

Summary
The antagonistic pleiotropy (AP) theory posits that aging occurs because alleles that are detrimental in older organisms are beneficial to growth early in life and thus are maintained in populations. Although genes of the insulin signaling pathway likely participate in AP, the insulin‐regulated cellular correlates of AP have not been identified. The mitochondrial quality control process called mitochondrial autophagy (mitophagy), which is inhibited by insulin signaling, might represent a cellular correlate of AP. In this view, rapidly growing cells are limited by ATP production; these cells thus actively inhibit mitophagy to maximize mitochondrial ATP production and compete successfully for scarce nutrients. This process maximizes early growth and reproduction, but by permitting the persistence of damaged mitochondria with mitochondrial DNA mutations, becomes detrimental in the longer term. I suggest that as mitochondrial ATP output drops, cells respond by further inhibiting mitophagy, leading to a further decrease in ATP output in a classic death spiral. I suggest that this increasing ATP deficit is communicated by progressive increases in mitochondrial ROS generation, which signals inhibition of mitophagy via ROS‐dependent activation of insulin signaling. This hypothesis clarifies a role for ROS in aging, explains why insulin signaling inhibits autophagy, and why cells become progressively more oxidized during aging with increased levels of insulin signaling and decreased levels of autophagy. I suggest that the mitochondrial death spiral is not an error in cell physiology but rather a rational approach to the problem of enabling successful growth and reproduction in a competitive world of scarce nutrients.
Keywords: aging, Foxo, insulin signaling, mitophagy, reactive oxygen species, Tor
Abbreviations
- AP
antagonistic pleiotropy
- IIS
insulin/insulin growth factor signaling
- ROS
reactive oxygen species
In the long run we are all dead. Economists set themselves too easy, too useless a task, if in tempestuous seasons they can only tell us, that when the storm is long past, the ocean is flat again.
John Maynard Keynes, 1923
The death spiral, pros and cons
A death spiral, also known as a vicious circle, is a specific form of positive feedback in which steps taken to handle a particular problem, while successful in the short term, exacerbate the problem in the long term. The classic example of a death spiral is a company with debt trouble that must borrow to pay for operating expenses. Although the operating expenses can get paid (short‐term success), the additional borrowing worsens the company's debt problem (long‐term exacerbation). Insurance companies can face death spirals when, as a consequence of adverse selection, claims increase unexpectedly, which necessitate premium increases, which increase the adverse selection, etc.
The death spiral is generally viewed unfavorably because the end result of a death spiral is generally catastrophic failure. However, in comparison with the alternative, the death spiral offers critical advantages. For example, when a debt‐afflicted company borrows money to pay operating costs, it survives longer – perhaps not forever, but longer at least than it would have in the absence of this activity. Reaping benefits in the long term first requires survival through the short term, as is indicated above in the quote from John Maynard Keynes, and the death spiral can, at least, promote this short‐term survival.
Aging as a form of death spiral
From an evolutionary perspective, aging has been difficult to understand. Natural selection increases organismal fitness, and yet aging, which clearly decreases fitness, is not only observed, but also appears to be nearly universal within multicellular (and even some single‐celled) organisms. To address this dilemma, it was proposed that aging occurs and is fixed in populations because alleles that have deleterious effects in old age benefit growth, survival, and reproduction in youth. This theory is called antagonistic pleiotropy (AP) theory (Williams, 1957). In this view, aging occurs because alleles that in the short term are beneficial in solving problems in growth and reproduction serve to exacerbate the problem in the long run. Therefore, aging can be viewed as a form of death spiral.
Evidence that the genes of the insulin signaling (IIS) pathway mediate AP
If this premise is accepted, the next step is to identify the alleles that mediate AP, understand the nature of these alleles, how they might exert AP, and finally identify and define the critical cellular processes affected by AP.
Alleles of genes in the insulin/insulin growth factor signaling (IIS) pathway are the likeliest candidates for AP alleles (Walker et al., 2000; Blagosklonny, 2010). Loss‐of‐function alleles in the IIS pathway slow aging and increase lifespan in a variety of invertebrate and vertebrate systems (Kenyon, 2010), which means that wild‐type alleles of the IIS pathway promote aging and decrease lifespan. In C. elegans, loss‐of‐function alleles in the insulin receptor (daf‐2), PI3K (age‐1) and Akt (two redundant genes, in double mutant), the D. melanogaster insulin receptor InR and insulin receptor substrate chico, and the mouse growth hormone‐releasing hormone gene GHRH and the insulin growth factor receptor IGFR1 each delay aging (Clancy et al., 2001; Flurkey et al., 2002; Holzenberger et al., 2003; Kenyon, 2010). Thus, wild‐type alleles of this pathway, by promoting aging and impairing longevity, fulfill the requirement that AP alleles are deleterious to organisms in old age. In addition, members of the Foxo transcription factor family, which are inhibited by IIS, slow aging in a number of systems (Martins et al., 2016).
IIS genes also fulfill the requirement that AP alleles promote growth and reproduction at young ages. Loss‐of‐function mutations in IIS genes confer many deleterious effects to young organisms, including very slow growth, dwarfism, and deficient fecundity. It is unlikely that such mutants could reproduce or even survive in the wild. Taken together, these results indicate that wild‐type alleles of the IIS pathway promote growth and reproduction in young organisms at the expense of rapid aging.
IIS pathway activity increases protein translation and inhibits autophagy
The accelerated aging by IIS pathway activity is most likely mediated by one or more of the cellular outputs regulated by IIS. These outputs include autophagy, which is inhibited by IIS, and cell growth, which is activated by IIS (Kapahi et al., 2010). Two molecular targets of IIS that regulate each include the Tor kinase (Schmelzle & Hall, 2000), which is activated by IIS (Hay & Sonenberg, 2004), and the Foxo transcription factor, which is inhibited by IIS (Tang et al., 1999; Fig. 1). Both Tor and Foxo have been implicated in mediating the effects of IIS on aging; Foxo activity promotes longevity and Tor activity promotes aging (Kenyon, 2010). Consistent with these observations, Tor and Foxo regulate a strongly overlapping series of outputs, but in opposite directions. Tor inhibits autophagy by directly phosphorylating and inhibiting critical autophagy proteins such as ATG13 (Kamada et al., 2010) while simultaneously promoting protein synthesis by phosphorylating and inhibiting the translation inhibitor 4E‐BP (Hay & Sonenberg, 2004; Kim et al., 2011; Fig. 1). In contrast, Foxo activates autophagy by activating transcription of autophagy genes ATG8 and ATG12 while simultaneously inhibiting protein synthesis by activating 4E‐BP transcription (Jünger et al., 2003; Webb & Brunet, 2014; Fig. 1). These effects of Tor and Foxo on autophagy components are physiologically significant. Tor activation decreases autophagy (Kim et al., 2011), whereas loss of Foxo decreases autophagy in muscle and other tissues (Mammucari et al., 2007).
Figure 1.
