

Abstract
Duchenne muscular dystrophy (DMD) has been a major target for gene therapy development for nearly 30 years. DMD is among the most common genetic diseases, and isolation of the defective gene (DMD, or dystrophin) was a landmark discovery, as it was the first time a human disease gene had been cloned without knowledge of the protein product. Despite tremendous obstacles, including the enormous size of the gene and the large volume of muscle tissue in the human body, efforts to devise a treatment based on gene replacement have advanced steadily through the combined efforts of dozens of labs and patient advocacy groups. Progress in the development of DMD gene therapy has been well documented in Molecular Therapy over the past 20 years and will be reviewed here to highlight prospects for success in the imminent human clinical trials planned by several groups.
Keywords: muscular dystrophy, gene therapy, AAV, microdystrophin, dystrophin, mdx mice
Duchenne muscular dystrophy (DMD) is one of the most common human genetic disorders. Cloning of the DMD gene preceded the human genome project and established DMD as an early gene therapy target. We summarize progress using AAV vectors for bodywide dystrophin gene delivery, an approach rapidly moving into clinical trials.
Main Text
Background
Duchenne muscular dystrophy (DMD) was identified as a genetic disorder by several groups in the mid-19th century.1 The disease is inherited in an X-linked recessive pattern, and in-line with Haldane’s hypothesis, one-third of all cases arise from spontaneous, new mutations. Accordingly, genetic counseling or even curing all current cases will not greatly reduce the incidence. Individuals with DMD display a progressive loss of skeletal muscle mass, increasing weakness, and a later-onset cardiomyopathy. Approximately one-third of patients display varying degrees of cognitive dysfunction, and in some cases, smooth muscle manifestations lead to gastrointestinal issues.1 A milder and more slowly progressing variant of the disorder is termed Becker muscular dystrophy (BMD). While DMD typically arises from genetic null allele mutations, BMD generally results from mutations that allow production of lower levels of, or partially functional, dystrophin protein. Patients from families without a prior history of the disorder are typically diagnosed between the ages of 2 and 6 years, but a family history enables early diagnosis, with the possibility for carrier testing and prenatal diagnosis. Increasing use of respiratory and cardiac support has extended lifespans over the past 20 years from the late teens up to the mid-30s, but these interventions do not by themselves significantly improve muscle function. The one treatment to date that has slowed muscle loss and extended ambulation is the use of corticosteroids, such as prednisone and deflazacort.2
DMD is among the most common single-gene disorders in humans, affecting ∼1 in 5,000 newborn males.3 Despite this relatively low incidence in the general population, it is one of the most well-known genetic disorders and has attracted enormous interest in the scientific and patient advocate communities. Much of this interest grew from early work by the Muscular Dystrophy Association (USA) (MDA) and a high-profile telethon (the first of its kind) that raised money under the leadership of the entertainer Jerry Lewis. The interest in DMD led the MDA to direct significant funding in the 1980s to finding the gene responsible for DMD. Funding efforts were an enormous success and, in a seminal series of publications by the laboratory of Louis Kunkel, resulted in the identification of the gene in 1986.4 That work enabled highly accurate prenatal diagnosis and carrier detection, an understanding of the tissue-specific effect of mutations, and delineated the differences between DMD and BMD. Cloning of the DMD gene arguably represents the beginning of the human genome project, as the gene was isolated based on genetic studies that identified its chromosomal location on Xp21. The availability of the gene and the cDNA for the muscle isoform made DMD an early candidate for gene therapy.5
The DMD Gene
Despite early enthusiasm for the development of genetic therapies, many features of DMD presented important obstacles to development of a therapy. The gene is 2.2 Mb in size, and numerous isoforms are expressed in muscle and non-muscle tissues from seven different promoters and via alternative splicing. The enormous size of the locus is likely a major reason that the gene displays the highest known spontaneous mutation frequency of any human gene. Fortunately, a number of discoveries suggested approaches to gene therapy that were simpler than initially envisioned. One was the identification of rare patients with large deletions within the gene, in one case encompassing almost half the gene, that were associated with extremely mild cases of BMD.6 A second came from isolation of the muscle cDNA, which was 14 kb but had an 11.2-kb open reading frame. These initial observations led to a series of studies establishing transgenic mouse lines on the mdx background, a model for DMD. In one transgenic line it was found that expression of the full-length dystrophin cDNA in a muscle-specific manner eliminated virtually all known muscle aspects of the disorder.7 From these studies, it became clear that an effective therapy could be developed if a synthetic gene derived from the muscle cDNA could be delivered to striated muscle, thus avoiding the need to deliver the entire gene, multiple isoforms, or to target tissues that expressed many of the smaller dystrophin isoforms. Truncated versions of the cDNA (∼6 kb in size) based on genetic deletions in mildly affected BMD patients were subsequently shown to almost completely prevent disease in the mdx mouse models, which was accompanied by intensive efforts to understand the overall structure and function of the dystrophin protein.8, 9 Such studies enabled the design of smaller but highly functional mini- and micro-dystrophin cDNAs as short as 3.6 kb (Figure 1).10, 11, 12, 13, 14 The next and enormously challenging task was finding a means to deliver the synthetic gene to the striated muscles that make up nearly 40% of human body mass.
