

Abstract
Nicotinic acetylcholine receptors regulate the nicotine dependence encountered with cigarette smoking, and this has stimulated a search for drugs binding the responsible receptor subtypes. Studies link a gene cluster encoding for α3β4α5-D398N nicotinic acetylcholine receptors to lung cancer risk as well as link a second mutation in this cluster to an increased risk for nicotine dependence. However, there are currently no recognized drugs for discriminating α3β4α5 signaling. In this study we describe the development of homogenous HEK-293 cell clones of α3β4 and α3β4α5 receptors appropriate for drug screening and characterizing biochemical and pharmacological properties of incorporated α5 subunits. Clones were assessed for plasma membrane expression of the individual receptor subunits by mass spectrometry and immunochemistry and their calcium signaling was measured in the presence of a library of kinase inhibitors and a focused library of acetylcholine receptor ligands. We demonstrated an incorporation of two α3 subunits in approximately 98% of plasma membrane receptor pentamers indicating a 2/3 subunit expression ratio of α3 to β4 alone or to coexpressed β4 and α5. With prolonged nicotine exposure the plasma membrane expression of receptors with and without incorporated α5 increased. Whereas α5 subunit expression decreased the cell calcium response to nicotine and reduced plasma membrane receptor number, it partially protected receptors from nicotine mediated desensitization. Hit compounds from both libraries suggest the α5 and α5-D398N subunits allosterically modify the behavior of nicotine at the parent α3β4 nicotinic acetylcholine receptor. These studies identify pharmacological tools from two distinct classes of drugs, antagonists and modifiers that are α5 and α5-D398N subtype selective that provide a means to characterize the role of the CHRNA5/A3/B4 gene cluster in smoking and cancer.
Keywords: addiction, acetylcholine, allosteric, calcium, cancer, cigarettes, drug-abuse, kinase, nAChR, nicotine, receptor, signaling, smoking
Graphical Abstract
Introduction
Smoking remains a leading cause of death and morbidity in the developed world. Smokers exhibit common behaviors characterized by heavy, early morning cigarette use, tolerance to repeated cigarette use, and withdrawal symptoms from chronic tobacco use. For 60% of cigarette smokers their difficulty in quitting is a direct consequence of tobacco related nicotine dependence 1. The molecular targets for nicotine are peripheral and central nervous system (CNS) nicotinic acetylcholine receptors (nAChRs) 2. As the name implies, nAChRs are activated by both the endogenous neurotransmitter acetylcholine and the environmental toxin nicotine. These receptors are integral membrane proteins formed by combinations of five subunits that function as cationic ligand gated ion channels for Na+, K+, and Ca2+. Multiple nAChR subtypes occur in the CNS. The human brain expresses nine different α subunits and 3 types of β subunits that mediate the release of dopamine and other neurotransmitters in a subtype specific manner 3. As a consequence of the many nAChR combinations and permutations, determining the role of an individual subunit in the physiology of nicotine dependence remains a difficult problem as does developing subtype specific ligands. Thus, there are very few drugs available for smoking cessation therapy and FDA approved treatments are restricted to nicotine based gums, lozenges, and patches; varenicline, a nAChR partial agonist, and the antidepressant bupropion whose mechanism of action in smoking cessation is not fully understood 4.
Common acetylcholine and nicotine binding domains form at the interfaces of the subunit extracellular faces. Depending upon the subunit composition each receptor may have from two to five agonist and antagonist sites with differing affinities 5, 6. There are major differences in the pharmacokinetic properties of acetylcholine and nicotine and their effects on receptor function. Hydrolytic enzymes at nerve terminals inactivate acetylcholine within milliseconds whereas the functional half-life of nicotine is many hours. For α4β2 nAChRs, prolonged exposure to nicotine up-regulates their expression and may represent an early step in nicotine addiction 6–8.
A large-scale study of nAChR genetic variants associated with nicotine dependence in smokers identified the CHRNA5/A3/B4 gene cluster that is found on chromosome 15. This locus codes for α3, β4, and α5 receptor subunits. Of the many different nAChR subtypes considered for treating nicotine addiction, α3β4α5 receptors are intriguing targets because they provide the strongest evidence of a genetic association between nicotine mediated addictive behavior and lung cancer risk 1, 3, 9. Additionally, the CHRNA5 SNP rs16969968 represents a D398N substitution in α5 associated with lung cancer 10, 11, and a second SNP in the gene cluster correlates to nicotine dependence 1. Some other associations between this gene cluster and addictive smoking behaviors include the expression of the α3 subunit in dopaminergic neurons, the observation that deletion in mice of the β4 subunit prevents signs of withdrawal, and a presence of α3β4 subunits in the peripheral nervous system that suggests a correlation to craving behaviors 5.
How the α5 subunit functions in modulating the signaling properties of α3β4 nAChRs is not well understood either under normal physiological conditions or in cancer. Identifying sub-type specific small molecule ligands to contrast the functions of α3β4 and α3β4α5 nAChRs in nicotine addiction should help determine how α5 subunits modify receptor properties. This applies as well to identifying regulatory enzymes such as kinases that affect the on, off, and desensitized nAChR signaling states 12. Additionally, the large number of conotoxins that can discriminate between distinct nAChR subtypes provide biological evidence supporting comprehensive searches for conventional drugs with similar selective capabilities 13, as does the recent discovery of small molecules with selectivity between α4β2 and α3β4 receptors 14, 15. Conversely, the observation that cone snails disable prey by simultaneously targeting multiple subtypes of nAChRs with a mixture of different conotoxins suggests that a similar comprehensive approach against multiple subtypes of nAChRs may be needed for modifying the strong, diverse, and refractory nicotine-based behaviors in smokers.
It is common for cell-based assays characterizing nAChR ligands to have a degree of ambiguity. Attempts to express homogenous in cellulo nAChR populations using excesses of one transfected subunit often fail due to unrestricted subunit rearrangement that occurs in the endoplasmic reticulum 6, 16. Drug assays with nAChRs are therefore frequently based upon measurements of binding, calcium signaling, membrane potential changes, or electrophysiological properties in mixed populations of recombinant receptors 16–18. With this assay limitation in mind, correlations between biochemical and clinical data suggest partial α2β4 nAChR agonists like varenicline have anti-smoking efficacy as nicotine substitutes and prevent reinforcing behaviors. Despite some early promise, many of these compounds have significant side effects that lessen their utility 5, 19.
This manuscript presents results from our studies to develop and provide tools to characterize a role for the α5 subunit in nAChRs biology. Our results provide cell-based assays for high throughput α3β4 and α3β4α5 screening, the identification of novel subtype specific ligands they bind, and the construction of α3β4 and α3β4α5 activity profiles, i.e. “response fingerprints”, using a library of enzyme inhibitors that change their desensitization. We show from mass spectrometry and immunochemical data that a homogeneous population of (α3)2(β4)3 receptors can be established in a model system of HEK-293 cells to enable comparative studies of α5 subunit modulation of α3β4 signaling and expression. We utilize these cells to screen a directed library of putative nAChR antagonists and evaluate a library of kinase inhibitors in order to identify compounds that differentially bind or selectively modulate the two different receptor subunit combinations (α3)2(β4)3 and (α3)2(β4)2(α5). Our results show that inclusion of the α5 subunit in the fully formed plasma membrane (α3)2(β4)2(α5) receptor can modulate its membrane expression and signaling, alter the response to kinases, and protect the mature receptor from desensitization in the continued presence of nicotine. Thus, association of the α5 subunit with cancer and addiction may result from α5 allosteric modification of multiple receptor behaviors in the presence of sustained nicotine exposure, where the term allosterism is not limited simply to ligand binding sites but is used in the broader context of “indirect interaction between topographically distinct sites mediated by a conformational change of a protein molecule,” 20. Moreover, these behaviors can be probed by drugs that are selective antagonists or enzymatic regulators of the receptor subtypes.
Results
The first part of the results will describe the development and validation of labeled receptor subunits, their corresponding cell clones, and their cell surface expression. The second part of the results will present data using the validated clones to assess two classes of compound libraries in primary screens and further characterize some hit compounds in secondary functional screens.
Epitope tagging and expression of nicotinic acetylcholine receptor subunits
nAChRs are approximately 300 kDa pentamers formed by subunits composed of four transmembrane domains and extracellular N and C termini 21. The N terminal regions are critical to creating interfaces in each pentamer that form canonical agonist acetylcholine binding sites and alternative allosteric sites 22. Pharmacological properties of these sites are affected by the particular arrangement of the subunits, and despite the ability of subunits to undergo stochastic arrangement certain combinations are favored 23. In the case of α5β4α3, the most likely arrangement is with an unpaired α5 subunit performing an allosteric function rather than participating in the formation of a canonical site, though it is now recognized that the α5 subunit is not always restricted to the fifth position 2, 24. Thus, difficulties in characterizing the physiological roles of nAChRs can arise from the expression of the numerous subunit combinations and their overlapping in vivo distributions.
Various assay strategies have arisen to circumvent this combinatorial problem including using receptors composed of subunit concatamers, subunits labeled with GFP, subunit specific antibodies, and epitope tagging of subunits 6, 16, 25–27. For various reasons including low expression and loss of functionality of the pentamers, the first two methods did not work adequately for us when tested in multiple cell types. Therefore, our strategy for deconvoluting the composition of membrane bound receptors was insertion of minimally-disruptive N-terminal epitopes following the subunit signal sequences, yielding HAα3, c-MYCβ4, and V5α5 tagged receptor subunits as shown in the cartoon in figure 1A. As shown in figures 1B and C, immunofluorescence of live, non-permeabilized HEK-293 cells with a stable transfection of the subunits, and western blots of the corresponding cell lysates using monoclonal antibodies against the various epitopes, confirmed the plasma membrane and total cell expressions of the different receptor subunits.
Figure 1. Expression of components of the nAChR α3β4α5 in HEK 293 cells.

A. Diagram of the different subunits of the α3β4α5 nAChR showing position of the HA, MYC, and V5 epitope tags, relative positions of the transmembrane regions, and the location of the D398N mutation in the α5 subunit. B. Immunofluorescence localization of the expression of the α3, β4 subunits of the nAChR on the plasma membrane as components of the complete α3β4 receptor in a permanently transfected line of live, non permeabilized HEK-293 cells. A corresponding SDS-PAGE gel evaluated with the same antibodies also demonstrates the expression of both α3 and β4 subunits in these cells. C. Fluorescence images comparable to those in (B) of cells in which the α5 subunits are also expressed as part of the complete α3β4α5 receptor imaged using a primary anti V5 antibody. The corresponding western blot is shown below. Fluorescence images of secondary fluorescent antibodies were acquired using the fluorescein and rhodamine channels with a 40X plan apochromat NA 1.4 oil objective on a Zeiss LSM-510 confocal microscope.