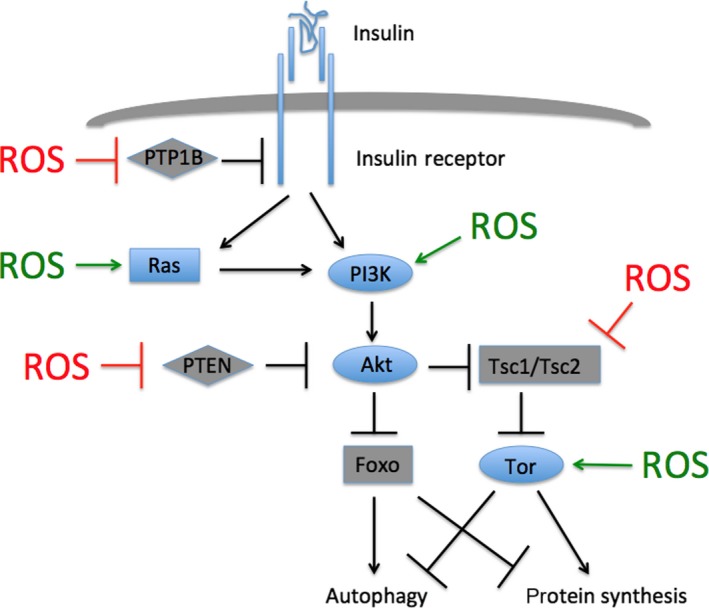
The insulin/insulin growth factor signaling pathway and its activation by reactive oxygen species (ROS). Insulin or other growth factors bind to and activate the insulin receptor or other receptor tyrosine kinases. This binding leads to PI3K and Akt activation either directly or via Ras. Akt phosphorylates and inhibits the activities of Foxo and the Tor inhibitor Tsc1/Tsc2. Activated Tor impairs autophagy and activates protein synthesis, whereas activated Foxo has the opposite effects. Phosphatases inhibit signaling either by catalyzing receptor dephosphorylation or by antagonizing PI3K activity. Pathway activators are shown in blue, and inhibitors in gray. ROS inhibits pathway inhibitors (red) and activates pathway activators (green).
Autophagy is likely to be an important process for control of aging (Rubinsztein et al., 2011; Tower, 2015). As a quality control mechanism that ensures adequate function of proteins and organelles over time, autophagy would enable cells to maintain viability over long periods. Indeed, inhibiting autophagy confers cellular deficits related to aging (Blagosklonny, 2010; Rubinsztein et al., 2011). Furthermore, autophagy declines during normal aging in Drosophila muscle (Demontis & Perrimon, 2010), mouse lung (Shirakabe et al., 2016), and human brain (Lipinski et al., 2010), and the mitochondrial autophagy (mitophagy) inducer PINK1 is transcriptionally downregulated during aging in mouse lung (Sosulski et al., 2015). It was previously proposed that IIS pathway activity is deleterious to old organisms via inhibition of autophagy (Blagosklonny, 2010; Gems & de la Guardia, 2012).
Although autophagy is responsible for degrading many types of damaged organelles or other macromolecular structures, mitophagy is likely to be the process most critical for aging. First, an early theory of aging posited that cellular damage caused by free radicals or reactive oxygen species (ROS; Harman, 1956) is a major cause of aging. ROS chemically modify a number of different functional groups on proteins, lipid, and DNA and thereby cause dysfunction. Mitochondria are a potent source of ROS generation and therefore would be expected to be particularly susceptible to ROS‐mediated damage. Second, alone among organelles and other macromolecular structures within animal cells, mitochondria possess DNA, which encodes several proteins critical for oxidative phosphorylation. Whereas every other macromolecular structure can be perfectly reconstructed with only nuclear genomic input, mitochondria are uniquely dependent on non‐nuclear DNA for continued activity. Thus, ROS‐mediated mitochondrial DNA damage, if allowed to persist, irreversibly impairs mitochondrial function. With time, the continuous accumulation of mitochondrial DNA mutations would continuously ratchet down mitochondrial ATP productive capacity, and be primarily responsible for the decline in cellular function over time.
IIS inhibits mitochondrial quality control by inhibiting mitophagy
Cells possess numerous mechanisms to enable maintenance of mitochondrial function and genome integrity over time, despite continuous generation of mitochondrial mutations. Most notably, cells possess mechanisms that enable the detection, segregation, and finally mitophagic destruction of dysfunctional mitochondria, in a process termed mitochondrial quality control. The importance of this quality control in cell physiology is demonstrated by experiments showing that mitophagy inhibition decreases bulk mitochondrial oxidative phosphorylation capacity and causes deficits in cell function (Twig et al., 2008).
As described above, IIS inhibits autophagy in general. This autophagy inhibition leads to long‐term declines in mitochondrial health: Long‐term (10‐week) Tor activation in the heart increases mitochondrial number, but decreases mitochondrial output, both phenotypes likely a consequence of impaired mitophagy (Grevengoed et al., 2015). Furthermore, IIS specifically inhibits transcription of the mitophagy inducer PINK1 (PTEN‐induced Kinase 1), which was originally identified as a gene transcriptionally upregulated by the IIS inhibitor PTEN (Unoki & Nakamura, 2001) (Fig. 1). This transcriptional induction is mediated by Foxo (Mei et al., 2009; Sengupta et al., 2011). This mitophagy inhibition has important physiological consequences, as mitophagy inhibition prevents the lifespan‐increasing effects of IIS inhibition in nematodes (Palikaras et al., 2015). In addition, increasing mitophagy genetically or pharmacologically can extend lifespan in several organisms (Rana et al., 2013; Ryu et al., 2016).
The observation that IIS inhibits autophagy and mitophagy in rapidly growing cells, despite deleterious long‐term consequences, suggests two conclusions. First, that mitochondrial ATP production is limiting, particularly under high growth conditions, suggesting further that rapidly growing cells operate under an ATP deficit. Protein synthesis requires a large expenditure of ATP; the observation that Tor induces mitochondrial protein translation to increase ATP production is consistent with this view (Morita et al., 2013). Second, that mitophagy decreases ATP production, at least in the short term; mitophagy requires several hours, and during this time, the engulfed mitochondrion is not able to contribute to ATP production. In this way, overzealous or precocious removal of mostly functional mitochondria will decrease peak mitochondrial ATP production in the short term (Fig. 2).
Figure 2.
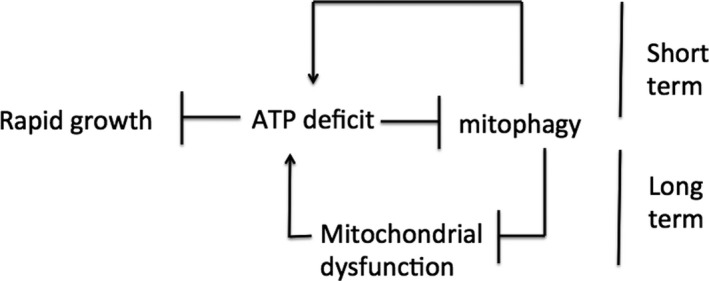
Short‐term and long‐term effects of impaired mitophagy. An ATP deficit impairs mitophagy by activating IIS. This mitophagy impairment prevents premature autophagic destruction of partially functional mitochondria. This impairment increases ATP production and thus facilitates growth in the short term. However, by allowing persistence of damaged mitochondria, this impairment leads to the accumulation of dysfunctional mitochondria and decreased ATP production in the long term. By combining short‐term benefit and long‐term detriment, I suggest that impaired mitophagy underlies antagonistic pleiotropy.
Experiments performed in invertebrates support both of these conclusions. Activation of mitophagy in nematodes decreases ATP levels in young worms (Ryu et al., 2016), and increasing mitophagy by PINK1 overexpression in the Drosophila eye decreases eye size (Koh et al., 2012). Similarly in Drosophila, ubiquitous expression of an activated, but not wild‐type, form of the mitophagy protein Parkin is lethal, and muscle‐specific expression of this activated Parkin decreases muscle function in adults. This result suggests that excessive mitophagy can be deleterious even in adulthood (Shiba‐Fukushima et al., 2014). I suggest that as damaged mitochondria accumulate during aging, organisms become increasingly dependent on these mitochondria for necessary ATP production. This increasing dependency, in fact, is what necessitates the decreasing mitophagy during aging. Consistent with this view, the effectiveness of decreased IIS on extending C. elegans lifespan progressively diminishes as the decreased IIS is initiated progressively later during aging (Dillin et al., 2002). I suggest that the abrupt increase in mitophagy caused by late‐in‐life IIS inhibition leads to a deleterious culling of damaged, but essential mitochondria.
