| Ovary | High estrogen | Reduced ovulation | Lack of ovulation |
| High testosterone | Sex-reversed follicles | ||
| Anovulatory | |||
| Uterus | Estrogen in sensitive | Normal | Estrogen insensitive |
| Mammary gland | No pubertal development | Normal | No pubertal development |
Unlike other estrogen target tissues, female bones were not significantly affected by depletion of either ERα (ERα−/−) or ERβ (ERβ−/−) or both (ERαβ−/−), and the bone phenotypes were quite mild (29, 30, 31, 32). It was unexpected, because OVX in the same mouse strains led to a decrease in bone mass with high bone turnover that resembled the bone phenotype in women with naturally occurring estrogen deficiency in a postmenopausal state (1, 3, 13, 33).
The Osteoporotic Phenotype Is Absent in Female Mice Deficient of ERs
Lower bone turnover and increased bone mass were seen in female ERα−/− mice, whereas OVX in the same mouse strain induced decreased bone mass due to increased bone resorption (30, 32). The lack of negative feedback regulation due to the absence of ERα in the pituitary in ERα−/− mice results in aberrantly high serum levels of testosterone, a major estrogen precursor. The absence of osteoporotic phenotypes in ER-deficient mice can be explained by the osteoprotective effects of enhanced androgen signaling by testosterone excess in the bone (33). Supporting this idea, OVX induces bone loss in the ERα−/− mice, whereas androgen administration has osteoprotective effects in OVX mutant females (34, 35).
ERβ−/− female mice also exhibit reduced bone resorption and increased trabecular bone volume without, however, alteration of circulating sex hormone levels (32). Because estradiol can still be effective at partially reversing bone loss in OVX ERα−/− females (35), it appears that ERβ also mediates the osteoprotective action of estrogen, but to a much lesser extent than ERα (11, 35).
In the double-knockout (ERαβ−/−) mice, levels of circulating sex steroid hormones were also elevated, similar to those in ERα−/−. However, unlike the ERα−/− or ERβ−/− mice, a marked bone mass decrease was observed in the ERαβ−/− mice (Table 2), but it was less pronounced than that observed in the OVX wild-type females (30, 32, 34). Although these findings suggest cooperation between ERα and ERβ in the osteoprotective action of estrogens, female mice deficient in both ERα and ERβ fail to recapitulate the osteoporotic bone phenotype of estrogen-deficient women. It has been proposed that at high levels of circulating sex steroids, androgen receptor (AR) may partially compensate for ER deficiency in these mice.
Table 2.
Summary of ERKO bone phenotypes and levels of sex steroid hormones
| ERαKO | ERβKO | ERαβKO | OcERαKO | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | Male | Female | |||||
| Trabecular BMD in young | ↓ | → | → | → | ↓ | ↓ | → | ↓ | ||||
| Trabecular BMD in old | ↑ | ↑ | → | ↑ | ↑ | → | → | ↓ | ||||
| Cortical BMD | ↓ | ↓ | → | → | ↓ | ↓ | → | → | ||||
| Trabecular BV/TV | ↑ | ↑ | → | ↑ | ↑ | ↓ | → | ↓ | ||||
| Bone formation | ↓ | ↓ | → | → | ↓ | ↓ | → | ↑ | ||||
| Bone resorption | ↓ | ↓ | → | ↓ | ↓ | → | → | ↑ | ||||
| 17β-Estradiol | → or ↑ | ⇈ | → | → | → | → or ↑ | → | → | ||||
| Testosterone | ⇈ | ↑ | → | → | ⇈ | → or ↑ | → | → | ||||
BMD, Bone mineral density; BV, bone volume; TV, trabecular volume.
Can Local Aromatase-Produced Estrogens Support Normal Bone Remodeling in Males?