Expression of plasma membrane receptors with chronic nicotine exposure
Studies have shown that nAChRs change their cellular expression in response to chronic nicotine exposure in culture and in vivo 28–30. In order to test the functionality of the epitope-tagged receptor clones and to measure nAChR plasma membrane expression we performed on-cell ELISA with non-permeabilized cells after overnight exposure of the cells to vehicle or 1 mM nicotine (fig. 2A). Nicotine produced a three-fold increase of cell surface receptor expression of the α3β4 clone as well as two-fold increases in expression of the α5 and α5D398N containing clones (fig. 2B). Additionally, the substitution of the α5 subunit into the α3β4 had minimal effect on the EC50 of the response (fig. 2C). The 95% confidence intervals for the EC50s were 22–47 μM (α3β4) and 11–18 μM (α3β4α5). These data indicate that the N-terminal epitope tags do not interfere with the chronic nicotine response and that both the α3β4 and α3β4α5 receptor populations undergo an analogous upregulation by nicotine that has been observed for the α4β2 28. To better define the initial homogeneity or composition of those untreated populations of receptors we performed mass spectrometry.
Figure 2. Plasma Membrane Expression of nAChR subunits in HEK-293 cells.
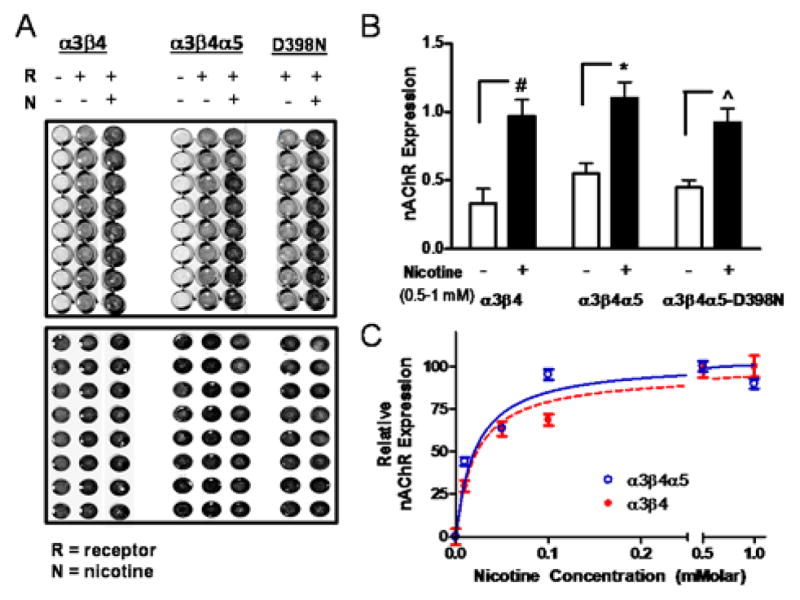
A. The expressions of the different subunit combinations were measured by on-cell ELISA in a 96 well plate format on live cells using a primary antibody against the MYC epitope for α3β4 receptors and the V5 epitope for α3β4α5 WT and D398N receptors (upper image). Data from individual wells were normalized by the well’s cell number, determined independently using the cell permeable fluorescent, infrared dye IR800CWNHS (lower image). B. The plasma membrane expression levels for each receptor type correspond to 0 nicotine exposure, white bars, and 1 mM nicotine exposure black bars. The receptor expression levels are presented as order pairs of values representing the mean ± STD and are as follows: α3β4, (0.33 ± 0.11, 0.971 ± 0.12); α3β4α5, (0.55 ± 0.07, 1.10 ± 0.11); α3β4α5-D398N, (0.45 ± 0.05, 0.92 ± 0.10). Individual experiments, N = 3, were performed in replicates of 8–16 wells C. Dose response of the change in receptor expression in the presence of various concentrations of nicotine. Background subtracted results were normalized by the response at 1 mM nicotine and data are presented as the mean ± SEM of the normalized response.
Composition of plasma-membrane expressed nAChRs
Receptors expressed in the permanent cell lines were purified using surface biotinylation followed by purification on avidin beads and SDS PAGE. The molecular weight of each subunit was determined by western blotting using anti-epitope antibodies (fig. 3A). Bands corresponding to their molecular weights were excised from the gel and the relative quantities of individual subunits (fig. 3B) were determined by mass spectrometry as described in Methods. Figure 3C presents plots of the relative expression of the α3 subunit versus either the β4 subunit alone (left) or the β4 subunit plus the α5 subunit (right), or a combination of the preceding data (lower). The relative amounts of each subunit can be determined in a straightforward manner by utilizing the slope of the regression line (see Methods - Determining fractional expression per pentamer of nAChR subunits). For the α5 not present β4/α3 = (2.93 ± 0.13)/(2.07 ± 0.13), and when the α5 is present (β4+ α5)/α3 = (2.99 ± 0.15)/(2.01 ± 0.15). Analyzing together both the previous scenarios in a pentamer model of two or three α3 subunits, we observe a probability p2α3 that on average 98% (1 ± 0.05) of pentamers contain precisely two α3 subunits (fig. 3C lower image). We similarly considered a model for the occurrence of α5 and β4 subunits in the α3β4α5 clone. If the α5 substitutes for the β4 subunit only once or not at all the percentages of pentamer receptors, p0α5 and p1α5 containing either zero or one α5 and correspondingly either three versus two β4 subunits are p0α5 and p1α5 0.57 ± 0.29 respectively (fig. 3 legend and Methods).
Figure 3. Expression of plasma membrane components of the nAChR α3β4 and α3β4α5 from HEK 293 cells as measured by mass spectrometry.

Stable cell lines expressing α3β4 or α3β4α5 were biotinylated on the cell surface and purified on avidin resin for use in mass spec analysis. A. Shown is a composite of western blots against the different subunits using anti epitope antibodies. B. Image of an Acqua-stained (a coomassie blue based protein reagent), SDS-Page gel containing biotinylated plasma membrane proteins from the cell lines. The material between 50–75 kDa designated by the hatched box was cut from the gel and eluted for mass spectrometry. C. Plotted are mass spectrometry ratio determinations with standard deviations (error bars) from seven (left plot) and six (right plot) independent preparations of the relative ratios of nAChR receptor subunits. Data were fit by linear regression. The red reference lines have slopes of 1.5. Results for the best fit lines describing the subunit ratios are in blue and are flanked by the 95% confidence intervals (hatched curves). From left to right image to below the slopes of the lines have the values as mean ± std error (1.41 ± 0.15, 1.49 ± 0.19, 1.47 ± 0.06). The ratio of α5 to β4 subunits, with the corresponding paired β4 measurement of the six determinations, ( , β4),were: (0.17, 8.9); (0.12, 24.3); (0.45, 4.7); (0.44, 3.4); (0.10, 10.8); (0.22, 17.1).
Restoration of the calcium response with loss of the α5 subunit
It is plausible that α5 subunits may play a role in Ca2+ permeability, which affects sensitivity to nicotine. When the D398N variant was associated with concatamers of α4β2 subunits in a Xenopus oocyte expression system, calcium ion permeability decreased. However, no changes were observed in that same system when α5 was coupled to the α3β4 using a 1:1:2 transfection ratio with the α5 in excess 31. To examine the influence of the α5 subunit on calcium flux in our HEK-293 cell model, we employed a knockdown strategy using RNA interference. Figure 4a shows the calcium response of the α3β4, α3β4α5, and α3β4α5-D398N clones in response to nicotine where the signal has been normalized by a total calcium cell response as described 32. Knockdown of the α5 subunit with siRNA had no effect on the α3β4 calcium response (fig.4B) but resulted in a two to four fold increase in the response of the WT and D398N α5 variants respectively (fig.4C and fig.4D). The effects of the siRNA knockdown on whole cell expression (western blot) versus plasma membrane expression (on-cell ELISA) is shown in figures 4E and 4F and indicates a preference for incorporating the α5 subunit into the mature receptor.
Figure 4. Subunit Dependence of the Calcium Response of the nAChR Permanent Cell Lines.

Receptor subunits were expressed in HEK-293 cells also permanently expressing a mitochondrial localized aequorin-based calcium reporter. Calcium response data per well were normalized by the total calcium response in that well determined upon cell lysis (see Methods). Measurements at given concentrations were performed at least in triplicate and the sigmoid concentration response curves of the different receptor variants were fit using GraphPad prism version 5.0 to determine the parameters basal response and maximal response (presented as mean ± sem), and additionally the EC50 95% CI. A. Results were respectively, α3β4 (0.0 ± 0.04, 0.9 ± 0.03, 11–21, N = 5), α3β4α5 (0.0 ± 0.03, 0.49 ± 0.06, 29–199, N = 6), α3β4α5-D398N (0.03 ± 0.02, 0.18 ± 0.11, 11–503, N = 3). Cells expressing only the reporter showed no response. Insert shows control demonstrating the effect of the antagonist mecamylamine on α3β4 signaling. B–D. Responses were measured in the presence of siRNA against the α5 subunit. (B) Data were fit with a shared parameter model (GraphPad Prism) for basal /maximal /EC50 95% CI, (0.01±0.04, 1.09±0.04, 23–53 μM, N=3). (C) The common fitting parameters were basal/EC50 95% CI of (0.06±0.06, 56–263 μM) and maximal responses of 1.69 ± 0.18, 0.91 ± 0.13, 0.97 ± 0.14, N=3. (D) The common fitting parameters were basal/EC50 95% CI of (0.20±0.06, 78–407 μM) and maximal responses of 2.4± 0.28, 0.51 ± 0.15, 0.74 ± 0.16, N=3. E. Shown in the bar graph are the effects of siRNA treatment on whole cell expression of α5 wild type and D398N subunits in stable cell line as determined by the corresponding subunit western blots. Data corresponding to control and siRNA knockdown plasmids are presented as mean ± STD. For the western blot they are, α3β4α5 (1.16 ± 0.02, 0.31 ± 0.16, N=3); α3β4α5-D398N (1.42 ± 0.31, 0.39 ± 0.17, N=3) and for the on-cell determination are, α3β4α5 (0.99 ± 0.047, 0.74 ± 0.084, N=4); α3β4α5-D398N (1.00 ± 0.017, 0.86 ± 0.026, N=4 with eight replicates analyzed for each condition). F. The lower section shows a representative western blot corresponding to the α5 subunit determinations (bands at 54 kDa) and normalizing GAPDH bands at 37 kDa. The right-hand upper image shows representative sections of a 96 well plate from a LiCor on-cell determination of α5 plasma membrane subunit expression as a function of siRNA treatment.
