

Abstract
Syndecans potently modulate agouti-related peptide (AgRP) signaling in the central melanocortin system. Through heparan sulfate moieties, syndecans are thought to anchor AgRP near its receptor, enhancing its orexigenic effects. Original work proposed that the N-terminal domain of AgRP facilitates this interaction. However, this is not compatible with evidence that this domain is posttranslationally cleaved. Addressing this long-standing incongruity, we used calorimetry and magnetic resonance to probe interactions of AgRP peptides with glycosaminoglycans, including heparan sulfate. We show that mature, cleaved, C-terminal AgRP, not the N-terminal domain, binds heparan sulfate. NMR shows that the binding site consists of regions distinct from the melanocortin receptor-binding site. Using a library of designed AgRP variants, we find that the strength of the syndecan interaction perfectly tracks orexigenic action. Our data provide compelling evidence that AgRP is a heparan sulfate-binding protein and localizes critical regions in the AgRP structure required for this interaction.
Keywords: G protein-coupled receptor (GPCR), glycosaminoglycan, heparan sulfate, heparin-binding protein, neuropeptide, AgRP, mc4r, syndecan
Introduction
AgRP2-releasing neurons play a central role in regulating hunger and body weight homeostasis (1). With cell bodies located in the arcuate nucleus of the hypothalamus, AgRP neurons detect signals from circulating hormones such as leptin, ghrelin, and insulin and respond through axonal projections to several distinct brain regions, including the paraventricular nucleus and the lateral hypothalamus (2). These orexigenic signals are opposed by the release of α-melanocyte-stimulating hormone (α-MSH) in overlapping brain regions by pro-opiomelanocortin neurons. AgRP neurons are essential for stimulating feeding in response to food deprivation, whereas ablation of these neurons leads to stress-induced anorexia (3). AgRP neuron activity is transient and returns quickly to basal levels when feeding commences (4). However, compared with NPY and GABA, which are also released from AgRP neurons, AgRP neuropeptide is uniquely responsible for sustaining long-term feeding (5).
AgRP signaling acts through the central melanocortin system, a critical hypothalamic pathway that consists of two G protein-coupled receptors, melanocortin receptors 3 and 4 (MC3/4R), and their peptide ligands. At MC4R, the receptor most directly linked to body weight regulation, AgRP acts as an antagonist and inverse agonist to stimulate feeding and promote positive energy balance (6–8). The antagonizing action of AgRP inhibits binding of agonist α-MSH, also expressed in the arcuate nucleus. Recently, AgRP has been demonstrated to modulate inward-rectifying potassium channels through MC4R in a G protein-independent manner (9). In humans, genetic disruptions of MC4R function lead to severe obesity (10), whereas plasma levels of AgRP correlate with body mass (11, 12). In rodents, centrally administered AgRP stimulates feeding above baseline for up to 7 days (13). The ability of this system to regulate energy balance to such a dramatic extent gives it potential to treat debilitating waste disorders, and AgRP and its mimetics are considered prime therapeutic leads in the treatment of cachexia (14).
Produced initially as a proprotein, AgRP is posttranslationally cleaved by proprotein convertases to release the mature, disulfide-rich, 50-amino acid AgRP(83–132) (15). The 10 cysteines within this core form five disulfide bonds, three of which adopt an inhibitor cystine knot (ICK) fold found most commonly in invertebrate toxins (16). A miniaturized variant of the peptide, AgRP(87–120), retains full affinity and cAMP-inhibitory action at MC4R as AgRP(83–132) (17). We have demonstrated previously that regions of AgRP outside of the ICK core, although not required for receptor binding, are necessary for increasing feeding and appetite in rats and contain positively charged residues well conserved among mammalian sequences. Fig. 1 summarizes these structural features of AgRP. Strikingly, we found that truncations or mutations that reduce the positive charge in this non-ICK region (Arg/Lys → Gln) attenuate feeding, whereas mutations that increase the charge significantly increase feeding. The most positively charged variant extended the number of days of feeding above baseline well past that of AgRP(83–132) and led to a 25% greater increase in body mass over 5 days (18).
Figure 1.
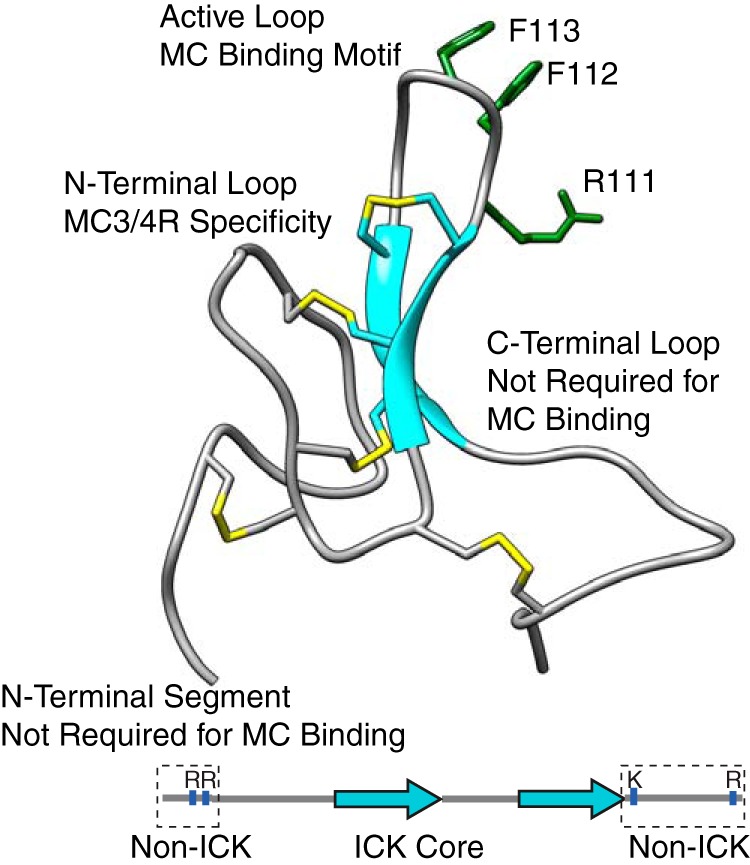
AgRP (83–132) NMR structure (PDB code 1HYK) and schematic indicating ICK and non-ICK regions. The structure of AgRP includes the functional domains shown here and their contribution to MC4R binding. The disulfide bonds are shown in yellow. The schematic highlights the N-terminal segment and C-terminal loop, which are conserved in mammalian sequences. The active loop possesses an RFF triplet (residues 111–113) necessary for melanocortin receptor binding. Positively charged residues within these non-ICK domains are indicated.