Male mice deficient of one (ERα−/− or ERβ−/−) or both (ERαβ−/−) of the ER subtypes exhibited neither clear bone mass decrease nor impaired bone development. Moreover, ERα−/− males displayed an increase in trabecular bone and decrease in bone resorption with elevated levels of testosterone (30, 32, 34) (Table 3). It has been suggested that the higher level of circulating testosterone in ERα−/− males compensates for impaired estrogen action by enhancing AR function in the bone (35, 36). These observations are, however, inconsistent with severe osteoporosis, and unfused epiphyses seen in men with nonfunctional ERα owing to hereditary mutations and despite high (albeit normal for men) levels of circulating testosterone (7, 9, 10, 37) (Table 3). Consistently, these male patients have failed to respond to estrogen treatment. The idea that ERα-mediated estrogen signaling is involved in the maintenance of the male skeleton is further supported by clinical observations that bone mass is low in aromatase-deficient male patients (8, 38, 39) (Table 3). Aromatase (CYP19) is the enzyme catalyzing conversion of androgens into estrogens (Fig. 2). Because circulating estrogen levels are low in males, the local conversion of testosterone into estrogen by aromatase appears to be physiologically significant for osteoprotective estrogen action in male bones. Reduced bone mass, high bone turnover, and unfused epiphyses, similar to those in ERα-deficient men (7), have also been observed in aromatase-deficient male patients (8, 38, 39). Significantly, such osteoporotic bone defects could be rescued by estrogen treatment only in aromatase-deficient, but not ERα-deficient, patients (40). Bone mass decrease was experimentally recapitulated in male mice deficient of aromatase (41, 42), whereas femur growth acceleration due to unclosed epiphyses in human male patients was not detected in male aromatase-deficient mice, presumably owing to innate absence of epiphyseal plate closure in mice of both sexes (8, 37, 42). Thus, the bone defects seen in naturally occurring aromatase-deficient men as well as experimentally generated aromatase-deficient male mice suggest that estrogens locally produced by aromatase are beneficial for bone health in males.
Table 3.
Serum hormone levels and bone phenotypes in male ERα- and aromatase-deficient patients
| ERα deficiency | Aromatase deficiency | |
|---|---|---|
| Estradiol | High | Low |
| Testosterone | Normal | Normal |
| FSH | High | High |
| LH | High | Normal |
| Bone mineral density | Low | Low |
| Bone formation marker | High | Normal |
| Bone resorption marker | High | Normal |
| Estrogen sensitivity | No | Yes |
| Others | Tall stature, unfused epiphysis | Tall stature, unfused epiphysis |
Indirect Osteoprotective Action of Estrogens in Males and Females
Absence of the expected osteoporotic defects in male and female mice lacking ERs suggests an indirect mode of the osteoprotective action of estrogens. Namely, bone marrow and some extraskeletal estrogen target cells and tissues secrete endocrine or paracrine factors known to support bone development and remodeling (Fig. 3). It has been proposed that these estrogen-induced antiresorptive factors may mediate the osteoprotective effects of estrogens. IGFs, known to stimulate osteoblastogenesis, are secreted by the liver. They modulate GH action in bone development, particularly during growth stages, and in cultured bone cell systems (37, 43, 44). Thus, hepatic IGF production stimulated by estrogens has been assumed to account for the osteoprotective effects of estrogen. However, the molecular basis of the estrogen-induced IGF regulation remains to be determined.
Fig. 3.
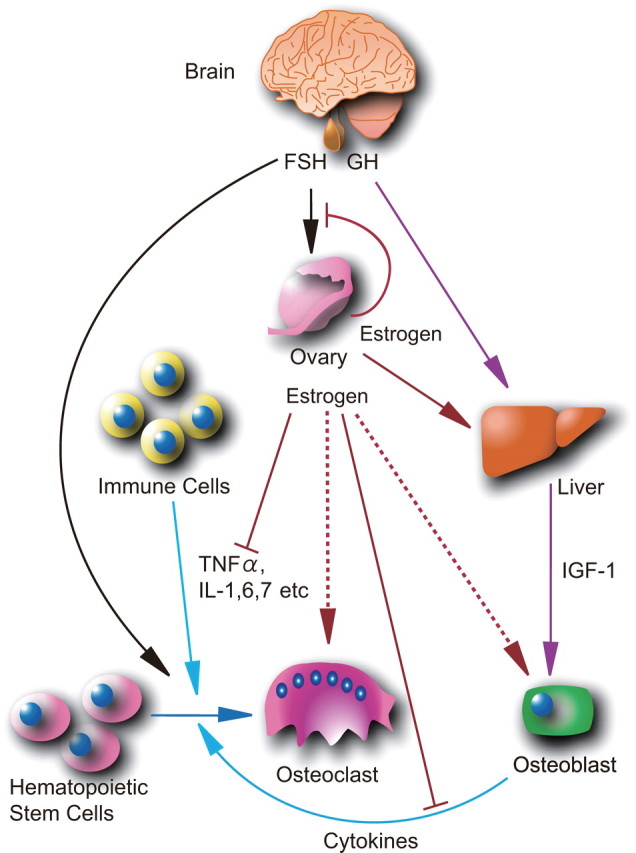
Indirect actions of estrogen control bone remodeling. Estrogen production by the ovaries is stimulated by FSH secreted by the pituitary gland. Estrogens inhibit FSH secretion through negative feedback regulation mediated by the pituitary ER. Estrogen deficiency increases FSH secretion and stimulates production by bone marrow and immune cells of cytokines, such as TNFα and several ILs. These cytokines facilitate osteoclast differentiation from hematopoietic stem cells. As an opposing action, ovarian estrogens and GH from the pituitary gland induce hepatic production of IGF-I that stimulates osteoblast differentiation.