Assay validation through screening of a kinase inhibitor library
The trafficking, desensitization and binding of ligandgated ion channels like nAChRs are subject to regulation by serine, threonine, and tyrosine kinases, and this type of phosphorylation plays a role in many pathologies including nicotine addiction 12, 33. Moreover, phosphorylation of the α5 subunit could provide a novel mechanism in which to regulate pentamer signaling. Therefore, because kinase inhibitors could provide tool compounds or become the basis of novel pharmacophores to regulate nAChRs in vivo, we screened a 96 compound kinase inhibitor library to assess the responses of the α3β4(α5) cell lines, to further validate the assay, and importantly to produce a kinase fingerprint for each of the subtypes (figure 5A). Compounds 22 through 32 serve as controls. Even compound numbers in this range correspond to nicotine only and odd numbered ones correspond to nicotine plus mecamylamine. The ability of mecamylamine to block the nicotine response is shown by an area contained within the rose shaded box. Compounds were designated as hits if they were able to inhibit the response to nicotine and fell outside three standard deviations of the mean response (blue shaded area). In order determine whether the kinase inhibitors could discriminate between the different nAChR forms, all three receptors were assessed in the primary calcium assay on the same day in parallel. The results, sorted by potency against the α3β4 nAChR, are presented as a heat map summarized in Table I. Hit compounds included inhibitors of PKC (tamoxifen), PKA (H-89), and tyrosine kinases (tyrphostin). What is evident from the initial portion of the table is the generally greater reduction in signaling for receptors without α5, and in some cases a complete lack of desensitization in the presence of inhibitor, for example with tyrphostin 25 or zaprinast. As a further validation of the primary assay, a dose response was performed on a representative sample of compounds showing activity in the primary screen (figure 5B).
Figure 5. Screening of a Kinase Inhibitor Library against Cell Lines Expressing nAChR.
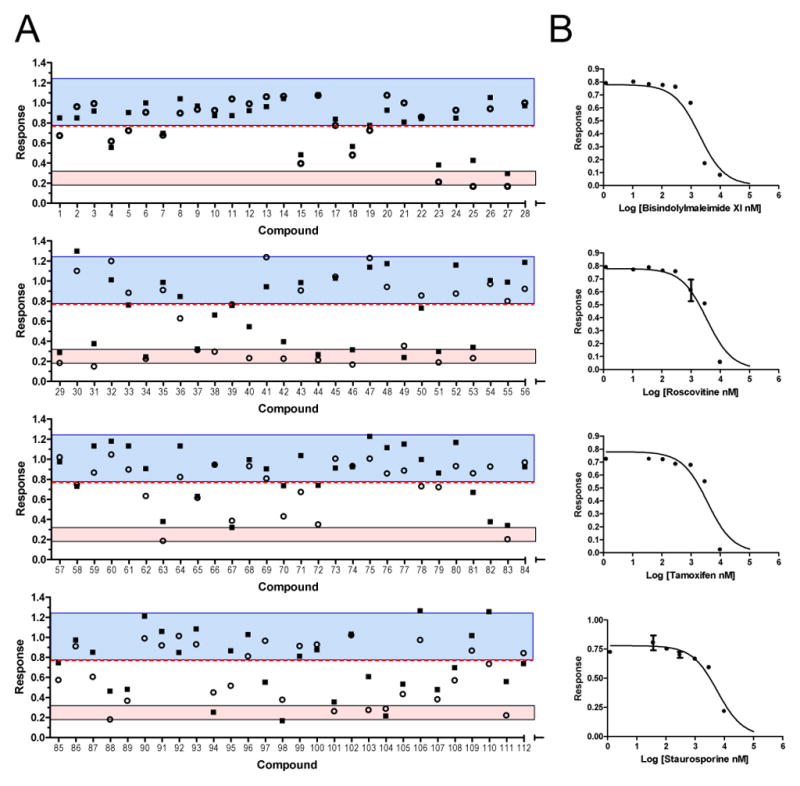
A. Cells were treated with 20 μM nicotine in the presence of 12.5 μM library compound and analyzed in the aequorin calcium assay as described above. The blue shaded area represents compounds lying within three standard deviations of the normalized mean response in the presence of nicotine plus vehicle (compounds 22, 24, 26, 28, 30, 32; mean ± std/1.00±0.07, n = 12). The red shaded area represents compounds within the 95% confidence interval of the nAChR antagonist mecamylamine (compounds 23, 25, 27, 29, 31; mean ± std/0.26±0.02, n=10). The square and circular points represent the results of two independent assays. Data were analyzed using GraphPad Prism ver. 5. B. The four panels are representative dose responses for triplicate wells in the primary calcium assay of a subset of hits from the primary screen that were chosen as controls for validation purposes. The IC50s for the 95% confidence intervals were: bisindolylmaleimide XI (0.81 to 3.1 μM), roscovitine (2.3 to 6.2 μM), tamoxifen (2.5 to 7.9 μM), and staurosporine (2.2 to 6.8 μM) (N = 3).
Table I.
Aequorin-Mediated Calcium Screen of Small Molecule Kinase Inhibitors
| α3β4 | α3β4α5 | -D398N | Compound | Inhibitor of | |
|---|---|---|---|---|---|
| 1.000 | 1.000 | 1.000 | nicotine | ||
| 0.002 | 0.578 | 0.640 | 10 | Nα-Tosyl-L-lysine chloromethyl ketone hydrochloride | Nuclear factor κB (NF-κB) |
| 0.003 | 1.103 | 0.848 | 12 | Tyrphostin 25 | EGFR tyrosine kinase inhibitor |
| 0.004 | 0.584 | 0.642 | 6 | Tyrphostin 1 | EGFR tyrosine kinase |
| 0.004 | 0.155 | 0.157 | 18 | Tamoxifen citrate salt | Protein kinase C |
| 0.006 | 0.277 | 0.180 | 2 | Tyrphostin AG 1433 | PDGFβ receptor tyrosine kinase |
| 0.009 | 0.143 | 0.324 | 4 | Tamoxifen | Protein kinase C |
| 0.010 | 0.157 | 0.130 | 15 | Tyrphostin SU 1498 | VEGF receptor kinase, Flk-1 |
| 0.014 | 1.298 | 1.189 | 21 | Zaprinast | cGMP-specific phosphodiesterases V and VI |
| 0.023 | 0.178 | 0.314 | 19 | U0126 monoethanolate | MEK1 and MEK2 |
| 0.023 | .826 | .217 | 58 | rac-2-Ethoxy-3-hexadecanamido-1-propylphosphocholine | Protein kinase C |
| 0.026 | 0.165 | 0.062 | 105 | Phloretin | Blocks L-type Ca2+ channels |
| 0.029 | 0.261 | 0.501 | 7 | Tyrphostin AG 490 | Jak-2 protein tyrosine kinase |
| 0.038 | 0.300 | 0.347 | 9 | Tyrphostin AG 1295 | Ttyrosine kinase in platelet-derived growth factor |
| 0.043 | 0.237 | 0.173 | 51 | ML-9 | Insulin-induced translocation of GLUT4 and GLUT1 |
| 0.045 | 0.405 | 0.588 | 8 | Tyrphostin 23 | EGFR tyrosine kinase |
| 0.048 | 0.738 | 0.684 | 13 | Tyrphostin AG 1478 | Epidermal growth factor receptor |
| 0.055 | 0.171 | 0.218 | 53 | Compound 52 | Cyclin-dependent kinases |
| 0.057 | 0.228 | 0.371 | 86 | Myricetin | Flavonol with antioxidant properties |
| 0.057 | 0.269 | 0.259 | 37 | (+)-Isocorydine | Inhibitor of eukaryote protein kinases |
| 0.058 | 0.145 | 0.313 | 36 | Butein | EGFR and Src tyrosine kinase |
| 0.058 | 0.308 | 0.188 | 67 | GF 109203X | Protein kinase C, glycogen synthase kinase-3 |
| 0.065 | 0.237 | 0.175 | 83 | ML-7 | Myosin light chain kinase |
| 0.074 | 0.362 | 0.250 | 89 | KN-92 | Negative control for KN-93 |
| 0.075 | 0.191 | 0.216 | 107 | Quercetin dihydrate | Mitochondrial ATPase and phosphodiesterase |
| 0.079 | 0.174 | 0.315 | 40 | Bisindolylmaleimide IV | Protein kinase C |
| 0.087 | 2.249 | 3.178 | 88 | rac-2-Methoxy-3-hexadecanamido-1-propylphosphocholine | Protein kinase C |
| 0.089 | 0.132 | 0.196 | 34 | H-89 dihydrochloride hydrate | cAMP-dependent protein kinase |
| 0.090 | 0.210 | 0.243 | 63 | Dequalinium chloride hydrate | Blocker of apamin-sensitive K+ channels |
| 0.092 | 0.278 | 0.313 | 104 | Roscovitine | Cyclin-dependent kinases |
| 0.094 | 0.393 | 0.430 | 11 | Tyrphostin AG 1296 | Platelet-derived growth factor (PDGF) receptor |
| 0.101 | 0.264 | 0.237 | 44 | Bisindolylmaleimide VI | Protein kinase C |
| 0.104 | 0.163 | 0.104 | 98 | Ro 32-0432 hydrochloride | GRK-5 (G protein-coupled receptor kinase) |
| 0.106 | 0.317 | 0.215 | 46 | Bisindolylmaleimide VII | Protein kinase C |
| 0.109 | 0.206 | 0.196 | 70 | Miltefosine | Protein kinase C, phosphatidylcholine synthesis |
| 0.113 | 0.154 | 0.253 | 78 | Indirubin-3′-oxime | Cyclin-dependent kinase inhibitor |
| 0.132 | 0.151 | 0.077 | 111 | Ro 31–8220 methanesulfonate salt | GRK-5; PKC; MAPKAP kinase 1β, and p70 S6 kinase |
| 0.134 | 0.197 | 0.266 | 50 | L-threo-Dihydrosphingosine | Sphingosine kinase inhibitor; protein kinase C alpha |
| 0.144 | 0.164 | 0.208 | 94 | NPC-15437 dihydrochloride hydrate | Protein kinase C |
| 0.149 | 0.242 | 0.207 | 103 | Palmitoyl-DL-carnitine chloride | Suppress the intracellular calcium signal transduction |
| 0.154 | 0.140 | 0.087 | 49 | Bisindolylmaleimide XI hydrochloride | Protein kinase C |
| 0.165 | 0.231 | 0.149 | 108 | Staurosporine from Streptomyces sp. | Phospholipid/calcium-dependent protein kinase |
| 0.166 | 0.813 | 0.814 | 17 | Tyrphostin AG 126 | Blocks production of TNF-α and NO in macrophages |
| 0.176 | 0.248 | 0.189 | 48 | Bisindolylmaleimide X hydrochloride | Protein kinase C |
| 0.176 | 0.386 | 0.294 | 100 | Radicicol Diheterospora chlamydosporia | Protein tyrosine kinase |
| 0.218 | 0.821 | 0.727 | 20 | Wortmannin | Protein kinase C |
| 0.225 | 0.261 | 0.171 | 38 | Bisindolylmaleimide II | Protein kinase C |
| 0.233 | 0.191 | 0.251 | 72 | (Z)-4-Hydroxytamoxifen | Mtabolite of tamoxifen |
| 0.305 | 0.212 | 0.211 | 82 | LY-294,002 hydrochloride | Cell permeable phosphatidylinositol 3-kinase |
| 0.312 | 0.287 | 0.207 | 112 | SB 203580 | MAPKAP kinase-2 |
| 0.316 | 1.159 | 0.393 | 73 | Genistein | Ttyrosine protein kinase |
| 0.428 | 0.448 | 0.405 | 85 | H-7 | PKA) and protein kinase C |