Syndecans, a family of heparan sulfate proteoglycans, have been suggested to act as accessory molecules to enhance the efficacy of AgRP at MC4R (19). Membrane-bound syndecans are involved in many physiological processes via interactions with other proteins through negatively charged heparan sulfate glycosaminoglycan (GAG) chains attached to the core protein (20). Via GAGs, syndecans tether and present proteins at specific locations, often near other receptor proteins, and also provide protection from proteolysis (21–24). The tethering of chemokines by GAGs is crucial for the formation of gradients, which directs leukocyte trafficking to damaged tissues. Mounting evidence suggests that, in the melanocortin system, syndecans significantly potentiate the effects of AgRP action. Ectopic expression of syndecans leads to severe obesity and type II diabetes in mice (25), with the phenotype being similar to that of the MC4R knockout and lethal yellow phenotypes. Similarly, transgenic mice that produce constitutively shed syndecan-1 are resistant to obesity. The regulation of syndecan-3, expressed in the hypothalamus, was found to be controlled by energy balance, where fed mice possess more syndecan-3 in its cleaved form, and fasted mice contain more cell surface-bound syndecan-3. Directly testing the effects on the melanocortin system in cell culture revealed that syndecan-3 co-transfected with MC4R in HEK cells potentiates the cAMP-inhibitory actions of AgRP. In comparison with syndecan overexpression, genetic disruption of hypothalamic syndecan-3 leads to a lean phenotype and reduced sensitivity to food deprivation (26). This same work demonstrated that syndecans directly potentiate the action of AgRP at MC4R. Furthermore, AgRP peptide localization near MC4R was significantly reduced compared with wild-type mice without reductions in neuropeptide Y (NPY) localization, which is expressed by the same neurons. This result was observed without any alterations in AgRP, NPY, or α-MSH expression levels. Adding to this hypothesis, heparanase was recently found to be an important regulator of body mass in an MC4R-dependent manner, more generally implicating the role of GAGs in MC4R (27).
Not only do these results provide compelling evidence for a role for syndecans in MC4R signaling, but they also point to a mechanism for increased feeding found previously for our charged AgRP variants. Despite this, questions remain regarding whether the mature form of AgRP possesses the molecular features necessary to interact with syndecans via GAGs. Although the unstructured N-terminal domain of AgRP has been suggested to drive this interaction (25), subsequent work by Creemers et al. (15) and our lab (28) demonstrated that this region is posttranslationally cleaved before secretion and decreases the efficacy of the peptide's inhibitory action. These results are in opposition to the paradigm above, as suggested by physiological experiments, that AgRP interacts with syndecans.
Here we describe the interaction of AgRP(83–132), the cleaved N-terminal domain, and our designed AgRP variants with size-heterogeneous heparin and heparan sulfate along with defined heparin oligosaccharides. We find that AgRP(83–132) binds with high affinity to heparin and heparan sulfate, whereas a synthetic N-terminal domain of AgRP fails to show in vitro binding. Remarkably, our truncated and designed AgRP mutants display a differential binding affinity to GAG polymers that perfectly matches the feeding trend we observed in rats. The differences in these peptides reside solely in the non-ICK domains. We then show evidence for general electrostatic complementarity rather than a specific GAG-binding epitope that is driven by these non-ICK domains. This study defines AgRP (83–132) as a heparan sulfate-binding protein and provides mechanisms for variants that significantly alter in vivo feeding, addressing a long-standing discrepancy between models of heparan sulfate proteoglycan modulation of MC4R and its molecular basis.
Results
Only AgRP(83–132) binds heparin, not the unstructured N-terminal domain
There exists virtually no literature examining the interaction of AgRP and either syndecans or protein-binding GAGs. Low-resolution experiments have identified the extended, unstructured N-terminal domain of AgRP (residues 21–82) as being responsible for syndecan binding, with AgRP(83–132) demonstrating no affinity (25). Proteins must be able to bind heparin in vitro to be classified as a heparan sulfate-binding protein (HSBP) (29). To directly test this proposed mechanism of GAG binding, we used isothermal titration calorimetry (ITC) to measure the thermodynamic properties of mixed-molecular-weight heparin (17–19 kDa) and AgRP peptides in solution. Heparin consists of repeating disaccharide units composed primarily of iduronic acid and glucosamine and possesses a high degree of sulfation. Heparin provides a convenient first test of GAG binding because of this high level of sulfation as well as the fact that heparin is commercially available in large quantities. We initially set out to measure the differences in binding affinity between heparin and AgRP(83–132) compared with full-length AgRP. Because of the difficulties in solubilizing full-length AgRP at concentrations appropriate for ITC, we used solid-phase peptide synthesis to chemically prepare the unstructured N-terminal domain separate from AgRP(83–132). We reasoned that if the N-terminal domain retained the molecular determinants of GAG binding, then it alone would display affinity by ITC.
We titrated heparin into a solution of the N-terminal domain to measure heparin affinity. To our surprise, the titrations produce no measurable heat of binding, indicating that this unstructured region does not bind heparin. Repeating the experiment with AgRP(83–132) produces a robust heat of binding and a dissociation constant of KD = 20 ± 6 nm, as shown in Fig. 2. The interaction is enthalpically driven and gives a stoichiometry of n = 15.19. This stoichiometry indicates that each heparin fragment of average molecular mass = 18,000 Da contains roughly 15 AgRP monomers.
Figure 2.

ITC results of size-heterogeneous heparin titrated into solutions of the Cys-rich AgRP(83–132) and the unstructured N-terminal domain. The AgRP Cys-rich domain (AgRP(83–132)) and the flexible N-terminal (N-term) domain (top panel) were synthesized as individual peptides. Heparin was titrated into solutions of each, and thermodynamic parameters, including dissociation constants, were obtained. Although the N-terminal domain exhibited no binding, AgRP(83–132) had a dissociation constant of KD = 20 ± 6 nm.
These data do not support the hypothesis that the N-terminal domain of AgRP, which is now known to be posttranslationally cleaved, interacts with the GAG components of syndecans. In direct contrast, the mature, structured C-terminal domain AgRP(83–132) displays a very high affinity to heparin in solution, comparable with other HSBPs (29).
