

Summary
Fungal polyketide synthases (PKSs) are large, multi-domain enzymes that biosynthesize a wide range of natural products. A hallmark of these megasynthases is the iterative use of catalytic domains to extend and modify a series of enzyme-bound intermediates. A subset of these iterative PKSs (iPKSs) contains a C-methyltransferase (CMeT) domain that adds one or more S-adenosylmethionine (SAM)-derived methyl groups to the carbon framework. Neither the basis by which only specific positions on the growing intermediate are methylated (“programming”) nor the mechanism of methylation are well understood. Domain dissection and reconstitution of PksCT, the fungal non-reducing PKS (NR-PKS) responsible for the first isolable intermediate in citrinin biosynthesis, demonstrates the role of CMeT-catalyzed methylation in precursor elongation and pentaketide formation. The crystal structure of the S-adenosyl-homocysteine (SAH) coproduct-bound PksCT CMeT domain reveals a two-subdomain organization with a novel N-terminal subdomain characteristic of PKS C-methyltransferase domains and provides insights into cofactor and ligand recognition.
Keywords: PKS, polyketide, structure, domain deconstruction, iterative biosynthesis, catalytic programming, citrinin, C-methylation
eTOC
C-methylation of growing polyketide intermediates represents the simplest example of programming in iterative polyketide synthase catalysis. Storm et al. show the importance of methylation in citrinin biosynthesis and present the crystal structure of a C-methyltransferase in complex with SAH.
Introduction
PKSs and fatty acid synthases (FASs) utilize common strategies to produce an enormous number of natural products with highly diverse chemical scaffolds, including pigments, environmental toxins, and pharmacologically active substances (Weissman and Leadlay, 2005);(Crawford and Townsend, 2010). Simple precursor acyl substrates are tethered to the acyl carrier protein (ACP) by thioesterification of a post-translational phosphopantetheine arm. They are then extended by the ketosynthase (KS) through a determined number of decarboxylative condensation reactions with ACP-bound malonyl extender units supplied by the malonyl-CoA:ACP transacylase (MAT). In many cases, the newly extended intermediate is delivered to additional tailoring domains that perform programmed modifications at the α- or β-carbon. Modular PKSs (modPKSs) generally have a distinct module for each round of extension and modification, whereas iPKSs use the same domains repeatedly a defined number of times. How iPKS domains control which tailoring domains are used after a given round of extension is still largely unanswered and poses a significant challenge to engineering efforts.
Perhaps the simplest case of programmed tailoring among PKSs occurs during iterative C-methylation of fungal non-reduced polyketides, exemplified by the group VI and VII, or Clade III, families where a tetra- or pentaketide is C-methylated one or more times (Ahuja et al., 2012); (Kroken et al., 2003). Each round of extension produces a potentially enolizable nucleophilic α-carbon, but only some such positions are methylated. A more complete understanding of how methylation is programmed and the structural basis by which methylation occurs will be essential for future efforts to rationally influence product methylation patterns. Site-selective polyketide C-methylation would be a powerful tool to engineer new biosynthetic pathways, and many methyltransferases can accept SAM analogs with larger alkyl donors, further expanding potential scaffolds (Struck et al., 2012).
We selected PksCT as an initial candidate to explore non-reducing PKS (NR-PKS) C-methylation (Shimizu et al., 2005). Responsible for the first isolable intermediate in citrinin (1) biosynthesis (He and Cox, 2016), PksCT produces a triply methylated pentaketide that is C2–C7 cyclized by the product template (PT) domain and released as the benzaldehyde (2) by the C-terminal reductase domain (R) (Figure 1). Previous efforts to understand NR-PKS programming have benefited greatly from a deconstruction approach that allows for the extension domains to be separated from the tailoring domains (Crawford et al., 2008);(Vagstad et al., 2013);(Newman et al., 2014). A growing number of excised PKS domains have been structurally characterized, providing the molecular details of catalysis and substrate selection (Keatinge-Clay, 2012). We sought to expand this experimental approach to PksCT in order to isolate CMeT domain activity and explore the structural basis for NR-PKS C-methylation programming.
Figure 1. Proposed biosynthesis of 2 by PksCT.
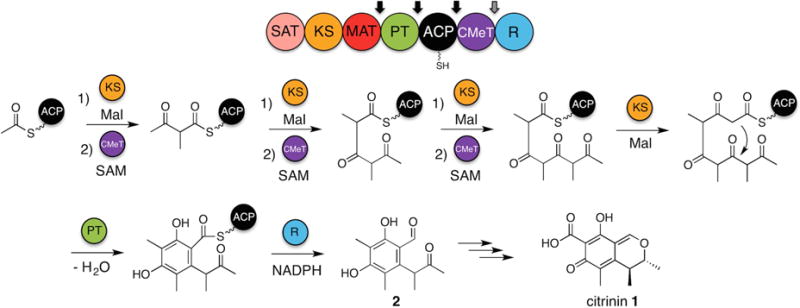
The domain architecture of PksCT is shown above, with arrows indicating points of domain dissection. Mal indicates a unit of ACP-bound malonyl used for decarboxylative condensation prior to methylation. Necessary domains and substrates are shown above and below the reaction arrows. See also Figure S1 and Item S1.
Results
Domain deconstruction and reconstitution of PksCT
Using the Udwary-Merski algorithm (Udwary et al., 2002) to recognize interdomain linker regions in a set of related proteins, a domain deconstruction approach was applied to PksCT and used to generate mono- and multidomain fragments (Figure S1A and Supplemental Procedures). All constructs gave soluble protein except for those containing the SAT domain in initial expressions. When multiple domain boundaries did not improve expression, we reassessed the reported exons using the FGENESH exon prediction suite (Solovyev et al., 2006). These results suggested alternate exon boundaries, which would include a conserved Trp195 in the SAT domain that was not present in the original annotated sequence. Total RNA was isolated from wild-type Monascus purpureus grown under citrinin producing conditions (Sakai et al., 2008),(Shimizu et al., 2005). The cDNA was synthesized, and a region containing the exon boundaries was amplified and subcloned. Five clones were sequenced and all verified that the revised prediction generated the correct exon boundaries (Figure S1B and Item S1). We propose an update to GenBank: AB167465, joining gene fragments 1197–1835 and 1892–9034. PksCT residue numbering used hereafter reflects this revision.
Earlier reports speculated that PksCT could use several potential starter units (Davison et al., 2012), but heterologous in vivo extracts suggested an acetyl starter (He and Cox, 2016). We confirmed this mode of initiation in vitro using a radiochemical assay (Foulke-Abel and Townsend, 2012). PksCT SAT was capable of loading an acetyl starter unit from [1-14C]-acetyl-CoA and transferring it to PksCT holo-ACP (Figure S1C). We then conducted a series of reconstitution reactions to identify the activity of the PksCT CMeT domain (Figures 2 and S2, Tables S2 and S4). Each individually expressed and purified fragment was combined as indicated with all of the expected substrates. Acetyl and malonyl units were included as the N-acetylcysteamine thioesters (SNAC), which serve as substitutes for the CoA thioesters used in vivo (Newman et al., 2014). Stock solutions of SAM and NADPH were freshly prepared.