Figure 1.

Domain Structure of Dystrophin
Comparative domain structures of full-length dystrophin (top), the mini-dystrophin expressed in a very mildly affected Becker muscular dystrophy (BMD) patient carrying a genomic deletion that removed exons 17–486 (middle) and the structure of a micro-dystrophin protein10 (bottom). Domains within dystrophin are abbreviated as follows: ABD, actin-binding domain; R, spectrin-like repeats; H, hinge domains; CR, cysteine-rich domain; CT, carboxy-terminal domain. Note that the exon 17–48 genomic deletion removes approximately two-thirds of the spectrin-like repeat 19 coding region. Numerous variants of micro-dystrophin structures have been described by different labs.
Development of Vectors for Dystrophin Gene Delivery
While transgenic animal studies led to important insights into dystrophin protein structure and function that informed the design of dystrophin expression cassettes needed for therapy, such technology is not directly applicable to human use without a way to administer the cassettes to patients. How could dystrophin mini-genes be delivered bodywide such that all muscles are rescued? The advent of mini-genes with a size of less than 7 kb allowed early studies of direct intramuscular gene transfer using retroviral and adenoviral vectors.14, 15, 16, 17 However, this route of administration resulted in localized, not systemic, gene delivery and did not appear amenable to whole-body therapy. Development of improved, helper-dependent adenoviral vectors overcame many immune-system-related barriers to vector delivery and allowed for delivery of cassettes expressing the full-length dystrophin protein, but these studies were also largely limited to administration via intramuscular injection.18, 19, 20, 21 A major advance came in 1997, when the lab of Hansell Stedman showed that the musculature of an entire limb could be transduced by infusing large quantities of adenoviral vectors into hindlimb blood vessels under elevated pressure.22 While this approach did not target muscles outside of the limb, such as the heart and respiratory muscles, it suggested that vasculature delivery of vectors might be adapted for bodywide gene delivery. Further testing of adenoviral vectors revealed numerous disadvantages for muscle gene transfer, including residual induction of an immune response, slow loss of gene expression, and difficulties targeting widespread muscles due to the large vector size and its high tropism for liver.18, 23 Many labs thus embarked on studies to identify more effective vectors, and several studies showed that recombinant vectors derived from adeno-associated virus (AAV) could lead to long-term expression following intramuscular injection.13, 24, 25
Adapting AAV vector technology for DMD still required significant work. AAV vectors have a carrying capacity of ∼5 kb, and the early studies all used vectors derived from serotype 2, which poorly transduced striated muscles and could only be administered by intramuscular injection. However, studies in transgenic mice had revealed that highly functional “micro-dystrophin” cassettes could be generated with a size less than 4 kb.10, 11, 12, 26 A major breakthrough occurred when it was discovered that improved vectors could be generated from newly discovered AAV serotypes (such as AAV6, 8 and 9) which, when injected into the vasculature at high dose (in the range of 1014 vector genomes [vg] per kilogram) could transduce all the striated muscles in adult mice.27 This led to the demonstration that dystrophy could be almost entirely halted and largely reversed in an adult mammal via systemic deliver of AAV/micro-dystrophin vectors (Figure 2).27, 28 Refinement of the gene delivery cassette through miniaturization of muscle-restricted gene regulatory cassettes provided greater vector functionality.29, 30
Figure 2.