Mitophagy inhibition as the cellular correlate of antagonistic pleiotropy
An organism that slows its growth through excessive mitophagy will allow out‐competition for scarce nutrients by other organisms. Thus, under rapid growth conditions, cells attain a short‐term selective advantage by inhibiting mitophagy. However, this mitophagy inhibition also allows persistence of mitochondria with damaged DNA, which will eventually lead to decreased mitochondrial ATP production as damaged mitochondria accumulate. Accumulation of damaged mitochondria has been proposed to promote aging (Dutta et al., 2012; Palikaras & Tavernarakis, 2012; Carnio et al., 2014; Diot et al., 2016). Thus, cells attain a long‐term selective disadvantage by inhibiting mitophagy (Fig. 2). The combination of short‐term advantage and long‐term disadvantage suggests that mitophagy inhibition acts as a cellular correlate with AP.
As mitophagy inhibition continues and mitochondrial dysfunction increases, ATP output will decline, exacerbating the ATP deficit. I suggest that as this ATP deficit increases, cells respond by further inhibiting mitophagy in order to salvage higher ATP production. This response eventually leads to a further decrease in mitochondrial ATP production, a further increase in the ATP deficit, and so on, in a classic death spiral (Fig. 2). Ultimately, a catastrophic collapse in ATP production ensues.
In this view, evolution selects for rapid growth as well as slow aging. However, because of the specific biology of mitochondria, organisms cannot simultaneously grow rapidly and age slowly. Organisms will balance these contradictory alternatives to maximize lifetime reproduction. Different species may choose to emphasize either rapid growth or slow aging, and evolutionary niches are available for many different growth/aging strategies. The house mouse combines extremely rapid growth (~20‐day gestation period) with extremely rapid aging (~3‐year lifespan) and presumably low levels of mitophagy, whereas the naked mole rat combines extremely slow growth (~70‐day gestation period) with extremely slow aging (~30‐year lifespan; Roelling et al., 2011) and presumably high levels of mitophagy. In addition, organisms are capable of modulating growth rate vs. aging rate upon changes in nutrient availability; this is accomplished by modulating IIS activity and hence mitophagy (Kenyon, 2010). However, the trade‐off between rapid growth and slow aging is never eliminated.
A proposed mechanism for the mitochondrial death spiral
As mitochondrial output begins to decline during aging, cellular demand for ATP outstrips the ability of mitochondria to produce the required ATP. I suggest that this inadequacy of ATP supply is communicated to the cytoplasm by an increase in mitochondrial ROS production. For example, mouse cardiac cells under metabolic load and Drosophila muscle cells with genetically impaired complex I function increase ROS generation (Sundaresan et al., 2009; Owusu‐Ansah et al., 2013). Other studies indicate increased ROS generation from mitochondria defective in oxidative phosphorylation (Turrens, 2003; Kregel & Zhang, 2007; Murphy, 2009, 2013; Tal et al., 2009; West et al., 2011; Raimundo et al., 2012). Finally, mitophagy impairment is sufficient to increase ROS generation in yeast (Kurihara et al., 2012; Bin‐Umer et al., 2014) and human monocytes (Zhou et al., 2011). How mitochondrial dysfunction increases ROS generation is not clear. Taken together, these observations indicate that cells respond to initial declines in mitochondrial ATP production by increasing ROS generation, which I suggest signals the cell that mitochondrial ATP output has become inadequate to meet cellular requirements.
I suggest that this ROS increase inhibits mitophagy via IIS activation. ROS has been shown to increase IIS pathway activity at several steps (Fig. 1; Okoh et al., 2013; reviewed in Sullivan and Chandel, 2014). First, several intermediates of IIS are activated by ROS either produced endogenously or supplied exogenously. In particular, Ras is activated when the thiol group of cysteine 118 is oxidized (Sawyer et al., 2002; Kuster et al., 2005; Sundaresan et al., 2009). This mechanism might underlie the observation that the activation of Erk by hydrogen peroxide requires Ras activity (Guyton et al., 1996). In addition, hydrogen peroxide activates PI3K (Wang et al., 2000; Qin & Chock, 2003; Stone & Yang, 2006), and ROS directly activates Tor (Sarbassov & Sabatini, 2005; Reiling & Sabatini, 2006) in part by inducing disulfide bond formation at the C‐terminus, which stabilizes the protein (Dames et al., 2005). Second, several IIS inhibitors are themselves inhibited by ROS. PTP1B, a phosphatase that deactivates receptor tyrosine kinases, is inhibited by oxidation, which enables activation of both the insulin and epidermal growth factor receptors (Knebel et al., 1996; Denu & Tanner, 1998; Lee et al., 1998; Finkel & Holbrook, 2000). In addition, PTEN, which removes the 3’ phosphate from PIP3 and thus opposes PI3K activity, is likewise inhibited by ROS (Lee et al., 2002; Leslie et al., 2003; Connor et al., 2005). Finally, ROS inhibits the Tsc1/Tsc2 complex, thereby relieving Tor from upstream inhibition (Yoshida et al., 2011). Thus, ROS acts through a variety of targets to activate IIS and Tor. This IIS activation is predicted to inhibit Foxo.
Not only does increased ROS activate IIS, but activated IIS also increases ROS (Irani et al., 1997; Trachootham et al., 2006; Nogueira et al., 2008; Silva et al., 2011; reviewed in Dolado & Nebreda, 2008). This increase in ROS likely occurs at least in part via mitophagy inhibition, which as described above is sufficient to activate ROS, although mitophagy‐independent, IIS‐dependent ROS increases might also occur. The ability of IIS and ROS to activate each other supports the notion that a ROS/IIS positive feedback can be generated. Such a positive feedback, once initiated, is anticipated to progressively impair mitophagy, accelerate mitochondrial dysfunction, and irreversibly decrease cellular ATP production.
Based on these molecular events, I propose the following model for the mitochondrial death spiral (Fig. 3). As deficit of mitochondrial ATP production continues to rise, the consequent rise in mitochondrial ROS production progressively oxidizes the cytoplasm and increases IIS pathway activity. Mitophagy thus becomes progressively attenuated, further exacerbating mitochondrial decline and thus the ATP supply deficit, in a positive feedback loop.
Figure 3.
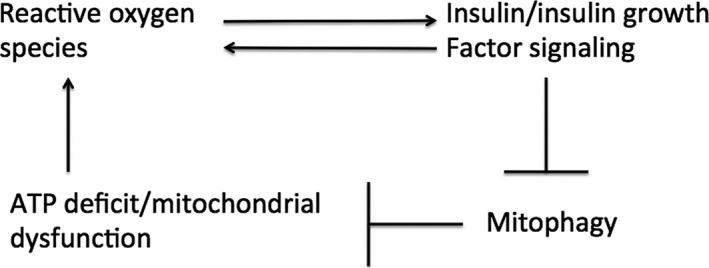
The mitochondrial death spiral. Cellular ATP deficit or mitochondrial dysfunction causes increased production of ROS. This increased ROS activates IIS, which in turn activates ROS by inhibiting mitophagy and thus promoting further mitochondrial dysfunction and exacerbating the cellular ATP deficit. IIS might also increase ROS through mitochondria‐independent mechanisms.