| 0.494 | 0.324 | 0.344 | 42 | Bisindolylmaleimide V | Negative control for protein kinase C-inhibitory activity |
| 0.495 | 0.504 | 4.086 | 74 | Hypocrellin A | Protein kinase C |
| 0.556 | 0.403 | 0.457 | 80 | N9-Isopropylolomoucine | Cyclin-dependent kinases |
| 0.596 | 0.453 | 0.641 | 3 | Tyrphostin AG 879 | Receptor tyrosine kinase |
| 0.607 | 0.196 | 0.395 | 1 | Tyrphostin A9 | Calcium release-activated calcium (CRAC) channels |
| 0.611 | 1.187 | 0.864 | 55 | Chelerythrine chloride | Translocation of PKC from cytosol to plasma membrane |
| 0.675 | 0.300 | 0.348 | 95 | Leflunomide | Immunosuppressive |
| 0.700 | 0.373 | 0.510 | 106 | DL-Stearoylcarnitine chloride | |
| 0.752 | 0.201 | 0.130 | 60 | rac-2-Ethoxy-3-octadecanamido-1-propylphosphocholine | Protein kinase C |
| 0.770 | 0.479 | 0.537 | 79 | HA-1077 dihydrochloride | Intracellular Ca2+ antagonist |
| 0.783 | 0.227 | 0.391 | 62 | Emodin | NF-κB activation and Casein Kinase 2 (CK2) |
| 0.794 | 0.813 | 1.129 | 16 | Tyrphostin 51 | EGFR tyrosine kinase |
| 0.833 | 1.026 | 0.817 | 39 | Rp-Adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt hydrate | Blocks cAMP-mediated effects |
| 0.850 | 0.488 | 0.509 | 81 | HA-100 | Inhibitor of PKA, PKC and myosin light chain kinase |
| 0.856 | 0.989 | 0.931 | 5 | D-α-Tocopherol succinate | Vitamin E supplement |
| 0.867 | 0.361 | 0.443 | 97 | Piceatannol | Kinases Syk and Lck |
| 0.870 | 0.567 | 0.790 | 43 | Apigenin | Arreststhe cell cycle at the G2/M phase |
| 0.873 | 0.680 | 0.718 | 68 | Hispidin | Protein kinase Cβ |
| 0.890 | 0.615 | 0.730 | 101 | Purvalanol A | Cyclin-dependent protein kinase (cdk) inhibitor |
| 0.895 | 0.456 | 0.199 | 109 | Rapamycin | Molecular target of rapamycin (mTOR) |
| 0.899 | 0.660 | 0.619 | 87 | H-7 dihydrochloride | PKA and protein kinase C |
| 0.902 | 0.517 | 0.538 | 110 | D-Sphingosine | Protein kinase C |
| 0.915 | 0.789 | 0.646 | 93 | Lavendustin A | Cell-permeable tyrosine kinase inhibitor |
| 0.930 | 1.072 | 1.028 | 14 | Tyrphostin 47 | EGFR tyrosine kinase inhibitor |
| 0.932 | 0.611 | 0.577 | 54 | Daidzein | A phytoestrogen |
| 0.957 | 0.950 | 0.541 | 75 | GW8510 | Cyclin kinase 2 |
| 0.962 | 0.763 | 0.491 | 76 | Hypocrellin B | Protein kinase C |
| 0.966 | 1.021 | 0.859 | 35 | 5-Iodo-2′-deoxyuridine | Thymidine kinase and thymidylate synthetase |
| 0.986 | 0.581 | 0.552 | 65 | HA-1004 hydrochloride | Intracellular calcium antagonist |
| 0.988 | 0.996 | 0.854 | 33 | 5-(2,5-Dihydroxybenzylamino)-2-hydroxybenzoic acid | Tyrosine kinase inhibitor |
| 0.999 | 0.825 | 0.660 | 84 | Melittin from honey bee venom | Inhibits Na+-K+-ATPase |
| 1.009 | 1.069 | 0.879 | 41 | A-134974 dihydrochloride hydrate | Adenosine kinase |
| 1.027 | 0.969 | 0.790 | 66 | Rp-8-Hexylaminoadenosine 3′,5′-monophosphorothioate | cAMP-dependent protein kinase |
| 1.041 | 1.833 | 0.912 | 90 | rac-2-Methoxy-3-octadecanamido-1-propylphosphocholine | Protein kinase C |
| 1.050 | 0.892 | 0.934 | 59 | 2,5-Dihydroxycinnamic acid methyl ester | EGF receptor-associated tyrosine kinase |
| 1.057 | 0.952 | 0.720 | 99 | PD 98,059 | Mitogen-activated protein kinase kinase |
| 1.066 | 1.138 | 0.893 | 91 | Lavendustin C | Protein tyrosine kinases |
| 1.069 | 0.960 | 0.832 | 71 | GW5074 | cRaf1 kinase |
| 1.073 | 0.725 | 0.733 | 96 | Olomoucine | Cyclin-dependent kinases and induces G arrest |
| 1.076 | 1.196 | 1.025 | 56 | Ellagic acid | Glutathione S-transferase |
| 1.081 | 0.924 | 0.841 | 45 | Amiloride hydrochloride hydrate | T-type calcium channel blocker |
| 1.089 | 0.741 | 0.598 | 69 | Geldanamycin Streptomyces hygroscopicus | Potent antitumor antibiotic |
| 1.099 | 0.925 | 0.918 | 52 | 7,8-Dihydroxycoumarin | Protein kinases |
| 1.105 | 0.970 | 0.780 | 64 | Genistin | Inactive analog of genistein |
| 1.106 | 1.490 | 1.156 | 57 | 8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt | Membrane permeable cAMP analog |
| 1.109 | 1.239 | 0.833 | 102 | Rottlerin | Activator large conduct volt and Ca activated K+ channel |
| 1.113 | 0.968 | 0.871 | 92 | H-8 | cAMP and cGMP-dependent protein kinase |
| 1.116 | 0.931 | 0.840 | 77 | H-9 dihydrochloride | Protein kinase C, cAMP and cGMP depend prot kinases |
| 1.125 | 1.093 | 1.204 | 47 | Adenosine | Endogenous neurotransmitter at adenosine receptors |
| 1.129 | 0.688 | 0.749 | 61 | D-erythro-Dihydrosphingosine | Protein kinase C |
Color Key

Primary screen against a nAChR directed library to identify biased compounds
We adopted a repurposing strategy similar to that described by Chong and Sullivan 34. We had Specs Chemicals assemble a directed library of 275 potential nAChR antagonists from an algorithm to identify likely candidates. The results of that screen are presented as supplemental data figure 1S-A. Compounds were binned for agonist and antagonist activity versus nicotine exposure of the different subunit combinations as above and also organized as a color map in order to more easily identify compounds that discriminated between the α3β4 and the presence of the α5 variants (supplemental figure 1S-B). Thirty-eight leads selected for further study on the basis of their differential responses with receptors containing α5 subunits were reevaluated in the aequorin calcium assay over 3 logs of concentration using a 6 point dose response (figure 6 and supplemental figure 2S). Approximately ¾ of them failed to discriminate between the different receptor variants in the secondary screen (figure 6A and supplemental figure 2S) whereas eight compounds demonstrated an ability to distinguish between the α3β4 and one of the α5 forms α3β4α5 or α3β4α5-D398N (figure 6B). In particular, compounds 6, 8, 9 exhibited a differential response between receptors containing the α5 and α5-D398N.
Figure 6. Screening of a Directed Library against Cell Lines Expressing α3β4, α3β4α5 and α3β4α5D398N nAChRs.


Antagonist assays for compounds from the directed Specs Library that were considered hits in the primary screen were performed as described. Dose responses were calculated from triplicate wells in three independent experiments to determine the effects of compounds on inhibition of the whole cell calcium mediated nicotine response (100 μM) in HEK-293 cell-lines permanently transfected with one of the nAChRs. Data were analyzed using GraphPad Prism 5.0 and the results for the IC50’s and Efficacies (indicated as Bottom of the curve) are presented as 95% confidence intervals. Solid black line is the α3β4-nAChR, solid red line is the α3β4α5-nAChR and the solid green line represents the α3β4α5D398N-nAChR A. Representative compounds from the secondary screen that showed antagonist activity but that did not show any distinct bias for at least one of the three receptor subtypes. B. Compounds that displayed bias or preference in antagonizing the signaling of at least one of the receptor subtypes when compared to the other two.
All eight of the nonselective compounds in figure 6A are 2-bromo-5-substituted furans, as are six of the eight in figure 6B, with the exception of compounds 9 and 17. The furan compounds predominantly have an N-substituted carbamoyl group in the 5-position of the furan ring, with the exception of compounds 10 and 34 that have an alkoxycarbonyl group. Compounds 9 and 17, which can distinguish between α3β4 and one of the α5 forms and which also appear easier to synthesize than the other fourteen compounds, are also quite different from them. Compound 9 is a tertiary amine having two N-methyl groups and a large lypophilic third N-substituent. Compound 17, a quaternary salt, is a piperidine having two large lypophile N-substituents and is the only quaternary compound in this group of 16 hits. The concentrations of these leads that produce half-maximal responses in antagonist as-says are in the low to sub-micromolar range (see accompanying data with figure in 6B, for instance compound 9).
