

Summary
A major obstacle in predicting and preventing the development of autoimmune type 1 diabetes (T1D) in at‐risk individuals is the lack of well‐established early biomarkers indicative of ongoing beta cell stress during the pre‐clinical phase of disease. Recently, serum levels of the α cytoplasmic isoform of heat‐shock protein 90 (hsp90) were shown to be elevated in individuals with new‐onset T1D. We therefore hypothesized that hsp90α could be released from beta cells in response to cellular stress and inflammation associated with the earliest stages of T1D. Here, human beta cell lines and cadaveric islets released hsp90α in response to stress induced by treatment with a combination of pro‐inflammatory cytokines including interleukin‐1β, tumour necrosis factor‐α and interferon‐γ. Mechanistically, hsp90α release was found to be driven by cytokine‐induced endoplasmic reticulum stress mediated by c‐Jun N‐terminal kinase (JNK), a pathway that can eventually lead to beta cell apoptosis. Cytokine‐induced beta cell hsp90α release and JNK activation were significantly reduced by pre‐treating cells with the endoplasmic reticulum stress‐mitigating chemical chaperone tauroursodeoxycholic acid. The hsp90α release by cells may therefore be a sensitive indicator of stress during inflammation and a useful tool in assessing therapeutic mitigation of cytokine‐induced cell damage linked to autoimmunity.
Keywords: endoplasmic reticulum stress, heat‐shock protein 90, inflammation, Jun N‐terminal kinase, type 1 diabetes
Abbreviations
- DMOG
dimethyloxaloylglycine
- ER
endoplasmic reticulum
- HIF‐1α
hypoxia inducible factor 1α
- hsp
heat‐shock protein
- IFN‐γ
interferon‐γ
- IL‐1β
interleukin‐1β
- iNOS
inducible nitric oxide synthase
- JNK
c‐Jun N‐terminal kinase
- MAPK
mitogen‐activated protein kinase
- NO
nitric oxide
- T1D
type 1 diabetes
- TNF‐α
tumour necrosis factor‐α
- TUDCA
tauroursodeoxycholic acid
- UPR
unfolded protein response
Introduction
Type 1 diabetes (T1D) is a T‐cell‐mediated autoimmune disease that results in the targeted destruction of pancreatic beta cells. Host immune responses directly targeting beta cells for destruction in T1D simultaneously promote beta cell‐intrinsic stress responses that accelerate cell death and loss of insulin production.1 This latent period of beta cell stress and death has recently been defined as stage 1 in the three‐stage progression to T1D.2 During this stage, autoimmunity and beta cell loss have begun; however, sufficient insulin‐secreting beta cells remain to inhibit overt hyperglycaemia and other clinical symptoms. In some patients, this pre‐clinical phase occurs over a number of months, whereas in others it can last many years.2, 3
Limitations in detecting beta cell stress during this clinically latent stage of T1D constitute a major obstacle in the implementation of effective disease prevention strategies.3 Serum levels of the α cytoplasmic isoform of heat‐shock protein 90 (hsp90) were recently shown to be elevated in individuals with new‐onset T1D compared with control subjects.4 Consistent with this observation, individuals with T1D had higher levels of circulating IgG1 and IgG3 class‐switched autoantibodies to hsp90. These antibody isotypes are most commonly produced during T helper type 1‐mediated immune responses, the predominant cell‐mediated immune response in T1D.5
Heat‐shock protein 90 is a member of the heat‐shock family of molecular chaperones. Within the cell, these chaperones assist their respective client proteins in folding and intracellular transport. The hsp90 clients are involved in cellular processes including signal transduction, protein trafficking, receptor maturation and immunity.6 The release of cytosolic hsp90α from certain cell types has been reported in response to specific stresses.7 Vascular smooth muscle cells release hsp90α in response to oxidative stress,8 while human fibroblasts release hsp90α in response to hypoxia and hypoxia inducible factor 1α (HIF‐1α) activation.9 Yet, whether or not hsp90α is released from pancreatic beta cells in response to cellular stresses associated with T1D remains untested.
Several markers of stress have been detected in beta cells during the latent period of T1D. Endoplasmic reticulum (ER) stress precedes the development of T1D in the non‐obese diabetic mouse model,10 and some ER stress markers are expressed in human islets from individuals with T1D.11 Before the onset of T1D, beta cells experience inflammatory stress brought on by insulitis, the infiltration of immune cells into the pancreatic islets of Langerhans. During insulitis, activated macrophages, natural killer cells, and T cells secrete pro‐inflammatory cytokines such as interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) that promote beta cell dysfunction and death.12, 13, 14 Beta cells are also sensitive to reactive oxygen species‐mediated oxidative stress, which can result from glucotoxicity.15
Here, human beta cell lines and cadaveric islets released elevated levels of hsp90α in response to a combination of pro‐inflammatory cytokines, IL‐1β, TNF‐α and IFN‐γ. Release of hsp90α was not linked to cellular inducible nitric oxide synthase (iNOS) or HIF‐1α activity. Rather, ER stress mediated by c‐Jun N‐terminal kinase (JNK) appeared to play a key role in hsp90α release. Beta cell hsp90α release was attenuated by pre‐treatment with tauroursodeoxycholic acid (TUDCA), which protects human beta cells against JNK‐mediated, pro‐inflammatory cytokine‐induced apoptosis.16 TUDCA treatment reduced beta cell JNK phosphorylation in response to cytokine stress. Pharmacological inhibition and small interfering RNA (siRNA)‐mediated knockdown of JNK attenuated hsp90α release in response to cytokine stress. Although p38 mitogen‐activated protein kinase (MAPK) was also activated by cytokine stress, inhibition of this kinase did not impact cellular hsp90α release. Hence, studies here provide mechanistic evidence supporting a role for extracellular hsp90α as a non‐invasive marker of human beta cell stress in response to inflammation, which may be useful in gauging therapeutic interventions to mitigate stress in these cells.
Materials and methods
Cell culture
The human beta cell lines βLox5 and 1.1B4 17 were kindly provided by Dr Clayton E. Mathews (University of Florida) and cultured as described elsewhere.18, 19 Human cadaveric islets were obtained from the Integrated Islet Distribution Program (City of Hope). Donors were adults ranging 28–48 years of age with no history of type 2 diabetes (Table 1). Intact islets were maintained in Connaught Medical Research Laboratories 1066 media (Gibco, Grand Island, NY) with 10% fetal bovine serum (HyClone, Logan, UT), 50 U/ml penicillin and 50 μg/ml streptomycin (Corning, Corning, NY), and 2 mm l‐glutamine (Corning).
Table 1.
Human islet donor profiles
| UNOS ID No. | Sex | Age | BMI | Race | Islet purity (%) | Islet viability (%) | Experimental use of islets |
|---|---|---|---|---|---|---|---|
| AAJY035 | M | 40 | 38·91 | Caucasian | 95 | 99 | ELISA, qRT‐PCR |
| AAKE155 | M | 36 | 33·8 | Caucasian | 95 | 95 | ELISA, Viability, qRT‐PCR |
| ABAF490 | F | 39 | 45·2 | Caucasian | 80 | 98 | ELISA, Viability, qRT‐PCR |
| ABDT221 | M | 28 | 22·9 | Caucasian | 85 | 94 | ELISA, Viability |
| ABEF251 | M | 48 | 31·2 | Caucasian | 90 | 95 | ELISA, Viability, qRT‐PCR |
The United Network for Organ Sharing (UNOS) identification number is specified for each organ donor. Organ donors were characterized by sex, age, body mass index (BMI), and race. Percentages indicate the purity and viability of cells in human islet preparations. The specific experimental use of islets is indicated for each donor.