Designed AgRP mutants differentially bind heparin and heparan sulfate
As our initial experiments showed that AgRP(83–132) binds heparin with high affinity, we next explored whether the positively charged amino acid residues in the non-ICK regions of the peptide had any influence on this binding. These regions were found previously to be critical for feeding in rats without having any effect on melanocortin receptor binding or cAMP inhibition, and it was hypothesized that this could be due to regulation by GAGs via heparan sulfate proteoglycan presentation (18). We synthesized variants of AgRP from that study with truncations of these non-ICK regions (AgRP87–132, AgRP83–120, and AgRP87–120) along with designed mutants where a positive charge was added as lysine residues (AgRP4K) or existing positively charged residues were mutated to glutamine (AgRP4Q). All sequences are summarized in Fig. 3.
Figure 3.

Sequences of AgRP variants used in this study. Mature AgRP(83–132) is displayed first. AgRP(87–120) represents the minimal ICK domain required for high-affinity in vitro binding. Non-ICK positive charges are highlighted in blue, and the removal of positively charged amino acids in AgRP4Q is shown in red.
We subjected each of the peptides to the same ITC experiments with heparin as described in the previous section and found dramatic differences in affinities among them. Raw isotherms from ITC data of heparin titrations are shown in Fig. 4. As shown in Fig. 5, of the three truncated peptides, the ability to bind heparin is abolished in AgRP(87–132) and AgRP(87–120). AgRP(83–120) retains some affinity but has more than a 4-fold higher dissociation constant, KD = 80 ± 20 nm. We found substantial differences in AgRP4Q and AgRP4K as well. AgRP4Q is absent of any heparin affinity, showing no appreciable heat of binding in the ITC experiment. The peptide lacks two arginines from the four-amino acid N-terminal segment as well as a lysine and arginine in the C-terminal loop, all of which are mutated to glutamine. AgRP4K displays significantly enhanced affinity for heparin. This peptide substitutes four non-charged amino acids in the N-terminal segment and C-terminal loop for lysines, which gives a dissociation constant of KD = 4.6 ± 0.5 nm.
Figure 4.

Raw isotherms of heparin and heparan sulfate ITC titrations into AgRP peptides. Size-heterogeneous heparin and heparan sulfate were titrated into AgRP variants as indicated. A–C, AgRP(83–132), AgRP(83–120), and AgRP4K isotherms are shown after titration with heparin (left panels) and heparan sulfate (right panels). D, AgRP(87–120), AgRP(87–132), and AgRP4Q isotherms after titration with heparin. As shown, these peptides do not bind heparin and also do not bind heparan sulfate.
Figure 5.

ITC results with AgRP variants. Each AgRP variant was titrated with either size-heterogeneous heparin or heparan sulfate. Thermodynamic data were measured, including dissociation constants and stoichiometry. GAG binding appears to be directly correlated to the non-ICK positive charge. As shown in the % Feeding Above Saline column, taken from our previous rat feeding study, this trend correlates with 24-h feeding. Four ITC experiments were repeated with each peptide two to four times, and the reported error is the standard deviation of each set of measurements. Feeding data are presented as percent feeding above saline and standard deviation.
Although heparin binding is a requirement for HSBPs, the polysaccharide does not accurately represent the physiologically relevant GAG found on syndecans and other heparan sulfate proteoglycans. All cells possess syndecans, and heparan sulfate (HS) is their primary GAG component. Structurally, HS varies most from heparin regarding its degree of sulfation at C2 of the uronic acids and C3/C6 at the glucosamine residues. In general, HS possess fewer stretches of polymer that are highly sulfated and negatively charged and thus would be expected to bind proteins with less affinity in electrostatically driven interactions. HSBPs should still display an affinity for HS in vitro. We used HS derived from bovine kidney with an average molecular mass of 14 kDa to test whether AgRP(83–132) and the same panel of truncates and designed variants retained their differential GAG-binding properties. The data are summarized in Fig. 5. As expected, there is a general trend of reduced affinity for HS compared with heparin, presumably because of the reduced global negative charge inherent to HS. AgRP(83–132) binds to HS with a dissociation constant of KD = 8.2 ± 0.6 μm, which is comparable with the affinity of other HSBPs for heparin. As with heparin, AgRP(87–132), AgRP(87–120), and AgRP4Q show no binding by ITC, whereas AgRP(83–120) displays a reduced affinity compared with AgRP(83–132) (KD = 15 ± 3 μm). AgRP4K retains a remarkably high affinity for HS compared with AgRP(83–132), with KD = 0.54 ± 0.1 μm. The stoichiometry for AgRP(83–132) is n = 7, indicating that each HS fragment contained roughly seven AgRP monomers and predicted a minimum protein-binding region to be an HS heptamer. The stoichiometry measurements did not differ greatly among the peptides exhibiting HS binding.
As seen in in the last column of Fig. 5, these calorimetry results show a remarkable relationship with our previous feeding studies using this panel of AgRP variants (18). AgRP4Q, AgRP(87–132), and AgRP(87–120) exhibit no affinity toward either heparin or HS and are the variants that produced the smallest 24-h feeding response in our previous feeding study (18). Interestingly, in that study, truncates AgRP(87–132) and AgRP(83–120) did not produce equivalent feeding responses, and our data here show that they similarly do not produce equivalent responses to heparin/HS binding. AgRP(83–120) has a high affinity for both GAG ligands, matching the increase in feeding it produced compared with the other truncated versions of AgRP. We find similar trends with AgRP(83–132), AgRP4Q, and AgRP4K. AgRP4Q has a significantly smaller effect on 24-h feeding compared with the other designs and here did not bind heparin or HS. The effect of AgRP4K on feeding is dramatic and significantly higher than that of AgRP(83–132), again matching the trend for GAG binding. Our previous study (18) also examined long-term effects on energy balance and change in body mass, although these were only measured in the non-truncate peptides. Consistent with 24-h feeding, body mass increased in the order AgRP4Q, AgRP(83–132), and AgRP4K, again matching the trend for increased GAG binding.
AgRP GAG-binding sites do not appear to be specific
We used NMR spectroscopy to determine the contact surface responsible for the AgRP/GAG interaction. Recombinant expression in Escherichia coli produced 15N-labeled versions of AgRP(83–132) suitable for chemical shift perturbation heteronuclear single quantum coherence (HSQC) experiments. Initially, we attempted to titrate size-heterogeneous heparin chains into solutions of the peptide but experienced severe solubility issues with substoichiometric amounts of heparin. Turning to our ITC experimental results, we calculated that AgRP monomers would interact with GAG heptamers in a roughly 1 to 1 manner. After optimizing the experimental conditions for several candidate GAG ligands, we found that heparin hexasaccharides (dp6) were most suitable for NMR. We confirmed the relevance of dp6 by ITC, titrating the hexasaccharide into solutions of AgRP at a physiologically relevant pH value. We determined that the interaction is roughly 1:1 with an n = 0.8 and dissociation constant of KD = 6.4 ± 0.5 μm (data not shown).