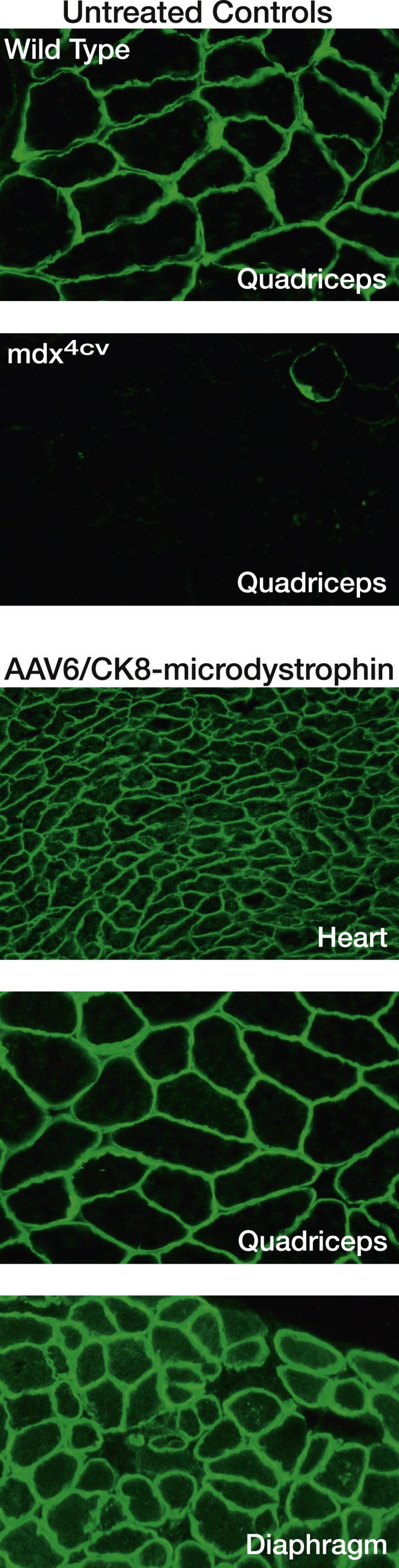
Systemic Delivery of AAV/Micro-dystrophin to Adult Mouse Muscles
Shown are images of muscle cryosections immunostained using an N-terminal antibody against dystrophin. Control quadriceps cryosections from wild-type (C57BL/6) or mdx4cv mice (top). Representative cryosections (heart, quadriceps, and diaphragm muscle) from mdx4cv mice infused with 4 × 1014 vector genomes per kilogram of AAV6/CK8-microdystrophin (bottom). Vector was administered via retro-orbital injection at ∼2 months of age, and mice were analyzed at 6 months.
Large Animal Studies
Since the murine studies showed significant promise, AAV/micro-dystrophin studies moved to testing in the larger canine model for DMD (CXMD). The dystrophic dog models not only allowed for evaluating scalability in a larger animal, but also enabled more sensitive assessment of potential immune responses against the vector. Several early studies using AAV6 suggested that T cell immune responses against the vector were limiting in this model, but other studies with AAV8 and 9 revealed minimal immune reactivity.31, 32, 33, 34, 35 It remains unclear whether this reflected differences in vector properties, vector formulation, or lab protocols. However, following extensive testing using various AAV vectors carrying reporter genes or variants of microdystrophin, systemic delivery protocols have been established in canine models that support the potential for whole-body gene delivery to human muscle.35, 36, 37, 38, 39, 40, 41
These encouraging results have led to considerable interest in developing clinical gene therapy protocols involving administration of AAV/micro-dystrophin vectors for DMD. To date only one clinical trial has been completed, involving intramuscular injection of an AAV2.5 vector. This study, performed collaboratively between the laboratories of Jerry Mendell and R. Jude Samulski, involved six patients, and while the vector injection proved safe, none of the patients expressed significant levels of micro-dystrophin (only two had any detectable exogenous dystrophin).42 Two of the six patients displayed a low-level T cell immune response (assayed by ELISpot) against dystrophin, and one other patient displayed a clear T cell response against the AAV vector (defined as greater than 3× background on the ELISpot assay).42, 43 These results indicated that immune responses against dystrophin and/or the AAV capsid were responsible for the poor transduction, but given the limited data available and the relative insensitivity of ELISpot assays, it is difficult to make firm conclusions. All the patients developed high-titer neutralizing antibodies against the vector, and two had pre-existing neutralizing antibodies.43 This, combined with a potential suboptimal vector serotype could have led to poor vector uptake by the injected muscle. In this study dystrophin expression was regulated by the ubiquitously active cytomegalovirus (CMV) immediate early enhancer plus promoter, which could have facilitated an immune response against dystrophin (through expression in immune effector cells) and/or resulted in loss of expression due to promoter shutdown. Finally, intramuscular injection tends to induce more inflammation and is better able to elicit a T cell immune response than is a vascular delivery method. These results suggested that better vectors (especially alternate capsid serotypes) coupled with a vascular delivery system and a gene regulatory cassette that is inactive in immune cells might lead to improved expression. Consequently, most current efforts to develop human clinical trials revolve around the use of AAV8 or 9 and a muscle-specific promoter/enhancer.