Stress induction
For stress induction, human βLox5 and 1.1B4 cells were cultured at 90% confluency and cadaveric islets at a density of 133 islets/ml. As a control, cells were treated with media. For classical ER stress, cells were treated with 0·1 μm thapsigargin (Cayman, Ann Arbor, MI), an inhibitor of sarcoendoplasmic reticulum Ca2+ ATPases, for 24 hr at 37°. To induce inflammatory stress, cells were treated with 5 ng/ml human recombinant IL‐1β (eBioscience, San Diego, CA), 10 ng/ml human recombinant TNF‐α (PeproTech, Rocky Hill, NJ), and 100 ng/ml human recombinant IFN‐γ (PeproTech). To examine glucotoxicity, cells were treated with medium with a final d‐glucose (Sigma‐Aldrich, St Louis, MO) concentration of 33·3 mm. Cell viability and plasma membrane integrity were determined during stress and other treatments by trypan blue exclusion and a lactate dehydrogenase cytotoxicity assay kit (Pierce, Waltham, MA).
Stress mitigating agents
βLox5 cells were treated with pharmacological agents for 6 hr before 24 hr pro‐inflammatory cytokine exposure. Optimal drug concentrations were selected based on cell viability and drug efficacy. Cells were treated with 100 μm 1400W (Cayman) to inhibit iNOS activity, 10 nm chetomin (Cayman) to inhibit HIF‐1α activity, 100 μm dimethyloxaloylglycine (DMOG) (Sigma‐Aldrich) to stabilize HIF‐1α, 0·2 and 1 mm TUDCA (Millipore, Billierica, MA) to mitigate ER stress, 10 μm SP600125 (Santa Cruz Biotechnology, Inc., Dallas, TX) to inhibit JNK signalling, or 5 and 10 μm SB202190 (Sigma‐Aldrich) to inhibit p38 MAPK activation.
ELISA
An ELISA kit for human hsp90α (Enzo, Exeter, UK) was used per manufacturer's instructions. To detect hsp90α in exosomes, vesicles were isolated from cytokine stressed beta cells using the ExoQuick‐TC kit (Systems Biosciences, Palo Alto, CA) per the manufacturer's instructions. Levels of hsp90α were detected in exosomes and non‐exosomal fractions of spent beta cell media by ELISA. Interleukin‐6 levels were quantified by ELISA using anti‐human IL‐6 capture and biotin‐conjugated antibodies (Invitrogen, Carlsbad, CA).
Immunoblotting
Cell lysates were prepared in 10 mm Tris–HCl pH 6·8, 150 mm NaCl and 1% Triton‐X 100 with protease (Sigma‐Aldrich) and phosphatase (Cell Signaling Technology, Danvers, MA) inhibitors. Lysates (20–80 μg of protein per lane) were resolved by SDS–PAGE and immunoblotted for protein detection.20
Antibodies
Antibodies to hsp90α were purchased from Enzo (9D2), while antibodies to phospho‐SAPK/JNK (Thr183/Tyr185) (81E11), total SAPK/JNK (rabbit polyclonal), phospho‐p38 MAPK (Thr180/Tyr182) (D3F9), or total p39 MAPK (rabbit polyclonal) were purchased from Cell Signaling Technology. Actin antibody was from Thermo Fisher Scientific (Waltham, MA) (ACTN05), and GAPDH antibody was obtained from Millipore (6C5). For immunoblots, primary antibodies were detected via peroxidase‐conjugated AffiniPure F(ab’)2 fragment goat anti‐mouse, anti‐rabbit, or anti‐rat IgG (H+L) (Jackson ImmunoResearch Laboratories, Inc., Bar Harbor, ME).
Quantitative RT‐PCR
Total cellular RNA was isolated using an RNeasy Mini Kit (Qiagen, Hilden, Germany), and cDNA was synthesized using a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) per manufacturers’ protocols. Amplification of cDNA was achieved by quantitative RT‐PCR using the 7500 Fast Real‐Time PCR System (Applied Biosystems). Commercial TaqMan gene expression assays were used to detect human ATF3, ATF4, B2M, DDIT3, GAPDH, HIF1A, HSPA5, HSP90B1, IL6, NOS2, and VEGFA, while a custom assay for human XBP1s was used to detect this transcript (Forward primer: 5′ CCTGGTTGCTGAAGAGGAG 3′, reverse primer: 5′ AGTCAATACCGCCAGAATCC 3′, and probe: 5′ CCTGCACCTGCTGCGGACTC 3′) (Applied Biosystems). Gene expression was presented as target mRNA levels detected relative to GAPDH levels (delta C T method).
siRNA transfection
βLox5 cells were grown in antibiotic‐free medium and transfected with 100 nm SignalSilence® non‐targeted control or SAPK/JNK siRNA (Cell Signaling Technology) for 72 hr using Lipofectamine 2000 (Invitrogen). Protein knockdown was confirmed by immunoblotting.
Statistics
For comparisons between two groups, statistical significance was determined using a two‐tailed, unpaired t test. For quantitative RT‐PCR experiments in which two treatments were tested on samples from the same human donor, a two‐tailed, ratio paired t‐test was used. To determine statistical significance among three or more groups, a one‐way analysis of variance was used with Dunnett's test to correct for multiple comparisons, each experimental mean being compared with the control mean. In experiments with two independent variables, statistical significance was determined using a two‐way analysis of variance with Tukey's test to correct for multiple comparisons between groups. A P‐value < 0·05 was considered statistically significant. Statistics were calculated using prism 6·04 (GraphPad Software, Inc., San Diego, CA).
Results
Beta cell hsp90α release in response to stress
Beta cell stress is a major factor contributing to the development of T1D, compromising the integrity of pancreatic islets and often leading to beta cell loss during the latent period of disease. To test whether overt stress could promote hsp90α release from human pancreatic beta cells, βLox5 and 1.1B4 cells were exposed to several agents known to induce established cellular stress pathways including thapsigargin, pro‐inflammatory cytokines, and culture media containing 33·3 mm glucose. Interestingly, both βLox5 and 1.1B4 cells released hsp90α in response to cytokine stress but not with exposure to thapsigargin or high glucose (Fig. 1a,b). Moreover, human cadaveric islets also released hsp90α in response to cytokine stress (Fig. 1c). These results suggest that hsp90α may be released from human beta cells in response to inflammatory stress. Assays to detect changes in cell viability and plasma membrane integrity indicated that hsp90α was not passively released as a result of cell death at 24 hr (Fig. 1d–g).
Figure 1.