We titrated heparin dp6 at increasing concentrations into solutions of 50 μm AgRP(83–132) at pH 6 to observe the highest number of resonances. Fig. 6A shows regions of this 15N-1H HSQC spectrum of AgRP(83–132) overlaid with that of AgRP-dp6 complexes at ratios of 1:0.5, 1:1, 1:2, 1:4, and 1:7 AgRP:dp6. All peaks from unbound AgRP have corresponding ligand-bound peaks, indicating the preservation of the structure of the peptide. Peaks from the numerous arginine side chains were aliased and could not be definitively assigned. Inspection of the titration data shows a number of peaks that shift in a manner consistent with fast exchange on a chemical shift timescale. Importantly, peaks from both non-ICK regions, the N-terminal segment and C-terminal loop, appear to shift with the addition of heparin dp6.
Figure 6.

HSQC chemical shift perturbation data from titration of heparin into AgRP peptides. A, top panels, HSQC data of AgRP(83–132) titrated with heparin dp6 showing residues from both C-terminal loop (Lys-121, Leu-122, and Ala-125) and N-terminal segment (Arg-86) shifting. A and B, bottom panels, data plotted as Δ chemical shift. A Δδ value is considered significant when it exceeds 1 S.D. over the mean chemical shift. Non-ICK regions, which are important for GAG binding by ITC, are boxed. B, top panel, HSQC data of AgRP4K titrated with heparin dp2. Residues from both the C-terminal loop (Lys-121, Leu-122, Lys-123, and Lys-125) and N-terminal segment (Arg-85 and Arg-86) shift systematically with added dp2. Non-ICK regions are boxed. We were unable to observe Lys-83 and Lys-84 because of solvent exchange.
We calculated average chemical shifts and noted which residues shifted more than the mean and more than 1 S.D. and considering these to be significant shifts (30). This analysis is shown in Fig. 6A. A number of significantly shifted residues appear within the C-terminal loop and N-terminal segment, including the positively charged Arg-86 and Lys-121. We also noted that regions between the C-terminal loop and N-terminal segment display dramatic shifts, including Leu-90 and His-91. Interestingly, Gly-123 and Ala-125 both exhibit significant perturbation as part of the C-terminal loop. This observation is noteworthy, as it is where two of the four lysine mutations occur in AgRP4K. A relatively smaller distribution of shifted residues occurs within the MC4R-binding ICK core. None of the positively charged residues in this domain display perturbation, including Arg-111, part of the RFF triplet required for high-affinity receptor binding. We were unable to assign resonances for the N-terminal Ser-83 and Ser-84, presumably because of solvent exchange. Taken together, our findings explain the relative GAG affinities of the AgRP analogs in Fig. 5. Specifically, despite carrying a positive charge, the C-terminal loop alone is not capable of conferring a strong GAG interaction. However, the AgRP fold organizes this loop to be proximal to the N-terminal extension, synergistically forming an extended positive patch capable of high-affinity GAG binding.
We next set out to characterize the binding surfaces of AgRP4Q and AgRP4K, the variants with the most extreme differences in feeding and HS binding, in the same manner as with AgRP(83–132). Both peptides were expressed in E. coli using recombinant techniques and 15N-labeled in a minimal growth medium. The 15N-1H HSQC spectra of both peptides were well dispersed and overlapped with AgRP(83–132) to a large extent, indicative of the peptide adopting a similar fold. Not unexpectedly, titration of heparin dp6 into a 50 μm solution of AgRP4Q did not yield any appreciable chemical shift perturbations of the peptide's spectrum (data not shown).
We were unable to perform the same NMR analysis with AgRP4K because of persistent solubility issues, likely arising from a much higher affinity for heparin dp6 compared with AgRP(83–132). As smaller heparin fragments can be employed to obtain structural information about HSBPs (31), we turned to heparin dp2 disaccharides. We first titrated heparin dp2 into solutions of AgRP(83–132) to determine binding differences between it and dp6. Although there are differences in perturbations, the general trends observed from the experiments with dp6 are consistent with the smaller oligosaccharide. Using heparin dp2 with AgRP4K, we found no issues with solubility as we did with dp6. As seen in Fig. 6B, heparin dp2 titration leads to multiple regions of the AgRP4K shifting in fast exchange as with AgRP(83–132). A large concentration of these shifting peaks occurs in the non-ICK regions, including the positively charged residues Arg-885, Arg-86, Lys-121, and Arg-131. As with AgRP(83–132), position 123 also shifts significantly, here where it has been mutated to lysine from glycine. Overall, the data suggest that, rather than relying on a specific region, AgRP(83–132) relies on general electrostatics to bind GAGs in a manner that is distinct from melanocortin receptor binding.
AgRP(83–132) and AgRP4K GAG-binding sites are similar to their electrostatic surfaces
Linear stretches of amino acids typically do not make up GAG-binding sites on HSBPs. Often, two or three elements define this site, and electrostatic surface potential can be a better predictor of GAG-binding regions. As our NMR data did not reveal a linear binding sequence for heparin oligosaccharides, we sought to determine the electrostatic topology of AgRP(83–132) and compare this with these NMR experimental results.
We first visualized our NMR data by plotting residues exhibiting shifts of more than the average and 1 S.D. onto a surface representation of AgRP(83–132). Inspection of the surface map for AgRP(83–132) confirms that there is not a specific site or GAG-binding epitope, although there are areas that seem to be less affected by heparin dp6, as revealed by the relative lack of perturbed residues upon 180° rotation. Using the built-in feature in Chimera, we solved the linearized Poisson-Boltzmann equation for the structure of AgRP(83–132) to determine the electrostatic surface properties. Fig. 7A shows the comparison of the surface potential map and NMR chemical shift data. AgRP(83–132) displays a mostly positive charge density throughout. By far the highest concentration of positive charge density occurs in the C-terminal loop. Comparing the electrostatics with our experimental results, we see that the regions most affected are mostly contained in the areas with a concentrated positive charge, particularly in the C-terminal loop. Furthermore, the majority of the positively charged surface is contained on one face of the peptide in a similar manner as residues perturbed by dp6.