Assessment of compound 9 activity in secondary functional assays
Compound 9 was evaluated for its ability to affect α3β4 and α3β4α5 upregulation by 0.5 mM nicotine (fig. 7A, B). It was compared to two compounds that were both active in the kinase inhibitor library screen, the PKC inhibitor bisindolylmaleimide (Bis) XI and the phosphodiesterase inhibitor zaprinast, the parent compound of sildenafil (Viagra). Each compound alone had no effect on the expression of either receptor subtype (legend fig. 7). However, in the presence of nicotine Bis XI was able to reverse the increased surface expression of the receptors due to nicotine exposure (fig. 7A), whereas both the zaprinast and compound 9 acted similarly and had no effect on upregulated receptor expression (fig. 7B). In contrast, compound 9 and zaprinast demonstrated activity in dopamine transporter knockout mice (DAT-KO), a model for increased brain dopaminergic tone 35. When administered i.p., they reduced the typical hyperlocomotion behavior associated with the DAT-KO phenotype by approximately one-half and 1/3 respectively (fig. 7C).
Figure 7. Secondary Screening of Representative Hit Compounds.
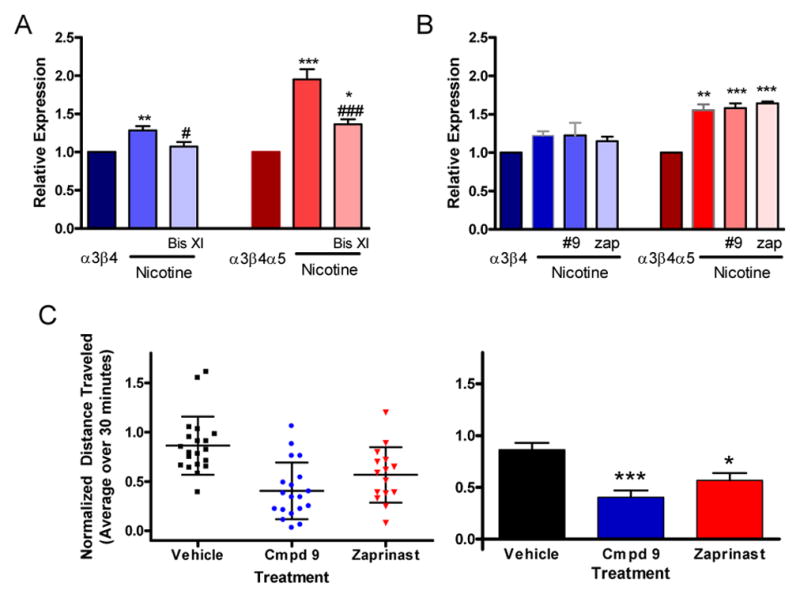
Relative expression of α3β4 and α3β4α5 nAChRs upon upregulation by 0.5 mM nicotine, and (A) 0.5 mM nicotine plus 1.0 μM bisindolylmaleimide XI or (B) 0.5 mM nicotine plus 10 μM compound 9 and 0.5 mM nicotine plus 10 μM zaprinast. Results expressed as mean ± sem were normalized to the surface expression of the respective vehicle treated parent receptor and assessed by repeated measure anova. A. α3β4 plus vehicle = 1, plus nicotine alone (1.28 ± 0.057) and with nicotine plus Bis XI (1.07 ± 0.059), vs. α3β4 ** p < 0.01, vs. α3β4 plus nicotine # p < 0.05; α3β4α5 plus vehicle = 1, plus nicotine alone (2.05 ± 0.16) and with nicotine plus Bis XI (1.36 ± 0.065), vs. α3β4α5 * p < 0.05 and *** p < 0.001, vs. α3β4α5 plus nicotine ### p < 0.001, (N = 6) B. α3β4 plus vehicle = 1, plus nicotine alone (1.22 ± 0.053) and with nicotine plus #9 (1.23 ± 0.16), plus nicotine plus zap (1.16 ± 0.06); α3β4α5 plus vehicle = 1, plus nicotine alone (1.56 ± 0.076) and with nicotine plus #9 (1.58 ± 0.063), plus nicotine plus zap (1.64 ± 0.025), vs. α3β4α5 ** p < 0.01 and *** p < 0.001, (N = 3). The compounds added alone at the same concentration as specified above without nicotine present had no effect on receptor expression upregulation, α3β4 plus vehicle = 1, plus #9 (1.05 ± 0.10), plus zap (1.03 ± 0.03); α3β4α5 plus vehicle = 1, plus #9 (0.97 ± 0.04), plus zap (0.95 ± 0.01), (N = 3). Data were analyzed by repeated measures anova with Tukey’s multiple comparison test C. Scatter plots of experimental values (left image) and bar graphs summarizing these results (right image) of locomotion data for DAT-KO mice. The plots show relative average locomotion for the 30 minute period following the various treatments. Values for each condition are normalized by the pre-treatment average of that group for 20 minutes prior to injection.. Error bars in the scatter plot represent mean ± std. In the column graph the error bars represent mean ± sem and these results are: for the vehicle group (0.86 ± 0.066, N = 20), for compound 9 (0.40 ± 0.066, N = 19), and for zaprinast (0.57 ± 0.073, N = 15). Data were analyzed by one way anova with Tukey’s multiple comparison test, * p < 0.05 and *** p < 0.001.
Discussion
In this study we describe model cell systems expressing homogeneous populations of (α3)2(β4)3 and (α3)2(β4)2(α5) receptors practical for drug discovery and biochemical characterization, determine α5 subunit regulation of their signaling and expression profiles, generate a kinase inhibitor fingerprint for them, and identify cognate antagonists of them as proof-of-principle that small molecule inhibitors can discriminate between the different α3β4(α5) pentamers. Considerable previous effort has gone into identifying compounds targeting the relatively large number of nAChR subunit combinations because of their physiological importance and relationship to nicotine addiction and cigarette smoking. By screening a small but focused nAChR library, we relatively quickly found candidate compounds that exhibit selectivity in inhibiting different variations of α3β4 and α3β4α5 based pentamers, supporting the repurposing approach suggested by Chong and Sullivan in their seminal article concerning drug screening 34.
Some common structural features shared by ligand gated ion channels and G protein coupled receptors, for example trans-membrane domains bounded by extracellular determinants, influenced our approach in modifying the nAChR subtypes and in developing a high throughput screen for them. Ideally, α3β4 pentameters should express sufficiently well in model cell systems so that plasma membrane expression level is not a limiting factor preventing their characterization or use in signaling assays. Identifying plasma membrane α3β4 subunits at levels commonly observed with stably expressed GPCRs, however, has been problematic, and a lack of reliable antibodies that recognize properly incorporated receptor subunits in the mature receptors may be a contributing factor 36. Additionally, despite reports of nAChRs successfully labeled with fluorescent proteins in their intracellular loops 37, we failed to develop sufficient, functional plasma membrane pentamers whose identity or plasma membrane expression could be validated using this strategy. In contrast, the solution of inserting distinct, short, high affinity epitopes following cleavable signal sequences of the individual subunits produced functional receptors for Ca2+ signaling whose identities and locations were easily determined using commercially available and proven anti c-MYC, HA, and V5 antibodies.
Population inhomogeneity remains a limiting factor in characterizing nAChRs. Fortunately, the composition of expressed nAChRs may be highly constrained despite the large number of available types of subunits 38. Commonly expressed forms for mature α3β4 pentamers have been reported as two α3 and three β4 per pentamer and three α3 and two β4 39–41. In Xenopus oocytes, which form predominates depends on the relative amounts of injected mRNA corresponding to the different subunits 39. Difficulties in obtaining homogeneous, functional receptor populations has led to work-arounds including designing receptor concatamers that express in Xenopus at low levels but are responsive in electro-physiology measurements 16, 42. This concatamer strategy also failed as we were unable to express visible amounts of functional plasma membrane α3β4 pentamers, despite western blots demonstrating that the protein was being made. Even though HEK-293 cells will express varying ratios of α3 and β4 subunits after transient transfection 39, it was unexpected that our stably transfected lines are upwards of 98% homogeneous in the expression of only two α3 subunits per pentamer as determined by mass spectrometry. This ratio of α3 expression also occurs in our cell clones with an average replacement in approximately one-half of the pentamers of one β4 subunit by one α5 subunit, in agreement with Xenopus observations 43. Moreover, the limited knockdown of surface α5 to 0.74 in the on-cell ELISA of figure 4E compared to the much greater α5 knockdown in the whole cell western to 0.31 suggests that the plasma membrane expression of α3β4α5 subtype is more than two fold ( ) preferred over the α3β4.
Studies show that exposure to nicotine upregulates the number of α4β2 nAChRs in distinct areas of the brain, and in rat models the addition of the α5 subunit to either this subtype or to the α3β4 subtype makes it more resistant to upregulation 38. The mechanism employed in either case remains unclear, but for α3β4 nicotine induced increases may be a consequence of increased protein synthesis, receptor stability, and trafficking of assembled receptors to the plasma membrane 38, 40. Increases in the membrane expression of GPCRs like the dopamine receptor also occur in the presence of cell permeant ligands presumably through chaperoning, and the mechanisms responsible here may be similar 44. Our data in figure 2 indicate that addition of either the α5 or α5-D398N does not prevent the upregulation of α3β4 surface receptor by nicotine.
We observed that nAChR cell clones containing stably expressed α5 subunits lose mature receptor expression over time. This plasma membrane down regulation suggests a role for the α5 in limiting channel activity. Supporting this notion are cell studies by Tammimaki et al 45 and our siRNA α5 knockdown observations (figure 4) that the reduction of calcium flux coinciding with α5 expression can be reversed by α5 suppression. The data additionally suggest that the D398N subunit may be better than wild type α5 in reducing the calcium flux. However, our experiments do not distinguish the relative contributions in changing calcium flux between a direct suppressive role for α5 subunits or whether a reduction of α5 subunits simply allows better incorporation of β4 subunits to maintain the calcium response.
Src and tyrosine kinases regulate nAChRs, but the functional consequences of phosphorylation may vary between receptors and even kinase members of the same family 46. The amount of nicotine exposure in one cigarette is sufficient to desensitize nAChRs and will affect diverse aspects of smoking involving withdrawal and reinforcement of environmental cues, indicating that nicotine can reward smokers by mechanisms that involve both activating and desensitizing nAChRs (for a comprehensive review see Picciotto et al 47). PKC phosphorylation of α4β2 nAChRs can promote recovery from nicotine induced desensitization 12, 48. Our finding that the PKC inhibitor Bis XI blocks nicotine mediated upregulation is consistent with their observation. Additionally, our kinase inhibitor data for the α3β4 in Table I support phosphorylation as a mechanism of α3β4 receptor recovery. For example, the data show a lack of α3β4 calcium signaling in the presence of the tyrosine phosphorylation inhibitors tyrphostin 1 and tyrphostin 25, PKC propylphosphocholine compound inhibitors, and the PI3 kinase inhibitor wortmanin. Interestingly, the α5 subunit and the α5-D398N may have a role in maintaining nicotine signaling because in many instances as outlined in Table I signaling of the α3β4α5 is maintained versus the α3β4. Thus, in contrast to its role in reducing expression of the nAChR pentamer, the α5 subunit may act by an alternative allosteric mechanism to preserve the ability of α3β4α5 pentamers to recover from sustained nicotine exposure.