Figure 2. In vitro reconstitution of PksCT.
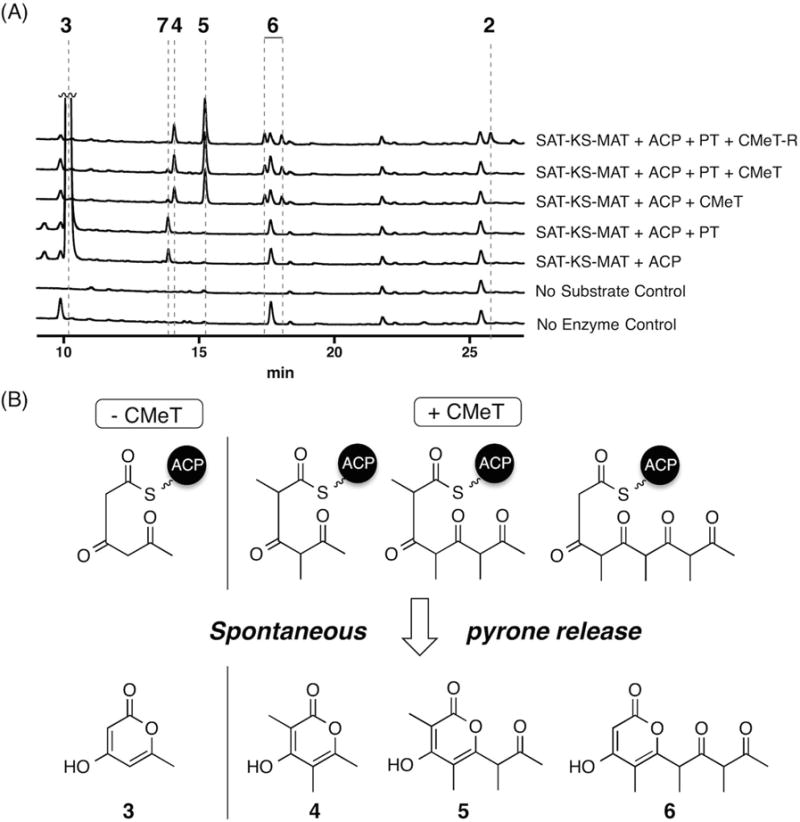
(A) HPLC absorbance traces are shown at 280 nm for the combination of domains indicated to the right. Traces are vertically offset and peak 3 is truncated for clarity. See also Figure S1 and Table S2.
(B) Spontaneous pyrone release of intermediates as tri-, tetra-, and pentaketide pyrones. High resolution UPLC-ESI-MS and UV-Vis data can be found in Table S4 and Figure S2.
The minimal PKS, SAT—KS—MAT and holo-ACP, with acetyl and malonyl substrates primarily generated triketide 3. Upon addition of PT, the product profile was not substantially different and no C2–C7 cyclized product was observed, even after attempts to chemically release any potentially cyclized intermediate with base or cysteamine (Belecki and Townsend, 2013). Addition of CMeT and SAM to the minimal PKS, however, resulted in the loss of 3 but the appearance of four new species corresponding to spontaneous pyrone release of methylated tri-, tetra-, and pentaketides 4, 5, and 6. The pentaketide 6 eluted as two peaks, identical by mass, that appear to be diastereomers as each collected peak equilibrates to the same two components upon isolation and reinjection. The minimal PKS reactions also contained small amounts of 7, having a mass consistent with a singly methylated form of 3. The disappearance of 7 in reactions containing CMeT suggests that it results from an on-pathway intermediate to 4, 5, and 6, and is discussed in detail below.
Full extension of the polyketide chain appears to rely upon the correct methylation of the growing linear intermediate, a feature seen previously in studies of the highly-reduced polyketide lovastatin (Cacho et al., 2015; Fisch et al., 2011; Ma et al., 2009). This series of intermediates indicates that methylation occurs at the thioester α-carbon iteratively following each extension and not processively on a fully elongated pentaketide. These methylated derailment products are observed in the presence of the PT and R domains as well. With all domains, AcSNAC, MalSNAC, SAM, and NADPH present, the expected post-PKS aldehyde 2 was observed also, albeit in low yield.
Several efforts to improve the efficiency of reconstituted PksCT were attempted, including addition of methylthioadenosine nucleosidase (MTAN) (Kitagawa et al., 2005) to hydrolyze the potentially inhibitory SAH co-product or inclusion of Mg2+, a cofactor used by some MTases (Liscombe et al., 2012). Previous work has demonstrated that NR-PKS deconstruction reduces the yield of post-PKS product and increases the abundance of derailment products (Newman et al., 2014);(Huitt-Roehl et al., 2015). In the case of deconstructed PksCT, it is possible that transfer of methylated polyketides from the KS active site to the CMeT active site allows for racemization of the methyl groups. Whether the KS, PT, or R domain active sites are structured for a specific methyl stereochemistry, and whether polyketides with scrambled methyl stereochemistry are inhibitory, is currently unknown. Additionally, in vivo experiments of citrinin biosynthesis indicate that co-expression of an adjacent gene, encoding the hydrolase CitA, significantly improves titres of 2 (He and Cox, 2016), though the specific role of CitA is still unclear.
Structure of PksCT CMeT in complex with SAH
The 1.65 Å crystal structure of the CMeT domain reveals an organization of two subdomains with the active site located at their interface (Figure 3A). PksCT CMeT was co-crystallized with the stable co-product S-adenosylhomocysteine (SAH) in rhombohedral space group H3 containing one protomer per asymmetric unit (Table S3), the structure was solved by SAD phasing and refined to R/Rfree: 0.19/0.22.
Figure 3. Crystal structure and ligand binding site of CMeT.