Systemic Delivery Clinical Trials for DMD
With considerable advances in the types of AAV vectors available, muscle-specific gene regulatory cassettes, and production/purification protocols, several groups are now planning human clinical trials involving vascular delivery of AAV/micro-dystrophin to patients. These plans are supported by extensive new data involving large-scale vector delivery to CXMD dogs and, for safety studies, to wild-type non-human primates.44, 45, 46 While most of these new data remain unpublished or proprietary, several trends are emerging that are being used to support upcoming clinical trial applications to the US Food and Drug Administration (FDA) or the European Medicines Agency (EMA). Planned trials have many similarities but differ in details including vector serotype, micro-dystrophin design, gene regulatory cassette usage, and the age of the patients. Depending on regulatory agency approval, some of these trials could begin within the next year.
These impending phase 1 clinical trials will answer several critical questions about the long-term feasibility of AAV/micro-dystrophin gene therapy for DMD. While safety will be the primary focus, the systemic gene delivery approaches will also enable important data to be gathered on efficacy. Outcome measurements should provide initial data on the functional capacity of the different micro-dystrophin designs. For example, will such miniaturized proteins improve muscle physiology similarly to what has been observed in mice and dogs? If improvement is obtained, how soon can this be observed and will the effect be sustained? Will DMD patients tolerate high-dose vector delivery as has been observed in canine and non-human primate studies? Will age and disease progression be a factor in transduction efficiency? Will T and/or B cell immune responses be observed against either dystrophin or the vector capsid proteins? Finally, can these immune responses be avoided or controlled, such that vector could be readministered years later if dystrophin expression drops below the threshold needed for therapy (see below)?
A difficulty with conducting these studies involves measuring vector distribution and dystrophin expression in widespread muscles throughout the body. The only direct method to measure dystrophin expression requires analysis of a muscle biopsy, and due to the invasive nature of obtaining biopsy specimens, very few can be obtained from any patient, limiting information on expression in multiple muscles or in one muscle over extended periods of time. Consequently, there is enormous interest in developing serum biomarkers that can be monitored non-invasively, and using imaging techniques, such as magnetic resonance imaging (MRI), to follow muscle structure over time.47, 48, 49, 50 The clearest indication of benefit will come from functional measurements, many of which have been developed for other DMD trials such as the 6-min walk test or newer, more informative tests.51
The potential for vector readministration is an important issue. Without some form of transient immune suppression, high-dose administration of AAV vectors can elicit production of neutralizing antibodies that preclude the ability to administer a second dose of vector.52 If the results from animal studies translate well to humans, it may be possible to obtain sufficient levels of dystrophin expression from a single dose to stabilize all striated muscles. However, the half-life of this expression is unknown in human muscles. Studies in dogs and non-human primates suggest that the episomal AAV vector genome is fairly stable over a period of at least 5 years in normal skeletal muscle, but a shorter half-life is likely in treated dystrophic muscles that might display a partially mosaic pattern of micro-dystrophin expression.53 Even moderate exercise can cause focal damage that would presumably be accompanied by partial vector loss. If immune responses against the vector can be avoided, such as with a transient immune suppression protocol, then readministration might be possible.37, 54, 55
Future Prospects
It has been 31 years since the dystrophin gene was cloned, 24 years since muscle-specific dystrophin expression was shown to eliminate muscular dystrophy in transgenic mice, and 13 years since systemic delivery of AAV/micro-dystrophin vectors was demonstrated.4, 7, 27 Progress in therapeutics for DMD has been accelerating, and the field is now at a critical juncture where the feasibility and efficacy of AAV-mediated systemic therapies for DMD will be tested in the clinic. Upcoming clinical trials should provide data on whether current technology can be adapted for widespread use or whether refinements are needed. Such refinements could include testing alternate AAV serotypes; using different regulatory cassettes; varying the route of delivery, dose, or age at treatment; and testing immune-suppression strategies. AAV-mediated delivery of dystrophin genes appears to have enormous potential for therapy, as it enables correction of the fundamental cause of DMD and BMD: failure to produce functional levels of the dystrophin protein. If successful, this approach could be applicable to any patient with DMD or BMD.
In addition to AAV/micro-dystrophin, several other gene therapies approaches are being contemplated for DMD. Many of these were summarized in a recent review, but a few comments are relevant here.56 Due to the possibility of an immune response against dystrophin, various groups are testing delivery of a surrogate gene that could partially substitute for dystrophin. Candidate genes include the paralog utrophin, GALGT2, or alpha7-integrin.57, 58, 59 Another emerging technology that might be tested in coming years involves the use of gene editing using the CRISPR/Cas9 system.60
Gene editing is attractive as a therapy as it has the potential to directly modify the mutant DMD gene to enable production of the dystrophin protein. In cases where the mutation is small, such as a point mutation or small deletion, this approach could lead to production of a nearly full-length protein.61 The potential for this strategy was demonstrated using AAV vectors to deliver CMV-Cas9 and guide RNA cassettes to bypass the premature stop codon in the genomes of mdx mice,62, 63, 64 and more recently, multiple strategies were shown to have potential for muscle-specific editing in the more complex mutational context in mdx4cv mice.56, 65 However, with large deletions, editing would only enable production of smaller dystrophins, similar to approaches using antisense oligonucleotides (below). A method to circumvent this issue by using editing to introduce a portion of the dystrophin cDNA into the mutant gene to restore production of larger and more functional dystrophins has recently been suggested.66 Overall, multiple strategies will be needed for treating the wide variety of mutations found in DMD patients.