The results of screening the different nAChRs with a directed nAChR antagonist library may provide a second example of allosteric modulation of receptor pentamers. The primary screen identified eight lead compounds using receptor subtype-based calcium flux assays. The most potent lead, compound 9, exhibited sub-micromolar activity and a 75% efficacy difference between the α3β4 and α3β4α5-D398N isotypes. To further investigate their properties, we searched the PubChem, Molecular Libraries database and found that compound 9 was the only one of the eight that had received any secondary screening validation in the Molecular Libraries program. Compound 9 was listed as a weak antagonist of the Kir2.1 inward rectifying potassium channel (PubChem 23723102, AID 743120). If these eight ligands directly bind the acetylcholine/nicotine binding sites at the two α3/β4 sub-unit interfaces, then the most straightforward explanation why their response profiles can differentiate between receptor pentamers lacking α5 subunits, or containing α5 or α5-D398N subunits, is that binding site conformations depend on the identity of the subunit occupying the fifth position. Moreover, while the DAT-KO studies cannot alone prove compound 9 has a mechanism of action solely involving α3β4 and α3β4α5 receptors in vivo, the results provide proof-of-concept support for this subtype-based cell strategy to identify drugs with a potential to treat nicotine-related behaviors.
These compound response profiles together with the kinase inhibitor results indicate that signaling differences between the α3β4 and α3β4α5 receptor subtypes can be pharmacologically realized on the basis of allosteric changes to the binding sites that are sequence dependent or that occur as the result of post translational modifications of regulatory phosphorylation sites in the subunit cytosolic loops. For treating nicotine addiction, modulating the conformations of α3β4α5 nAChRs and consequently their interactions with nicotine by use of non-selective kinase inhibitors in preference to selective nicotine binding site antagonists may at first seem counterintuitive. However, we propose that a strategy of targeting post translational nicotine regulatory sites either expressed or modulated by α5 subunits may provide substantial advantages over ligands that compete nicotine because it could repurpose drugs approved for treating other diseases including cancer. Moreover, this notion is testable in animal models using addiction-related behavioral paradigms.
Methods
Materials
Human cDNAs for CHRNA3 (BC001642), CHRNB4 (BC096080.1), and CHRNA5 (BC033639.1) were obtained from Open Biosystems (Thermo Fisher Scientific Pittsburg, PA), Plasmids pcDNA3.1 Hygro and pcDNA3.1 Zeo, and the reagents Lipofectamine 2000, Zeocin, G418, hygromycin, MEM, and HEPES were obtained from Life Technologies (Thermo Fisher Scientific). Plasmid EGFP-N3 was from Takara/Clontech (Mountain View, CA). Coelenterazine H was obtained from Promega (Madison, WI). Nicotine-ditatrate was obtained from Tocris, (Minneapolis, MN). Mecamylamine was purchased from Sigma (St Louis, MO). Tritiated-epibatidine, #NET1102, was purchased from Perkin Elmer (Waltham, MA). The Ambion Silencer siRNA, #AM4613, and CHRNA5 siRNA, ID118985; AM51331, were obtained from Ambion/Thermofisher. Mouse monoclonal anti-HA antibody (clone 12CA5) was purchased from Sigma, rabbit polyclonal anti-MYC (#9106) from Ab-Cam (Cambridge, MA), mouse monoclonal anti-V5 (#46-0705) from Life technologies, and Alexa Flour secondary antibodies from Molecular Probes/Thermo Fisher.
Cloning of Nicotinic Acetylcholine Receptors
An HA epitope tag (YPYDVPDYA) was added after the CHRNA3 N-terminal signal sequence and the product was sub-cloned with stop codon intact into an EGFP-N3 plasmid containing a Neomycin selection cassette. The CHRNB4 expression plasmid with a MYC epitope tag (EQKLISEEDL) after the N terminus signal sequence was sub-cloned into pcDNA3.1 ZEO. A CHRNA5 expression plasmid was constructed with a V5 epitope tag (GKPIPNPLLGLDST) after the signal sequence and sub-cloned into pcDNA3.1 Hygro. CHRNA5-D398N was constructed using a Quick Change Kit from Agilent Technology (Cary, NC) by site-directed mutagenesis with the following primers, 5′-cacattggaagctgcgctcaattctattcgctacattac and 3′-gtaatgtagcgaatagaattgagcgcagcttccaatgtg.
An Aequorin Reporter Cell Line of Calcium Activity
An aequorin expression plasmid was constructed by inserting the MYC epitope tag onto the N-terminus of the mitochondrial-targeting Apo-aequorin expression vector p47, (a gift of Dr. Stanely Thayer, Department of Pharmacology, and University of Minnesota) and sub-cloned into pcDNA 3.1 Puro. HEK-293 cells were transfected with the plasmid using Lipofectamine 2000 according to the manufacturer’s protocol, and a stable cell line selected using puromycin at 2 μg/ml. These cells were maintained in DMEM with high glucose supplemented with 10% FCS and 2 μg/ml of puromycin.
Stable Cell Lines Containing nAChR subtypes
Cells were transfected with plasmids using Lipofectamine 2000 as above. α3β4 receptor expressing cells were selected with 50 μg/ml Zeocin, 250 μg/ml G418, and 2 μg/ml puromycin; and α3β4α5 cells were derived from the α3β4 parent line with 400 μg/ml hygromycin. Ten days post selection distinct receptor clones were plated into separate wells of 96 well plates by limiting dilutions. Clones were screened for ligand binding with 3H-epibatidine as follows: 2x105 cells of each clone or control HEK-293 cells were seeded into 24 well plates. The following day the media was replaced with 0.5 ml of clear MEM-HEPES containing 5 nM 3H-epibatidine, the cells were incubated at room temperature for 15 minutes, then washed twice with clear MEM-HEPES, lysed on ice for 15 minutes with 50 μl of 0.1 N NaOH, and the suspension neutralized with 50 μl of 0.1 N HCl. Neutralized lysates were added to vial containing 2 ml of Biosafe II scintillation fluid and counted. Cell lines expressing the wild type clone of α3β4α5 (clone B8) and the mutant clone α3β4α5-D398N (clone E6 ) were sorted by FACS analysis using a Beckman Coulter MoFlo Astrios cell sorter for expression of the three epitope tags that were labeled with primary antibodies against the respective epitopes. Cells were further sub-cloned based on their cell surface expression of the V5 epitope determined by On-Cell ELISA assay, below and as described in 49.
On-Cell ELISA Assay
Individual wells of 96 well, poly-D-lysine treated plates seeded with 3x104 nAChR-expressing cells were stained on ice for 45 minutes with a 1/2000 dilution of a primary anti-epitope antibody in clear MEM/HEPES/2% FBS, washed 3X with cold HHBS, fixed for 15 minutes at room temperature with 4% paraformaldehyde in PBS, and rewashed. Prior to secondary antibody treatment the cells were exposed for 20 minutes at room temperature to a 1/50,0000 dilution in PBS of the general cell stain IRDye 800CW (LiCor, Lincoln, NE ) in order to enable immunofluorescence data to be normalized by cell number. The cells were next washed, treated at room temperature for 45 minutes with a 1/2000 dilution of secondary antibody 680 (Molecular Probes/Thermo Fisher ), rewashed 3x, placed in PBS, and imaged on a LI-COR Odyssey with the 700-nm and 800 nm channels using a LI-COR focal offset setting of 1.5. HEK293 stained control cells were used to determine a background signal and background corrected data were normalized by the IRDye 800CW staining determined in the 800 nm channel.
Fluorescence Imaging of Surface nAChRs
Cells were plated onto fibronectin coated 35 mM glass bottomed dishes (#P35G-0-10-C, MatTek Corporation, Ashland, MA) stained live on ice for 45 minutes with a 1/500 dilution of primary antibody in clear MEM/HEPES/5% BSA, washed 3 times with cold HBSS, fixed with 4% paraformaldehyde in room temperature PBS for 15 minutes, washed 3 times in HBSS, and counterstained for 45 minutes at room temperature with a 1/500 dilution of secondary antibody in MEM/HEPES/5% BSA. Primary and secondary antibody pairs were: CHRNA3, mouse monoclonal anti HA antibody and Alexa Fluor goat anti mouse 488; CHRNB4, rabbit polyclonal anti MYC and Alexa Fluor goat anti rabbit 568; CHRNA5, mouse monoclonal anti V5 and AlexaFluor goat anti mouse 568. Images were acquired with a Zeiss LSM510 microscope using a PlanApo, 40X, 1.4 NA oil-objective.
Determination of Receptor Stoichiometry by Mass Spectrometry
For each determination of receptor expression, 4x107 cells were washed in ice cold PBS, and with mild rocking they were surface-treated with Sulfo-NHS-Biotin (kit #89881, Pierce/ThermoFisher) for 30 minutes at 4°C 40. The biotin conjugation reaction was quenched and the cells lysed using the proprietary kit reagents per the kit protocol in buffer also containing 1X protease inhibitors (#11873580001, Roche Indianapolis, IN). The cells were then sonicated on ice with a Fisher Scientific 550 Sonic Dismembrator using 5 one second bursts at 30% power, incubated for 30 minutes on ice, centrifuged at 10,000 x g for 2 minutes, and the biotin-labeled surface proteins were recovered using a NeutrAvidin gel provided with the kit. The enriched biotinylated proteins were eluted at 95°C for 5 minutes in SDS PAGE sample buffer containing 50 mM DTT and protease inhibitors.