Testing predictions of this model
Mitochondrial dysfunction increases with age
Many lines of evidence indicate that mutations in mitochondrial DNA accumulate and mitochondrial function declines during aging (reviewed in Payne & Chinnery, 2015). Mitochondrial DNA damage increases in aging rodents (Hamilton et al., 2001; Genova et al., 2004; Hagen et al., 2004) and humans (Taylor et al., 2003), and these increases in mutation can lead to reduced flow through the electron transport chain during aging (Wanagat et al., 2001; Hagen et al., 2004; Short et al., 2005; reviewed in Golden & Melov, 2001; Ikeda et al., 2014).
Cells become more oxidized with age
The model shown in Fig. 3 suggests that as the mitochondrial death spiral progresses, cells should become progressively more oxidized. This possibility is supported by investigations by several groups (reviewed in Droge, 2003). In particular, older animals generate more oxidation products than younger animals in response to radiation (Beckman & Ames, 1998). In addition, levels of reduced glutathione decline with age both in plasma and in multiple tissues (Maher, 2005; Jones, 2006), perhaps as a consequence of age‐dependent decreases in glucose 6 phosphate dehydrogenase activity (Beckman & Ames, 1998). Finally, it was shown in yeast, nematodes, and Drosophila that the cytoplasm or mitochondria become increasingly oxidized during aging (Liu et al., 2012; Brandes et al., 2013; Kirstein et al., 2015; Knieß & Mayer, 2016). This cellular oxidation might be responsible for the observation that very old Drosophila display strikingly similar changes in gene expression to Drosophila placed under oxidative stress (Landis et al., 2004).
ROS production increases with age
The increase in oxidation state during aging is most likely a consequence, at least in part, of increased generation of ROS from mitochondria. In Drosophila, hydrogen peroxide production significantly increases during aging (Cochemé et al., 2011; Sohal & Orr, 2012; Orr et al., 2013) and increased ROS release during aging was observed from rodent muscles, heart, liver, and brain (Sohal et al., 1994; Bejma & Ji, 1999; Bejma et al., 2000; Driver et al., 2000; Vasilaki et al., 2006; reviewed in Hekimi et al., 2011).
IIS becomes more activated with age
Several lines of evidence suggest that Foxo activity diminishes during aging in the rat muscle and kidney (Edström et al., 2006; Kim et al., 2008, 2014). In addition, the transcription of several autophagy genes decreases during aging in the Drosophila flight muscle. This decrease is mostly likely due to decreased Foxo activity, as ectopic overexpression of Foxo is sufficient to rescue this transcriptional decrease (Demontis & Perrimon, 2010). Finally, transcription of the Foxo‐dependent mitophagy gene PINK1 is downregulated during aging in the mouse lung (Sosulski et al., 2015).
The effect of aging on Tor activity is less clear. Although Tor activity was reported to increase with age in muscle, liver, lung, and stem cells (Chen et al., 2009; Sandri et al., 2013; Leontieva et al., 2014; reviewed by Nacarelli et al., 2015; Romero et al., 2016; White et al., 2016), other studies failed to confirm some of these findings (Baar et al., 2016). It appears that phosphorylation of various Tor substrates is affected differentially during aging. Unfortunately, phosphorylation status of autophagy components is difficult to evaluate due to lack of phospho‐specific antibodies. In addition, Tor activity is less effective in regulating autophagy when Foxo activity is low (Mammucari et al., 2007), most likely because of the low expression of autophagy proteins.
Autophagy declines with age
Many lines of evidence demonstrate that autophagy declines with aging (reviewed in Keller et al., 2004; Bergamini et al., 2004; Massey et al., 2006; Cuervo, 2008; Rubinsztein et al., 2011; Kroemer, 2015; Romero et al., 2016). Autophagy declines during normal aging in Drosophila muscle (Demontis & Perrimon, 2010), rat liver (Del Roso et al., 2003); mouse lung (Shirakabe et al., 2016), and human brain (Keller et al., 2004; Lipinski et al., 2010). Finally, the mitophagy inducer PINK1 is transcriptionally downregulated during aging in mouse lung (Sosulski et al., 2015).
The declines in mitophagy during aging might be causally related to declines in mitochondrial biogenesis also observed during aging (Vina et al., 2009; Seo et al., 2010). Declines in mitochondrial biogenesis are likely caused in part by decreased levels of transcription factors such as PGC‐1α and NRF1 that increase expression of mitochondrial genes (Baker et al., 2006; Finley & Haigis, 2009). In addition, PGC‐1α activity is inhibited by Akt‐dependent phosphorylation (Li et al., 2007), which might link the observed increase in IIS during aging with attenuated mitochondrial biogenesis. Thus, cells combine attenuated mitophagy with attenuated mitochondrial biogenesis, which enables total mitochondrial mass to be held within controlled limits.
Greatly elevated cytoplasmic oxidation late in life: implications for oxidative stress, mitohormesis, and insulin resistance
Different levels of ROS confer distinct cellular effects. Low levels of ROS induce growth, protein synthesis, and proliferation (Antunes & Cadenas, 2001; Kwon et al., 2003; Cadenas, 2004). I suggest that these ROS levels are generated as the mitochondrial death spiral progresses. In contrast, higher ROS levels can induce an oxidative stress response that involves JNK activation and Tor inhibition (Reiling & Sabatini, 2006; Takimoto and Kass, 2007). Activated JNK, in turn, activates Foxo by phosphorylation, which overcomes the Foxo nuclear import barrier induced by IIS (Oh et al., 2005; Tzivion et al., 2011) and enables Foxo activity despite Akt‐dependent phosphorylation (Wang et al., 2005). This Foxo activation is necessary for activated JNK to increase lifespan (Wang et al., 2005). Although the mechanism underlying the Tor inhibition that occurs under high ROS is not completely clear, it is possible that a role is played by the ROS‐dependent activation of AMPK (Cardaci et al., 2012), which inhibits Tor both by phosphorylating and activating the Tor inhibitor Tsc1/Tsc2 (Fig. 1; Inoki et al., 2003) and by phosphorylating and inhibiting the Tor‐associated scaffold Raptor (Gwinn et al., 2008). JNK is activated during aging in the rodent brain and liver and in the gut of very old Drosophila (Suh, 2001; Hsieh et al., 2003; Williamson et al., 2003; Biteau et al., 2008; Zhou et al., 2009); this gut JNK activation is at least partly responsible for the Foxo activation that occurs in these very old Drosophila (Guo et al., 2014). Taken together, these results raise the possibility that very late in life, the cytoplasm can become oxidized sufficiently to induce an oxidative stress response that reactivates Foxo and inhibits Tor. These late‐stage effects on Foxo and Tor are predicted to induce a cellular switch to mitochondrial protection late in life. Mitochondrial protection triggered by oxidative stress is termed ‘mitohormesis’ (Tapia, 2006).