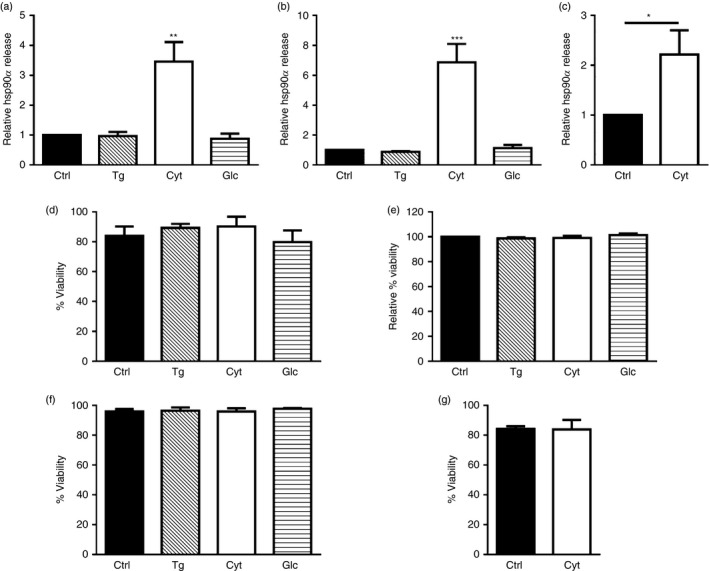
Heat‐shock protein 90α (hsp90α) was released by human beta cells in response to cytokine stress. Human beta cell lines βLox5 (a) and 1.1B4 (b) were treated for 24 hr with media alone (Ctrl); thapsigargin (Tg); interleukin 1β (IL‐1β), tumour necrosis factor‐α (TNF‐α), and interferon‐γ (IFN‐γ (Cyt); or culture media containing 33·3 mm glucose (Glc), while primary human islets were treated 24 hr with pro‐inflammatory cytokines only (c). Extracellular hsp90α released into the culture media was detected by ELISA. Data for extracellular hsp90α release are presented as relative to cells cultured under control conditions. Hsp90α values were detected between 0·7–4·9 ng/ml for βLox5, 0·9–6·4 ng/ml for 1·1B4, and 0·9–20·1 ng/ml for human islets. Changes in cell viability (%) were determined by trypan blue exclusion for βLox5 cells (d), 1·1B4 cells (f), or human cadaveric islets (g). Plasma membrane integrity or leakiness was monitored by detecting cellular release of cytoplasmic lactate dehydrogenase, and calculated as a relative value comparing cells cultured under control or stress conditions (e). Data are mean + SEM of n = 3 experiments or n = 4 islet donors. *P < 0·05, **P < 0·01, ***P < 0·001.
Synergistic effects of cytokines on beta cell hsp90α release
In studies with human islets, full induction of beta cell stress requires exposure to IL‐1β, TNF‐α and IFN‐γ in concert.21 To determine if IL‐1β, TNF‐α or IFN‐γ alone or in combination was sufficient to promote beta cell hsp90α release, βLox5 cells were treated with individual or combinations of cytokines for 24 hr at 37°. Although IL‐1β alone along with combinations of two different cytokines induced a slight elevation in beta cell hsp90α release relative to control‐treated cells, by far the most robust release of hsp90α was observed when cells were treated with all three cytokines (Fig. 2). These results are consistent with previous studies showing that this combination of cytokines induces the most stress in human islets.21
Figure 2.
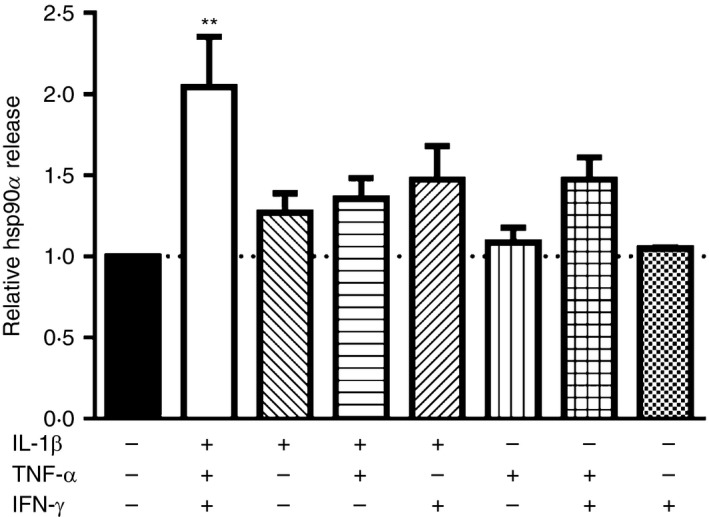
Pancreatic beta cells optimally release heat‐shock protein 90α (hsp90α) in response to a combination of interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ). Human βLox5 cells were treated with 5 ng/ml IL‐1β, 10 ng/ml TNF‐α and 100 ng/ml IFN‐γ alone or in various combinations for 24 hr at 37°. Media were harvested after incubation, and hsp90α levels were measured by ELISA. Data are presented as relative to control cells without cytokine. Extracellular hsp90α values ranged between 3·4 and 30·9 ng/ml. Data are mean + SEM of n = 3 experiments. **P < 0·01 one‐way analysis of variance with Dunnett's correction for multiple comparisons, each mean compared with no cytokine control.
Beta cell hsp90α association with exosomes
Several studies have shown that heat‐shock proteins including hsp90 are associated with extracellular vesicles known as exosomes.22, 23 Here, a small percentage of the extracellular hsp90α released by cytokine‐stressed beta cells was associated with exosomes, yet most (> 90%) of the released hsp90α was soluble and not exosome‐associated (Fig. 3a). Hsp90α and exosome release by beta cells were detected after 24 hr of cytokine stress, before any decreases in cell membrane integrity (Fig. 3b). Hsp90α is abundantly expressed by cells, and intracellular levels of this chaperone were only marginally altered by cytokine stress of beta cells. No change in hsp90α protein levels was observed in cytokine‐treated compared with control βLox5 cells (Fig. 3c,d). Cytokine‐treated 1.1B4 cells exhibited a slight decrease in hsp90α protein levels compared with control cells (Fig. 3e,f).
Figure 3.
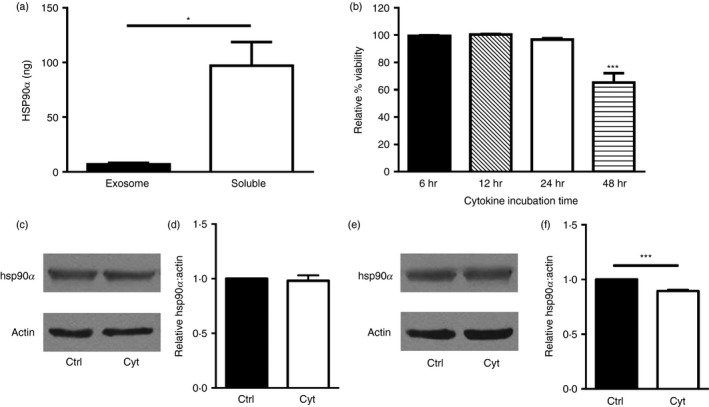
Mechanisms of heat‐shock protein 90α (hsp90α) release. Human βLox5 cells were treated with pro‐inflammatory cytokines and after 24 hr the extracellular medium was collected and fractionated to separate exosomes and the residual soluble media fraction, for analysis of hsp90α distribution. Total hsp90α levels in each fraction were determined by ELISA (a). βLox5 cells were treated with or without pro‐inflammatory cytokines for various times, and cellular release of cytoplasmic lactate dehydrogenase was monitored for up to 48 hr (b). Human beta cells βLox5 (c, d) and 1.1B4 (e, f) were treated for 24 hr with media alone (Ctrl) or interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) (Cyt). Total cell hsp90α and actin protein levels in each sample were assessed by immunoblotting. (c, e) Representative images of Western blots, (d, f) densitometry results indicating relative cellular hsp90 levels compared with actin expression from n = 3 experiments. Data are mean + SEM of n = 3 experiments. *P < 0·05, ***P < 0·001.