Targeted Liquid Chromatography Mass Spectrometry with Multiple Reaction Monitoring for nAChR Subunit Stoichiometry
SDS-PAGE separation of receptor enriched samples was performed and gel bands corresponding to the entire molecular weight region of nAChR subunits α3, β4, and α5 (approximately 45 kDa to 55 kDa) were excised and subjected to an ingel trypsin digestion at 37°C for 18 hours. Extracted peptides were split into two aliquots and lyophilized to dryness. Each aliquot was then resuspended in 2% acetonitrile/0.1% formic acid containing either 1.66 fmol/μL (5 fmol total on column) or 16.6 fmol/ μL (50 fmol total on column) stable-isotope labeled (SIL) SpikeTide TQL peptides (C13/N15 at C-terminal Arg or Lys residues, JPT Corporation, Germany) corresponding to two unique peptides within subunit α3, β4, and α5 protein sequences. Injections of 3 μL of each sample were acquired in triplicate on a Waters NanoAcquity UPLC equipped with a 75 um x 150 mm BEH C15 1.8 um column running a linear gradient of 5% to 40% acetonitrile/0.1% for-mic acid over 30 min at 400 nL/min at 55°C. Eluting peptides were analyzed on a Waters Xevo TQ-S triple quadrupole mass spectrometer through an electrospray ionization interface operating at 2.5 kV. The instrument was operated in a positive ionization targeted MRM mode with a cone voltage of 35.0 V, a charge state dependent normalized collision energy of 30–42, and the auto-dwell time feature set to acquire at least 10 points across a 20s peak width. Retention time windows were set to 4 min and with 4 unique transitions chose per targeted precursor ion. Calculation of measured MRM ratios between endogenous and SIL spiked standards was performed within the Skyline (MacCoss Laboratory, University of Washington) software package. Endogenous peptide quantities were determined by the calculated slope between triplicate high (50 fmol) and triplicate low (5 fmol) SIL peptide quantities. Endogenous peptide fmol quantities were then calculated based on fitting the measured intensity to the corresponding SIL peptide slope. Protein molar stoichiometry was determined based on the lowest empirical stoichiometry of the protein components within that particular sample.
Determining fractional expression per pentamer of nAChR subunits
The relative amounts of the α3, β4, and α5 subunits were determined by linear regression as follows. Each separate experiment j defined a point ( ) representing the subunit expressions. For Nj receptor pentamers there are 5Nj total components represented by the and subunits and the subunits have experimental fractional expressions per pentamer of , and . A regression line that best fits all the points ( ) goes through the origin since when Nj = 0. Therefore each term should provide an estimate of the regression line slope, defined as slopereg, where . Solving for the fractional expressions and using the relationships and we find for each experiment and , and by fitting that and .
Probability of occurrence of the α3 subunit
The probability of occurrence pα3 of an α3 sub-unit in a formed pentamer is . Let p2α3 and p3α3 be the respective probabilities of incorporating either 2 or 3 α3 subunits in the pentamer, those two forms being the only alternatives that we consider possible. Then p2α3 + p3α3 and the expectation value (fractional expression) pα3 · 5 = f3 = p2α3 · 2 + p3α3 ·3 so that . The probability of occurrence in a pentamer of either a β4 or α5 subunit is . Thus, for the α5 subunit not being a replacement for the α3, the relative probability of an α5 subunit replacing a β4 subunit is . Let p0α5 and p1α5 be the respective probabilities of incorporating either 0 or 1 α5 subunits by replacing a β4 subunit, those two forms being the only alternatives that we consider possible. Then p0α5 + p1α5 = 1 and the expectation value (fractional expression) is: so that .
Aequorin-Based Whole Cell Calcium Assay
In stable cell lines the intracellular calcium activation of plasma membrane nAChRs was measured using mitochondrial localized aequorin as described 32. Two to three million cells were first plated in 60 or 100 mm tissue culture dishes in standard media. Two days later the cell media was replaced with 2–4 ml DMEM (a 60 or 100 mm dish respectively) containing 10% serum and 2.5 μM Coelenterazine H. The cells were incubated 2–3 hours at 37°C, and gently scraped and transferred to 15 ml tubes where an equal volume of HBSS containing and excess of 100 mM calcium chloride was added for 10 minutes at room temperature. This brief calcium incubation eliminates a nonspecific calcium spike associated with calcium diffusion into non-viable cells during the start of the assay. The cells were next pelleted by centrifugation at 4°C, washed with cold assay buffer (13 mM NaCl, 5.4 mM KCl, 10 mM CaCl2, 5 mM glucose, 25 mM HEPES at pH 7.5), suspended in 4–6 ml of fresh assay buffer, and held at 4°C until use.
Agonist Measurements
Agonists were diluted in assay buffer and 50 μL aliquots were added to each well of a 96 well plate with opaque walls. Luminescence was measured using a Berthold LB940 plate reader with multiple injection ports. For calcium flux measurements, 50 μL of stirred cells that were protected from light were injected into a well and readings were obtained at 1 second intervals for 10–15 seconds. After reading a plate the remaining luminescent signal for each well was determined by injecting 100 μL of lysis buffer (0.1% Triton X-100 in deionized water with 100 mM CaCl2) into each well and recording for 4 seconds. Results were normalized as the (peak agonist-induced signal)/ (peak agonist induced signal + peak cell-lysis signal). A Z factor between 0.46–0.76 was obtained for the 96/384 well plate assay using nicotine (60 μM) as the control. DMSO had no effect on the assay when present between 0–1.6 %.
Kinase Library Screen Agonist Assay
Measurements were conducted using 96 well plates, and compounds were tested at 30 μM final concentration for agonist activity as above in a 0.2% DMSO/Ca assay buffer and with 30 μL of cells.
Kinase Library Screen Antagonist Assay
The plates with compound were first read and the compounds were then allowed to incubate with the cells for a further 25 minutes at room temperature before 30 μL of nicotine was added to a final concentration of 20 μM for α3β4 cells and 60 μM for α3β4α5 cells. Positive controls contained only assay buffer in 1% DMSO and negative controls also contained 20 μM of the non-selective, non-competitive nAChR antagonist mecamylamine in 1% DMSO. Following these measurements the cells were lysed as above for normalization of the signals.
Directed Specs-Library Screen of Lead Compounds
Potential lead compounds were selected by Specs Chemicals based on predicted agonist and antagonist activity against nAChRs using their proprietary software and the algorithm (PASS, version 1.41 professional) applied to their screening libraries containing over five-million compounds. The group contained 266 potential antagonist compounds and 11 potential agonist compounds. Compounds were provided as 10 mM stocks in DMSO and screened as above for agonist activity and antagonist activity at a concentration of 33 μM.
Knockdown of α5 Receptor Subunits by siRNA expression
A day before transfection, 100 mM dishes were plated with approximately 5 x 106 cells each and the following day they were transfected with RNAi MAX (#13778-075, Life technologies/ Thermo Fisher Scientific) using the manufacturers protocol. Plates received either of the following: no RNA, 10 pmol of negative control, or 10 pmol siRNA specific to the α5 subunit. The cells were incubated for 48 hours, and then assayed for calcium activity as described. The signal from the secondary antibody in the on-cell assay was measured as above on a LiCor Odyssey reader.
SDS PAGE of α5 Knockdown Cells
Cells were washed in PBS, resuspended in 200 ml of ice cold 1% SDS/10 mM Tris/pH 7.4 containing protease inhibitors, sonicated three times for 3 seconds each on ice and centrifuged at 13,000g for 5 minutes at 4°C. The total protein was determined using a BCA Protein Assay Kit (# 23225, Pierce/ Thermo Fisher Scientific), with equal amounts of protein loaded into each well of 4–12% Nu PAGE gels (NOVEX, Lifetechnologies, Thermo Fisher Scientific). Proteins were transferred and probed for the presence of the α5 subunit using a mouse anti-V5 epitope-tagged antibody and developed with an anti-mouse 680 secondary antibody. Blots were probed for GAPDH as a loading control.
Locomotion of Dopamine Transporter Knockout Mice
Horizontal locomotion activity was evaluated by an open field automated activity monitor (21 × 21 × 30 cm; AccuScan Instruments, Columbus, OH) 50. DAT knockout mice, generated as previously described 35, were acclimated to the open field for 30 min, intraperitoneally injected with either vehicle (4%DMSO-Saline) or 20 mg/kg of compound 9 or 10 mg/kg of zaprinast (Sigma #Z0878), and immediately returned to the chamber for 2 hours after injection. Mice were reused three times within a 10-day interval to allow drug wash out. Total number of beam breaks seen in 5 min segments assessing all movements including running and turning behaviors were recorded in terms of the total distance traveled. All experiments were conducted within the guidelines of the Duke University Institutional Animal Care and Use Committee.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH parent grant and supplement P30DA029925 (M.G.C., L.S.B and C.R.) and DA12001 (F.I.C.).
The authors have no other competing financial interests related to this work. We thank J. Snyder for discussions and review of this manuscript. Materials used in this study including plasmids and cell lines will be provided to interested investigators as described on https://web.duke.edu/gpcr-assay/ContactFormF.html.
ABBREVIATIONS
- CNS
central nervous system
- nAChR
nicotinic acetylcholine receptor
Footnotes
Author Contributions
The manuscript was written through contributions of all authors and all authors have given approval to the final version of the manuscript.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Figure legends 1S and 2S, screening data, and dose response data (PDF)
References
- 1.Bierut LJ. Convergence of genetic findings for nicotine dependence and smoking related diseases with chromosome 15q24–25. Trends Pharmacol Sci. 2010;31:46–51. doi: 10.1016/j.tips.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrosi P, Becchetti A. Targeting neuronal nicotinic receptors in cancer: new ligands and potential side-effects. Recent patents on anti-cancer drug discovery. 2013;8:38–52. doi: 10.2174/15748928130105. [DOI] [PubMed] [Google Scholar]
- 3.Decker MW, Sullivan JP, Arneric SP, Williams M. Neuropsychopharmacology: the fifth generation of progress -Neuronal Nicotinic Acetylcholine Receptors: Novel Targets for CNS Therapeutics. Lippincott Williams & Wilkins; Philadelphia: 2002. [Google Scholar]
- 4.Administration, F.-U. S. F. a. D. FDA 101: Smoking Cessation Products. 2016 http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm198176.htm.