I suggest that induction of mitohormesis by high ROS production explains at least in part the well‐established observation that pharmacologically or genetically crippling ATP production is capable of increasing lifespan (Lee et al., 2003; Schulz et al., 2007; Copeland et al., 2009; Owusu‐Ansah et al., 2013; Sun et al., 2014). For example, Owusu‐Ansah et al. (2013) reported that the increased lifespan caused by knockdown of the complex I subunit ND75 is accompanied by, and requires, a ROS increase, followed by JNK activation, transcriptional induction of several Foxo target genes, including 4E‐BP, InR, and ImpL2, and increased mitophagy (Owusu‐Ansah et al., 2013). In addition, Schulz et al. (2007) reported that impaired glycolysis extended lifespan by the ROS‐dependent activation of AMPK (Schulz et al., 2007). In a similar manner, feeding superoxide generators can increase lifespan in C. elegans (Yang & Hekimi, 2010). This lifespan increase requires Foxo (Heidler et al., 2010) and thus might be due to JNK‐dependent mitohormesis as well. Taken together, these results indicate that high ROS levels, beginning early in life, enable cells to bypass the mitochondrial death spiral and proceed directly to the late‐stage mitohormetic state, and that this phenomenon is responsible for the increased lifespan observed.
Furthermore, cytoplasmic oxidation sufficient to promote an oxidative stress response might also be relevant to understanding the insulin resistance (IR) that often develops in the elderly. Oxidative stress and JNK are implicated in IR (Salmon, 2012); JNK phosphorylates the insulin receptor substrate 1 (IRS‐1) and attenuates the ability of ligand‐bound insulin receptor to activate IRS‐1 (Aguirre et al., 2000). A JNK deletion at least partly restores insulin sensitivity in a mouse obesity model (Hirosumi et al., 2002), indicating that this JNK‐dependent phosphorylation is functionally relevant. Given the observation that oxidative stress is increased during aging (Mendoza‐Núñez et al., 2011), these results suggest that age‐dependent IR, like the late‐stage mitohormetic state, occurs at least in part when the mitochondrial death spiral‐induced cytoplasmic oxidation progresses sufficiently to activate JNK.
Increased ROS also promotes mitochondrial protection through activation of the transcription factor Nrf2, which triggers expression of a number of antioxidant genes (Ristow & Schmeisser, 2014). Increased expression of antioxidants is expected to attenuate the ROS‐mediated positive feedback (Fig. 3), and the observation that Nrf2 activity shows dose‐dependent effects on lifespan (An et al., 2005) is consistent with this attenuation. However, Nrf2 activity declines with age (Suh et al., 2004), which likely occurs at least in part by increased Tor‐dependent inhibition of Nrf2 activity (Robida‐Stubbs et al., 2012; Lerner et al., 2013). Loss of Nrf2 activity with age will thus weaken the ability of Nrf2 to attenuate the positive feedback and might play a part in permitting the positive feedback shown in Fig. 3 to accelerate during aging.
Role of antioxidants in lifespan
The hypothesis proposed here predicts that antioxidant administration, if applied before the mitohormetic state develops, should extend lifespan. However, data on the effects of antioxidant administration have been difficult to interpret. Although ectopic overexpression of the peroxiredoxin Prx5 increases lifespan in Drosophila (Radyuk et al., 2009), indicating that decreasing ROS production can attenuate the mitochondrial death spiral as expected, the effects of feeding antioxidants on lifespan have been inconsistent. Difficulties in enabling antioxidant access to the cytoplasm might represent one issue, as the effectiveness of specific antioxidants can vary depending on the precise method of antioxidant presentation (Shibamura et al., 2009; in data interpretation include the difficulty in determining the Desjardins et al., 2017). Additional difficulties extent to which the antioxidant feeding actually reduces the cytoplasm. Fluorescent ROS indicators that monitor light production from the whole organism are problematic as these combine signal from the various subcellular and extracellular compartments, including the cytoplasm, mitochondria, peroxisomes, ER, and extracellular space, which all possess different redox states. This complicates ability to isolate redox changes specific to the cytoplasm. In addition, it is often difficult to distinguish lifespan effects due to antioxidant properties from beneficial or toxic effects of the compounds distinct from antioxidant properties. From these results, I suggest that the ability of antioxidants to increase lifespan remains an unresolved question.
Future work and limitations and extensions of this model
Causality has not yet been determined for several proposed events. Thus, for example, it is not yet known whether the increased oxidation state of the aging cytoplasm is causal for increased IIS. Further studies will be needed to address this issue. Second, this analysis deals specifically with only one mechanism proposed to underlie AP. It is likely that AP is also driven by other mechanisms. In addition, it is also likely that processes independent of AP drive aging. Such non‐AP aging processes could arise as a consequence of decreased selective pressure in old age. Finally, the ROS‐IIS positive feedback system described here is likely to advance aging through processes in addition to loss of mitochondrial ATP production. For example, the age‐dependent activation of Tor and loss of Foxo described above are predicted to inhibit autophagy in general. This progressive loss of autophagy, combined with increased ROS and thus ROS‐mediated oxidative damage, might be responsible in part for the loss of proteostasis that occurs during aging and likely plays a critical role in the aging process. This loss of proteostasis is manifested by the accumulation of protein aggregates, inclusion bodies, and other damaged macromolecules, which are degraded via autophagy (Yao, 2010). The possibility that Tor activation during aging might be partly responsible for the accumulation of these damaged macromolecules has led to the suggestion that rapamycin administration might be helpful in reversing this accumulation. In addition, Foxo plays a critical role in inducing expression of components of the proteasome as well as components of the autophagosome (Webb & Brunet, 2014). Thus, loss of Foxo activity during aging is likely to contribute to loss of proteostasis through multiple outputs.
You cannot get there from here
The mitochondrial death spiral should not be viewed as an error in cell physiology. Rather, this death spiral should be viewed as a deliberate, sensible, in fact necessary, approach by cells to solve the difficult problem of successfully growing and reproducing in a competitive world of scarce nutrients. This problem is exacerbated by the fact that cells have not yet been able to relocate every mitochondrial gene to the nucleus. This deficit means that the simultaneous demand for both high ATP production and high mitochondrial quality control becomes contradictory. The mitochondrial death spiral represents a cellular attempt to resolve these contradictory demands.
It might seem plausible to dispose of the mitochondrial death spiral after growth and reproduction are complete. Thus, when very high levels of ATP production are no longer needed, perhaps an aging cell could rejuvenate by greatly amplifying the few pristine mitochondria that might remain and degrading the rest. However, in practice, this course of action might require as a temporary intermediate a lethal decline in ATP production. Extrication from a death spiral once initiated is not easy. If a path from one point to another requires transit through a lethal state, then this path cannot be taken, regardless of the attractiveness of the destination (see John Maynard Keynes’ quote above). Evolution has not found a way to overcome this drawback. That is not to say that this problem is intractable. Perhaps this problem could be solved by organisms possibly more ingenious and certainly more motivated than evolution.
Funding
No funding information provided.
Conflict of interest
I have no conflicts of interest to declare.
Acknowledgments
I am grateful to James McNew for comments on the manuscript.