FSH reportedly mediates indirect estrogen action in bone mass maintenance as a negative bone remodeler (13, 45). FSH is an upstream pituitary hormone that stimulates estrogen production by the ovary. FSH secretion is under negative feedback control of circulating estrogens that activate ERs in the pituitary to suppress FSH production. Estrogen deficiency in postmenopausal women is often associated with increased levels of circulating FSH. Zaidi et al. (45) applied this epidemiological observation to a mouse genetic approach using mice deficient in either FSH or FSH receptor. Female mice in which FSH signaling was defective displayed increased bone mass and low bone turnover without significant alteration of serum estrogen levels. Because this group also showed FSH’s ability to stimulate osteoclastogenesis, they inferred that increased FSH levels stemming from estrogen deficiency might cause a decrease in bone mass through enhancement of bone resorption by increased osteoclastogenesis (45). However, it is problematic to experimentally assess the significance of FSH’s action in the overall osteoprotective action of estrogens under normal physiological levels of the sex steroid. Moreover, it has been reported that GnRH agonist-induced estrogen deficiency causes dramatic bone loss despite significant suppression of FSH production (46). Therefore, it appears unlikely that FSH exerts beneficial effects on the bone at optimal levels of estrogens, or in an estrogen-sufficient state.
Estrogen deficiency-induced osteoporosis associates with persistent imbalance between bone resorption and bone formation activities that leads to progressive bone mass reduction. This suggests that osteoclastogenesis and/or osteoclastic function are augmented under suboptimal estrogen concentrations. Because inflammatory cytokines are potent inducers of osteoclastogenesis in vitro, a number of cytokines expressed by nonbone cells have been tested for possible contributions to estrogen deficiency-induced osteoclastogenesis (12, 47, 48) (Fig. 3). In this respect, hematopoietic cells in the bone marrow have emerged as modulators of bone remodeling through secretion of various pro- and antiresorptive cytokines (12). Among circulating blood cells, T cells are drawing much interest as potential regulators of bone resorption. Estrogen deficiency activates adaptive immune responses leading to stimulation of IL-7 and IGF-I production by activated T cells residing in bone that further induces secretion of interferon-γ (49, 50). Interferon-γ acts as an osteoclastogenic factor in concert with locally produced receptor activator of nuclear factor-κB ligand and tumor necrosis factor (51, 52). In cell culture systems, these cytokines activate transcriptional factors activation protein-1 and nuclear factor-κB that are known to promote osteoclastogenesis. Thus, loss of bone mass during estrogen deficiency can be attributed, at least in part, to a consequent increase of proresorptive cytokine production (12). However, these cytokine effects only reflect enhancement of osteoclast activity subsequent to estrogen deficiency and do not provide insight into the molecular basis of the beneficial action of estrogens on bone remodeling.
Osteoclastic ERα Mediates the Osteoprotective Action of Estrogens in Females
As outlined above, studies of systemic estrogen deficiency and impaired estrogen signaling in experimental animal models failed to indentify the mechanisms of osteoprotective actions of estrogens. Indeed, complete elimination of estrogen signaling in the whole organism (e.g., by conventional knockout of the ER genes) causes systemic imbalance in the endocrine system as well as possible nonphysiological events like aberrant cytokine production (12, 13). Thus, it became apparent that a bone cell-specific disruption of the ER genes is required to directly assess the role of estrogen signaling in the bone without interference from its systemic action or potential secondary defects.
