

Abstract
Bioinformatic analysis can not only accelerate drug target identification and drug candidate screening and refinement, but also facilitate characterization of side effects and predict drug resistance. High-throughput data such as genomic, epigenetic, genome architecture, cistromic, transcriptomic, proteomic, and ribosome profiling data have all made significant contribution to mechanism-based drug discovery and drug repurposing. Accumulation of protein and RNA structures, as well as development of homology modeling and protein structure simulation, coupled with large structure databases of small molecules and metabolites, paved the way for more realistic protein-ligand docking experiments and more informative virtual screening. I present the conceptual framework that drives the collection of these high-throughput data, summarize the utility and potential of mining these data in drug discovery, outline a few inherent limitations in data and software mining these data, point out news ways to refine analysis of these diverse types of data, and highlight commonly used software and databases relevant to drug discovery.
Keywords: Drug target, Drug candidate, Drug screening, Genomics, Epigenetics, Transcriptomics, Proteomics, Structure
1. INTRODUCTION
Drug discovery starts with diagnosis of a disease with well characterized symptoms that reduce the quality of life. Conventionally, a desirable drug is a chemical (which could be a simple chemical or a complicated protein) or a combination of chemicals that reduces the symptoms without causing severe side effects in the patient. Other properties of a desirable drug include affordability and profit for drug companies [1, 2], low chance of drug resistance [3] leading to dramatic decrease in the commercial value of the drug, low deleterious effect on the environment, e.g., no re-activation by bacterial species after human use [4]. Thus, a desirable drug is one that not only is efficacious with little side effects, but also has minimal long-term negative effect on the patient, the society and the environment.
This review will focus on how bioinformatics can facilitate the discovery of such desirable drugs. Bioinformatics is an interdisciplinary science spanning genomics, transcriptomics, proteomics, population genetics and molecular phylogenetics. Bioinformaticians in drug discovery use high-throughput molecular data (Fig. 1) in comparisons between symptom-carriers (patients, animal disease models, cancer cell lines, etc.) and normal controls. The key objectives of such comparisons are to 1) connect disease symptoms to genetic mutations, epigenetic modifications, and other environmental factors modulating gene expression, 2) identify drug targets that can either restore cellular function or eliminate malfunctioning cells, e.g., cancer cells, 3) predict or refine drug candidates that can act upon the drug target to achieve the designed therapeutic result and minimize side effects, and 4) assess the impact on environmental health and the potential of drug resistance.
Fig. (1).
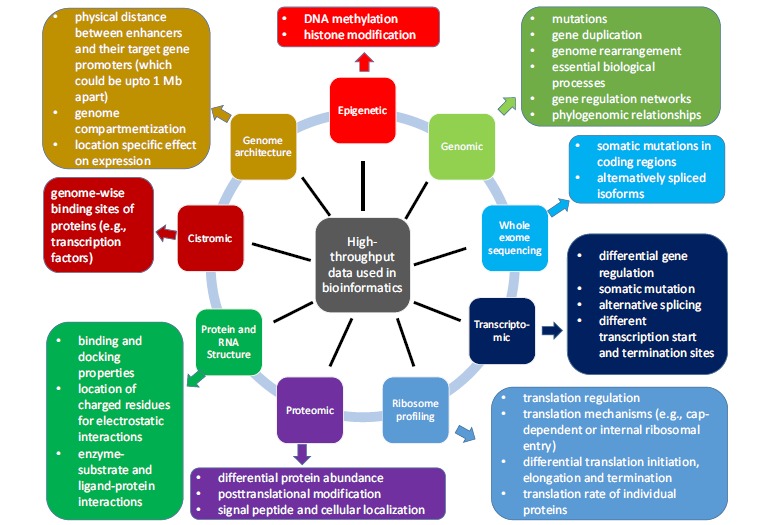
Major types of high-throughput data and their key information relevant to drug discovery. Metabolomic data belong to cheminformatics and are not included.
2. GENOMIC SEQUENCE AND EXOME DATA IN DRUG DISCOVERY
One of the early contributions from bioinformatics to drug target discovery is the identification of sequence homology between simian sarcoma virus onc gene, v-sis, and a platelet-derived growth factor (PDGF) by simple string matching [5, 6]. This finding not only resulted in PDGF being used as a cancer drug target [7-9], but also led to two new lines of thinking. First, the viral transforming factor may work simply by changing transient expression of a growth factor to constitutive expression, suggesting growth factors as targets for anti-cancer drug development. Second, any factors modulating gene expression patterns can potentially contribute to cancer. This new conceptual framework of cancer biology contributed to the progress of mechanism-based anti-cancer drug development in the following years [10-12].