Figure 7.

Comparison of GAG binding and electrostatic surfaces of AgRP peptides. A, top panel, NMR chemical shift perturbation data from heparin dp6 titration depicted on the surface of AgRP(83–132). Residues that shift above the average chemical shift and 1 S.D. are colored as noted. Bottom panel, the same perspective of AgRP(83–132), with surface electrostatics as calculated by APBS. This comparison shows similarities in the most positively charged regions of the peptide to regions perturbed by heparin titration. The dashed red ovals highlight the non-ICK segments and the region between them, and the RFF triplet is indicated by solid red ovals. B, top panel, NMR chemical shift perturbation data from heparin dp2 titration depicted on the surface of AgRP4K with the same color code as in A. Bottom panel, the same perspective of AgRP4K, with surface electrostatics as calculated by APBS.
We then visualized the binding surface of AgRP4K for heparin dp2 from NMR in a similar fashion and also determined its surface electrostatics. The electrostatic surface of AgRP4K is very similar to that of AgRP(83–132). The differences are seen primarily through an even greater concentration of positive charge in the C-terminal loop of AgRP4K. A comparison of the electrostatic surface with NMR data from heparin dp2 titration is shown in Fig. 7B. Again, the perturbed regions of the peptide overlap well with the most positively charged surfaces of AgRP4K, especially in the C-terminal loop. Compared with AgRP(83–132), AgRP4K possesses a positive charge density on both halves of the peptide, and the NMR data mimic this difference. The calculation of the surface electrostatics of AgRP4Q produced little positive charge density, which was in agreement with the lack of any chemical shifts resulting from titration with heparin dp6.
These comparisons of the peptide electrostatic surface and NMR perturbation surface are in agreement with the hypothesis that AgRP peptides rely on general electrostatic complementarity to bind GAGs rather than containing a defined epitope. The region with the most positive charge density is in the C-terminal loop, a region not required for high-affinity melanocortin receptor interactions. AgRP4K increases the charge density by adding two lysine mutations in this loop, further increasing the high concentration of positive charge, leading to enhanced heparin binding and increased feeding.
Discussion
Heparan sulfate-binding proteins typically rely on electrostatic complementarity to bind GAGs (32–34), although linear sequences of positively charged amino acids do not necessarily predict these interactions. They range from being mediated by specific, GAG-binding epitopes to nonspecific global electrostatic patches. All types are found in nature and can be equally important in their relevant systems (29). For a protein to be considered an HSBP, it has been suggested that it should bind heparin, a commercially available surrogate for more relevant GAGs, in vitro; bind heparan sulfate, the major sugar component of syndecans; and be present in a biologically relevant context, most commonly in the extracellular space. The work of Reizes et al. (19, 25, 35) has clearly demonstrated this third criterion through a thorough set of animal and cell culture experiments (26). To this point, however, the mechanism of syndecan involvement in AgRP action has been contingent on the flexible N-terminal domain binding protein-bound glycosaminoglycans. This model is not consistent with literature demonstrating that this region is posttranslationally cleaved away. The evidence for this N-terminal domain-GAG interaction is extremely limited, and a thorough examination of AgRP(83–132) GAG-binding potential is needed to address this discrepancy in the literature.
Our data provide compelling evidence to contradict the hypothesis that syndecan binding requires the N-terminal domain of AgRP. Our calorimetry data demonstrate this clearly with heparin, a paradigm that strongly favors showing even modest peptide-GAG interactions using a technique that is quite sensitive. AgRP(83–132) binds heparin with a very high affinity, comparable with other established HSBPs (29). We also show that the peptide binds to the less negatively charged heparan sulfate with an affinity still comparable with the affinity of other peptides for heparin. These experiments reveal the physiologically relevant C-terminal domain of AgRP to be an HSBP. This result is significant in that it is compatible with both the extensive physiological basis for syndecan involvement in melanocortin signaling and the current model for AgRP processing and pharmacology. Although the C- and N-terminal domains together may act synergistically to bind GAGs, our results clearly demonstrate that mature C-terminal AgRP alone is sufficient.
We have also gained insight into divergent molecular aspects of AgRP compared with agouti-signaling peptide. Agouti-signaling peptide utilizes regions outside of its ICK- and MC3/4R-binding core to recognize MC1R and attractin as an accessory molecule. Although these equivalent domains of AgRP are non-essential for its function in vitro, our biophysical data reveal that these regions contribute to form a GAG-binding surface on the peptide. This provides an explanation for the observation that these non-ICK regions are essential for maximal in vivo feeding effects. We hypothesize that these regions allow AgRP to be locally recruited by heparan sulfate proteoglycans near its cognate receptor, increasing its local concentration as well as potentially increasing the half-life of AgRP (Fig. 8). Our results do not indicate that there is an overlap of HS- and MC4R-binding sites. Therefore, the possibility remains that syndecans may also act as a co-receptor to facilitate MC4R binding, although AgRP possesses a low nanomolar affinity in the absence of syndecans in cell culture binding assays. This model is in agreement with the significant reduction of peptide localization in the paraventricular nucleus, a critical site for MC4R function, in mice lacking syndecan-3. Using surface electrostatics to examine potential heparan-sulfate binding sites has been suggested as current best practice to identify GAG-binding regions (29). Generating a surface representation of the electrostatics of AgRP reveals a general agreement between the regions of highest positive charge and amino acids influenced by heparin titration. Large patches of positive charge, rather than a single site, characterize the GAG-binding site. These large patches do not include positively charged amino acids within the MC4R-binding ICK core.
Figure 8.
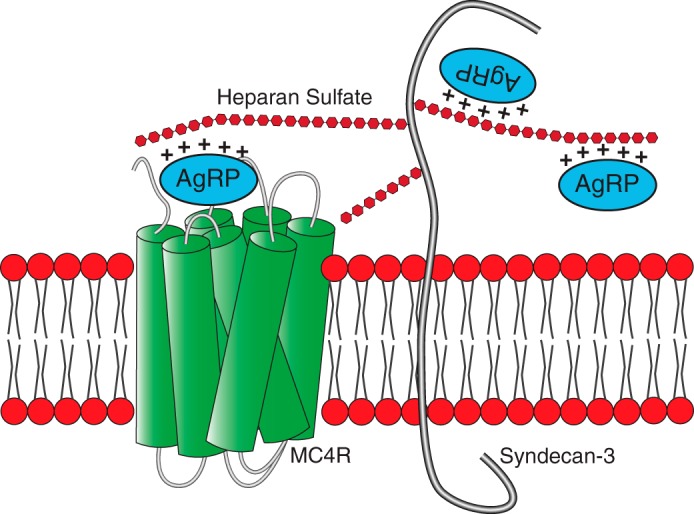
Schematic of a proposed mechanism for MC4R modulation by syndecans. Full-length AgRP is posttranslationally cleaved to release AgRP(83–132). This mature form of AgRP (represented here) interacts with both heparan sulfate proteoglycans, via GAGs, and MC4R. The positively charged non-ICK regions of the peptide are dispensable for MC4R binding and activity but required for high-affinity GAG binding. Our results suggest that syndecans either concentrate AgRP near its receptor or act as a co-receptor.