References
- Aguirre V, Uchida T, Yenush L, Davis R, White MF (2000) The c‐Jun NH2‐terminal kinase promotes insulin resistance during association with insulin receptor substrate‐1 and phosphorylation of Ser307 . J. Biol. Chem. 275, 9047–9054. [DOI] [PubMed] [Google Scholar]
- An JH, Vranas K, Lucke M, Inoue H, Hisamoto N, Matsumoto K, Blackwell TK (2005) Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN‐1 by glycogen synthetase kinase‐3. Proc. Natl Acad. Sci. USA 102, 16275–16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes F, Cadenas E (2001) Cellular titration of apoptosis with steady state concentrations of H(2)O(2): submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic. Biol. Med. 30, 1008–1018. [DOI] [PubMed] [Google Scholar]
- Baar EL, Carbajal KA, Ong IM, Lamming DW (2016) Sex‐and tissue‐specific changes in mTOR signaling with age in C57/BL6J mice. Aging Cell 15, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Betik AC, Krause DJ, Hepple RT (2006) No decline in skeletal muscle oxidative capacity with aging in long‐term calorically restricted rats: effects are independent of mitochondrial DNA integrity. J. Gerontol. A Biol. Sci. Med. Sci. 61, 675–684. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol. Rev. 78, 547–581. [DOI] [PubMed] [Google Scholar]
- Bejma J, Ji LL (1999) Aging and acute exercise enhance free radical generation in rat skeletal muscle. J. Appl. Physiol. 87, 465–470. [DOI] [PubMed] [Google Scholar]
- Bejma J, Ramires P, Ji LL (2000) Free radical generation and oxidative stress with ageing and exercise: differential effects in the myocardium and liver. Acta Physiol. Scand. 169, 343–351. [DOI] [PubMed] [Google Scholar]
- Bergamini E, Cavallini G, Donati A, Gori Z (2004) The role of macroautophagy in the ageing process, anti‐ageing intervention and age‐associated diseases. Int. J. Biochem. Cell Biol. 36, 2392–2404. [DOI] [PubMed] [Google Scholar]
- Bin‐Umer MA, McLaughlin JE, Butterly MS, McCormick S, Tumer NE (2014) Elimination of damaged mitochondria through mitophagy reduces mitochondrial oxidative stress and increases tolerance to trichothecenes. Proc. Natl Acad. Sci. USA 111, 11798–11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biteau B, Hochmuth CE, Jasper H (2008) JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell 3, 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV (2010) Revisiting the antagonistic pleiotropy theory of aging: Tor‐driven program and quasi‐program. Cell Cycle 9, 3151–3156. [DOI] [PubMed] [Google Scholar]
- Brandes N, Tienson H, Lindemann A, Vitvitsky V, Reichmann D, Banerjee R, Jakob U (2013) Time line of redox events in aging postmitotic cells. Elife 2, e00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E (2004) Mitochondrial free radical production and cell signaling. Mol. Aspects Med. 25, 17–26. [DOI] [PubMed] [Google Scholar]
- Cardaci S, Filomeni G, Ciriolo MR (2012) Redox implications of AMPK‐mediated signal transduction beyond energetic clues. J. Cell Sci. 125, 2115–2125. [DOI] [PubMed] [Google Scholar]
- Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M (2014) Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 8, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu Y, Zheng P (2009) mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L (2001) Extension of life‐span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292, 104–106. [DOI] [PubMed] [Google Scholar]
- Cochemé HM, Quin C, McQuaker SJ, Cabreiro F, Logan A, Prime TA, Abakumova I, Patel JV, Fearnley IM, James AM, Porteous CM, Smith RA, Saeed S, Carré JE, Singer M, Gems D, Hartley RC, Partridge L, Murphy MP (2011) Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 13, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre‐Ghiso J, Flores SC, Melendez JA (2005) Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280, 16916–16924. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW (2009) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. [DOI] [PubMed] [Google Scholar]
- Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet. 204, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dames SA, Mulet JM, Rathgeb‐Szabo K, Hall MN, Grzesiek S (2005) The solution structure of the FATC domain of the protein kinase Target of Rapamycin suggests a role for redox‐dependent structural and cellular stability. J. Biol. Chem. 280, 20558–20564. [DOI] [PubMed] [Google Scholar]
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E (2003) Ageing‐related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp. Gerontol. 38, 519–527. [DOI] [PubMed] [Google Scholar]
- Demontis F, Perrimon N (2010) FOXO/4E‐BP signaling in Drosophila muscles regulates organism‐wide proteostasis during aging. Cell 143, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM, Tanner KG (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642. [DOI] [PubMed] [Google Scholar]
- Desjardins D, Cacho‐Valadez B, Liu JL, Wang Y, Yee C, Bernard K, Khaki A, Breton L, Hekimi S (2017) Antioxidants reveal an inverted U‐shaped dose‐response relationship between reactive oxygen species levels and the rate of aging in Caenorhabditis elegans . Aging Cell 16, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C (2002) Timing requirements for insulin/IGF‐1 signaling in C. elegans . Science 298, 830–834. [DOI] [PubMed] [Google Scholar]
- Diot A, Moroten K, Poulton J (2016) Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 27, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolado I, Nebreda AR (2008) AKT and oxidative stress team up to kill cancer cells. Cancer Cell 14, 427–429. [DOI] [PubMed] [Google Scholar]
- Driver AS, Kodavanti PRS, Mundy WR (2000) Age‐related changes in reactive oxygen species production in rat brain homogenates. Neurotoxicol. Teratol. 22, 175–181. [DOI] [PubMed] [Google Scholar]
- Droge W (2003) Oxidative stress and aging. Adv. Exp. Med. Biol. 543, 191–200. [DOI] [PubMed] [Google Scholar]
- Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E (2012) Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ. Res. 110, 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edström E, Altun M, Hägglund M, Ulfhake B (2006) Atrogin‐1/MAFbx and MuRF1 are downregulated in aging‐related loss of skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 61, 663–674. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. [DOI] [PubMed] [Google Scholar]
- Finley LWS, Haigis MC (2009) The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res. Rev. 8, 173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE (2002) Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl Acad. Sci. USA 98, 6736–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, de la Guardia Y (2012) Alternative perspectives on aging in Caenorhabditis elegans: reactive oxygen species or hyperfunction? Antioxid. Redox Signal. 19, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genova ML, Pich MM, Bernacchia A, Bianchi C, Biondi A, Bovina C, Falasca AI, Formiggini G, Castelli GP, Lenaz G (2004) The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann. N. Y. Acad. Sci. 1011, 86–100. [DOI] [PubMed] [Google Scholar]
- Golden TR, Melov S (2001) Mitochondrial DNA mutations, oxidative stress, and aging. Mech. Ageing Dev. 122, 1577–1589. [DOI] [PubMed] [Google Scholar]
- Grevengoed TJ, Cooper DE, Young PA, Ellis JM, Coleman RA (2015) Loss of long‐chain acyl‐CoA synthetase isoform 1 impairs cardiac autophagy and mitochondrial structure through mechanistic target of rapamycin complex 1 activation. FASEB J. 29, 4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Karpac J, Tran SL, Jasper H (2014) PGRP‐SC2 promotes gut immune homeostasis to limit commensal dysbiosis and extend lifespan. Cell 156, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ (1996) Activation of mitogen‐activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 271, 4138–4142. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Schackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of Raptor mediates a metabolic checkpoint. Mol. Cell 30, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen JL, Krause DJ, Baker DJ, Fu MH, Tarnopolsky MA, Hepple RT (2004) Skeletal muscle aging in F344BN F1‐hybrid rats: I. Mitochondrial dysfunction contributes to the age‐associated reduction in VO2max. J. Gerontol. A Biol. Sci. Med. Sci. 59, 1099–1110. [DOI] [PubMed] [Google Scholar]
- Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A (2001) Does oxidative damage to DNA increase with age? Proc. Natl Acad. Sci. USA 98, 10469–10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- Heidler T, Hartwig K, Daniel H, Wenzel U (2010) Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS‐generator is dependent on DAF‐16 and SIR‐2.1. Biogerontology 11, 183–195. [DOI] [PubMed] [Google Scholar]
- Hekimi S, Lapointe J, Wen Y (2011) Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y (2003) IGF‐1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182–187. [DOI] [PubMed] [Google Scholar]