The osteoprotective action of estrogens is associated with attenuation of bone resorption and restoration of normal bone remodeling. We therefore selectively disrupted the ERα gene in mature osteoclasts (53). Because cathepsin K is expressed only in the developed osteoclasts (54), the Cre gene was knocked into the cathepsin K gene locus, and the resulting Cre knock-in transgenic line (CatK-Cre) was shown to express Cre at detectable levels only in mature osteoclasts. Osteoclast-specific ERαKO mice (ERαΔOC/ΔOC) were generated by crossing mice from the CatK-Cre line with mice from the floxed ERα gene line that was previously used to obtain the complete ERαKO mice (ERα−/−) (26). As expected, neither clear alterations of circulating sex steroids nor FSH nor phenotypic abnormalities in growth and reproduction were detectable in male or female ERαΔOC/ΔOC mice. At 8 wk of age, significant bone loss in the trabecular bone area with high bone turnover was observed in the ERαΔOC/ΔOC females, but not males (Table 2). OVX caused only a negligible bone loss in the ERαΔOC/ΔOC mutants when compared with the bone loss in OVX wild-type females. In ERαΔOC/ΔOC females, 17β-estradiol effectively restored bone mass in the cortical area but not in the trabecular area. Considering that bone loss becomes evident first in the trabecular area in estrogen-deficient female rodents and in women, the osteoporotic features observed in the ERαΔOC/ΔOC female appear to support the idea that the osteoclastic ERα mediates, at least in part, the osteoprotective action of estrogens in females (53). Because estrogens can trigger apoptosis in osteoclasts through induction of the Fas ligand (FasL) gene, we suggest that the proapoptotic actions of estrogens in mature osteoclasts underlie the antiresorptive effects of estrogens in the bone (53) (Fig. 4). These findings are consistent with earlier observations that OVX-induced estrogen deficiency in mice resulted in decreased apoptosis and extended lifespan of mature osteoclasts (55).
Fig. 4.

Proposed model of the osteoclastic ERα-mediated osteoprotective action of estrogens through attenuation of bone resorption by inducing osteoclastic cell death. Estrogens (17β-estradiol) activate ERs in functionally opposing types of bone cells: osteoblasts, which derive from mesenchymal stem cells, and osteoclasts, multinucleated giant cells derived from hematopoietic stem cells. The estrogen-dependent induction of FasL gene expression in these bone cells triggers apoptosis in mature osteoclasts.
Male ERαΔOC/ΔOC Mice Display No Apparent Bone Defects
Contradictory to expectations raised by previous studies in rodents and male patients (8, 29, 41, 42), no abnormality has been observed in either cortical or trabecular bone areas in ERαΔOC/ΔOC males (53). It is difficult to ascertain the reason why ERαΔOC/ΔOC males do not exhibit bone loss, and here we can only speculate about a possible difference in mechanisms of osteoprotective estrogen actions in males. First, high levels of androgens, and consequently, activated AR may critically impact bone formation and resorption even in the absence of ERα. Second, the CatK-Cre transgenic line is limited to disrupt a given gene only at late stages of osteoclastic life, whereas the activated ERα may play a critical role at earlier stages. There may be stage-specific differences between males and females in the ERα action during osteoclast differentiation and maturation. This idea is supported by previous reports that estrogens may be inhibitory for osteoclastogenesis and osteoclastic function in vitro (56, 57). Generation of differentiation stage-specific osteoclastic ERα knockout mice may be able to clarify this hypothesis. Thirdly, osteoblastic ERα may also mediate the osteoprotective effects of estrogens. Earlier, estrogens were shown to stimulate osteoblastogenesis in vitro (58). More recently, Brown’s group provided evidence that the FasL gene is a direct ER target in cultured osteoblasts (59). It is thus conceivable that estrogens support apoptosis of mature osteoclasts via stimulation of FasL expression in osteoclasts and osteoblasts (60). Reported earlier antiapoptotic effects of estrogens on osteoblasts (61, 62) suggest the existence of differential mechanisms or even pathways of estrogen action in these two cell types. Thus, it is conceivable that, in contrast to proapoptotic effects of estrogens on osteoclasts, their antiapoptotic effects on osteoblasts are similar in males and females. Obviously, osteoblast-specific ablation of ERs in male mice is required to challenge or prove this idea. Likewise, there are several cell types, including osteocytes and bone marrow cells, which remain to be tested for the impact of their ERs on the osteoprotective estrogen actions in males.