Beta cell hsp90α release in response to cellular iNOS activity
Release of hsp90α by vascular smooth muscle cells can be prompted by oxidative stress,8 and reactive oxygen species such as nitric oxide (NO) are detected in human islets exposed to cytokines.24 Furthermore, cytokine‐induced apoptosis of primary rat beta cells and the rat beta cell line INS‐1E is dependent on NO production.25 Cytokine exposure increased transcript expression for the NO‐generating enzyme iNOS in human βLox5 cells and cadaveric islets (Fig. 4a,b). To determine if beta cell NO production was linked to hsp90α release, βLox5 cells were pre‐treated with an iNOS inhibitor 1400W, which blocks cytokine‐induced iNOS production as shown with the rat beta cell line INS‐1.26 Treatment with 1400W failed to affect hsp90α release by cytokine‐stressed beta cells (Fig. 4c), indicating NO production is not responsible for cellular hsp90α release.
Figure 4.
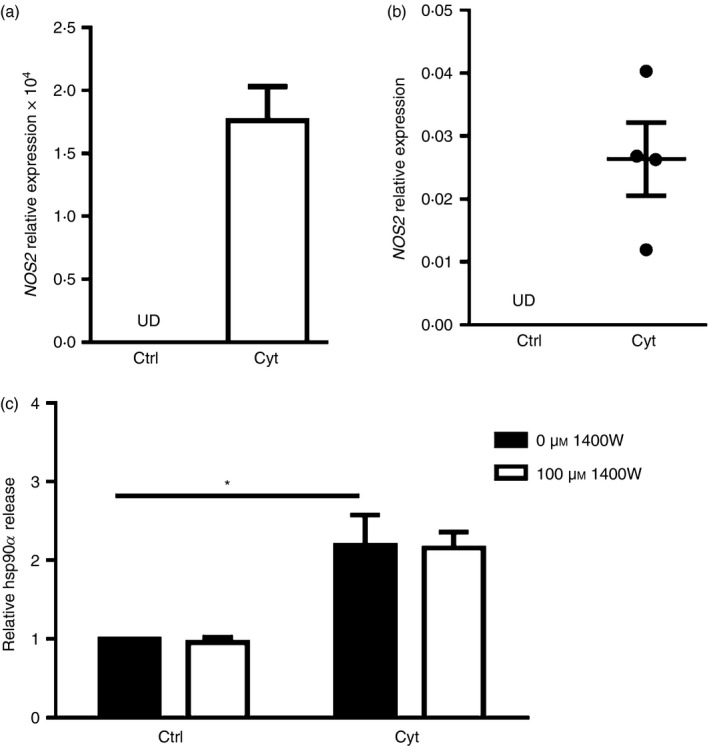
Heat‐shock protein 90α (hsp90α) was not released in response to inducible nitric oxide synthase (iNOS) activity. Human βLox5 (a) and human islets (b) were treated for 24 hr with media alone (Ctrl) or with interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) (Cyt). NOS2 expression was detected only after cytokine treatment as measured by quantitative RT‐PCR (UD, undetected). Human βLox5 cells were treated for 6 hr at 37° with medium alone or 100 μm 1400W, an iNOS inhibitor, followed by 24 hr treatment with pro‐inflammatory cytokines. Following these treatments, extracellular hsp90α levels were detected by ELISA. hsp90α release by beta cells was increased in response to cytokine exposure irrespective of iNOS inhibitor addition (c). Data are presented as relative to control cells cultured without cytokines or 1400W. Extracellular hsp90α values detected ranged between 10·0 and 34·6 ng/ml. Data are mean + SEM of n = 3 experiments or mean ± SEM of n = 4 islet donors. *P < 0·05.
Beta cell hsp90α release in response to HIF‐1α activity
The transcription factor HIF‐1α is known to be important for regulating beta cell function.27 Activation of HIF‐1α has been linked to hsp90α release by fibroblasts during hypoxia‐induced stress associated with wound healing.9 Reactive oxygen species can stabilize HIF‐1α even under normoxic conditions,28 whereas pro‐inflammatory cytokines induce nuclear factor‐κB activation leading to increased HIF‐1α gene expression.29 Pro‐inflammatory cytokine treatment prompted increased expression levels of HIF1A and the HIF‐1α target VEGFA in βLox5 (Fig. 5a,b) and islets (Fig. 5c,d), indicating an increase in HIF‐1α activity. βLox5 cells were treated with chetomin, a HIF‐1α inhibitor, before pro‐inflammatory cytokine exposure. This inhibitor promoted even greater hsp90α release by cytokine‐stressed beta cells (Fig. 5e). Moreover, pre‐treating βLox5 cells with DMOG, a prolyl hydroxylase inhibitor that stabilizes HIF‐1α, had no effect on beta cell hsp90α release in response to cytokines (Fig. 5f). Together these results suggest that activation of HIF‐1α or its downstream targets, is not responsible for beta cell hsp90α release upon cytokine stress.
Figure 5.
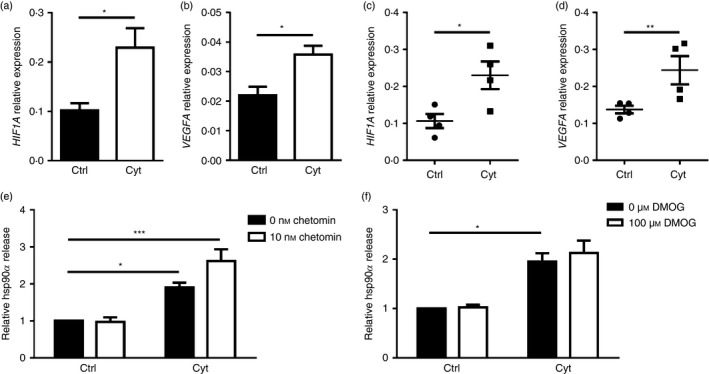
Heat‐shock protein 90α (hsp90α) was not released in response to altered hypoxia inducible factor 1α (HIF‐1α) activity. Human βLox5 and islets were treated for 24 hr with media alone (Ctrl) or interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) (Cyt). Expression levels of HIF1A and VEFGA transcripts were measured in βLox5 (a, b) and islets (c, d) by quantitative RT‐PCR. Human βLox5 cells were treated for 6 hr at 37° with media alone, 10 nm chetomin (e), a HIF‐1α inhibitor, or 100 μm DMOG (f), a HIF‐1α stabilizer, followed by 24 hr treatment with pro‐inflammatory cytokines. Extracellular hsp90α levels were detected by ELISA. Data are presented as relative to control cells without drug treatment or cytokines. Extracellular hsp90α values ranged between 1·4 and 46·5 ng/ml (e) and 3·0 and 23·9 ng/ml (f). Data are mean + SEM of n = 3 or n = 4 experiments or mean ± SEM of n = 4 islet donors. *P < 0·05, **P < 0·01, ***P < 0·001.