2.1. Genetic Diseases
Genomic and whole exome sequencing of patients with inherited disorders have recovered many somatic mutations which are associated with genetic diseases [13-15] and could be potential drug targets. The main difficulty concerning bioinformatic research on somatic mutations lies in the identification of disease-causing mutations among many observed genetic differences between matched patient and normal control [16]. Some diseases such as cancer exhibit high genetic heterogeneity [17], even among cells within a single tumor [18]. Many of these somatic mutations could be the consequence rather than the cause of cellular malfunction [16].
Effort must be made to distinguish three types of somatic mutations: 1) those that cause the disease and may serve as drug targets, 2) those that are closely linked to the disease gene and consequently are associated with the disease, and 3) those not associated with the disease but happen to occur in the patient group and not in the control group. The second type of mutations can be used for disease diagnosis, but not as drug target. The third type can be excluded in two ways. The first is by increasing sample size. If thousands of breast cancers all share the same somatic mutation, then the relevance of the mutation to breast cancer is high relative to a somatic mutation occurring in only one breast cancer [19]. The second is by collecting longitudinal data, recognizing that many diseases may have a genetic determinant long before the manifestation of the disease [20]. Suppose mutation X predisposes a person to Alzheimer’s disease (AD). If we compare one groups of AD patients with a non-AD control group, and if the control group has people who already have mutation X but have not developed AD yet, we may fail to recognize the importance of mutation X simply because it is not unique in the AD group. Only if we follow patients or relevant animal models over time can we come to the conclusion that whoever has mutation X eventually develop AD.
It is much more difficult to distinguish between the first and second type of genetic differences between patient and control without an understanding of disease mechanism. A loss-of-function mutation can happen in the coding sequence (CDS), in the regulatory motif (e.g., response elements for ligand-activated nuclear receptors) or in an enhancer that could be up to 1 million bases away from the CDS. Bioinformaticians will typically take three approaches to check if the mutation has major impact on gene function: 1) whether the mutation replaces an amino acid by a very different one (e.g., non-polar uncharged glycine by a positively charged arginine) at a typically conserved site, 2) whether the mutation occurs in a highly conserved non-coding sequence (which is typically done by comparing genomes between human and non-human primates.), and 3) whether the mutation occurs in a known signal (e.g., regulatory motif, splice sites, transcription initiation and termination sites) for cellular machinery (e.g., ribosome, spliceosome, degradosome). The last approach is facilitated by the availability of extensively compiled and curated databases of known regulatory motifs [21-23]. Bioinformatic tools are often used to scan genomes for regulatory motifs. Such tools include position weight matrix (PWM) to find the genomic location of a known motif, Gibbs sampler for de novo motif discovery [24, 25] and support vector machines [26, 27] that can be used to extract differences between two groups of sequences (e.g., motif-present and motif-absent) and to use the resulting information to detect/scan motifs in genomes. The regulatory motifs could be response elements of nuclear receptors whose identification often leads to refinement of drug targets [28]. Such studies are facilitated by software such as DAMBE [29] which, when given an annotated genomic sequence, can extract coding sequences, rRNAs, tRNAs, introns, exons, 5’ and 3’ splice sites, upstream or downstream sequences of gene features, etc., with just a few mouse clicks. In addition to functions for PWM, Gibbs sampler, and minimum folding energy estimation, DAMBE can also compute protein isoelectric point and indices of protein translation efficiency.
If a deleterious mutation is identified to be a loss-of-function mutation, then bioinformatics can help identify a paralogous gene or an alternative cellular pathway that can compensate for the mutation effect. Functional redundancy or partial redundancy is common in mammals, e.g., the function of paralogous genes USP4 and USP15 in mice are partially redundant [30]. Human adrenoleukodystrophy (ALD) is caused by partial deletion of the 10-exon gene ABCD1 resulting in the accumulation of very long chain fatty acids [31], which suggests not only diet limitation of very long chain fatty acids (VLCFA) in disease management, but also activation of alternative metabolic pathways for VLCFA through regulating another gene involved in fatty acid metabolism (ABCD2) and suppression of the activity of elongase involved in generating VLCFA [32]. Another example of activating alternative biological pathways or genes with partial functional redundancy involves sickle-cell anemia [33] caused by a single amino acid replacement in human beta-globin gene [34, 35]. Fetal hemoglobin gene (HbF) is a promising drug target because HbF reduces hemoglobin polymerization and clumping. A drug that could revive the silenced HbF would alleviate the symptoms of sickle-cell anemia and thalassemia in adults [36, 37]. Interestingly, some β-thalassemia patients have the correct version of the β-globin gene but the gene is not expressed because of mutations that occurred far away from it [38, 39]. Such long-range gene regulation will be addressed later on epigenetic modification and genome architecture.