A number of issues need to be resolved before gene editing can be tested in the clinic. One is the obvious problem of low efficiency observed to date. A second is the issue of expressing the bacterial Cas9 enzyme for extended periods in muscle. As noted above for micro-dystrophin, the studies that expressed Cas9 from the CMV enhancer/promoter are likely to lead to an immune response against the bacterial Cas9.23, 62, 63, 64, 67 Muscle-specific expression could reduce this concern but would still lead to long-term nuclease expression, albeit only in post-mitotic cells.61 Safety issues related to off-target editing also need to be addressed. While gene editing has been suggested as a method that could lead to permanent correction of dystrophin deficiency, such a goal will require targeting the gene in myogenic stem cells. This latter issue arises because even normal muscles cells display a low rate of turnover. At present, it is unclear whether AAV vectors are capable of transducing myogenic stem cells in vivo at an efficiency needed for significant gene editing, but future studies may clarify this issue.63, 68
Non-viral genetic approaches include systemic administration of morpholino antisense oligonucleotides that can induce skipping of mutant exons from pre-mRNAs to restore dystrophin production in muscles. Such a strategy recently resulted in FDA approval for exondys 51 (tradename of eteplirsen).69, 70 The EMA has given conditional approval for the use of ataluren,71 a small molecule designed to suppress use of premature stop codon mutations found in ∼10% of DMD patients. Other strategies are aimed at testing small molecules that could target aspects of muscle pathology resulting from dystrophin deficiency. These include drugs designed to upregulate expression of utrophin, reduce muscle inflammation, reduce fibrosis, enhance regeneration by stimulating myogenic stem cell function, or even induce new muscle cell formation via myogenic stem cell transplantation. Ultimately, an optimal treatment might result from the use of several approaches in combination. For example, one can envision gene therapy to restore dystrophin expression coupled with small-molecule therapies to reduce fibrosis.
Conclusions
The prospects of gene therapy using systemic delivery of AAV/micro-dystrophin vectors appears increasingly feasible and will soon be tested in clinical trials. The method as originally developed in mdx mice was shown to be safe and largely eliminates dystrophic pathophysiology for the lifespan of the mice. Recent and ongoing studies suggest that similar results are observed in canine models for DMD, and various types of AAV vectors have been shown to be safe in non-human primate studies and clinical trials for other genetic disorders. The general approach is amenable to significant modification if initial human studies do not achieve satisfactory results. While DMD was once viewed as an incurable disease, progress in the field suggests that successful gene therapies may soon be available.
Conflicts of Interest
J.S.C. is a member of the scientific advisory boards for Solid GT and Akashi.
Acknowledgments
This study was supported by grants U54AR065139 and RO1HL122332 from the National Institutes of Health and the Muscular Dystrophy Association (USA).
References
- 1.Emery A.E.H. Oxford Medical Publications; 1993. Duchenne Muscular Dystrophy. [Google Scholar]
- 2.Griggs R.C., Miller J.P., Greenberg C.R., Fehlings D.L., Pestronk A., Mendell J.R., Moxley R.T., 3rd, King W., Kissel J.T., Cwik V. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology. 2016;87:2123–2131. doi: 10.1212/WNL.0000000000003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 4.Monaco A.P., Neve R.L., Colletti-Feener C., Bertelson C.J., Kurnit D.M., Kunkel L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 5.Chamberlain J.S., Caskey C.T. Duchenne muscular dystrophy. In: Appel S.H., editor. Volume 10. Yearbook Medical Publishers; 1990. pp. 65–103. (Current Neurology). [Google Scholar]
- 6.England S.B., Nicholson L.V., Johnson M.A., Forrest S.M., Love D.R., Zubrzycka-Gaarn E.E., Bulman D.E., Harris J.B., Davies K.E. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–182. doi: 10.1038/343180a0. [DOI] [PubMed] [Google Scholar]
- 7.Cox G.A., Cole N.M., Matsumura K., Phelps S.F., Hauschka S.D., Campbell K.P., Faulkner J.A., Chamberlain J.S. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- 8.Phelps S.F., Hauser M.A., Cole N.M., Rafael J.A., Hinkle R.T., Faulkner J.A., Chamberlain J.S. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum. Mol. Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- 9.Wells D.J., Wells K.E., Asante E.A., Turner G., Sunada Y., Campbell K.P., Walsh F.S., Dickson G. Expression of human full-length and minidystrophin in transgenic mdx mice: implications for gene therapy of Duchenne muscular dystrophy. Hum. Mol. Genet. 1995;4:1245–1250. doi: 10.1093/hmg/4.8.1245. [DOI] [PubMed] [Google Scholar]