Conclusion
Estrogens exert osteoprotective actions in females and males. Key features of the estrogen deficiency-induced osteoporosis observed in postmenopausal women, such as bone loss and high-turnover bone metabolism, has been recapitulated in osteoclast-specific ERα knockout female mice (53). This is in striking contrast to the phenotype of mice with conventional or general ER knockout that exhibited increased bone mass. It appears that in females, osteoclastic ERα mediates estrogen-dependent attenuation of bone resorption through stimulation of apoptosis in osteoclasts. Although primary cultured bone cells from males and females equally respond to estrogen, the osteoclast-selective ablation of ERα in male mice caused neither bone defects nor unclosed epiphyses (53) that had been consistently observed in male patients with impaired estrogen signaling (7, 8). This discrepancy suggests sex specificity in mechanisms of osteoprotective action of estrogens in vivo and raises a hypothesis that in male bones, beneficial effects of estrogens are predominantly mediated by the osteoblastic ERα, rather than through the antiresorptive action of osteoclastic ERα, which is more critical in females. This idea can be tested by cell type-specific ablation of ERα in males as well as in females to decipher molecular and cellular mechanisms of the anabolic action of estrogens in the skeleton. At the same time, a compensatory action of AR at high concentrations of circulating testosterone may account for different physiological consequences of osteoclast-targeted ERα ablation in male mice. A double osteoclast-specific AR and ERα knockout may clarify this possibility.
NURSA Molecule Pages:
Ligands: 17β-estradiol;
Nuclear Receptors: ER-α | ER-β.
Footnotes
This work was supported by priority areas from the Ministry of Education, Culture, Sports, Science, and Technology (to S.K.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online November 12, 2009
Abbreviations: AR, Androgen receptor; ER, estrogen receptor; ERαKO, ERα knockout; FasL, Fas ligand; OVX, ovariectomy.
References
- 1.Nelson HD2008. Menopause. Lancet 371:760–770 [DOI] [PubMed] [Google Scholar]
- 2.Ebeling PR2008. Clinical practice. Osteoporosis in men. N Engl J Med 358:1474–1482 [DOI] [PubMed] [Google Scholar]
- 3.Harada S, Rodan GA2003. Control of osteoblast function and regulation of bone mass. Nature 423:349–355 [DOI] [PubMed] [Google Scholar]
- 4.Harman SM2006. Estrogen replacement in menopausal women: recent and current prospective studies, the WHI and the KEEPS. Gend Med 3:254–269 [DOI] [PubMed] [Google Scholar]
- 5.Draper MW2003. The role of selective estrogen receptor modulators (SERMs) in postmenopausal health. Ann NY Acad Sci 997:373–377 [DOI] [PubMed] [Google Scholar]
- 6.Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD2002. Postmenopausal hormone replacement therapy: scientific review. JAMA 288:872–881 [DOI] [PubMed] [Google Scholar]
- 7.Jones ME, Boon WC, Proietto J, Simpson ER2006. Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol Metab 17:55–64 [DOI] [PubMed] [Google Scholar]
- 8.Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS1994. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 331:1056–1061 [DOI] [PubMed] [Google Scholar]
- 9.Couse JF, Korach KS1999. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 20:358–417 [DOI] [PubMed] [Google Scholar]
- 10.Syed F, Khosla S2005. Mechanisms of sex steroid effects on bone. Biochem Biophys Res Commun 328:688–696 [DOI] [PubMed] [Google Scholar]
- 11.Windahl SH, Andersson G, Gustafsson JA2002. Elucidation of estrogen receptor function in bone with the use of mouse models. Trends Endocrinol Metab 13:195–200 [DOI] [PubMed] [Google Scholar]
- 12.Pacifici R2008. Estrogen deficiency, T cells and bone loss. Cell Immunol 252:68–80 [DOI] [PubMed] [Google Scholar]
- 13.Zaidi M2007. Skeletal remodeling in health and disease. Nat Med 13:791–801 [DOI] [PubMed] [Google Scholar]
- 14.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM1995. The nuclear receptor superfamily: the second decade. Cell 83:835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O1998. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci USA 95:15677–15682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato S, Tora L, Yamauchi J, Masushige S, Bellard M, Chambon P1992. A far upstream estrogen response element of the ovalbumin gene contains several half-palindromic 5′-TGACC-3′ motifs acting synergistically. Cell 68:731–742 [DOI] [PubMed] [Google Scholar]