2.2. Human Diseases Caused by Pathogens
Well annotated genomes are essential for target-based drug discovery against pathogens. The general bioinformatic approach involves three essential steps. The first is to identify essential genes in the pathogen as drug targets. A genome, especially a well-annotated one, can facilitate identification of such essential genes. For example, genes involved in nucleotide synthesis are well known, but are often missing in pathogenic species because they use salvage pathway instead of de novo synthesis pathway to procure nucleotides. In, Trypanosoma brucei, genes for de novo synthesis of ATP, GTP and TTP have gone missing, but the pathogen retains limited capacity for de novo synthesis of CTP [40], presumably because CTP generally has much lower centration than the other three nucleotides in the cell and cannot be reliably obtained through salvage. This points to CTP synthesis pathway as a drug target. Indeed, inhibiting CTP synthesis arrests the growth and replication of the pathogen [40]. Essential genes are often highly conserved and can be revealed by genomic comparisons between pathogens and their phylogenetic relatives. Sometimes they may also inferred from experimental data from model organisms such as Escherichia coli, Bacillus subtilis or Saccharomyces cerevisiae whose genes have been systematically and individually knocked out. Genes essential for the two bacterial species are likely to be essential in another bacterial species.
The second step in developing drugs against pathogen is to check if such essential genes have homologues in the host. If they do, then inhibiting such essential genes in the pathogen may have adverse effect on the function of the host homologue, and we consequently need to perform sequence and structural comparisons between the pathogen and host homologues to identify unique part in the pathogen homologue to assist in the design of pathogen-specific drugs.
Third, to minimize the chance of pathogen developing drug resistance, it is important for the drug to target at specific pathogen and not its phylogenetic relatives that are not pathogenic. For this reason, pathogenicity islands that are unique in pathogenic bacteria but not in their non-pathogenic relatives have increasingly become the preferred source of drug targets [41-43].
Bioinformatic analysis revealed a glutamate transport system that is present in the pathogen Clostridium perfringens but absent in mammals and birds [44]. Drugs developed against such a transport system will protect not only humans, but also domesticated mammals and fowls. In the human parasite Giardia intestinalis, the phosphoinositide-3 kinase (PI3K) signaling pathways are essential and could serve as a drug target. However, the PI3K pathway is also essential in many eukaryotes so it is important to identify what is unique in the PI3K homologues (Gipi3k1 and Gipi3k2) in G. intestinalis relative to mammals. Sequence comparisons revealed a unique insertion only in the parasite that can serve as a pathogen-specific drug target [45]. The same approach is used in targeting Pseudomonas aeruginosa [46]. Similarly, in developing anti-HIV-1 drugs, one can target genes involved in reverse transcription and protease digestion of its translated polyprotein because these processes not only are essential for viral survival and transmission, but also have no close homologues in human so their inhibition should have minimal side effect on human.
Genomic analysis can also help in repurposing existing drugs against other pathogens. Galactofuranose (Galf) is an important constituent on the cell surface of a variety of bacterial pathogens [47, 48], and its synthesis requires UDP-galactopyranose mutase (UGM). Because Galf is absent in human [44], UGM has been used as a desirable drug target [49]. UGM coded by gene GLF was later found in several eukaryotic unicellular pathogens [50] as well as in nematodes [51]. Can we repurposing drugs developed against bacterial pathogens to fight eukaryotic unicellular pathogens [50]? Drug repurposing is cost-effective in drug development [52]. Genomic analysis shows that eukaryotic UGMs, while similar to each other, is quite different from prokaryotic UGMs, suggesting difficulty in drug repurposing from bacterial pathogen to eukaryotic pathogens. However, if one develops an effective drug against one eukaryotic UGM, the drug would have a very good chance of being repurposed for another eukaryotic pathogen.
Genomics has also contributed to understanding drug actions. The venom protein PcFK1 of spider Psalmopoeus cambridgei was able to inhibit the growth of Plasmodium falciparum, but the mechanism was unknown. A sequence analysis revealed sequence homology between PcFK1 and the protein substrate of P. falciparum enzyme PfSUB1, leading to the hypothesis that PcFK1 is an antagonist of PfSUB1. Subsequent docking prediction and in vitro experiments confirm this hypothesis, pointing to PfSUB1 as a drug target [53].