- 10.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., Chamberlain J.S. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 11.Crawford G.E., Faulkner J.A., Crosbie R.H., Campbell K.P., Froehner S.C., Chamberlain J.S. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J. Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rafael J.A., Cox G.A., Corrado K., Jung D., Campbell K.P., Chamberlain J.S. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J. Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B., Li J., Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuasa K., Miyagoe Y., Yamamoto K., Nabeshima Y., Dickson G., Takeda S. Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett. 1998;425:329–336. doi: 10.1016/s0014-5793(98)00251-8. [DOI] [PubMed] [Google Scholar]
- 15.Clemens P.R., Krause T.L., Chan S., Korb K.E., Graham F.L., Caskey C.T. Recombinant truncated dystrophin minigenes: construction, expression, and adenoviral delivery. Hum. Gene Ther. 1995;6:1477–1485. doi: 10.1089/hum.1995.6.11-1477. [DOI] [PubMed] [Google Scholar]
- 16.Fassati A., Wells D.J., Walsh F.S., Dickson G. Transplantation of retroviral producer cells for in vivo gene transfer into mouse skeletal muscle. Hum. Gene Ther. 1996;7:595–602. doi: 10.1089/hum.1996.7.5-595. [DOI] [PubMed] [Google Scholar]
- 17.Decrouy A., Renaud J.M., Davis H.L., Lunde J.A., Dickson G., Jasmin B.J. Mini-dystrophin gene transfer in mdx4cv diaphragm muscle fibers increases sarcolemmal stability. Gene Ther. 1997;4:401–408. doi: 10.1038/sj.gt.3300407. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y., Haecker S.E., Su Q., Wilson J.M. Immunology of gene therapy with adenoviral vectors in mouse skeletal muscle. Hum. Mol. Genet. 1996;5:1703–1712. doi: 10.1093/hmg/5.11.1703. [DOI] [PubMed] [Google Scholar]
- 19.Kochanek S., Clemens P.R., Mitani K., Chen H.H., Chan S., Caskey C.T. A new adenoviral vector: Replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta-galactosidase. Proc. Natl. Acad. Sci. USA. 1996;93:5731–5736. doi: 10.1073/pnas.93.12.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DelloRusso C., Scott J.M., Hartigan-O’Connor D., Salvatori G., Barjot C., Robinson A.S., Crawford R.W., Brooks S.V., Chamberlain J.S. Functional correction of adult mdx mouse muscle using gutted adenoviral vectors expressing full-length dystrophin. Proc. Natl. Acad. Sci. USA. 2002;99:12979–12984. doi: 10.1073/pnas.202300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert R., Nalbantoglu J., Howell J.M., Davies L., Fletcher S., Amalfitano A., Petrof B.J., Kamen A., Massie B., Karpati G. Dystrophin expression in muscle following gene transfer with a fully deleted (“gutted”) adenovirus is markedly improved by trans-acting adenoviral gene products. Hum. Gene Ther. 2001;12:1741–1755. doi: 10.1089/104303401750476249. [DOI] [PubMed] [Google Scholar]
- 22.Greelish J.P., Su L.T., Lankford E.B., Burkman J.M., Chen H., Konig S.K., Mercier I.M., Desjardins P.R., Mitchell M.A., Zheng X.G. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat. Med. 1999;5:439–443. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- 23.Hartigan-O’Connor D., Kirk C.J., Crawford R., Mulé J.J., Chamberlain J.S. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol. Ther. 2001;4:525–533. doi: 10.1006/mthe.2001.0496. [DOI] [PubMed] [Google Scholar]
- 24.Kessler P.D., Podsakoff G.M., Chen X., McQuiston S.A., Colosi P.C., Matelis L.A., Kurtzman G.J., Byrne B.J. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl. Acad. Sci. USA. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao X., Li J., Samulski R.J. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakamoto M., Yuasa K., Yoshimura M., Yokota T., Ikemoto T., Suzuki M., Dickson G., Miyagoe-Suzuki Y., Takeda S. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 2002;293:1265–1272. doi: 10.1016/S0006-291X(02)00362-5. [DOI] [PubMed] [Google Scholar]
- 27.Gregorevic P., Blankinship M.J., Allen J.M., Crawford R.W., Meuse L., Miller D.G., Russell D.W., Chamberlain J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gregorevic P., Allen J.M., Minami E., Blankinship M.J., Haraguchi M., Meuse L., Finn E., Adams M.E., Froehner S.C., Murry C.E., Chamberlain J.S. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X., Eastman E.M., Schwartz R.J., Draghia-Akli R. Synthetic muscle promoters: activities exceeding naturally occurring regulatory sequences. Nat. Biotechnol. 1999;17:241–245. doi: 10.1038/6981. [DOI] [PubMed] [Google Scholar]