Expression of ER stress markers in cytokine‐treated beta cells
Signature features of ER stress have been detected in the pancreatic islets of non‐obese diabetic mice10 and individuals with T1D.11 Given thapsigargin, an inducer of classical ER stress, failed to promote beta cell hsp90α release (Fig. 1a,b), changes in the ER stress‐associated genes XBP1s, ATF4, ATF3, DDIT3, HSPA5 and HSP90B1 were compared in human βLox5 cells exposed to thapsigargin or pro‐inflammatory cytokines (Fig. 6a). Beta cell cytokine treatment resulted in a modest increase in the expression levels of XBP1s, ATF3 and HSP90B1 relative to control; however, changes in XBP1s and HSP90B1 were not significant when compared with the more robust responses of thapsigargin‐treated cells. HSP90B1 encodes GRP94, an ER‐retained chaperone with structural homology to cytoplasmic hsp90α.6 In contrast with thapsigargin, no differences were observed in expression levels of ATF4, DDIT3 and HSPA5 in cytokine‐exposed beta cells. In human islets, expression levels of ATF3, HSPA5 and HSP90B1 were significantly increased in response to cytokine stress (Fig. 6b), with some variation observed in individual islet donors. These results suggest that pro‐inflammatory cytokines may induce a mild or non‐canonical ER stress in pancreatic beta cells after 24 hr. Others have shown that some of these transcripts can be induced to a greater extent in human pancreatic beta cells after 48 hr of cytokine stress, although such prolonged treatments are often associated with reduced beta cell viability.16 In the present study, a significant drop in human beta cell viability was not detected until 48 hr post‐cytokine treatment (Fig. 3b). Hence, full induction of a classical ER stress response in human beta cells may require prolonged exposure to cytokines. Still even after 24 hr of cytokine exposure, human beta cells display signs of a mild or non‐canonical ER stress coinciding with hsp90α release.
Figure 6.
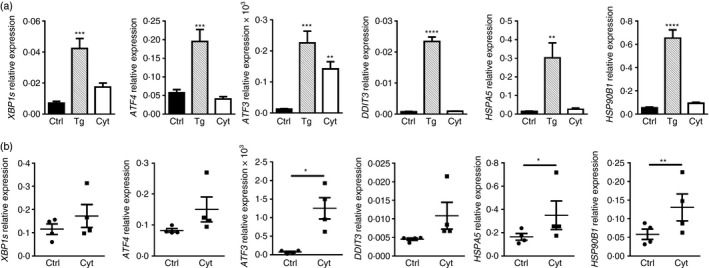
Pro‐inflammatory cytokine treatment induced a mild endoplasmic reticulum (ER) stress response in pancreatic beta cells after 24 hr. Human βLox5 cells were treated with media alone (Ctrl), thapsigargin (Tg), or interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) (Cyt) for 24 hr (a). Human islets were treated for 24 hr with pro‐inflammatory cytokines only (b). Expression levels of XBP1s, ATF4, ATF3 and DDIT3, the last of which encodes the CCAAT/enhancer‐binding protein homologous protein (CHOP), HSPA5, which encodes the binding immunoglobulin protein (BiP), and HSP90B1, which encodes the ER chaperone GRP94, were measured by quantitative RT‐PCR. Data represent the mean + SEM of n = 5 experiments or the mean ± SEM results from n = 4 islet donors. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Effects of TUDCA on cytokine‐induced hsp90α release and JNK activation in beta cells
TUDCA is a bile acid that functions as a chemical chaperone and reduces ER stress.30, 31, 32 This compound has potent anti‐apoptotic effects both in vitro and in vivo.32, 33, 34 Administration of TUDCA during the pre‐diabetic stage of T1D dramatically reduced diabetes incidence in two mouse models of disease.35 Pre‐treatment of βLox5 cells with TUDCA substantially inhibited hsp90α release in response to cytokine stress (Fig. 7a). At higher concentrations, TUDCA partially blocked up‐regulation of the ER stress marker ATF3 in cytokine‐treated cells (Fig. 7b), consistent with TUDCA mitigation of beta cell ER stress. Yet, TUDCA treatment did not block all pro‐inflammatory cytokine‐induced changes in beta cells as shown by the sustained induction of transcripts for β 2‐microglobulin (Fig. 7c). Recent studies with human beta cells have shown that cytokine‐induced apoptosis can be mediated by JNK activation downstream of the unfolded protein response (UPR) sensor IRE1, rather than NO production, as has been reported for rat beta cells. This cytokine‐induced JNK activation could be mitigated by pre‐treatment with TUDCA.16 Hence, levels of JNK phosphorylation were examined in βLox5 cells when TUDCA pre‐treatment was provided before cytokine exposure. TUDCA treatment of beta cells inhibited JNK phosphorylation at 30 min (Fig. 7e,f) and 24 hr (Fig. 7g,h) post‐cytokine treatment. Notably, thapsigargin failed to induce JNK phosphorylation in human beta cells (Fig. 7d). These results suggest that mitigating cytokine‐induced cell stress with TUDCA inhibits hsp90α release by pancreatic beta cells, potentially through alterations in the UPR and JNK activation.
Figure 7.