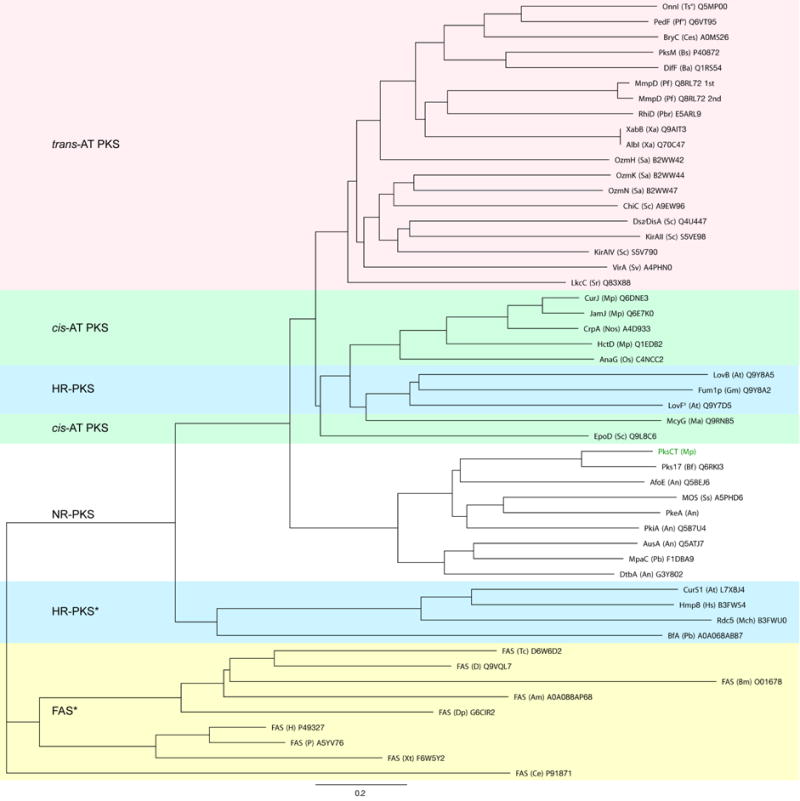
(A) CMeT is organized into an N-terminal linker (grey), an N-terminal subdomain (violet) and a C-terminal subdomain (yellow). The active site is located at the subdomain interface, Fo-Fc omit difference density at 2.5 σ level is shown for SAH. See also Table S3 and Figure S4.
(B) Topology of CMeT highlighting domain organization and substrate binding sites. α-Helices are numbered, β-strands are numbered relative to their position in the respective β-sheet B1 or B2. Helix 17 (dotted) is a 310 helix.
(C) Substrate interactions and ligand binding tunnel. The C-terminal subdomain laterally binds SAH and forms an active site tunnel (grey surface) along SAH. The back of this tunnel is lined with hydrophobic residues of the palm helix region and is closed by the N-terminal subdomain. The invariant residues Tyr1955 as well as His2067 together with Glu2093 face the ligand binding tunnel from opposite sides and are involved in catalysis. SAH contributes to the formation of an extended cavity for binding larger substrates. Difference density is depicted as in A. See also Figure S5.
(D) Schematic active site representation. Hydrogen bonds are indicated by dotted lines. Cα atoms are shown as spheres in the color of their respective subdomain.
The 202 amino acid (aa) C-terminal subdomain (residues 1959–2160) reveals a class l methyltransferase fold (Martin and McMillan, 2002), which is the most common methyltransferase fold in natural product biosynthetic enzymes, despite diverse acceptor substrates and low sequence conservation (Liscombe et al., 2012) (Figure 3B). The closest structural homologs are the SAM-dependent methyltransferase from Aquifex aeolicus (unpublished, PDB:3DH0), the methyltransferase domain of bacterial-AvHen1-CN (PDB: 3JWJ), a putative methyltransferase from Sulfolobus solfataricus (unpublished, PDB: 3I9F), NodS from Bradyrhizobium japonicum WM9 (PDB: 3OFK) and the methyltransferase domain of bacterial-CtHen1-C (PDB: 3JWG) with a Cα rmsd of 2.0–2.4 Å and 160–195 aligned residues. The catalytically inactive pseudo-methyltransferase (ψCMeT) of the human fatty acid synthase (FAS) aligns with a Cα rmsd of 2.5 Å (158 aligned aa) (Hardwicke et al., 2014) and is the closest structural homolog in carrier protein-dependent multienzymes. The C-terminal subdomain integrates into the N-terminal subdomain through a region around helix 15 that forms the base of the ligand binding tunnel, resembling an open palm (“palm helix region”), and is structurally conserved in mammalian FAS (Hardwicke et al., 2014; Maier et al., 2008), but deleted in insect FAS and some highly reducing-PKSs (HR-PKS) (Figure S3).
The 144 aa N-terminal CMeT subdomain (residues 1815–1958) shares an interface of 1,515 Å2 with the C-terminal subdomain and adopts an uncharacterized helical fold (Figure 3B). Such N-terminal subdomains of Class I methyltransferases that cap the SAH/SAM-binding pocket often serve a role in acceptor substrate selection (Peng et al., 2011). In the related mammalian FAS multienzymes, which comprise catalytically inactive ΨCMeTs domains and provide the only structural depiction of CMeT integration into a multienzyme, this region is considerably truncated and disordered (Hardwicke et al., 2014; Maier et al., 2008). Nevertheless, the N-terminal CMeT subdomains in FASs reveal helices at equivalent positions to helix 4, 7 and 9/10 with a Cα rmsd of 2.5 Å over 42 aligned aa, indicating a common evolutionary origin (Figure S4A). The N-terminal subdomain is connected to the preceding protein regions in PksCT by a 30 aa long linker containing two helices and wraps around the C-terminal subdomain in a groove with an interface area of 1,461 Å2. In mammalian FASs (Hardwicke et al., 2014; Maier et al., 2008) the N-terminal linker leading into ΨCMeT contacts this domain by appending a β-strand to the central β-sheet of the C-terminal subdomain (Figure S4B). In PksCT, however, the N-terminal linker (residues 1785–1814) wraps around the C-terminal subdomain in a prominent surface groove (Figure S4C). Consequently, in contrast to mammalian FAS, the N- and C-terminal linker ends are located in close proximity to each other (Figure S4C), indicating an alternate integration of CMeT in the PksCT multienzyme.
The active site of PksCT CMeT is located at the interface of the N- and C-terminal subdomains. Most of the active site is formed by the C-terminal subdomain, which binds the SAH parallel to the ligand binding tunnel, which is lined with hydrophobic residues of the palm helix region. Helix 9 and 10 of the N-terminal subdomain contributes four residues to the distal region of the ligand binding tunnel to form the opposite face (Figure 3C and 3D), and the highly conserved Tyr1955 splits the active site into a tunnel for the SAM co-substrate and the polyketide substrate. Remarkably, the homocysteine moiety of the SAH contributes to the formation of a pocket for extended substrates, which could accommodate varying polyketide chain lengths during elongation cycles 1–3. The SAH is tightly bound at an interface of 356 Å2 along one side of the co-substrate binding pocket via highly conserved residues: A GxGxGG (residues 1992–1997) motif forms hydrogen bonds with the homocysteine moiety, Asp2019 binds the ribose through two hydrogen bonds and the adenine moiety is positioned in a hydrophobic pocket formed by Leu2020 and Ile2045 with a backbone hydrogen bond to Ile2045.
The active site tunnel reveals a funnel like shape and branches off into the extended substrate cavity at its inner end, providing a central channel for Cα substrate alignment with regard to different polyketide chain length (Figures 3C and S5). The central channel is surrounded by a patch of hydrophobic residues (Figure 3C and 3D); the highly conserved Phe1942 as well as Leu1938 line the substrate entrance region and the conserved residues Leu2108 and Trp2111 and residues Trp2100, Val2101, Phe2105 of the palm helix would likely interact with the linear polyketide substrates (Figure S3). Together, these residues form a hydrophobic patch along which the hydrophobic backbone of substrates at various elongation states might slide into the active site. Interestingly, the residues involved in the hydrophobic patch are highly conserved among other NR-PKS CMeT domains – such as AusA, PkeA, MpaC, and DtbA – that install different methylation patterns than PksCT (Ahuja et al., 2012); (Regueira et al., 2011);(Yeh et al., 2013) and are functionally conserved in both cis- and trans-AT PKSs (Figure S3).