- 30.Hauser M.A., Robinson A., Hartigan-O’Connor D., Williams-Gregory D.A., Buskin J.N., Apone S., Kirk C.J., Hardy S., Hauschka S.D., Chamberlain J.S. Analysis of muscle creatine kinase regulatory elements in recombinant adenoviral vectors. Mol. Ther. 2000;2:16–25. doi: 10.1006/mthe.2000.0089. [DOI] [PubMed] [Google Scholar]
- 31.Arruda V.R., Stedman H.H., Nichols T.C., Haskins M.E., Nicholson M., Herzog R.W., Couto L.B., High K.A. Regional intravascular delivery of AAV-2-F.IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood. 2005;105:3458–3464. doi: 10.1182/blood-2004-07-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z., Kuhr C.S., Allen J.M., Blankinship M., Gregorevic P., Chamberlain J.S., Tapscott S.J., Storb R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 2007;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 33.Koo T., Okada T., Athanasopoulos T., Foster H., Takeda S., Dickson G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J. Gene Med. 2011;13:497–506. doi: 10.1002/jgm.1602. [DOI] [PubMed] [Google Scholar]
- 34.Kornegay J.N., Li J., Bogan J.R., Bogan D.J., Chen C., Zheng H., Wang B., Qiao C., Howard J.F., Jr., Xiao X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. 2010;18:1501–1508. doi: 10.1038/mt.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vulin A., Barthélémy I., Goyenvalle A., Thibaud J.L., Beley C., Griffith G., Benchaouir R., le Hir M., Unterfinger Y., Lorain S. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther. 2012;20:2120–2133. doi: 10.1038/mt.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bish L.T., Sleeper M.M., Forbes S.C., Wang B., Reynolds C., Singletary G.E., Trafny D., Morine K.J., Sanmiguel J., Cecchini S. Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skipping. Mol. Ther. 2012;20:580–589. doi: 10.1038/mt.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z., Storb R., Halbert C.L., Banks G.B., Butts T.M., Finn E.E., Allen J.M., Miller A.D., Chamberlain J.S., Tapscott S.J. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol. Ther. 2012;20:1501–1507. doi: 10.1038/mt.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin J.H., Pan X., Hakim C.H., Yang H.T., Yue Y., Zhang K., Terjung R.L., Duan D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol. Ther. 2013;21:750–757. doi: 10.1038/mt.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yue Y., Pan X., Hakim C.H., Kodippili K., Zhang K., Shin J.H., Yang H.T., McDonald T., Duan D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015;24:5880–5890. doi: 10.1093/hmg/ddv310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yue Y., Ghosh A., Long C., Bostick B., Smith B.F., Kornegay J.N., Duan D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol. Ther. 2008;16:1944–1952. doi: 10.1038/mt.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao C., Li J., Zheng H., Bogan J., Yuan Z., Zhang C., Bogan D., Kornegay J., Xiao X. Hydrodynamic limb vein injection of adeno-associated virus serotype 8 vector carrying canine myostatin propeptide gene into normal dogs enhances muscle growth. Hum. Gene Ther. 2009;20:1–10. doi: 10.1089/hum.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendell J.R., Campbell K., Rodino-Klapac L., Sahenk Z., Shilling C., Lewis S., Bowles D., Gray S., Li C., Galloway G. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowles D.E., McPhee S.W., Li C., Gray S.J., Samulski J.J., Camp A.S., Li J., Wang B., Monahan P.E., Rabinowitz J.E. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012;20:443–455. doi: 10.1038/mt.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toromanoff A., Adjali O., Larcher T., Hill M., Guigand L., Chenuaud P., Deschamps J.Y., Gauthier O., Blancho G., Vanhove B. Lack of immunotoxicity after regional intravenous (RI) delivery of rAAV to nonhuman primate skeletal muscle. Mol. Ther. 2010;18:151–160. doi: 10.1038/mt.2009.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodino-Klapac L.R., Montgomery C.L., Bremer W.G., Shontz K.M., Malik V., Davis N., Sprinkle S., Campbell K.J., Sahenk Z., Clark K.R. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol. Ther. 2010;18:109–117. doi: 10.1038/mt.2009.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum. Gene Ther. Clin. Dev. 2015;26:57–69. doi: 10.1089/humc.2015.