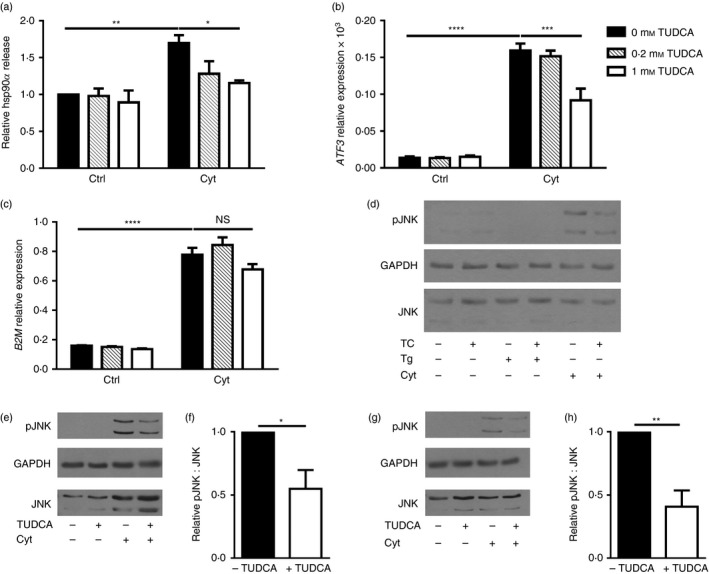
Pre‐treatment with TUDCA inhibited beta cell heat‐shock protein 90α (hsp90α) release, ATF3 up‐regulation, and c‐Jun N‐terminal kinase (JNK) phosphorylation in response to cytokine stress. Human βLox5 cells were treated with media alone, 0·2, or 1 mm TUDCA for 6 hr followed by 24 hr of stimulation with interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) (Cyt). Extracellular hsp90α release was measured by ELISA and calculated relative to control cells cultured without cytokines or TUDCA (a). Extracellular hsp90α values ranged between 5·0 and 20·5 ng/ml. ATF3 expression levels were measured by quantitative RT‐PCR (b). Gene expression levels of B2M, which encodes β2‐microglobulin, a component of the MHC class I, were measured by quantitative RT‐PCR (c). For (a–c), data are mean + SEM of n = 3 experiments. Phospho‐JNK, total JNK, and GAPDH levels were examined in βLox5 cells pre‐treated with or without TUDCA (TC) for 6 hr followed by 24 hr of thapsigargin (Tg) or pro‐inflammatory cytokine (Cyt) exposure (d). Shown is a representative Western blot image from n = 3 blots. Phospho‐JNK and total JNK levels were examined in βLox5 cells pre‐treated with or without TUDCA (TC) for 6 hr followed by 30 min (e, f) or 24 hr (g, h) of pro‐inflammatory cytokine (Cyt) exposure. JNK and phosphor‐JNK migrate as doublets on SDS–PAGE, while GAPDH protein levels were assessed in samples as a loading control. (e, g) Representative images, (f, h) densitometry results of phospho‐JNK to total JNK ratios in cytokine‐treated cells with and without TUDCA pre‐treatment for n = 3 or n = 4 blots. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Beta cell hsp90α release in response to JNK activity
To confirm that JNK activation was important in regulating hsp90α release from cytokine‐treated beta cells, βLox5 cells were pre‐treated with a JNK inhibitor, SP600125, before pro‐inflammatory cytokine exposure. JNK activation is required for the synthesis and secretion of cytokines such as IL‐6, and here JNK inhibition was demonstrated by analysis of beta cell IL‐6 production (Fig. 8a). The JNK inhibitor SP600125 attenuated hsp90α release in response to cytokine stress, suggesting that JNK activation is, at least in part, required for hsp90α release in pancreatic beta cells in response to cytokine stress (Fig. 8b). To further confirm that beta cell release of hsp90α was mediated by JNK activity, siRNA‐mediated knockdown of JNK was performed in βLox5 cells. Decreased JNK protein expression was observed in beta cells transfected with JNK siRNA relative to cells transfected with control siRNA (Fig. 8c,d). Here also, hsp90α release in response to cytokine stress was markedly reduced in beta cells transfected with JNK siRNA relative to cells transfected with control siRNA (Fig. 8e). Cytokine‐induced stress can also trigger the activation of another cellular kinase, p38 MAP kinase, which also promotes beta cell IL‐6 release. Indeed, beta cell IL‐6 secretion in response to pro‐inflammatory cytokines was in part due to the activation of p38 kinase, as demonstrated by the reduction in beta cell IL‐6 release upon inclusion of a p38 inhibitor (Fig. 8f). In contrast, treatment of beta cells with this p38 kinase inhibitor resulted in greater hsp90α release by βLox5 cells treated with pro‐inflammatory cytokines (Fig. 8g). These results support a role for JNK, but not p38, activity in promoting hsp90α release from pancreatic beta cells in response to cytokine‐induced stress.
Figure 8.
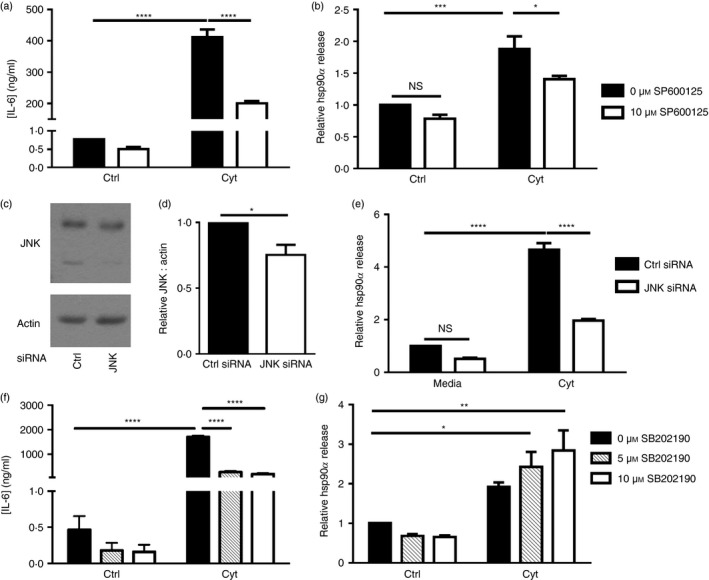
Cytokine‐induced heat‐shock protein 90α (hsp90α) release by pancreatic beta cells was mediated by c‐Jun N‐terminal kinase (JNK) and not p38 mitogen‐activated protein (MAP) kinase. Human βLox5 cells were treated with media alone or a JNK inhibitor (SP600125) for 6 hr before 24 hr stimulation with interleukin‐1β (IL‐1β), tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) (Cyt). Extracellular IL‐6 (a) and hsp90α (b) levels were measured by ELISA. Extracellular hsp90α levels were calculated relative to hsp90α release by control cells cultured without SP600125 or cytokines. Extracellular hsp90α values ranged between 6·8 and 77·1 ng/ml. Human βLox5 cells were transfected for 72 hr with control or JNK siRNA. JNK knockdown was assessed by immunoblotting (c, d). Transfected cells were then treated with media alone or IL‐1β, TNF‐α, and IFN‐γ (Cyt) for 24 hr. Extracellular hsp90α levels were analysed by ELISA (e). Data are presented as relative to control siRNA‐transfected cells treated with media alone. Hsp90α values ranged between 13·1 and 178·5 ng/ml. Human βLox5 cells were treated with media alone or the p38 MAP kinase inhibitor (SB202190) for 6 hr before 24 hr stimulation with IL‐1β, TNF‐α, and IFN‐γ (Cyt). Extracellular IL‐6 (f) and hsp90α (g) levels were measured by ELISA. Extracellular hsp90α levels were indicated relative to control cells treated without SB202190 or cytokines. Extracellular hsp90α values ranged between 2·8 and 34·1 ng/ml. Data are mean + SEM of n = 3 or n = 4 experiments. *P < 0·05, ***P < 0·001, ****P < 0·0001 (NS, not significant).
Discussion
The identification of a panel of accessible biomarkers to identify patients early during T1D development remains an important objective on the path to implementing new disease interventions and therapies. Understanding the connections between these markers and beta cell stress is also important as they may reveal new approaches to disease prediction and prevention. Sustained elevation of serum pro‐insulin and hsp90α were recently detected in children at the onset and during the honeymoon initiation period of T1D.4 The premature release of pro‐insulin by beta cells suggests defects in protein biosynthesis, post‐translational processing, and sorting associated with beta cell stress during the earliest stages of T1D. Given these data, we postulated that elevated levels of serum hsp90α may also be indicative of beta cell stress associated with inflammation.