Although the non-ICK regions of AgRP were previously considered dispensable with respect to MC4R binding, both our previous results and the work here provide evidence of an important in vivo function for the short N-terminal segment and C-terminal loop of AgRP. We had hypothesized that manipulation of these regions and the resultant differential feeding in the AgRP variants could arise from differential binding to heparan sulfate proteoglycans. Along with characterizing AgRP(83–132) as an HSBP, we established that this differential binding among AgRP variants exists, from a biophysical standpoint, with both heparin and heparan sulfate. The alignment between the trends in GAG binding and feeding adds weight to the hypothesis of MC4R modulation by syndecans, although this must next be directly tested physiologically. AgRP4K contained mutations at Gly-123 and Ala-125, as they were unlikely to play a structural role. This study shows that, in AgRP(83–132), both of these amino acids are perturbed by GAGs and lie in a region of AgRP that is highly dense in positively charged surface. Lysine substitution was therefore advantageous both in terms of GAG perturbation and increasing the positive charge density in an area already concentrated in it. The combination of NMR and electrostatics may be useful to guide further peptide designs that increase both the affinity for GAGs and appetite.
The implications of AgRP possessing the molecular determinants of GAG binding are intriguing. Critically, this reconciles the physiological importance of heparan sulfate proteoglycans in MC4R signaling with AgRP posttranslational processing, strengthening the current hypothesis and providing a new context for pursuing this paradigm further. Considering how heparan sulfate proteoglycans act on other peptides, these data also propose a mechanism for the robust long-term effects of AgRP on energy balance, a characteristic recently corroborated at a neurobiological level (5). Finally, considering that AgRP has therapeutic potential for wasting disorders such as cachexia, our data provide mechanistic explanations to describe how variants may differentially stimulate feeding in a manner distinct from direct manipulation of MC4R binding. As central control of feeding and energy balance regulation continues to be a rich area of research, deciphering the molecular details that underpin this circuitry and expanding our understanding of the increasingly complex biology of MC4R stands to grow our understanding of energy homeostasis and the diseases that arise from its disruption.
Experimental procedures
Solid-phase peptide synthesis and purification
Chemically synthesized peptides were prepared using Fmoc solid-phase peptide chemistry on an automated microwave Liberty 1 System (CEM Corp., Matthews, NC). Peptides were assembled on a ChemMatrix H-rink amide resin from PCAS BioMatrix Inc. (PCAS). Amino acids were purchased through Aapptec (Louisville, KY), HOBt hydrate and DIC were obtained from Oakwood Chemical (Estill, SC), and piperidine from Spectrum (Gardena, CA), and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO). Fmoc deprotection was achieved using a 20% piperidine solution in N,N-dimethylformamide. All amino acids were double-coupled with microwave cycles using four equivalents of Fmoc-amino acid in HOBt/DIC, with arginine couplings also subjected to extended coupling. Coupling cycles concluded with a capping step using 10% acetic anhydride in N,N-dimethylformamide. AgRP(87–132) and AgRP(87–120) were acetylated at the N terminus by reacting with the same acetic anhydride solution for 5 min. Fully synthesized peptide resins were split into two reaction vessels, washed with dichloromethane and dried. A solution of 12 ml of TFA containing 0.5 ml each of triisopropylsilane (TIPS)/EDT/liquefied phenol (as scavengers) was added to each reaction vessel of dry peptide resin and incubated for 1.5 h. The resin was filtered and washed with 1 ml of TFA, and the combined filtrate and wash was then added to 90 ml of cold, dry diethyl ether for precipitation. The precipitate was collected by centrifugation, and the ether was discarded. The pellet was dissolved in 15 ml of 1:1 H2O/acetonitrile (0.1% TFA) and lyophilized. Peptides were purified by RP-HPLC on Vydac (Hesperia, CA) preparative C18 columns, and fractions were collected and analyzed by ESI-MS on a Micromass (Wythenshawe, UK) ZMD mass spectrometer. Peptides corresponding to the correct molecular weight were pooled together and lyophilized.
Peptide folding with disulfide bond formation was accomplished by incubation in an oxidative folding buffer (2.0 m GuHCl/0.1 m Tris, 3 mm GSH, and 400 μm GSSG (pH 8.5)) at a peptide concentration of 0.1 mg/ml. Peptides were stirred gently overnight. Folding was monitored by RP-HPLC on a C18 analytical column. All peptides folded to one major peak with a mass corresponding to the formation of the correct number of disulfide bonds. The folded product was purified by RP-HPLC on a C18 semipreparative column. We assessed purity by reinjecting a small amount of folded peptide on an analytical RP-HPLC. Sample purity was determined to be >95%. Quantitative analysis of the peptide concentrations was done by UV absorption.
Protein expression and purification
We purchased plasmids from DNA 2.0 (Newark, CA) using the pJ411 vector and containing the sequence corresponding to MKd5-AgRP(25–132). The sequence included an N-terminal His10 tag and an internal TEV protease recognition site, ENLYFQS, with cleavage occurring after Gln, allowing the release of AgRP(83–132). AgRP4Q and AgRP4K were constructed in the same manner, with appropriate amino acid mutations. Protein expression was done in E. coli BL21Star (DE3) (Invitrogen). The 15N-labeled proteins were grown in M9 minimal medium supplemented with 1 g/liter [15N]ammonium chloride. Cells were grown at 37 °C until they reached an A600 of 0.6–0.8, at which point protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside overnight at room temperature. The full-length constructs were solubilized from inclusion bodies in 8 m urea and run over a Ni2+-charged immobilized metal affinity column on an AKTA purifier system (GE Healthcare). Peptides were then purified further using RP-HPLC and subsequently identified by mass spectrometry and lyophilized.