Essential cellular processes are often functionally redundant, and understanding such functional redundancy is crucial in developing effective drugs against pathogens. In Mycobacterium tuberculosis, arabinofuranosyltransferases Mt-EmbA and Mt-EmbB contribute to the synthesis of cell wall mycolyl-arabinogalactan-peptidoglycan complex and are targeted by the drug ethambutol. Bioinformatic analyses revealed another arabinofuranosyltransferase, Mt-AftA, which is not inhibited by ethambutol and consequently would serve as a drug target [54]. A combination of drugs against all three arabinofuranosyltransferases will not only be more effective against the pathogen, but also reduce the chance of the pathogen developing drug resistance. Activating alternative biological pathways to satisfy the need of growth and survival has been known in bacterial species since the discovery of the lac operon and the glucose/lactose genetic switch [55], and a drug cannot be effective against a pathogen or a cancer cell unless we know how cells do things with alternative pathways that can be activated in response to the drug.
Bioinformatics, with its inherent evolutionary perspective and its integration of molecular phylogenetics [56, 57], can often contribute to resolving controversies on molecular mechanisms. One such example involves the causal interpretation of CpG methylation causing CpG deficiency through subsequent C→T mutation mediated by spontaneous deamination. A controversy arose when both Mycoplasma genitalium and M. pneumoniae genomes were found to lack DNA CpG methyltransferase, yet M. genitalium genome exhibits much stronger CpG deficiency than M. pneumoniae genome, suggesting a conclusion that the difference in CpG deficiency between the two species is irrelevant to CpG methylation [58, 59]. Such a conclusion from genomic studies without an evolutionary perspective is often wrong. A comprehensive phylogenetic study using software DAMBE [29] showed that the ancestors of the two species should have multiple CpG methyltransferases because M. pulmonis and other relatives that branch off earlier than M. genitalium and M. pneumoniae have multiple CpG methyltransferases. After the loss of the CpG methyltransferases in the ancestor of M. genitalium and M. pneumoniae, both species began to gain CpG frequency, but M. pneumoniae evolved much faster (with a much longer branch) and regained CpG much faster than M. genitalium [60]. These findings restored the validity of causal relationship between CpG-specific DNA methylation and CpG deficiency, and illustrate the importance of having an evolutionary perspective in understanding biological processes. Because many such studies involve highly diverged bacterial or viral species, and because it is often difficult to obtain reliable multiple sequence alignment with highly divergent sequences, a new phylogenetic method based on pairwise sequence alignment has recently been developed [61] to facilitate phylogenomic studies involving highly diverged species.
3. EPIGENETICS, GENOME ARCHITECTURE AND CISTROMES IN DRUG DISCOVERY
Monozygotic twins carrying the same deleterious mutations such as the aforementioned ALD mutation often differ much in phenotype [62-65]. Such observations serve to highlight the relationship between epigenetic modifications and human diseases [66, 67]. Epigenetic modification includes two interrelated events, DNA methylation and histone modification. The maintenance of DNA methylation pattern in mammals is accomplished by the mammalian DNA methyltransferase 1 (DNMT1) whose CatD domain recognizes hemi-methylated CpG sites [68] so that DNA methylation pattern can be maintained from parental to daughter cells. In mammals, the methylated CpG recruits proteins with a methyl-CpG binding domain such as MBD1, MBD2, MBD3 and MeCP2 which then recruit histone deacetylase to remove the acetyl group and restore the positive charge of lysine residues (or histone N-terminal) in histone so that the negatively charged backbone of DNA can wrap tightly around the positively charged histone to silence the gene [69]. A silenced gene is in many ways equivalent to a loss-of-function mutation. Because some cancers appear to be caused by permanent silencing of genes involved in apoptosis pathway through DNA methylation and histone deacetylation [70-71], histone deacetylase has been used as a drug target with its inhibitors aiming to reactivate the apoptosis pathway [72]. The main problem in this approach is specificity because deacetylase inhibitors often have profound effect on the regulation of many other genes, which may explain why such drugs often do not enter clinical trials [73]. Methods for precise editing of the epigenome, involving components for DNA-binding and specific sequence recognition and modification are currently being developed [74].