Studies here clearly illustrate that selective release of the chaperone hsp90α by beta cells coincides with the initiation of other stress‐associated changes brought about by pro‐inflammatory cytokine exposure. Beta cell release of hsp90α was detected 24 hr post‐treatment (Fig. 1a–c) along with mild induction of NOS2 gene expression (Fig. 4a,b). However, inhibition of iNOS function had no effect on hsp90α release in response to cytokine stress (Fig. 4c). Interestingly, recent studies have shown that iNOS inhibition does not protect human islet cells from cytokine‐induced apoptosis,16 suggesting that this pathway may not be a major contributor to human beta cell stress, as had been demonstrated previously with rat beta cells.25
HIF‐1α is a transcription factor that regulates cellular stress responses to hypoxia and is known to play an important role in maintaining beta cell function.27 Here pro‐inflammatory cytokine exposure increased gene expression levels of HIF1A and the HIF target VEGFA in human βLox5 cells (Fig. 5a,b) and whole islets (Fig. 5c,d), suggesting that the HIF pathway is also induced in response to beta cell inflammatory stress. However, this pathway was found not to contribute to hsp90α release by beta cells (Fig. 5e,f), contrasting with the pathway for hsp90α release by human fibroblasts during hypoxia‐associated stress in wound healing.9
Beta cell hsp90α release in response to pro‐inflammatory cytokine exposure did coincide with increases in the expression levels of certain UPR‐associated genes (Fig. 6). Moreover, pre‐treatment with the chemical chaperone TUDCA inhibited βLox5 cell hsp90α release (Fig. 7a) and ATF3 (Fig. 7b) up‐regulation in response to cytokine stress, suggesting that hsp90α release from pancreatic beta cells may be linked to ER stress and induction of the UPR. However, exposure to thapsigargin, a classical inducer of ER stress, did not promote hsp90α release by beta cells (Fig. 1a,b) even though marked induction of UPR‐associated genes was observed with this sarcoendoplasmic reticulum Ca2+ ATPase inhibitor (Fig. 6a). Classically, the UPR is thought to increase ER protein‐folding capacity by up‐regulating expression of ER chaperones, such as BiP and GRP94, while simultaneously blocking translation of proteins not involved in alleviating ER stress.36, 37, 38 However, recent studies are identifying new roles for the UPR in regulating a variety of cellular processes, including metabolism, inflammation and apoptosis.39 One such ‘non‐canonical’ UPR or ER stress pathway is the activation of JNK.39
In the context of the UPR, JNK activation initiates inflammatory responses via AP‐1 activation and also promotes survival by initiating autophagy. JNK activation can also induce apoptosis in cells with unsurmountable levels of ER stress.36, 37 Interestingly, recent studies have shown that pro‐inflammatory cytokine‐mediated apoptosis of human beta cells is facilitated, at least in part, by activation of JNK and can be inhibited by the chemical chaperone TUDCA.16 In the current study, beta cell exposure to pro‐inflammatory cytokines activated JNK as detected by phosphorylation, which could be inhibited by pre‐treatment with TUDCA, similarly to hsp90α release (Fig. 7a,e,f). Supporting this key role for JNK in sensing beta cell stress, hsp90α release in response to pro‐inflammatory cytokines was also linked to JNK. Both pharmacological inhibition (Fig. 8b) and siRNA‐mediated knock‐down of JNK (Fig. 8e) inhibited hsp90α release from βLox5 cells in response to pro‐inflammatory cytokine exposure, suggesting that hsp90α release was indeed related to JNK activity. Exposure to thapsigargin did not induce JNK phosphorylation, consistent with the lack of hsp90α release in response to this classical inducer of ER stress (Figs 1a,b, 7d). These results point to a specific role for cytokine‐induced stress in promoting hsp90α release from cells.
Cellular release of hsp90α has been demonstrated in a number of studies; however, the underlying molecular mechanisms remain elusive, as hsp90α does not express an N‐terminal classic secretion signal peptide;40, 41 however, the possibility remains that hsp90α may contain a cryptic signal sequence that has not yet been identified. Others have proposed that hsp90α secretion is regulated by post‐translational modifications such as cleavage, phosphorylation and acetylation.40, 41 Several proteins involved in inflammation, cytoprotection and tissue repair are released by cells through an unconventional, rather than ER/Golgi‐dependent, release mechanism that requires cleavage by caspase‐1.42 Release of hsp70 from prostate cancer cells is mediated by a similar mechanism and is sensitive to weak bases.43 Yet treatment of beta cells here with weak bases failed to block hsp90α release (data not shown).
Several heat‐shock proteins including hsp70 and hsp90 are associated with exosomes.22, 23 However, analysis of the hsp90α released by βLox5 cells revealed only about 10% of the extracellular released chaperone was associated with exosomes (Fig. 3a). Yet, this does not preclude cellular release of exosomes as a conduit pathway for extracellular hsp90α. Given its cytoplasmic localization, hsp90α may associate loosely with protein ligands on the cytoplasmic face of exosomal vesicles. Upon vesicle release, hsp90α may be released as a chaperone from the surface of exosomes as ATP concentrations drop, resulting in increased levels of free hsp90α detected outside the cell. Alternatively, cytoplasmic hsp90α may be packaged or sequestered from the cytoplasm within autophagosomes or mutivesicular bodies during formation of these organelles, resulting in the release of hsp90α into the extracellular environment upon vesicle fusion with the plasma membrane. hsp90α has been identified in autophagosomes isolated from several types of human cells following starvation stress.44, 45 However, more work is undoubtedly needed to understand the specific secretory mechanism by which hsp90α is released from the cell.
Studies here are the first to demonstrate an association between cellular hsp90α release and inflammatory stress responses linked to the eventual apoptosis of human pancreatic beta cells.16 Given the evidence of beta cell stress before the onset of T1D in mice and humans, the current studies are also consistent with the observed elevated serum levels of hsp90α in patients with new‐onset T1D.4 Equally important, the current work establishes hsp90α as a readily detectable and non‐invasive indicator of beta cell stress, which may be useful in the testing of pharmacological agents to mitigate beta cell stress in vitro and in pre‐clinical studies.
Disclosure
The authors have no financial or commercial conflicts of interest to disclose.
Acknowledgements
G.J.O. researched data, analysed data and wrote the manuscript. L.P. researched data, analysed data and reviewed the manuscript. L.G, and S.N.D. researched data and reviewed the manuscript. C.E.M and D.C.T. provided critical reagents and protocols, contributed to discussion and reviewed the manuscript. J.S.B. conceived the study and edited the manuscript. J.S.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. This work was supported by the National Institutes of Health (RO1A1079065 and T32HL007910), the Leona M. and Harry B. Helmsley Charitable Trust (47‐2013‐523), and the JDRF (2‐SRA‐2014‐299).