Although no obvious delineation amongst the NR-PKS CMeTs correlated with observed product methylation pattern, the number of available sequence–product pairs may be insufficient to identify specific programming motifs. Furthermore, a phylogenetic analysis of CMeT domains across multiple PKS/FAS families clearly demonstrated a branching between active CMeT and inactive ΨCMeTs (Figure 4). Active CMeTs clustered into three groups: trans-AT PKSs, cis-AT and HR-PKSs, and NR-PKSs. Active HR-PKS CMeT domains are nested among cis-AT CMeTs, perhaps reflecting similar embedding amongst reductive domains not present in NR-PKSs.
Figure 4. Phylogenetic analysis of 51 CMeT domains of PKS and FAS.
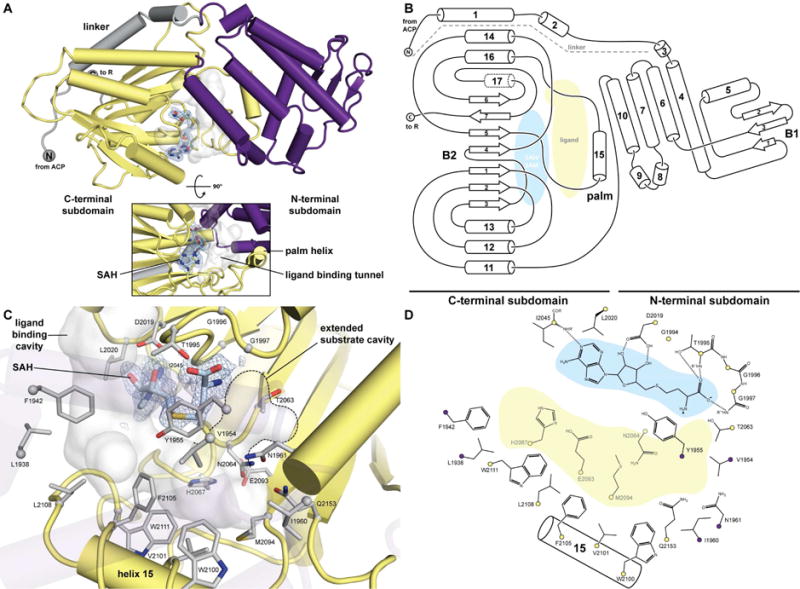
38 active CMeT domains of PKS and 13 inactive ΨCMeT domains of HR-PKS and FAS were aligned and phylogenetically analyzed (see Figure S4). Multienzyme family classifications are indicated in colored groups. Units are given as amino-acid substitutions per site. All sequences are labelled as “protein name (organism abbreviation) Uniprot number”. The sequence of PksCT corresponds to Item S1 (°, endosymbiont of this org anism; ‡, diketide synthase; *, inactive ΨCMeT domain)
Catalysis by CMeT
For catalysis, the α-carbon of the acceptor substrate presumably must be deprotonated to generate the enolate nucleophile for SN2-like attack at the methyl donor. The invariant residues Tyr1955 and His2067 together with Glu2093 face the ligand binding tunnel from opposite sides and are well positioned to act as catalytic residues. His2067 forms a hydrogen bridge with Glu2093 and might act as catalytic dyad for enolization, as previously proposed for the homocitrate synthase (Qian et al., 2008)(Figure 5B).
Figure 5. PksCT CMeT His2067 is essential for methyl transfer and positioned to act as the catalytic base.
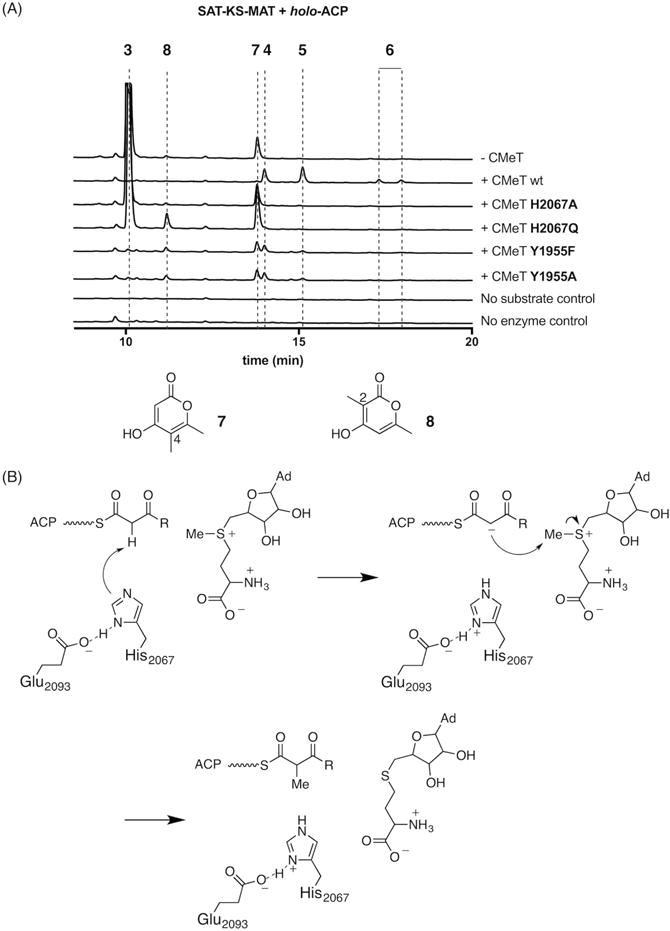
(A) In vitro reconstitution of PksCT SAT-KS-MAT and holo-ACP with CMeT mutants show that methylated products 4, 5, and 6 are not produced by His2067 mutants. Tyr1955 mutants are capable of generating methylated triketides, but not pentaketides, suggesting a role for this residue in acceptor substrate binding. Absorbance traces are shown at 280 nm for the CMeT variant indicated on the right and vertically offset; peak 3 is truncated for clairty. For mass and UV-Vis absorption data, see Table S4 and Figure S2.
(B) Proposed mechanism for methyl transfer. His2067 forms a catalytic dyad with Glu2093 and deprotonates the α-carbon to generate an enolate nucleophile capable of SN2-like attack at the methyl donor. Completion of the catalytic cycle by loss of the removed proton to solvent is not explicitly shown. R indicates the potential chain lengths described in Figure 1.
Reconstitution of the minimal PksCT with the CMeT His2067Ala mutant primarily gave triketide 3, matching the profile of the minimal PKS without CMeT (Figure 5A). This experimental observation demonstrated the essential role of His2067 for efficient methyl transfer and supports its likely role as the catalytic base. The more conservative His2067Gln mutant (Khaleeli et al., 2000);(Kyte and Doolittle, 1982) also produced 3 as a major product in addition to smaller amounts of two different triketides 7 and 8 identical by mass, but singly methylated at C-4 and C-2, respectively. Tyr1955Phe and Tyr1955Ala mutations appeared to have a lesser effect on catalysis. Neither produced significant amounts of 3, but instead yielded small amounts each of 4, 5, 7, and 8. The reduced amounts of 5 and the absence of 6, compared to wild-type, suggest that Tyr1955 may be especially important for methylation of tetra- and pentaketide substrates.