- Hsieh C‐C, Rosenblatt JI, Papaconstantinou J (2003) Age‐associated changes in SAPK/JNK and p38 MAPK signaling in response to the generation of ROS by 3‐nitropropionic acid. Mech. Ageing Dev. 124, 733–746. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Sciarretta S, Nagarajan N, Rubattu S, Volpe M, Frati G, Sadoshima J (2014) New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxid. Med. Cell Longe. 2014, 210934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt‐Clermont PJ (1997) Mitogenic signaling mediated by oxidants in Ras‐transformed fibroblasts. Science 275, 1649–1652. [DOI] [PubMed] [Google Scholar]
- Jones DP (2006) Extracellular redox state: refining the definition of oxidative stress in aging. Rejuvenation. Res. 9, 169–181. [DOI] [PubMed] [Google Scholar]
- Jünger MA, Rintelen F, Stocker H, Wasserman JD, Végh M, Radimerski T, Greenberg ME, Hafen E (2003) The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J. Biol. 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y (2010) Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 30, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L (2010) With TOR, less is more: a key role for the conserved nutrient‐sensing TOR pathway in aging. Cell Metab. 11, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q (2004) Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 36, 2376–2391. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ (2010) The genetics of aging. Nature 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Kim DH, Kim JY, Yu BP, Chung HY (2008) The activation of NF‐kappaB through Akt‐induced FOXO1 phosphorylation during aging and its modulation by calorie restriction. Biogerontology 9, 33–47. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Park MH, Chung KW, Kim MJ, Jung YR, Bae HR, Jang EJ, Lee JS, Im DS, Yu BP, Chung HY (2014) The essential role of FoxO6 phosphorylation in aging and calorie restriction. Age 36, 9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Morito D, Kakihana T, Sugihara M, Minnen A, Hipp MS, Nussbaum‐Krammer C, Kasturi P, Hartl FU, Nagata K, Morimoto RI (2015) Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 34, 2334–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebel A, Rahmsdorf HJ, Ullrich A, Herrlich P (1996) Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 15, 5314–5325. [PMC free article] [PubMed] [Google Scholar]
- Knieß RA, Mayer MP (2016) The oxidation state of the cytoplasmic glutathione redox system does not correlate with replicative lifespan in yeast. NPJ Aging Mech. Dis. 2, 16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh H, Kim H, Kim MJ, Park J, Lee HJ, Chung J (2012) Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN‐induced kinase 1 (PINK1) null mutant. J. Biol. Chem. 287, 12750–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kregel KC, Zhang HJ (2007) An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R18–R36. [DOI] [PubMed] [Google Scholar]
- Kroemer G (2015) Autophagy, a druggable process that is deregulated in aging and human disease. J. Clin. Invest. 125, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D (2012) Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 287, 3265–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuster GM, Pimentel DR, Adachi T, Ido Y, Brenner DA, Cohen RA, Liao R, Siwik DA, Colucci WS (2005) Alpha‐adrenergic receptor‐stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin‐1‐sensitive oxidative modification of thiols on Ras. Circulation 111, 1192–1198. [DOI] [PubMed] [Google Scholar]
- Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS (2003) H(2)O(2) regulates cardiac myocyte phenotype via concentration‐dependent activation of distinct kinase pathways. J. Mol. Cell. Cardiol. 35, 615–621. [DOI] [PubMed] [Google Scholar]
- Landis GN, Abdueva D, Skvortsov D, Yang J, Rabin BE, Carrick J, Tavare S, Tower J (2004) Similar gene expression patterns characterize aging and oxidative stress in Drosophila melanogaster . Proc. Natl Acad. Sci. USA 101, 7663–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S‐R, Kwon K‐S, Kim S‐R, Rhee SG (1998) Reversible inactivation of protein‐tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372. [DOI] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342. [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RYN, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Leontieva OV, Paszkiewicz GM, Blagosklonny MV (2014) Fasting levels of hepatic p‐S6 are increased in old mice. Cell Cycle 13, 2656–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner C, Bitto A, Pulliam D, Nacarelli T, Konigsberg M, Van Remmen H, Torres C, Sell C (2013) Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 12, 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP (2003) Redox regulation of PI 3‐kinase signalling via inactivation of PTEN. EMBO J. 22, 5501–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ (2007) Akt/PKB regulates hepatic metabolism by directly inhibiting PGC‐1α transcriptional coactivator. Nature 447, 1012–1017. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J (2010) Genome‐wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc. Natl Acad. Sci. USA 107, 14164–14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Celotto AM, Romero G, Wipf P, Palladino MJ (2012) Genetically encoded redox sensor identifies the role of ROS in degenerative and mitochondrial disease pathogenesis. Neurobiol. Dis. 45, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P (2005) The effects of stress and aging on glutathione metabolism. Ageing Res. Rev. 4, 288–314. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M (2007) FoxO3 controls autophagy in skeletal muscle in vivo . Cell Metab. 6, 458–471. [DOI] [PubMed] [Google Scholar]
- Martins R, Lithgow GJ, Link W (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AC, Kiffin R, Cuervo AM (2006) Autophagic defects in aging: looking for an “emergency exit”? Cell Cycle 5, 1292–1296. [DOI] [PubMed] [Google Scholar]
- Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H (2009) FOXO3a‐dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc. Natl Acad. Sci. USA 106, 5153–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza‐Núñez VM, Rosado‐Pérez J, Santiago‐Osorio E, Ortiz R, Sánchez‐Rodríguez MA, Galván‐Duarte RE (2011) Aging linked to type 2 diabetes increases oxidative stress and chronic inflammation. Rejuvenation. Res. 14, 25–31. [DOI] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St‐Pierre J, Topisirovic I, Sonenberg N (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E‐BP‐dependent translational regulation. Cell Metab. 18, 698–711. [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2013) Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 18, 145–146. [DOI] [PubMed] [Google Scholar]
- Nacarelli T, Azar A, Sell C (2015) Aberrant mTOR activation in senescence and aging: a mitochondrial stress response? Exp. Gerontol. 68, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA (2005) JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF‐16. Proc. Natl Acad. Sci. USA 102, 4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr WC, Radyuk SN, Sohal RS (2013) Involvement of redox state in the aging of Drosophila melanogaster . Antioxid. Redox Signal. 19, 788–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu‐Ansah E, Song W, Perrimon N (2013) Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Tavernarakis N (2012) Mitophagy in neurodegeneration and aging. Front. Genet. 3, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans . Nature 521, 525–528. [DOI] [PubMed] [Google Scholar]
- Payne BAI, Chinnery PF (2015) Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim. Biophys. Acta 1847, 1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Chock PB (2003) Implication of phosphatidylinositol 3‐kinase membrane recruitment in hydrogen peroxide‐induced activation of PI3K and Akt. Biochemistry 42, 2995–3003. [DOI] [PubMed] [Google Scholar]
- Radyuk SN, Michalak K, Klichko VI, Benes J, Rebrin I, Sohal RS, Orr WC (2009) Peroxiredoxin 5 confers protection against oxidative stress and apoptosis and also promotes longevity in Drosophila . Biochem. J. 419, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo N, Song L, Shutt TE, McKay SE, Cotney J, Guan MX, Gilliland TC, Hohuan D, Santos‐Sacchi J, Shadel GS (2012) Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW (2013) Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics and extends lifespan. Proc. Natl Acad. Sci. USA 110, 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling JH, Sabatini DM (2006) Stress and mTORture signaling. Oncogene 25, 6373–6383. [DOI] [PubMed] [Google Scholar]