The conventional view that DNA methylation and histone deacetylation mainly serve the purpose of permanent gene silencing has now been replaced by a more general conceptual framework of epigenetic modification and gene regulation (Fig. 2). This conceptual shift demands integrated analysis of several types of high-throughput data: methylation pattern from bisulfite sequencing [75-76], DNA/protein binding data (cistrome) from ChIP-on-chip and ChIP-Seq [77], and genome architecture data from Hi-C [78] or its derivatives. DNA methylation alters DNA/protein binding which in turn alters genome architecture, i.e., two DNA segments far apart along the linear DNA can be brought together. Genome architecture data pave the way for studying spatial interaction between enhancers and promoters that can be up to one million bases apart. That gene expression depends on gene location on the genome is known since 1930 through studies of translocation [79], but empirical evidence accumulated much later to demonstrate that protein/DNA interactions resulted in nucleosome reconfiguration and interaction between enhancer and promoter [80-84]. This had spawned the formulation of the enhancer hub model of gene regulation [85, 86]. That is, the hub contains one or more enhancers and a gene with its promotor looping close to the hub will be expressed; deletion of such a hub will silence the expression of all genes that depends on their physical proximity to the hub to be expressed.
Fig. (2).

A general framework of epigenetic effects on gene expression, through 1) DNA methylation and histone acetylation/deacetylation, 2) alteration of DNA-binding proteins and consequent protein-DNA and protein-protein interactions, and 3) alteration of long-distance interactions such as enhancer-promotor interactions. LM – laboratory method, BQ: sample bioinformatic questions.
From a bioinformatics point of view, the key question concerns what is the methylation signal on DNA and whether it is possible to modulate such a signal to alter epigenetic modifications. I have mentioned before that some β-thalassemia patients have the correct version of the β-globin gene but the gene is not expressed because of mutations that occurred far away from it [38, 39]. One may formulate two hypotheses. First, the enhancer that controls the expression of β-globin gene is mutated or deleted in the patient [38, 87]. Second, the enhancer that is brought close to the promotor of β-globin gene in normal genome architecture is relocated somewhere else due to abnormal epigenetic modifications and protein/DNA binding. Testing these hypotheses, which has become possible only with the availability of high-throughput data of genome architecture, methylation patterns and cistromes (the set of all protein/DNA binding sites), would shed light on how we can reposition the enhancer and the β-globin promotor so that the gene will be expressed [88-90]. Similarly, if the β-globin gene is silenced through DNA methylation, then the knowledge of how to modulate the signal to modify the methylation pattern would bring us closer to reactivating the silenced β-globin gene. Along the same line of reasoning, if the fetal globin genes are silenced by methylation, and if reactivation of these fetal globin genes can alleviate the problem caused by mutations in adult globin genes, then the knowledge of site-specific demethylation would be highly desirable [74].
Given that some CpG are methylated and some are not in mammalian genomes, one straightforward bioinformatic analysis would be to compare the flanking sites of these two groups of CpG dinucleotides to detect if flanking nucleotides contributes to methylation signals. Equivalent analyses of splice sites have revealed strong splice signal in flanking sequences of the 5’ and 3’ splice sites [91, 92], but such comparisons of flanking regions between methylated and unmethylated CpG, although done in a limited scale [93-95], have not yielded clear-cut results. Equally disappointing is that, while the concept of imprinting center (IC) has been known for many years [96], the physical basis of IC, either at the sequence level or structural level, remains elusive.
Because monozygotic twins carrying the same genetic defect often differ much in manifestation of the associated disease [62-65], one naturally wishes to identify environmental contributions such as diet to epigenetic modification [97, 98]. As methylation needs S-adenosyl L-methionine (SAM) as the methyl donor, a deficiency in methionine most likely will, and indeed has been confirmed to, affect DNA methylation [99, 100]. Similarly, one would predict that any major perturbation on methionine, such as the deletion of methylthioadenosine phosphorylase (MTAP) crucial in the methionine salvage pathway, would also affect DNA methylation, gene regulation and cancer. Indeed, MTAP deletion is common in cancer cells [101]. Thus, all genes that affect methionine metabolism could be drug targets, and bioinformatics, with databases such as KEGG [102-104] can identify such genes effectively.