Because 7 was observed also in the absence of CMeT, but appears to be on-path to 4, 5, and 6, we speculate that some SAT–KS–MAT may be purified with a methylacetoacetyl unit bound to Cys555 in the KS active site. Such a substrate could arise from direct loading from methylacetoacetyl-CoA, an intermediate in isoleucine catabolism (Conrad et al., 1974), or perhaps homologation of KS-bound acetyl by endogenous methylmalonyl-CoA during heterologous expression [for closely related biochemistry see: (Steyn and Vleggaar, 1984; Steyn et al., 1981)]. A single round of extension of methylacetoacetyl, followed by spontaneous pyrone formation would result in 7, methylated at C-4. The decreased activity of the CMeT His2067Gln mutant could allow for a relative increase in 7 and the appearance of 8, methylated at C-2, where the first potential methylation is skipped but the second methylation is successful. It is known that polyketide extension is exceedingly rapid in NR-PKSs (Crawford et al., 2008);(Vagstad et al., 2012). Kinetic competition between chain elongation and programmed methylation where the latter is impaired in the His2067Gln mutant could lead to expected alkylation at C-4 to be missed.
Discussion
Methylation is a common and powerful strategy to diversify the structures of microbial natural products. iPKSs incorporate C-methyl groups into their product scaffolds early in the biosynthetic pathway because the pKa of the methylated position is generally never lower or more accessible to enzymatic chemistry than immediately following extension to the β-ketothioester. Early indications of the interplay between KS and CMeT domains were observed in LovB, where the lack of SAM or CMeT resulted in the accumulation of truncated intermediates (Cacho et al., 2015; Ma et al., 2009). Recently, it was shown that the LovB CMeT domain outcompetes the LovB KR activity by virtue of its higher catalytic efficiency (Cacho et al, 2015). Non-cognate substrates, due to incorrect modifications in previous cycles, caused significant attenuation of the highly specific CMeT leading to off-loading reactions. This finding suggested a role of CMeT as gatekeeper, which has to be passed after three of six cycles of polyketide chain extension. Unlike for LovB CMeT, PksCT CMeT is not competing with a KR domain and methylates multiple substrates. Therefore, PksCT CMeT appears to have a similar function by adding methyl groups as check-point tags, which are recognized by PksCT KS, such that a lack of methylation causes release of immature products at the triketide stage. Abortive release of an off-pathway intermediate may be especially important for NR-PKSs that lack a C-terminal thioesterase domain to serve an editing role (Vagstad et al., 2012).
Any future efforts to modulate the pattern of C-methylation in iPKSs may require perturbation of both CMeT and KS substrate binding sites. The highly conserved residues of the acceptor binding pocket also indicate that methylation programming is determined by other features of the active site or acceptor:CMeT complex. That programmed methylation is retained even after domain dissection, however, points towards control residing in intrinsic features of the CMeT. Additional studies will be needed to identify and manipulate these features to control programmed methylation. The existence of gem-dimethylating CMeTs in trans-AT PKSs (Helfrich and Piel, 2016) combined with the iterative action of NR-PKS CMeTs show that considerable flexibility is possible, potentially allowing for engineered products.
Sequence motifs have been identified in other iPKS modification domains that allow for prediction of the stereochemical outcome, notably the A- and B-type ketoreductase (KR) domains (Keatinge-Clay, 2012). A similar dichotomy for CMeTs, pro-R or pro-S, has not been identified. The stereochemistry of methylation could not be inferred from our data, and in the context of NR-PKSs this information is lost upon aromatization. Future work should provide greater detail about the methyl transfer mechanism.
Structure determination of the PksCT CMeT domain revealed a characteristic organization of the N-terminal subdomain in PKS C-Met domains and a detailed description of residues lining the substrate binding site. Structural alignments to mammalian FAS and sequence comparison to modular cis- and trans-AT PKS demonstrate a conserved organization of inactive ΨCMeT domains, commonly observed in HR-PKS and FAS and active CMeT domains in all systems. The PksCT CMeT structure thus provides a blueprint for the systematic analysis and variation of CMeT functions across all PKS systems. Despite a high degree of conservation at the level of subdomain organization and active site assembly, the organization of N-terminal linkers in the CMeT domain of the NR-PKS PksCT clearly deviates from those observed for the ΨCMeT domain of fully-reducing mammalian FAS, indicating distinct modes of integration in the two systems. The exact modes of domain integration (and the extent of their conservation) of CMeT domains in systems other than FAS, namely iterative and modular cis- and trans-AT PKS, remains to be explored. First indications may be obtained from a careful comparison of flanking regions in excised CMeT domains.
In connection with these considerations, while this manuscript was under review, a structure of an excised CMeT from a modular PKS was determined and closely agrees with the present data on a comparable domain from an iterative PKS (Skiba et al., 2016).
Significance
Iterative PKSs reuse in a “programmed” manner their complement of catalytic domains to selectively synthesize a defined product. The non-reducing iterative PKSs have no tailoring domains that carry out the reduction and dehydration steps during polyketide chain extension characteristic of FASs and other PKS subclasses. In the absence of tailoring domains, the number of cycles of ketide homologation (chain length control) is dictated principally by the ketosynthase domain. C-methyltransferase domains distinguish the Clade III NR-PKSs and afford the simplest systems to study programmed events occurring during iterative polyketide extension. Revealed clearly here is a defined interplay between correctly patterned methylations, which take place only during certain chain elongation cycles, and successful ketide extension. The strong interdependence of ketosynthase and C-methyltransferase behavior will be a constraint in the re-programming of iterative PKSs to new synthetic tasks. To guide these efforts and to help understand how methylation impacts substrate elongation and programmed product formation, we obtained the crystal structure of a non-reducing PKS C-methyltransferase domain in complex with SAH, providing a structural foothold for analyzing programmed methyl transfer. A previously uncharacterized N-terminal subdomain is involved in substrate binding, and sequence comparisons show this subdomain is an indicator of an active C-methyltransferase. Several conserved features are present across functionally and biologically diverse PKS families, including an active site histidine necessary for activity and aromatic residues lining the substrate binding pocket. Systematic variation of C-methyltransferase domains may be explored for the bioproduction of unnatural polyketides with programmable methylation or alternate alkyl donors.
Experimental Procedures
Materials
Reagents were purchased from Sigma Aldrich (St. Louis, MO) unless stated otherwise. Standard molecular biology procedures were used for nucleic acid isolation and assembly of expression constructs. Sequencing was done by the Johns Hopkins University Synthesis and Sequencing Facility (Baltimore, MD). M. purpureus NRRL 1596 was received from the ARS, USDA (Peoria, IL) and cultured on PDA plates. Genomic DNA was extracted from mycelia using the DNeasy Plant Mini kit (Qiagen, Hilden, Germany). Target sequences were amplified by polymerase chain reaction using the primers listed in the Supplemental Information (Table S1).