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Triplett W.T., Baligand C., Forbes S.C., Willcocks R.J., Lott D.J., DeVos S., Pollaro J., Rooney W.D., Sweeney H.L., Bönnemann C.G. Chemical shift-based MRI to measure fat fractions in dystrophic skeletal muscle. Magn. Reson. Med. 2014;72:8–19. doi: 10.1002/mrm.24917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hathout Y., Brody E., Clemens P.R., Cripe L., DeLisle R.K., Furlong P., Gordish-Dressman H., Hache L., Henricson E., Hoffman E.P. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA. 2015;112:7153–7158. doi: 10.1073/pnas.1507719112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinali M., Arechavala-Gomeza V., Cirak S., Glover A., Guglieri M., Feng L., Hollingsworth K.G., Hunt D., Jungbluth H., Roper H.P. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology. 2011;76:346–353. doi: 10.1212/WNL.0b013e318208811f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ayoglu B., Chaouch A., Lochmüller H., Politano L., Bertini E., Spitali P., Hiller M., Niks E.H., Gualandi F., Pontén F. Affinity proteomics within rare diseases: a BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014;6:918–936. doi: 10.15252/emmm.201303724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald C.M., Henricson E.K., Abresch R.T., Florence J.M., Eagle M., Gappmaier E., Glanzman A.M., Spiegel R., Barth J., Elfring G., PTC124-GD-007-DMD Study Group The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48:343–356. doi: 10.1002/mus.23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halbert C.L., Rutledge E.A., Allen J.M., Russell D.W., Miller A.D. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J. Virol. 2000;74:1524–1532. doi: 10.1128/jvi.74.3.1524-1532.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schnepp B.C., Clark K.R., Klemanski D.L., Pacak C.A., Johnson P.R. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol. 2003;77:3495–3504. doi: 10.1128/JVI.77.6.3495-3504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lorain S., Gross D.A., Goyenvalle A., Danos O., Davoust J., Garcia L. Transient immunomodulation allows repeated injections of AAV1 and correction of muscular dystrophy in multiple muscles. Mol. Ther. 2008;16:541–547. doi: 10.1038/sj.mt.6300377. [DOI] [PubMed] [Google Scholar]
- 55.Corti M., Cleaver B., Clément N., Conlon T.J., Faris K.J., Wang G., Benson J., Tarantal A.F., Fuller D., Herzog R.W., Byrne B.J. Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in pompe disease: Preclinical to clinical planning. Hum. Gene Ther. Clin. Dev. 2015;26:185–193. doi: 10.1089/humc.2015.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bengtsson N.E., Seto J.T., Hall J.K., Chamberlain J.S., Odom G.L. Progress and prospects of gene therapy clinical trials for the muscular dystrophies. Hum. Mol. Genet. 2016;25(R1):R9–R17. doi: 10.1093/hmg/ddv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tinsley J.M., Potter A.C., Phelps S.R., Fisher R., Trickett J.I., Davies K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 58.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chicoine L.G., Rodino-Klapac L.R., Shao G., Xu R., Bremer W.G., Camboni M., Golden B., Montgomery C.L., Shontz K., Heller K.N. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther. 2014;22:713–724. doi: 10.1038/mt.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long C., McAnally J.R., Shelton J.M., Mireault A.A., Bassel-Duby R., Olson E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tabebordbar M., Zhu K., Cheng J.K., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young C.S., Hicks M.R., Ermolova N.V., Nakano H., Jan M., Younesi S., Karumbayaram S., Kumagai-Cresse C., Wang D., Zack J.A. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hIPSC-derived muscle cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chew W.L., Tabebordbar M., Cheng J.K., Mali P., Wu E.Y., Ng A.H., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arnett A.L., Konieczny P., Ramos J.N., Hall J., Odom G., Yablonka-Reuveni Z., Chamberlain J.R., Chamberlain J.S. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev. 2014;1:14038. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dowling J.J. Eteplirsen therapy for Duchenne muscular dystrophy: skipping to the front of the line. Nat. Rev. Neurol. 2016;12:675–676. doi: 10.1038/nrneurol.2016.180. [DOI] [PubMed] [Google Scholar]
- 70.Aartsma-Rus A., Krieg A.M. FDA approves eteplirsen for Duchenne muscular dystrophy: The next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bushby K., Finkel R., Wong B., Barohn R., Campbell C., Comi G.P., Connolly A.M., Day J.W., Flanigan K.M., Goemans N., PTC124-GD-007-DMD STUDY GROUP Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50:477–487. doi: 10.1002/mus.24332. [DOI] [PMC free article] [PubMed] [Google Scholar]