If wrong DNA methylation pattern has formed, then an ideal drug (or an epigenome-repairing nano-machine) should be able to specifically identify the wrong pattern and correct it [74]. To develop such a drug or nano-machine, we first have to know the correct methylation pattern or ideally discover a set of molecules that encode such a correct pattern. Experimental results have accumulated in support of RNA’s role in epigenetic modification [105]. Given that DNA in the zygote undergoes demethylation to regain pluripotency [106], the epigenomic code is perhaps not on DNA. As proteins do not seem to be good in writing code in and because most core histones are replaced by protamine in male germ cells [107], the epigenetic codes, especially the ones that specify de novo DNA methylation, is unlikely to be found in proteins. However, such codes may exist in a set of highly conserved and structurally stable RNA molecules that might be present as early as the oocyte and sperm stage. Long noncoding RNAs (lncRNAs) can participate in epigenetic modification and regulate chromatin state. Characterization of lncRNAs bound to DNA and protein by the ChiRP-seq method [108, 109] revealed numerous sequence-specific binding sites on DNA, and the binding of lncRNA such as HOTAIR [108, 110] and Kcnq1ot1 [111] to such sites facilitates the recruitment of Polycom Repressive Complex 2 (PRC2) for mediating histone H3 lysine-27 trimethylation. Short RNAs can also modulate epigenetic changes. Mature sperm contain a number of small RNA species [97, 98, 112, 113], and these small RNAs do affect offspring phenotype [113, 114]. Furthermore, these small RNAs on offspring appear to contribute to epigenetic modification [97, 98, 113, 114]. The ENCODE pilot project shows that “the genome is pervasively transcribed, such that the majority of its bases can be found in primary transcripts” [115]. Those non-coding transcripts may be a treasure trove for bioinformaticians to discover epigenome-modifying RNAs as drug targets.
Epigenetic modification has an early origin. Many bacterial species modify their own DNA by methylation to protect against endogenic type II restriction endonucleases. Some Bacteriophage have their own methyltransferase that can modify their own genome against host restriction digestion [116], and human viral pathogens such as HIV-1 can induce profound alteration in host epigenetic pattern [117]. It is now known that some of the host defense mechanisms against pathogens are implemented through epigenetic modifications [118, 119] and many pathogens can modify host epigenetic patterns in favour of their survival and reproduction in the host [118]. What is the eventual fate of such pathogen-mediated epigenetically modified host cells remains unclear. Do they defeat the pathogen invasion, restore the normal epigenetic pattern and reassume normal function again or do they initiate certain apoptosis pathway and perish? What epigeneticists need is a model organism or a cell line in which the epigenetic pattern can be perturbed by extrinsic factors and then restored back to normal.
4. TRANSCRIPTOMICS AND DRUG DISCOVERY
Transcriptomic data have been increasingly used to identify differentially regulated genes, alternatively spliced isoforms and different transcription start and termination sites between patient and matched control [120-125]. Transcriptomic data analysis contributes to drug discovery mainly in two ways, one in phenotypic screening to identify and refine drug candidates, and the other in drug target identification.
4.1. Phenotypic Screening
There has been debates on what constitutes phenotypic screening, but recently proposed definitions [12, 126] converge in five points: 1) the screening involves a large number of compounds (drug candidates) ideally chosen systematically, 2) phenotypic changes in response to each compound is monitored, 3) a criterion of desirability is formulated and used in ranking the compounds, 4) those compounds generating desirable biological effects (phenotypes) are kept as drug candidates for further testing and validation, and 5) the mechanism of action is unknown and not the focus of the screening. Phenotypic screening can be quite effective in identifying active ingredients in traditional medicine, with one of the success stories being the discovery of artemisinin which is the most effective drug against the malaria parasite Plasmodium falciparum [127].
While the target-based approach is effective in developing drugs against diseases with relatively simple mechanisms such as single-gene genetic diseases, phenotypic screening is more effective in drug development against diseases with multiple causes such as multi-gene genetic diseases [128-129]. Cancer is composed of heterogeneous genetic background [17], with extremely high genetic diversity among cells within a single tumor [18]. For such complex diseases, phenotypic screening designed specifically for cancer has been used widely in cancer drug development [11]. The identification of an efficacious chemical by screening often shed lights on the molecular mechanism of action [130].
Phenotypic screening of FDA-approved drugs for drug repurposing is cost-effective because these drugs have already gone through the difficult path of regulatory authorities. This approach has resulted in promising inhibitors against Enteroviruses [131], anti-aging therapeutics [132], anti-cancer drugs [133], and allosteric Bcr-Abl inhibitors in the fight against chronic myeloid leukemia [134].