References
- 1. Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D et al How does type 1 diabetes develop?: the notion of homicide or beta‐cell suicide revisited. Diabetes 2011; 60:1370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA et al Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015; 38:1964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watkins RA, Evans‐Molina C, Blum JS, DiMeglio LA. Established and emerging biomarkers for the prediction of type 1 diabetes: a systematic review. Transl Res 2014; 164:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Watkins RA, Evans‐Molina C, Terrell JK, Day KH, Guindon L, Restrepo IA et al Proinsulin and heat shock protein 90 as biomarkers of beta‐cell stress in the early period after onset of type 1 diabetes. Transl Res 2016; 168:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qin HY, Mahon JL, Atkinson MA, Chaturvedi P, Lee‐Chan E, Singh B. Type 1 diabetes alters anti‐hsp90 autoantibody isotype. J Autoimmun 2003; 20:237–45. [DOI] [PubMed] [Google Scholar]
- 6. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 2010; 11:515–28. [DOI] [PubMed] [Google Scholar]
- 7. Cheng CF, Li W. Secretion of Heat Shock Protein‐90 (Hsp90) by normal cells under stress or by tumor cells during invasion: why? Cancer Ther 2008; 6:765–72. [Google Scholar]
- 8. Liao DF, Jin ZG, Baas AS, Daum G, Gygi SP, Aebersold R et al Purification and identification of secreted oxidative stress‐induced factors from vascular smooth muscle cells. J Biol Chem 2000; 275:189–96. [DOI] [PubMed] [Google Scholar]
- 9. Li W, Li Y, Guan S, Fan J, Cheng CF, Bright AM et al Extracellular heat shock protein‐90α: linking hypoxia to skin cell motility and wound healing. EMBO J 2007; 26:1221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC et al Islet beta‐cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012; 61:818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marhfour I, Lopez XM, Lefkaditis D, Salmon I, Allagnat F, Richardson SJ et al Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012; 55:2417–20. [DOI] [PubMed] [Google Scholar]
- 12. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta‐cell loss in type 1 diabetes. Nature Rev Endocrinol 2009; 5:219–26. [DOI] [PubMed] [Google Scholar]
- 13. Allagnat F, Fukaya M, Nogueira TC, Delaroche D, Welsh N, Marselli L et al C/EBP homologous protein contributes to cytokine‐induced pro‐inflammatory responses and apoptosis in beta‐cells. Cell Death Differ 2012; 19:1836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meyerovich K, Fukaya M, Terra LF, Ortis F, Eizirik DL, Cardozo AK. The non‐canonical NF‐κB pathway is induced by cytokines in pancreatic beta cells and contributes to cell death and proinflammatory responses in vitro. Diabetologia 2016; 59:512–21. [DOI] [PubMed] [Google Scholar]
- 15. Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta‐cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003; 52:581–7. [DOI] [PubMed] [Google Scholar]
- 16. Brozzi F, Nardelli TR, Lopes M, Millard I, Barthson J, Igoillo‐Esteve M et al Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015; 58:2307–16. [DOI] [PubMed] [Google Scholar]
- 17. Lightfoot YL, Chen J, Mathews CE. Immune‐mediated beta‐cell death in type 1 diabetes: lessons from human beta‐cell lines. Eur J Clin Invest 2012; 42:1244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lightfoot YL, Chen J, Mathews CE. Role of the mitochondria in immune‐mediated apoptotic death of the human pancreatic beta cell line betaLox5. PLoS One 2011; 6:e20617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCluskey JT, Hamid M, Guo‐Parke H, McClenaghan NH, Gomis R, Flatt PR. Development and functional characterization of insulin‐releasing human pancreatic beta cell lines produced by electrofusion. J Biol Chem 2011; 286:21982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez L, McLetchie S, Gardiner GJ, Deffit SN, Zhou D, Blum JS. LAMP‐2C inhibits MHC class II presentation of cytoplasmic antigens by disrupting chaperone‐mediated autophagy. J Immunol 2016; 196:2457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rabinovitch A, Sumoski W, Rajotte RV, Warnock GL. Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J Clin Endocrinol Metab 1990; 71:152–6. [DOI] [PubMed] [Google Scholar]
- 22. Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z. Induction of heat shock proteins in B‐cell exosomes. J Cell Sci 2005; 118:3631–8. [DOI] [PubMed] [Google Scholar]
- 23. Buschow SI, van Balkom BW, Aalberts M, Heck AJ, Wauben M, Stoorvogel W. MHC class II‐associated proteins in B‐cell exosomes and potential functional implications for exosome biogenesis. Immunol Cell Biol 2010; 88:851–6. [DOI] [PubMed] [Google Scholar]
- 24. Oleson BJ, McGraw JA, Broniowska KA, Annamalai M, Chen J, Bushkofsky JR et al Distinct differences in the responses of the human pancreatic beta‐cell line EndoC‐βH1 and human islets to proinflammatory cytokines. Am J Physiol Regul Integr Comp Physiol 2015; 309:R525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M et al Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta‐cells. Diabetes 2005; 54:452–61. [DOI] [PubMed] [Google Scholar]
- 26. Ko SH, Ryu GR, Kim S, Ahn YB, Yoon KH, Kaneto H et al Inducible nitric oxide synthase‐nitric oxide plays an important role in acute and severe hypoxic injury to pancreatic beta cells. Transplantation 2008; 85:323–30. [DOI] [PubMed] [Google Scholar]
- 27. Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ et al Hypoxia‐inducible factor‐1α regulates beta cell function in mouse and human islets. J Clin Investig 2010; 120:2171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pouyssegur J, Mechta‐Grigoriou F. Redox regulation of the hypoxia‐inducible factor. Biol Chem 2006; 387:1337–46. [DOI] [PubMed] [Google Scholar]
- 29. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol 2009; 9:609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO et al Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006; 313:1137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab 2010; 12(Suppl 2):108–15. [DOI] [PubMed] [Google Scholar]
- 32. Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE et al Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress‐induced caspase‐12 activation. Hepatology 2002; 36:592–601. [DOI] [PubMed] [Google Scholar]
- 33. Gupta S, Li S, Abedin MJ, Noppakun K, Wang L, Kaur T et al Prevention of acute kidney injury by tauroursodeoxycholic acid in rat and cell culture models. PLoS One 2012; 7:e48950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci USA 2002; 99:10671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL et al Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med 2013; 5:211ra156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 2008; 8:663–74. [DOI] [PubMed] [Google Scholar]
- 37. Celli J, Tsolis RM. Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat Rev Microbiol 2015; 13:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol 2016; 16:469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arensdorf AM, Diedrichs D, Rutkowski DT. Regulation of the transcriptome by ER stress: non‐canonical mechanisms and physiological consequences. Front Genet 2013; 4:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X, Song X, Zhuo W, Fu Y, Shi H, Liang Y et al The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc Natl Acad Sci USA 2009; 106:21288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010; 10:537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keller M, Ruegg A, Werner S, Beer HD. Active caspase‐1 is a regulator of unconventional protein secretion. Cell 2008; 132:818–31. [DOI] [PubMed] [Google Scholar]
- 43. Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J immunol 2006; 177:7849–57. [DOI] [PubMed] [Google Scholar]
- 44. Dengjel J, Hoyer‐Hansen M, Nielsen MO, Eisenberg T, Harder LM, Schandorff S et al Identification of autophagosome‐associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol Cell Proteomics 2012; 11:M111–014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pallet N, Sirois I, Bell C, Hanafi LA, Hamelin K, Dieude M et al A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics 2013; 13:1108–20. [DOI] [PubMed] [Google Scholar]