Full-length AgRP proteins were first allowed to fold in an oxidative buffer. Folding was confirmed by ESI mass spectrometry. To release the disulfide-rich C-terminal domain, all full-length, folded proteins were dissolved in potassium phosphate buffer (pH 5.5) and incubated with TEV protease at a mass ratio of 5:1 peptide to protease. The reactions were allowed to proceed at room temperature overnight and then purified using semipreparative RP-HPLC. In all cases, peaks were observed for the folded C-terminal domain, the cleaved unstructured N-terminal domain, and TEV protease.
ITC
All heparins (H3393) and heparan sulfates (H7640) were purchased from Sigma-Aldrich. Heparin oligosaccharides (dp6 and dp2) were produced by Iduron and purchased from Galen Laboratory Supplies (Middletown, CT).
ITC experiments were conducted with a MicroCal VPITC calorimeter. In general, for all peptides, 25–45 μm AgRP peptide and 150–250 μm heparin/heparan sulfate were used. For AgRP(83–132)-heparin dp6 binding, the peptide concentration was 35 μm, and the dp6 concentration was 1.3 mm. Peptides were dialyzed overnight before the assay in a buffer containing 100 mm NaCl and 50 mm HEPES (pH 7.4). Data analysis was performed using the Origin calorimetry software package assuming a one site-binding model with n values, reflecting the stoichiometry of the peptide-GAG complex. Experiments were repeated two to four times in each case, and the reported error is the standard deviation of each set of measurements.
NMR spectroscopy
All samples were prepared in a buffer containing 50 mm MES monohydrate, free acid, 100 mm NaCl, 10% D2O, and 0.2 mm trimethylsilylpropanoic acid (TMSP) at pH 6. Protein was added to a final concentration of 50–100 μm, depending on the heparin oligosaccharide being used. Heparin dp6 or dp2 were titrated in increasing peptide:GAG ratios (1:0.5, 1:1, 1:2, 1:4, 1:7, and 1:10). Concentrations beyond this tended to cause peptide precipitation. 1H-15N HSQC spectra were recorded at 25 °C in-house on a Bruker Ascend 800-MHz spectrometer (Bruker-Biospin, Boston, MA). NMR spectra were analyzed using NMR Pipe and Sparky. Structural analysis was done with UCSF Chimera. Assignment of resonance peaks affected by mutation for AgRP4K was done using three-dimensional 15N total correlation spectroscopy (TOCSY) HSQC and 15N NOESY HSQC NMR spectra of peptide alone.
Electrostatic calculations
Electrostatic calculations were performed on AgRP(83–132) with the Adaptive Poisson-Boltzmann Solver (APBS) in Chimera (36). The mutations AgRP4Q and AgRP4K were introduced with the swapaa command in Chimera before calculating the surface electrostatics with APBS.
Author contributions
Conceptualization, R. P. and G. L. M.; methodology, R. P. and G. L. M.; investigation, R. P. and H. W. L; formal analysis, R. P. and H. W. L; writing—original draft, R. P.; writing—review and editing, R. P. and G. L. M.; visualization, R. P. and G. L. M., supervision, G. L. M.; funding acquisition, R. P. and G. L. M.
Acknowledgments
We thank Lauren Crowther and Charlotte Nixon for helping with the preparation of several of the mutants used in this study.
This work was supported by National Institutes of Health Fellowship F31 DK102343 (to R. P.) and National Institutes of Health Grants DK064265 and DK110403 (to G. L. M.). The 800-MHz NMR spectrometer was funded by National Institutes of Health Grant S10OD018455. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- AgRP
- agouti-related peptide
- α-MSH
- α-melanocyte-stimulating hormone
- NPY
- neuropeptide Y
- ICK
- inhibitor cystine knot
- GAG
- glycosaminoglycan
- RP
- reverse phase
- HSBP
- heparan sulfate-binding protein
- ITC
- isothermal titration calorimetry
- HS
- heparan sulfate
- HSQC
- heteronuclear single quantum coherence
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- TEV
- tobacco etch virus
- APBS
- Adaptive Poisson-Boltzmann Solver
- HOBt
- 1-hydroxybenzotriazole
- DIC
- N,N′-diisopropylcarbodiimide
- ESI
- electrospray ionization.
References
- 1. Liu T., Wang Q., Berglund E. D., and Tong Q. (2012) Action of neurotransmitter: a key to unlock the AgRP neuron feeding circuit. Front. Neurosci. 6, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Krashes M. J., Shah B. P., Madara J. C., Olson D. P., Strochlic D. E., Garfield A. S., Vong L., Pei H., Watabe-Uchida M., Uchida N., Liberles S. D., and Lowell B. B. (2014) An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Denis R. G., Joly-Amado A., Webber E., Langlet F., Schaeffer M., Padilla S. L., Cansell C., Dehouck B., Castel J., Delbès A.-S., Martinez S., Lacombe A., Rouch C., Kassis N., Fehrentz J.-A., et al. (2015) Palatability can drive feeding independent of AgRP neurons. Cell Metab. 22, 646–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen Y., Lin Y.-C., Kuo T.-W., and Knight Z. A. (2015) Sensory detection of food rapidly modulates arcuate feeding circuits. Cell 160, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krashes M. J., Shah B. P., Koda S., and Lowell B. B. (2013) Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab. 18, 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chai B.-X., Neubig R. R., Millhauser G. L., Thompson D. A., Jackson P. J., Barsh G. S., Dickinson C. J., Li J.-Y., Lai Y.-M., and Gantz I. (2003) Inverse agonist activity of agouti and agouti-related protein. Peptides 24, 603–609 [DOI] [PubMed] [Google Scholar]
- 7. Haskell-Luevano C., and Monck E. K. (2001) Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul. Pept. 99, 1–7 [DOI] [PubMed] [Google Scholar]
- 8. Ollmann M. M., Wilson B. D., Yang Y.-K., Kerns J. A., Chen Y., Gantz I., and Barsh G. S. (1997) Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278, 135–138 [DOI] [PubMed] [Google Scholar]
- 9. Ghamari-Langroudi M., Digby G. J., Sebag J. A., Millhauser G. L., Palomino R., Matthews R., Gillyard T., Panaro B. L., Tough I. R., Cox H. M., Denton J. S., and Cone R. D. (2015) G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature 520, 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farooqi I. S., and O'Rahilly S. (2005) Monogenic obesity in humans. Annu. Rev. Med. 56, 443–458 [DOI] [PubMed] [Google Scholar]