Sequence analysis, cloning, and protein production
To verify the mRNA sequence of pksCT, M. purpureus NRRL 1596 was grown in MC medium following reported methods for citrinin production (Shimizu et al., 2005). After 7 days of growth at 28 °C, the mycelia were harvested by centrifugation and lyophilized. The mycelial powder was ground under liquid nitrogen and total RNA was purified using the RNeasy Plant Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol with on-column DNA digestion. A cDNA library was prepared using 1 μg total RNA with 0.5 μg random hexamer (Applied Biosystems, Foster City, CA) and M-MLV reverse transcriptase (Promega, Madison, WI) at 37 °C for 1 h. Negative control reactions lacked any reverse transcriptase. PCR amplification of the desired exon boundaries was done using the cDNA library products directly (2 μL) with primers MpPksCTex1–5 and MpPksCT-SAT-3 and Phusion polymerase (New England Biolabs, Ipswich, MA). Primers for the constitutively expressed actin gene MpAct5 and MpAct3, based on a previous report (Shimizu et al., 2005), were used as positive controls. The PCR reactions were separated by 1 % agarose gel and the lone product band was ligated into pCR-blunt (Life Technologies, Carlsbad, CA) and transformed into DH5α by heat shock treatment. Five clones were sequenced using M13F and M13R primers.
Domain boundaries and expression constructs are detailed in Table S2. Each construct was transformed to E. coli BL21(DE3) and grown in LB media at 37 °C to OD600 of 0.6. The cultures were cooled in an ice bath for 30–60 min, and expression was induced with 1 mM IPTG (GoldBio, St. Louis, MO) overnight at 18 °C. Cell pellets were harvested by centrifugation for 15 min, at 4,000 × g, 4 °C. Pellets were either flash frozen in N2(l) and stored at −80 °C until use, or resuspended immediately in 5 mL/g cell pellet in buffer A (50 mM potassium phosphate, 300 mM NaCl, 10 % glycerol, pH 7.6). The slurry was sonicated on ice for 10 × 10 s at 40 % amplitude (Vibra-Cell Ultrasonic Processor, Sonics & Materials, Inc., Newtown, CT) and the lysate was cleared by centrifugation for 25 min at 27,000 × g, 4 °C. The cleared lysate was batch bound to Co2+-TALON (Clontech, Mountain View, CA) at 4 °C for 1 h. Purification was performed by gravity column and the applied resin was washed with buffer A (10 CV) and buffer A + 2 mM imidazole (5 CV) before eluting in buffer A + 100 mM imidazole (5 CV). Each domain fragment was dialyzed into reaction buffer (100 mM potassium phosphate, 10 % glycerol, pH 7.0) and used immediately or flash frozen in N2(l) and stored at −80 °C until use.
For crystallization, PksCT CMeT was expressed and initially purified as described above, but dialyzed into 25 mM Tris, 5 % glycerol, pH 7.5 (buffer B). A secondary purification was performed by applying 60 mg PksCT CMeT to 25 mL Q-Sepharose Fast-flow resin pre-equilibrated in buffer B. A series of 2 CV stepwise washes of buffer B + 0, 10, 50, 100, 150, 200, 300, and 500 mM KCl were then applied to the column, with the CMeT eluting in the 50 and 100 mM KCl fractions. These fractions were combined, dialyzed into 2 × 1 L buffer B, and concentrated to 10 mg/mL.
Selenomethionine-labeled (SeMet) CMeT was prepared by growth in M9 minimal media to OD600 = 0.6, at which point the media was supplemented with the following L-amino acids per 1 L: 100 mg each lysine, phenylalanine, and threonine; 50 mg each isoleucine, leucine, and valine; 30 mg selenomethionine (Van Duyne et al., 1993). The cultures were allowed to continue to grow at 37 °C for 15 min before being cooled and inoculated as decribed above. Purification of SeMet CMeT for crystallization was identical to that of the native CMeT.
In vitro reconstitution of PksCT and analysis of product formation
Protein concentration was determined by Bradford assay (BioRad, Hercules, CA) in duplicate using bovine serum albumin as a standard. Prior to in vitro reactions, ACP was activated by Sfp with CoASH and MgCl2 in reaction buffer for 1 h at room temperature as previously reported (Newman et al., 2014). PksCT reconstitution reactions contained 10 μM of each included domain with 0.5 mM AcSNAC, 2 mM MalSNAC, 2 mM SAM, 1 mM NADPH, and 1 mM TCEP in reaction buffer totaling 250 μL. AcSNAC was prepared synthetically and MalSNAC was purified from MatB reactions (Vagstad et al., 2012). After 4 h at room temperature, the reactions were quenched with 5 μL concentrated HCl and extracted into ethyl acetate 3 × 250 μL. The combined organic extracts were dried to a residue and resuspended in 250 μL of 20 % aqueous acetonitrile. Extracts were analyzed on an Agilent 1200 HPLC with autosampler by injecting 100 μL onto a Prodigy ODS3 column (4.5 × 250 mm, 5μ, Phenomenex, Torrence, CA) with 5–85 % MeCN/H2O, with 0.1 % formic acid, over 40 min at 1 mL/min. Mass spectrometric analysis was done using a Waters Acquity Xevo G-2 UPLC-ESI-MS in positive ion mode, with 5 μL injected on a BEHC C18 column and 10–90 % MeCN/H2O, with 0.1 % formic acid, over 10 min.
Radiolabel transfer assays were carried out with 10 μM each protein in the above reaction buffer. Reactions were initiated with the addition of [1-14C]-acetyl-CoA (American Radiolabeled Chemicals, St. Louis, MO) to 50 μM at room temperature for 5 min and quenched with the addition of 5× SDS loading buffer. The samples were separated by 16 % SDS-PAGE and dried. The dried gel was exposed to a phosphorimager screen (Amersham, Piscataway, NJ). Data was collected on a Typhoon 9410 Variable Mode imager (Amersham, Piscataway, NJ) and analyzed using ImageJ (Schneider et al., 2012).