- 5.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 6.Millar NS. A review of experimental techniques used for the heterologous expression of nicotinic acetylcholine receptors. Biochem Pharmacol. 2009;78:766–776. doi: 10.1016/j.bcp.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Govind AP, Vezina P, Green WN. Nicotine-induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem Pharmacol. 2009;78:756–765. doi: 10.1016/j.bcp.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopalakrishnan M, Buisson B, Touma E, Giordano T, Campbell JE, Hu IC, Donnelly-Roberts D, Arneric SP, Bertrand D, Sullivan JP. Stable expression and pharmacological properties of the human alpha 7 nicotinic acetylcholine receptor. Eur J Pharmacol. 1995;290:237–246. doi: 10.1016/0922-4106(95)00083-6. [DOI] [PubMed] [Google Scholar]
- 9.Bierut LJ. Nicotine dependence and genetic variation in the nicotinic receptors. Drug Alcohol Depend. 2009;104(Suppl 1):S64–69. doi: 10.1016/j.drugalcdep.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russo P, Cesario A, Rutella S, Veronesi G, Spaggiari L, Galetta D, Margaritora S, Granone P, Greenberg DS. Impact of genetic variability in nicotinic acetylcholine receptors on nicotine addiction and smoking cessation treatment. Current medicinal chemistry. 2011;18:91–112. doi: 10.2174/092986711793979715. [DOI] [PubMed] [Google Scholar]
- 11.Wen L, Jiang K, Yuan W, Cui W, Li MD. Contribution of Variants in CHRNA5/A3/B4 Gene Cluster on Chromosome 15 to Tobacco Smoking: From Genetic Association to Mechanism. Molecular neurobiology. 2016;53:472–484. doi: 10.1007/s12035-014-8997-x. [DOI] [PubMed] [Google Scholar]
- 12.Lee AM, Wu DF, Dadgar J, Wang D, McMahon T, Messing RO. PKCepsilon phosphorylates alpha4beta2 nicotinic ACh receptors and promotes recovery from desensitization. Br J Pharmacol. 2015;172:4430–4441. doi: 10.1111/bph.13228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM, McIntosh JM. alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci. 1998;18:8571–8579. doi: 10.1523/JNEUROSCI.18-21-08571.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khroyan TV, Yasuda D, Toll L, Polgar WE, Zaveri NT. High affinity alpha3beta4 nicotinic acetylcholine receptor ligands AT-1001 and AT-1012 attenuate cocaine-induced conditioned place preference and behavioral sensitization in mice. Biochem Pharmacol. 2015;97:531–541. doi: 10.1016/j.bcp.2015.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi B, Long S, Gonzalez-Cestari TF, Henderson BJ, Pavlovicz RE, Werbovetz K, Li C, McKay DB. Discovery of benzamide analogs as negative allosteric modulators of human neuronal nicotinic receptors: pharmacophore modeling and structure-activity relationship studies. Bioorg Med Chem. 2013;21:4730–4743. doi: 10.1016/j.bmc.2013.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carbone AL, Moroni M, Groot-Kormelink PJ, Bermudez I. Pentameric concatenated (alpha4)(2)(beta2)(3) and (alpha4)(3)(beta2)(2) nicotinic acetylcholine receptors: subunit arrangement determines functional expression. Br J Pharmacol. 2009;156:970–981. doi: 10.1111/j.1476-5381.2008.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitch RW, Xiao Y, Kellar KJ, Daly JW. Membrane potential fluorescence: a rapid and highly sensitive assay for nicotinic receptor channel function. Proc Natl Acad Sci U S A. 2003;100:4909–4914. doi: 10.1073/pnas.0630641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Xiao Y, Abdrakhmanova G, Wang W, Cleemann L, Kellar KJ, Morad M. Activation and Ca2+ permeation of stably transfected alpha3/beta4 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1999;55:970–981. doi: 10.1124/mol.55.6.970. [DOI] [PubMed] [Google Scholar]
- 19.Crunelle CL, Miller ML, Booij J, van den Brink W. The nicotinic acetylcholine receptor partial agonist varenicline and the treatment of drug dependence: a review. Eur Neuropsychopharmacol. 2010;20:69–79. doi: 10.1016/j.euroneuro.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Changeux JP. 50th anniversary of the word “allosteric”. Protein Sci. 2011;20:1119–1124. doi: 10.1002/pro.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itier V, Bertrand D. Neuronal nicotinic receptors: from protein structure to function. FEBS letters. 2001;504:118–125. doi: 10.1016/s0014-5793(01)02702-8. [DOI] [PubMed] [Google Scholar]
- 22.Kuo YP, Xu L, Eaton JB, Zhao L, Wu J, Lukas RJ. Roles for nicotinic acetylcholine receptor subunit large cytoplasmic loop sequences in receptor expression and function. J Pharmacol Exp Ther. 2005;314:455–466. doi: 10.1124/jpet.105.084954. [DOI] [PubMed] [Google Scholar]
- 23.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiological reviews. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin X, Bermudez I, Steinbach JH. The nicotinic alpha5 subunit can replace either an acetylcholine-binding or nonbinding subunit in the alpha4beta2* neuronal nicotinic receptor. Mol Pharmacol. 2014;85:11–17. doi: 10.1124/mol.113.089979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA. Subcellular trafficking, pentameric assembly, and subunit stoichiometry of neuronal nicotinic acetylcholine receptors containing fluorescently labeled alpha6 and beta3 subunits. Mol Pharmacol. 2008;73:27–41. doi: 10.1124/mol.107.039180. [DOI] [PubMed] [Google Scholar]
- 26.Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA. Assembly of alpha4beta2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci. 2003;23:11554–11567. doi: 10.1523/JNEUROSCI.23-37-11554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramarao MK, Cohen JB. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc Natl Acad Sci U S A. 1998;95:4007–4012. doi: 10.1073/pnas.95.7.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.St John PA. Cellular trafficking of nicotinic acetylcholine receptors. Acta pharmacologica Sinica. 2009;30:656–662. doi: 10.1038/aps.2009.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, Lindstrom J. Chronic nicotine treatment up-regulates human alpha3 beta2 but not alpha3 beta4 acetylcholine receptors stably transfected in human embryonic kidney cells. J Biol Chem. 1998;273:28721–28732. doi: 10.1074/jbc.273.44.28721. [DOI] [PubMed] [Google Scholar]
- 30.Corringer PJ, Sallette J, Changeux JP. Nicotine enhances intracellular nicotinic receptor maturation: a novel mechanism of neural plasticity? J Physiol Paris. 2006;99:162–171. doi: 10.1016/j.jphysparis.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 31.Kuryatov A, Berrettini W, Lindstrom J. Acetylcholine receptor (AChR) alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)(2)alpha5 AChR function. Mol Pharmacol. 2011;79:119–125. doi: 10.1124/mol.110.066357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karadsheh MS, Shah MS, Tang X, Macdonald RL, Stitzel JA. Functional characterization of mouse alpha4beta2 nicotinic acetylcholine receptors stably expressed in HEK293T cells. J Neurochem. 2004;91:1138–1150. doi: 10.1111/j.1471-4159.2004.02801.x. [DOI] [PubMed] [Google Scholar]
- 33.Talwar S, Lynch JW. Phosphorylation mediated structural and functional changes in pentameric ligandgated ion channels: implications for drug discovery. The international journal of biochemistry & cell biology. 2014;53:218–223. doi: 10.1016/j.biocel.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 34.Chong CR, Sullivan DJ., Jr New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 35.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 36.Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De Biasi M, Schroder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem. 2007;102:479–492. doi: 10.1111/j.1471-4159.2007.04498.x. [DOI] [PubMed] [Google Scholar]
- 37.Murray TA, Liu Q, Whiteaker P, Wu J, Lukas RJ. Nicotinic acetylcholine receptor alpha7 subunits with a C2 cytoplasmic loop yellow fluorescent protein insertion form functional receptors. Acta pharmacologica Sinica. 2009;30:828–841. doi: 10.1038/aps.2009.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colombo SF, Mazzo F, Pistillo F, Gotti C. Biogenesis, trafficking and up-regulation of nicotinic ACh receptors. Biochem Pharmacol. 2013;86:1063–1073. doi: 10.1016/j.bcp.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 39.Krashia P, Moroni M, Broadbent S, Hofmann G, Kracun S, Beato M, Groot-Kormelink PJ, Sivilotti LG. Human alpha3beta4 neuronal nicotinic receptors show different stoichiometry if they are expressed in Xenopus oocytes or mammalian HEK293 cells. PLoS One. 2010;5:e13611. doi: 10.1371/journal.pone.0013611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazzo F, Pistillo F, Grazioso G, Clementi F, Borgese N, Gotti C, Colombo SF. Nicotine-modulated subunit stoichiometry affects stability and trafficking of alpha3beta4 nicotinic receptor. J Neurosci. 2013;33:12316–12328. doi: 10.1523/JNEUROSCI.2393-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Millar NS, Harkness PC. Assembly and trafficking of nicotinic acetylcholine receptors (Review) Mol Membr Biol. 2008;25:279–292. doi: 10.1080/09687680802035675. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Y, Nelson ME, Kuryatov A, Choi C, Cooper J, Lindstrom J. Human alpha4beta2 acetylcholine receptors formed from linked subunits. J Neurosci. 2003;23:9004–9015. doi: 10.1523/JNEUROSCI.23-27-09004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Groot-Kormelink PJ, Boorman JP, Sivilotti LG. Formation of functional alpha3beta4alpha5 human neuronal nicotinic receptors in Xenopus oocytes: a reporter mutation approach. Br J Pharmacol. 2001;134:789–796. doi: 10.1038/sj.bjp.0704313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder JC, Pack TF, Rochelle LK, Chakraborty SK, Zhang M, Eaton AW, Bai Y, Ernst LA, Barak LS, Waggoner AS, Caron MG. A rapid and affordable screening platform for membrane protein trafficking. BMC biology. 2015;13:107. doi: 10.1186/s12915-015-0216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tammimaki A, Herder P, Li P, Esch C, Laughlin JR, Akk G, Stitzel JA. Impact of human D398N single nucleotide polymorphism on intracellular calcium response mediated by alpha3beta4alpha5 nicotinic acetylcholine receptors. Neuropharmacology. 2012;63:1002–1011. doi: 10.1016/j.neuropharm.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiesner A, Fuhrer C. Regulation of nicotinic acetylcholine receptors by tyrosine kinases in the peripheral and central nervous system: same players, different roles. Cellular and molecular life sciences: CMLS. 2006;63:2818–2828. doi: 10.1007/s00018-006-6081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, Lester RA. Regulation of alpha4beta2 nicotinic receptor desensitization by calcium and protein kinase C. Mol Pharmacol. 1999;55:432–443. [PubMed] [Google Scholar]
- 49.Snyder JC, Rochelle LK, Lyerly HK, Caron MG, Barak LS. Constitutive internalization of the leucine-rich G protein-coupled receptor-5 (LGR5) to the trans-Golgi network. J Biol Chem. 2013;288:10286–10297. doi: 10.1074/jbc.M112.447540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguiz RM, Wilkins JJ, Creson TK, Biswas R, Berezniuk I, Fricker AD, Fricker LD, Wetsel WC. Emergence of anxiety-like behaviours in depressive-like Cpe(fat/fat) mice. The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. 2013;16:1623–1634. doi: 10.1017/S1461145713000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.