How does bioinformatics contribute to phenotypic screening? The answer lies in the fact that many modern phenotypic screening studies, especially in screening for anti-cancer drugs, typically define phenotype, either implicitly or explicitly, as a gene expression (transcripts or protein) profile [11] or a metabolomic profile [135-137]. From this perspective, there are two alternative approaches to treat cancer cells. The first is to restore the gene expression of cancer cells to that of normal cells. The second, when the first is not achievable, is to kill cancer cells by inducing apoptosis [11-12]. These two approaches imply two criteria in phenotypic screening for anti-cancer drugs: 1) increased similarity in gene expression between cancer cells and normal cells, and 2) increased similarity in gene expression between cancer cells and apoptotic cells.
Bioinformatics can contribute to gene expression and drug discovery by formulating an objective and rational index of drug desirability (Idd) in phenotypic screening studies with gene expression profiles as phenotypes. Such an Idd would complement therapeutic indices [138, 139] based on various pharmacokinetic models for evaluating drug effects and safety under various drug concentrations [140-142]. The lack of an explicit Idd may have contributed to the low rate of successful drugs discovered through phenotypic screen [126]. For this reason, I will take a rare step in a review article to initiate the effort of developing an index of drug desirability integrating both symptom reduction and side effect.
Designate gene expression profile of a “patient” (which could be an animal disease model or cancer cell line) as Gp, that of a normal control as Gn, and that of a patient after the use of a candidate drug as Gd. It is now easy to compute a variety of pairwise distances [143] between Gn and Gp, between Gd and Gp and between Gn and Gd (designated Dnp, Ddp, and Dnd, respectively, Fig. 3). Dnp is a measure of severity of the symptoms, and (Dnp – Dnd) a measure of symptom reduction by the application of the candidate drug, equivalent to drug efficacy (Emax) in pharmacodynamics models [141-142]. Side effect could be measured by the difference between (Dnd + Ddp) and Dnp, i.e., (Dnd + Ddp - Dnp), which implies that the side effect is greater for Drug B in Fig. (3b) than for Drug A in Fig. (3a). With these definitions, we can formulate an index of drug desirability (Idd) as:
Fig. (3).
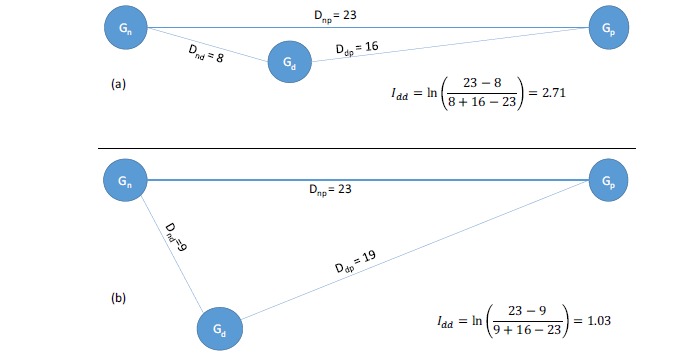
Numerical illustration of applying Idd in Eq. (1) in phenotypic screening to two sets of transcriptomic data (a) and (b). Gn, Gp and Gd refer to gene expression of normal cells, disease cells before drug application, and disease cells after drug application, respectively.
| (1) |
The application of Eq. (1) is illustrated in Fig. (3) where Drug A in Fig. (3a), with Idd = 2.71, is more desirable than Drug B, with Idd = 1.03, in Fig. (3b). One potential problem with Eq. (1) is that the denominator would be zero when Gd = Gn or Gd = Gp, although this is not expected to happen in practice. However, one may add a small pseudo number (c) to the equation so that;
| (2) |
The only requirement for c is that it should be small relative to (Dnp – Dnd) so that its effect on Idd is small. One may set c = 0.01*(Dnp – Dnd).
The application of Idd is not limited to gene expression or metabolomics profiles, but can be applied to any laboratory data in which the patient before the drug use, the normal control and the patient after the drug use can be represented by a vector of values such as blood ferritin and transferrin concentrations, calcium and iron levels, etc. It can be used not only to evaluate desirability of different drugs, but also to evaluate drugs applied at different concentrations or administered through different routes (e.g. oral, subcutaneous injection, etc.). Idd for the second criterion, i.e., how much can a drug induce apoptosis in cancer cells, can be obtained by simply replacing Gn by gene expression of apoptotic cells.
Effective application of the two criteria depends on accurate characterization of gene expression. Development of bioinformatic methods and software has followed the development of high-throughput technologies, such as microarray in the past [143, 144] and next-generation sequencing now [145-153]. Unfortunately, the fundamental problem encountered in allocating sequence reads to paralogous genes, which has previously plagued microarray data analysis, remains unsolved, with nearly all software offering two simple but unsatisfactory options, i.e., either ignoring sequence reads matching multiple genes or allocating such sequence reads equally among paralogous genes. Because a large number of genes are duplicated in multicellular eukaryotes, the lack of proper allocation of sequence reads to paralogous genes implies that the expression of a large number of genes cannot be properly characterized. The method implemented in the software Tuxedo [154], which I outline in the section on ribosomal profiling, may serve as a good starting point.