- 17.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P1995. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270:1491–1494 [DOI] [PubMed] [Google Scholar]
- 18.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M2005. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122:33–43 [DOI] [PubMed] [Google Scholar]
- 19.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38:1289–1297 [DOI] [PubMed] [Google Scholar]
- 20.Moggs JG, Orphanides G2001. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep 2:775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pettersson K, Delaunay F, Gustafsson JA2000. Estrogen receptor β acts as a dominant regulator of estrogen signaling. Oncogene 19:4970–4978 [DOI] [PubMed] [Google Scholar]
- 22.Hall JM, McDonnell DP1999. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566–5578 [DOI] [PubMed] [Google Scholar]
- 23.Lindberg MK, Movérare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C2003. Estrogen receptor (ER)-β reduces ERα-regulated gene transcription, supporting a “ying yang” relationship between ERα and ERβ in mice. Mol Endocrinol 17:203–208 [DOI] [PubMed] [Google Scholar]
- 24.Imamov O, Shim GJ, Warner M, Gustafsson JA2005. Estrogen receptor β in health and disease. Biol Reprod 73:866–871 [DOI] [PubMed] [Google Scholar]
- 25.Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, Bayard F, Arnal JF2002. The AF-1 activation-function of ERα may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci USA 99:2205–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M2000. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development 127:4277–4291 [DOI] [PubMed] [Google Scholar]
- 27.Antal MC, Krust A, Chambon P, Mark M2008. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERβ-null mutant. Proc Natl Acad Sci USA 105:2433–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deroo BJ, Korach KS2006. Estrogen receptors and human disease. J Clin Invest 116:561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt A, Seedor JG, Gentile MA, Gentile BL, Pennypacker BL, Rodan GA, Kimmel DB1999. Femoral bone density and length in male and female estrogen receptor-α (ERα) knockout mice. J Bone Miner Res 14(Suppl 1):S456
- 30.Lindberg MK, Alatalo SL, Halleen JM, Mohan S, Gustafsson JA, Ohlsson C2001. Estrogen receptor specificity in the regulation of the skeleton in female mice. J Endocrinol 171:229–236 [DOI] [PubMed] [Google Scholar]
- 31.Windahl SH, Vidal O, Andersson G, Gustafsson JA, Ohlsson C1999. Increased cortical bone mineral content but unchanged trabecular bone mineral density in female ERβ−/− mice. J Clin Invest 104:895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, Gaillard-Kelly M, Baron R2002. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-β in bone remodeling in females but not in males. Bone 30:18–25 [DOI] [PubMed] [Google Scholar]
- 33.Riggs BL, Khosla S, Melton 3rd LJ2002. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev 23:279–302 [DOI] [PubMed] [Google Scholar]
- 34.Couse JF, Yates MM, Walker VR, Korach KS2003. Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) null mice reveals hypergonadism and endocrine sex reversal in females lacking ERα but not ERβ. Mol Endocrinol 17:1039–1053 [DOI] [PubMed] [Google Scholar]
- 35.Sims NA, Clément-Lacroix P, Minet D, Fraslon-Vanhulle C, Gaillard-Kelly M, Resche-Rigon M, Baron R2003. A functional androgen receptor is not sufficient to allow estradiol to protect bone after gonadectomy in estradiol receptor-deficient mice. J Clin Invest 111:1319–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawano H, Sato T, Yamada T, Matsumoto T, Sekine K, Watanabe T, Nakamura T, Fukuda T, Yoshimura K, Yoshizawa T, Aihara K, Yamamoto Y, Nakamichi Y, Metzger D, Chambon P, Nakamura K, Kawaguchi H, Kato S2003. Suppressive function of androgen receptor in bone resorption. Proc Natl Acad Sci USA 100:9416–9421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C2004. Androgens and bone. Endocr Rev 25:389–425 [DOI] [PubMed] [Google Scholar]
- 38.Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C2004. Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab 89:61–70 [DOI] [PubMed] [Google Scholar]
- 39.Bouillon R, Bex M, Vanderschueren D, Boonen S2004. Estrogens are essential for male pubertal periosteal bone expansion. J Clin Endocrinol Metab 89:6025–6029 [DOI] [PubMed] [Google Scholar]