- 11. Shen C.-P., Wu K. K., Shearman L. P., Camacho R., Tota M. R., Fong T. M., and Van der Ploeg L. H. (2002) Plasma agouti-related protein level: a possible correlation with fasted and fed states in humans and rats. J. Neuroendocrinol. 14, 607–610 [DOI] [PubMed] [Google Scholar]
- 12. Hoggard N., Johnstone A. M., Faber P., Gibney E. R., Elia M., Lobley G., Rayner V., Horgan G., Hunter L., Bashir S., and Stubbs R. J. (2004) Plasma concentrations of α-MSH, AgRP and leptin in lean and obese men and their relationship to differing states of energy balance perturbation. Clin. Endocrinol. (Oxf.) 61, 31–39 [DOI] [PubMed] [Google Scholar]
- 13. Hagan M. M., Benoit S. C., Rushing P. A., Pritchard L. M., Woods S. C., and Seeley R. J. (2001) Immediate and prolonged patterns of agouti-related peptide-(83–132)-induced c-Fos activation in hypothalamic and extrahypothalamic sites. Endocrinology 142, 1050–1056 [DOI] [PubMed] [Google Scholar]
- 14. Krasnow S. M., and Marks D. L. (2010) Neuropeptides in the pathophysiology and treatment of cachexia. Curr. Opin. Support. Palliat. Care 4, 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Creemers J. W., Pritchard L. E., Gyte A., Le Rouzic P., Meulemans S., Wardlaw S. L., Zhu X., Steiner D. F., Davies N., Armstrong D., Lawrence C. B., Luckman S. M., Schmitz C. A., Davies R. A., Brennand J. C., and White A. (2006) Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83–132: interaction between AGRP83–132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology 147, 1621–1631 [DOI] [PubMed] [Google Scholar]
- 16. McNulty J. C., Thompson D. A., Bolin K. A., Wilken J., Barsh G. S., and Millhauser G. L. (2001) High-resolution NMR structure of the chemically synthesized melanocortin receptor binding domain AGRP(87–132) of the agouti-related protein. Biochemistry 40, 15520–15527 [DOI] [PubMed] [Google Scholar]
- 17. Jackson P. J., McNulty J. C., Yang Y.-K., Thompson D. A., Chai B., Gantz I., Barsh G. S., and Millhauser G. L. (2002) Design, pharmacology, and NMR structure of a minimized cystine knot with agouti-related protein activity. Biochemistry 41, 7565–7572 [DOI] [PubMed] [Google Scholar]
- 18. Madonna M. E., Schurdak J., Yang Y. K., Benoit S., and Millhauser G. L. (2012) Agouti-related protein segments outside of the receptor binding core are required for enhanced short- and long-term feeding stimulation. ACS Chem. Biol. 7, 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reizes O., Clegg D. J., Strader A. D., and Benoit S. C. (2006) A role for syndecan-3 in the melanocortin regulation of energy balance. Peptides 27, 274–280 [DOI] [PubMed] [Google Scholar]
- 20. Bishop J. R., Schuksz M., and Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 21. Duchesne L., Octeau V., Bearon R. N., Beckett A., Prior I. A., Lounis B., and Fernig D. G. (2012) Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol. 10, e1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lortat-Jacob H., Baltzer F., and Grimaud J.-A. (1996) Heparin decreases the blood clearance of interferon-γ and increases its activity by limiting the processing of its carboxyl-terminal sequence. J. Biol. Chem. 271, 16139–16143 [DOI] [PubMed] [Google Scholar]
- 23. Sadir R., Imberty A., Baleux F., and Lortat-Jacob H. (2004) Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 279, 43854–43860 [DOI] [PubMed] [Google Scholar]
- 24. Stanford K. I., Bishop J. R., Foley E. M., Gonzales J. C., Niesman I. R., Witztum J. L., and Esko J. D. (2009) Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Invest. 119, 3236–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reizes O., Lincecum J., Wang Z., Goldberger O., Huang L., Kaksonen M., Ahima R., Hinkes M. T., Barsh G. S., Rauvala H., and Bernfield M. (2001) Transgenic expression of syndecan-1 uncovers a physiological control of feeding behavior by syndecan-3. Cell 106, 105–116 [DOI] [PubMed] [Google Scholar]
- 26. Zheng Q., Zhu J., Shanabrough M., Borok E., Benoit S. C., Horvath T. L., Clegg D. J., and Reizes O. (2010) Enhanced anorexigenic signaling in lean obesity resistant syndecan-3 null mice. Neuroscience 171, 1032–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karlsson-Lindahl L., Schmidt L., Haage D., Hansson C., Taube M., Egeciouglu E., Egeciouglu E., Tan Y. X., Admyre T., Jansson J.-O., Vlodavsky I., Li J.-P., Lindahl U., and Dickson S. L. (2012) Heparanase affects food intake and regulates energy balance in mice. PLoS ONE 7, e34313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jackson P. J., Douglas N. R., Chai B., Binkley J., Sidow A., Barsh G. S., and Millhauser G. L. (2006) Structural and molecular evolutionary analysis of agouti and agouti-related proteins. Chem. Biol. 13, 1297–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 [DOI] [PubMed] [Google Scholar]
- 31. Murphy J. W., Cho Y., Sachpatzidis A., Fan C., Hodsdon M. E., and Lolis E. (2007) Structural and functional basis of CXCL12 (stromal cell-derived factor-1α) binding to heparin. J. Biol. Chem. 282, 10018–10027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Faller B., Mely Y., Gerard D., and Bieth J. G. (1992) Heparin-induced conformational change and activation of mucus proteinase inhibitor. Biochemistry 31, 8285–8290 [DOI] [PubMed] [Google Scholar]
- 33. Olson S. T., Halvorson H. R., and Björk I. (1991) Quantitative characterization of the thrombin-heparin interaction: discrimination between specific and nonspecific binding models. J. Biol. Chem. 266, 6342–6352 [PubMed] [Google Scholar]
- 34. Ori A., Wilkinson M. C., and Fernig D. G. (2008) The heparanome and regulation of cell function: structures, functions and challenges. Front. Biosci. 13, 4309–4338 [DOI] [PubMed] [Google Scholar]
- 35. Reizes O., Benoit S. C., Strader A. D., Clegg D. J., Akunuru S., and Seeley R. J. (2003) Syndecan-3 modulates food intake by interacting with the melanocortin/AgRP pathway. Ann. N.Y. Acad. Sci. 994, 66–73 [DOI] [PubMed] [Google Scholar]
- 36. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]