CMeT crystallization
Protein stock solutions were dialyzed (4×, 1:2000) against crystallization buffer (25 mM Tris-HCl, 5 % (v/v) glycerol, 2 mM DTT, pH 7.0). All crystallization experiments were performed using a robotic setup applying the sitting drop vapor diffusion method. Crystals in space group H3 were grown at 17 °C by mixing 0.5 μL of protein at 14.9 mg/mL (supplemented with 2 mM SAH) in crystallization buffer with 0.5 μL reservoir solution (0.1 M BIS-TRIS pH 6.57, 25% (w/v) PEG MME 2K) and grew to a final size of 0.4×0.2×0.1 mm3 within three days. Prior to harvesting, crystals were cryo-protected by slowly exchanging the drop solution (0.1 M BIS-TRIS 8.0 pH, 25 % (w/v) PEG MME 2K, 10 mM acetoacetyl-CoA, 20 % (v/v) ethylene glycol) followed by an incubation of 5 min and flash freezing in N2(l). SeMet CMet was crystallized in space group H3 at 17 °C by mixing 0.5 μL of protein at 14.3 mg/mL (supplemented with 2 mM SAH) in crystallization buffer with 0.5 μL reservoir solution (0.1 M BIS-TRIS pH 6.21, 21.4% (w/v) PEG MME 2K). Crystals were harvested after four days at a size of 0.4×0.3×0.05 mm3. Prior to harvesting and flash freezing in N2(l), crystals were dehydrated and cryo-protected (0.1 M BIS-TRIS pH 6.5, 30.0 % (w/v) PEG MME 2K, 2 mM SAH, 25 % (v/v) ethylene glycol) by carefully exchanging the drop solution over a period of 30–60 min.
Data collection, structure determination and analysis
Data sets were collected at the Swiss Light Source (SLS, Villigen, Switzerland) at beamline X06DA (native: λ = 0.99998 Å and 1.90747 Å; SeMet: λ = 0.97929 Å) and a temperature of 100 K. Data reduction was performed using DIALS (Waterman et al., 2013) and XDS (Kabsch, 2010), datasets were analyzed with Phenix (Adams et al., 2010). Resolution cutoffs were determined by CC1/2 criterion (Karplus and Diederichs, 2012). The structure of SeMet CMeT was solved by SAD phasing using SHELX (Sheldrick, 2008). The asymmetric unit contained one protomer, which was used for initial phasing of native crystals diffracting to higher resolution using molecular replacement in PHASER (McCoy et al., 2007). Initial maps were improved by density modification using PARROT (Cowtan, 2010), followed by automated rebuilding with BUCCANEER (Cowtan, 2008). The final model was obtained after iterative cycles of model building in COOT (Emsley et al., 2010) and TLS refinement in Phenix (Adams et al., 2010), yielding excellent geometry (Ramachandran favored/outliers: 99.48 %/0.00 %) and Rwork/Rfree values of 0.19/0.22 (Table S3). The final model covers residues 1785–2163. All structures of native and SeMet-labeled protein revealed weak, but significant Fo-Fc difference density for a partially occupied ligand in the ligand binding tunnel (Figure S5), which could not be displaced by acetoacetyl-CoA soaks (cryo solutions + 15 mM acetoacetyl-CoA). Anomalous data collection at λ = 1.90747 Å did not reveal significant anomalous signal as expected for a fully occupied sulfur or phosphorus atom as part of the ligand (Figure S5B). The observation was verified by two protein purifications and crystal treatments in the presence or absence of acetoacetyl-CoA. Despite the featured shape of the density, the nature and origin of the ligand, either from the crystallization conditions or protein purification, could not be unambiguously determined. Related folds were identified using the protein structure comparison service PDBeFold at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm), authored by E. Krissinel and K. Henrick (Krissinel and Henrick, 2004), using 40 % (query) and 70 % (target) as matching parameters, and interfaces were analyzed using QtPISA (Krissinel and Henrick, 2007). Bias-removal for Fo-Fc omit maps was achieved by applying a random perturbation to coordinates (Δ0.2 A) and B-factors (Δ20 % of the mean overall B-factor) using MOLEMAN2 (Kleywegt and Jones, 1997) prior to refinement. Figures were generated using PYMOL (Schrodinger, 2015).
Phylogenetic sequence analysis
51 sequences of CMeT domains from NR-, HR-, cis-AT- and trans-AT PKS as well as FAS were selected to analyze phylogenetic distances. Sequences of inactive pseudo CMeT domains were included for FAS and HR-PKS. Alignments were generated using Clustal Omega (Sievers et al., 2011) and phylogenetic trees were generated using the neighbor joining algorithm implemented in Geneious version 8.1.6 (http://www.geneious.com) (Kearse et al., 2012).
Supplementary Material
Highlights.
Domain deconstruction and reconstitution of a C-methylating iterative PKS
Processive methylation by the CMeT domain is required for pentaketide production
CMeT has a two-subdomain organization with a unique N-terminal subdomain
CMeT defines a common architecture of methyltransferase domains in PKS
Acknowledgments
Data were collected at beamlines PXIII of PSI; we acknowledge excellent support from the beamline teams. This work was supported by the Swiss National Science Foundation (SNF) project grants 159696 and R’equip grant 145023, as well as NIH grant ES001670. DAH acknowledges a fellowship by the Werner-Siemens Foundation. PAS thanks Anna Vagstad for providing the MTAN expression plasmid, as well as Adam Newman, Dan Marous, and Callie Huitt-Roehl for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
PAS carried out all molecular biology, protein production, in vitro reconstitution and functional analysis; DAH crystallized and determined the structure of CMeT and analyzed the structure and phylogeny; TM contributed to structure determination and analysis of CMeT; CAT designed the work and analyzed data. All authors contributed to manuscript preparation.