- 40.Oz OK, Zerwekh JE, Fisher C, Graves K, Nanu L, Millsaps R, Simpson ER2000. Bone has a sexually dimorphic response to aromatase deficiency. J Bone Miner Res 15:507–514 [DOI] [PubMed] [Google Scholar]
- 41.Fisher CR, Graves KH, Parlow AF, Simpson ER1998. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci USA 95:6965–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balasch J2003. Sex steroids and bone: current perspectives. Hum Reprod Update 9:207–222 [DOI] [PubMed] [Google Scholar]
- 43.Sjögren K, Bohlooly-Y M, Bohlooly YM, Olsson B, Coschigano K, Törnell J, Mohan S, Isaksson OG, Baumann G, Kopchick J, Ohlsson C2000. Disproportional skeletal growth and markedly decreased bone mineral content in growth hormone receptor −/− mice. Biochem Biophys Res Commun 267:603–608 [DOI] [PubMed] [Google Scholar]
- 44.Sims NA, Clément-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, Goffin V, Coschigano K, Gaillard-Kelly M, Kopchick J, Baron R, Kelly PA2000. Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest 106:1095–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, Zaidi S, Zhu LL, Yaroslavskiy BB, Zhou H, Zallone A, Sairam MR, Kumar TR, Bo W, Braun J, Cardoso-Landa L, Schaffler MB, Moonga BS, Blair HC, Zaidi M2006. FSH directly regulates bone mass. Cell 125:247–260 [DOI] [PubMed] [Google Scholar]
- 46.Carmina E, Janni A, Lobo RA1994. Physiological estrogen replacement may enhance the effectiveness of the gonadotropin-releasing hormone agonist in the treatment of hirsutism. J Clin Endocrinol Metab 78:126–130 [DOI] [PubMed] [Google Scholar]
- 47.Compston JE1994. Hormone replacement therapy for osteoporosis: clinical and pathophysiological aspects. Reprod Endocr Rev 3:209–224 [Google Scholar]
- 48.Rodan GA, Martin TJ2000. Therapeutic approaches to bone diseases. Science 289:1508–1514 [DOI] [PubMed] [Google Scholar]
- 49.Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, Pacifici R2001. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci USA 98:13960–13965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryan MR, Shepherd R, Leavey JK, Gao Y, Grassi F, Schnell FJ, Qian WP, Kersh GJ, Weitzmann MN, Pacifici R2005. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc Natl Acad Sci USA 102:16735–16740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, Wagner EF, Taniguchi T2002. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 416:744–749 [DOI] [PubMed] [Google Scholar]
- 52.Teitelbaum SL, Ross FP2003. Genetic regulation of osteoclast development and function. Nat Rev Genet 4:638–649 [DOI] [PubMed] [Google Scholar]
- 53.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S2007. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 130:811–823 [DOI] [PubMed] [Google Scholar]
- 54.Li YP, Chen W1999. Characterization of mouse cathepsin K gene, the gene promoter, and the gene expression. J Bone Miner Res 14:487–499 [DOI] [PubMed] [Google Scholar]
- 55.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF1996. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-β. Nat Med 2:1132–1136 [DOI] [PubMed] [Google Scholar]
- 56.Shevde NK, Bendixen AC, Dienger KM, Pike JW2000. Estrogens suppress RANK ligand-induced osteoclast differentiation via a stromal cell independent mechanism involving c-Jun repression. Proc Natl Acad Sci USA 97:7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srivastava S, Toraldo G, Weitzmann MN, Cenci S, Ross FP, Pacifici R2001. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-κB ligand (RANKL)-induced JNK activation. J Biol Chem 276:8836–8840 [DOI] [PubMed] [Google Scholar]
- 58.Okazaki R, Inoue D, Shibata M, Saika M, Kido S, Ooka H, Tomiyama H, Sakamoto Y, Matsumoto T2002. Estrogen promotes early osteoblast differentiation and inhibits adipocyte differentiation in mouse bone marrow stromal cell lines that express estrogen receptor (ER) α or β. Endocrinology 143:2349–2356 [DOI] [PubMed] [Google Scholar]
- 59.Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M2008. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J 27:535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krum SA, Brown M2008. Unraveling estrogen action in osteoporosis. Cell Cycle 7:1348–1352 [DOI] [PubMed] [Google Scholar]
- 61.Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC2001. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 104:719–730 [PubMed] [Google Scholar]
- 62.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC2002. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 298:843–846 [DOI] [PubMed] [Google Scholar]