- Ristow M, Schmeisser K (2014) Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose‐Response 12, 288–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robida‐Stubbs S, Glover‐Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann‐Haefelin E, Sabatini DM, Blackwell TK (2012) TOR signaling and rapamycin influence longevity by regulating SKN‐1/Nrf and DAF‐16/FoxO. Cell Metab. 15, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelling K, Drews B, Goeritz F, Hildebrand TB (2011) The long gestation of the small naked mole‐rat (Heterocephalus glaber Rüppell, 1842) studied with ultrasound biomicroscopy and 3D‐ultrasonography. PLoS ONE 6, e17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero Y, Bueno M, Ramirez R, Álvarez D, Sembrat JC, Goncharova EA, Rojas M, Selman M, Mora AL, Pardo A (2016) mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 15, 1103–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell 146, 682–695. [DOI] [PubMed] [Google Scholar]
- Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet‐Dit‐Félix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, Aebischer P, Sandi C, Rinsch C, Auwerx J (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 222, 879–888. [DOI] [PubMed] [Google Scholar]
- Salmon AB (2012) Oxidative stress in the etiology of age‐associated decline in glucose metabolism. Longev Healthspan 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, Mammucari C, Meskers CG, Pallafacchina G, Paoli A, Pion D, Roceri M, Romanello V, Serrano AL, Toniolo L, Larsson L, Maier AB, Muñoz‐Cánoves P, Musarò A, Pende M, Reggiani C, Rizzuto R, Schiaffino S (2013) Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1‐Akt‐mTOR‐FoxO pathway. Biogerontology 14, 303–323. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Sabatini DM (2005) Redox regulation of the nutrient‐sensitive raptor‐mTOR pathway and complex. J. Biol. Chem. 280, 39505–39509. [DOI] [PubMed] [Google Scholar]
- Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS (2002) Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 34, 379–388. [DOI] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN (2000) TOR, a central regulator of cell growth. Cell 103, 253–262. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE (2011) FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J. Biol. Chem. 286, 7468–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo AY, Joseph A‐M, Dutta D, Hwang JCY, Aris JP, Leeuwenburgh C (2010) New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 123, 2533–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba‐Fukushima K, Inoshita T, Hattori N, Imai Y (2014) PINK1‐mediate phosphorylation of Parkin boosts Parkin activity in Drosophila . PLoS Genet. 10, e1004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibamura A, Ikeda T, Nishikawa Y (2009) A method for oral administration of hydrophilic substances to Caenorhabditis elegans: effects of oral supplementation with antioxidants on the nematode lifespan. Mech. Ageing Dev. 130, 652–655. [DOI] [PubMed] [Google Scholar]
- Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J (2016) Aging and autophagy in the heart. Circ. Res. 118, 1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen‐Schiemke J, Raghavakaimal S, Nair KS (2005) Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl Acad. Sci. USA 102, 5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A, Gírio A, Cebola I, Santos CI, Antunes F, Barata JT (2011) Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR‐dependent IL‐7‐mediated viability of T‐cell acute lymphoblastic leukemia cells. Leukemia 25, 960–967. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Orr WC (2012) The redox stress hypothesis of aging. Free Radic. Biol. Med. 52, 539–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Ku H‐H, Agarwal S, Forster ML, Lal H (1994) Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech. Ageing Dev. 84, 121–133. [DOI] [PubMed] [Google Scholar]
- Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG (2015) Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFβ1. Aging Cell 14, 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Yang S (2006) Hydrogen peroxide: a signaling messenger. Antioxid. Redox Signal. 8, 243–270. [DOI] [PubMed] [Google Scholar]
- Suh Y (2001) Age‐specific changes in expression, activity and activation of the c‐Jun NH2‐terminal kinase and p38 mitogen‐activate protein kinases by methyl methanesulfonate in rats. Mech. Ageing Dev. 122, 1791–1811. [DOI] [PubMed] [Google Scholar]
- Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu R‐M, Hagen TM (2004) Decline in transcriptional activity of Nrf2 causes age‐related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl Acad. Sci. USA 101, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wheeler CT, Yolitz J, Laslo M, Alberico T, Sun Y, Song Q, Zou S (2014) A mitochondrial ATP synthase subunit interacts with TOR signaling to modulate protein homeostasis and lifespan in Drosophila . Cell Rep. 8, 1781–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP (2009) Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a‐dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Kass DA (2007) Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 49, 241‐248. [DOI] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yorda B, Shadel GS, Iwasaki A (2009) Absence of autophagy results in reactive oxygen species‐dependent amplification of RLR signaling. Proc. Natl Acad. Sci. USA 106, 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ED, Nuñez G, Barr FG, Guan KL (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741–16746. [DOI] [PubMed] [Google Scholar]
- Tapia PC (2006) Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: ‘Mitohormesis’ for health and vitality. Med. Hypotheses 66, 832–843. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC, Taylor GA, Plusa SM, Needham SJ, Greaves LC, Kirkwood TB, Turnbull DM (2003) Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 112, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J (2015) Mitochondrial maintenance failure in aging and role of sexual dimorphism. Arch. Biochem. Biophys. 576, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P (2006) Selective killing of oncogenically transformed cells through a ROS‐mediated mechanism by beta‐phenylethyl isothiocyanate. Cancer Cell 10, 231–242. [DOI] [PubMed] [Google Scholar]
- Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; Regulation by AKT and 14‐3‐3 proteins. Biochim. Biophys. Acta 1813, 1938–1945. [DOI] [PubMed] [Google Scholar]
- Unoki M, Nakamura Y (2001) Growth‐suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 20, 4457–4465. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, Mansouri A, Van Remmen H, van der Meulen JH, Larkin L, Richardson AG, McArdle A, Faulkner JA, Jackson MJ (2006) Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell 5, 109–117. [DOI] [PubMed] [Google Scholar]
- Vina J, Gomez‐Cabrera MC, Borras C, Froio T, Sanchis‐Gomar F, Martinez‐Bello VE, Pallardo FV (2009) Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 61, 1369–1374. [DOI] [PubMed] [Google Scholar]
- Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ (2000) Natural selection: evolution of lifespan in C. elegans . Nature 405, 296–297. [DOI] [PubMed] [Google Scholar]
- Wanagat J, Cao Z, Pathare P, Aiken JM (2001) Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 15, 322–332. [DOI] [PubMed] [Google Scholar]
- Wang X, McCullough KD, Franke TF, Holbrook NJ (2000) Epidermal growth factor receptor‐dependent Akt activation by oxidative stress enhances cell survival. J. Biol. Chem. 275, 14624–14631. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H (2005) JNK extends life span and limits growth by antagonizing cellular and organism‐wide responses to insulin signaling. Cell 121, 115–125. [DOI] [PubMed] [Google Scholar]
- Webb AE, Brunet A (2014) FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 39, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S (2011) Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White Z, White RB, McMahon C, Grounds MD, Shavlakadze T (2016) High mTORC1 signaling is maintained, while protein degradation pathways are perturbed in old murine skeletal muscles in the fasted state. Int. J. Biochem. Cell Biol. 78, 10–21. [DOI] [PubMed] [Google Scholar]
- Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411. [Google Scholar]
- Williamson D, Gallagher P, Harber M, Hollon C, Trappe S (2003) Mitogen‐activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle. J. Physiol. 547, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S (2010) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans . PLoS Biol. 8, e2000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TP (2010) The role of ubiquitin in autophagy‐dependent protein aggregate processing. Genes Cancer 1, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, Okada M, Guan KL, Inoki K (2011) Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2‐Rheb GTPase pathway. J. Biol. Chem. 286, 32651–32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lam PY, Han D, Cadenas E (2009) Activation of c‐Jun‐N‐terminal kinase and decline of mitochondrial pyruvate dehydrogenase activity during brain aging. FEBS Lett. 583, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]