Accession number
The atomic coordinates and structure factors have been deposited in the PDB (http://wwpdb.org) under the accession code PDB: 5MPT.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja M, Chiang YM, Chang SL, Praseuth MB, Entwistle R, Sanchez JF, Lo HC, Yeh HH, Oakley BR, Wang CC. Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans. J Am Chem Soc. 2012;134:8212–8221. doi: 10.1021/ja3016395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belecki K, Townsend CA. Biochemical determination of enzyme-bound metabolites: preferential accumulation of a programmed octaketide on the enediyne polyketide synthase CalE8. J Am Chem Soc. 2013;135:14339–14348. doi: 10.1021/ja406697t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacho RA, Thuss J, Xu W, Sanichar R, Gao Z, Nguyen A, Vederas JC, Tang Y. Understanding Programming of Fungal Iterative Polyketide Synthases: The Biochemical Basis for Regioselectivity by the Methyltransferase Domain in the Lovastatin Megasynthase. J Am Chem Soc. 2015;137:15688–15691. doi: 10.1021/jacs.5b11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad RS, Massey LK, Sokatch JR. D- and L-isoleucine metabolism and regulation of their pathways in Pseudomonas putida. J Bacteriol. 1974;118:103–111. doi: 10.1128/jb.118.1.103-111.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. Fitting molecular fragments into electron density. Acta Crystallogr D Biol Crystallogr. 2008;64:83–89. doi: 10.1107/S0907444907033938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr. 2010;66:470–478. doi: 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JM, Thomas PM, Scheerer JR, Vagstad AL, Kelleher NL, Townsend CA. Deconstruction of iterative multidomain polyketide synthase function. Science. 2008;320:243–246. doi: 10.1126/science.1154711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JM, Townsend CA. New insights into the formation of fungal aromatic polyketides. Nat Rev Microbiol. 2010;8:879–889. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison J, al Fahad A, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. Proc Natl Acad Sci U S A. 2012;109:7642–7647. doi: 10.1073/pnas.1201469109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch KM, Bakeer W, Yakasai AA, Song Z, Pedrick J, Wasil Z, Bailey AM, Lazarus CM, Simpson TJ, Cox RJ. Rational domain swaps decipher programming in fungal highly reducing polyketide synthases and resurrect an extinct metabolite. J Am Chem Soc. 2011;133:16635–16641. doi: 10.1021/ja206914q. [DOI] [PubMed] [Google Scholar]
- Foulke-Abel J, Townsend CA. Demonstration of starter unit interprotein transfer from a fatty acid synthase to a multidomain, nonreducing polyketide synthase. Chembiochem. 2012;13:1880–1884. doi: 10.1002/cbic.201200267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwicke MA, Rendina AR, Williams SP, Moore ML, Wang L, Krueger JA, Plant RN, Totoritis RD, Zhang G, Briand J, et al. A human fatty acid synthase inhibitor binds beta-ketoacyl reductase in the keto-substrate site. Nat Chem Biol. 2014;10:774–779. doi: 10.1038/nchembio.1603. [DOI] [PubMed] [Google Scholar]
- He Y, Cox RJ. The molecular steps of citrinin biosynthesis in fungi. Chem Sci. 2016;7:2119–2127. doi: 10.1039/c5sc04027b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich EJ, Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2016;33:231–316. doi: 10.1039/c5np00125k. [DOI] [PubMed] [Google Scholar]
- Huitt-Roehl CR, Hill EA, Adams MM, Vagstad AL, Li JW, Townsend CA. Starter unit flexibility for engineered product synthesis by the nonreducing polyketide synthase PksA. ACS Chem Biol. 2015;10:1443–1449. doi: 10.1021/acschembio.5b00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay AT. The structures of type I polyketide synthases. Nat Prod Rep. 2012;29:1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- Khaleeli N, Busby RW, Townsend CA. Site-directed mutagenesis and biochemical analysis of the endogenous ligands in the ferrous active site of clavaminate synthase. The His-3 variant of the 2-His-1-carboxylate model. Biochemistry. 2000;39:8666–8673. doi: 10.1021/bi000534c. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Model building and refinement practice. Methods Enzymol. 1997;277:208–230. doi: 10.1016/s0076-6879(97)77013-7. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci U S A. 2003;100:15670–15675. doi: 10.1073/pnas.2532165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A Simple Method for Displaying the Hydropathic Character of a Protein. Journal of Molecular Biology. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Liscombe DK, Louie GV, Noel JP. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat Prod Rep. 2012;29:1238–1250. doi: 10.1039/c2np20029e. [DOI] [PubMed] [Google Scholar]
- Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, et al. Complete reconstitution of a highly reducing iterative polyketide synthase. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AG, Vagstad AL, Storm PA, Townsend CA. Systematic domain swaps of iterative, nonreducing polyketide synthases provide a mechanistic understanding and rationale for catalytic reprogramming. J Am Chem Soc. 2014;136:7348–7362. doi: 10.1021/ja5007299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Sartini D, Pozzi V, Wilk D, Emanuelli M, Yee VC. Structural basis of substrate recognition in human nicotinamide N-methyltransferase. Biochemistry. 2011;50:7800–7808. doi: 10.1021/bi2007614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Khandogin J, West AH, Cook PF. Evidence for a catalytic dyad in the active site of homocitrate synthase from Saccharomyces cerevisiae. Biochemistry. 2008;47:6851–6858. doi: 10.1021/bi800087k. [DOI] [PubMed] [Google Scholar]
- Regueira TB, Kildegaard KR, Hansen BG, Mortensen UH, Hertweck C, Nielsen J. Molecular basis for mycophenolic acid biosynthesis in Penicillium brevicompactum. Appl Environ Microbiol. 2011;77:3035–3043. doi: 10.1128/AEM.03015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Kinoshita H, Shimizu T, Nihira T. Construction of a citrinin gene cluster expression system in heterologous Aspergillus oryzae. J Biosci Bioeng. 2008;106:466–472. doi: 10.1263/jbb.106.466. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8 2015 [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kinoshita H, Ishihara S, Sakai K, Nagai S, Nihira T. Polyketide synthase gene responsible for citrinin biosynthesis in Monascus purpureus. Appl Environ Microbiol. 2005;71:3453–3457. doi: 10.1128/AEM.71.7.3453-3457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skiba MA, Sikkema AP, Fiers WD, Gerwick WH, Sherman DH, Aldrich CC, Smith JL. Domain Organization and Active Site Architecture of a Polyketide Synthase C-methyltransferase. ACS Chem Biol. 2016 doi: 10.1021/acschembio.6b00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006;7(Suppl 1):S10, 11–12. doi: 10.1186/gb-2006-7-s1-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyn PS, Vleggaar R. Biosynthesis of Asteltoxin by Cultures of Emericella-Variecolor - the Role of Propionate in the Biosynthesis and Evidence for a 1,2-Bond Migration in the Formation of the Bistetrahydrofuran Moiety. Journal of the Chemical Society-Chemical Communications. 1984:977–979. [Google Scholar]
- Steyn PS, Vleggaar R, Wessels PL. Biosynthesis of the Aurovertin-B and Aurovertin-D - the Role of Methionine and Propionate in the Simultaneous Operation of 2 Independent Biosynthetic Pathways. Journal of the Chemical Society-Perkin Transactions. 1981;1:1298–1308. [Google Scholar]
- Struck AW, Thompson ML, Wong LS, Micklefield J. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem. 2012;13:2642–2655. doi: 10.1002/cbic.201200556. [DOI] [PubMed] [Google Scholar]
- Udwary DW, Merski M, Townsend CA. A method for prediction of the locations of linker regions within large multifunctional proteins, and application to a type I polyketide synthase. J Mol Biol. 2002;323:585–598. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagstad AL, Bumpus SB, Belecki K, Kelleher NL, Townsend CA. Interrogation of global active site occupancy of a fungal iterative polyketide synthase reveals strategies for maintaining biosynthetic fidelity. J Am Chem Soc. 2012;134:6865–6877. doi: 10.1021/ja3016389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagstad AL, Newman AG, Storm PA, Belecki K, Crawford JM, Townsend CA. Combinatorial Domain Swaps Provide Insights into the Rules of Fungal Polyketide Synthase Programming and the Rational Synthesis of Non-Native Aromatic Products. Angew Chem Int Ed Engl. 2013;52:1718–1721. doi: 10.1002/anie.201208550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Waterman DG, Winter G, Parkhurst JM, Fuentes-Montero L, Hattne J, Brewster A, Sauter NK, Evans G. The DIALS framework for integration software. CCP4 Newsletter on protein crystallography. 2013;49:16–19. [Google Scholar]
- Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- Yeh H, Chang SL, Chiang YM, Bruno KS, Oakley BR, Wu TK, Wang CC. Engineering Fungal Nonreducing Polyketide Synthase by Heterologous Expression and Domain Swapping. Org Lett. 2013;15:756–759. doi: 10.1021/ol303328t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.