4.2. Drug Target Identification
Transcriptomic data obtained from RNA-Seq can be used to identify alternative splicing isoforms and differential gene expression and regulation between patient and control. Alteration of spatial and temporal distributions of different splicing isoforms often leads to diseases [155] such as Alzheimer’s disease (AD) associated with abnormal splicing of the amyloid precursor protein (APP). Proteolytic processing of APP generates Amyloid β which contributes to the formation of the extracellular neuritic plaques commonly believed to be the causal factor of AD [156]. APP is a multi-exon gene with exon 7 (E7) encoding a Kunitz protease inhibitor. At least eight isoforms are formed by alternative splicing of APP pre-mRNA, with three isoforms expressed in mammalian brain (one lacking E7 and two others containing E7). The E7-lacking isoform (APP695) is normally prevalent in neurons while the E7-containing isoforms (APP770 and APP751) are expressed mainly in astrocytes and microglial cells [157]. The secreted E7-containing APPs form stable, non-covalent, inhibitory complexes with trypsin, whereas the secreted E7-lacking isoform does not [158]. Increased E7-containing isoforms is associated with AD symptoms in both human and mouse [159]. Expression of U2AF is down-regulated during cellular differentiation of neural tissues [160], which is likely responsible for the E7-skipping in APP695. However, a recent study [156] suggested that E7-skipping is directly linked to the RBFox1 protein with its binding motif (U)GCAUG found both upstream of E7 and within E7. RBFox1 [161] is a neuron- and muscle-specific splicing factor that induces exon skipping of several genes including APP [156]. Thus, both U2AF and RBFox1 could be potential drug targets for AD, i.e., a drug candidate that downregulates U2AF or upregulates RBFox1 specifically in neural tissues could reduce the risk of developing AD. These transcriptomic studies have significantly contributed to our understanding of pathology of not only AD, but many other human diseases associated with alternative splicing.
Abnormal changes in gene expression or regulation is often associated with diseases. The main difficulty is in the interpretation of cause and effects because a gene may have its disease-causing expression occurring at time t1, causing differential expression of many other genes at time t2, where t2 may be years away from t1. Thus, comparing gene expression patterns between a disease group and a control group almost always lead to many false positives [162]. Longitudinal data collected over time will help narrow down to the real culprit of the disease, which is illustrated well by a bioinformatic meta-analysis of 84 kidney transplant biopsies collected at different stage of kidney injury progression [163]. Unfortunately, while it is easy to take a piece of wood from a tree at regular time intervals, it is much more difficult to take a piece of liver out of the patient weekly or monthly.
Transcriptomic data analysis has revealed that most of the human genome is transcribed [115]. Because RNA interference can modulate many cellular processes and that RNA has been recognized as a new type of drugs [164-166], mining transcriptomic data may uncover many RNA molecules either as drugs or as drug targets. Among the numerous unannotated transcripts in human, which are functionally important in human biology? From an evolutionary point of view, a functionally important sequence is one that is expected to be conserved among related species, such as within apes or primates. One can identify functionally important RNAs among millions of different transcripts by checking sequence conservation with one of numerous bioinformatics tools. Any functionally important RNA species may be a potential drug target.
5. PROTEOMIC DATA AND DRUG DISCOVERY
Proteins are the workhorses in living cells and their abnormal abundance is often associated with diseases. A transcribed gene may be differentially translated [167, 168] or not translated [169], and different proteins have different degradation rate, so transcriptomic data is often not a good predictor of protein abundance. For this reason, characterizing and comparing proteomes between patient and control is often more effective in identifying drug targets than genomic or transcriptomic data. Proteomic data have been obtained from nearly all model organisms and deposited in public databases such as PaxDB [170]. Such data have greatly facilitated the development [168] and application of indices predicting translation efficiency [171-173].
Bioinformatic tools used for proteomic data analysis is similar to those in transcriptomic data, i.e., using proteomic data for phenotypic screening and for drug target discovery. Most proteomic data are used in comparisons either between treatment and control animals [174-176] or between patients and matched normal control [177]. For example, caffeine-treated rats differ in protein expression from control rats [175]. Numerous such relationships between drugs and protein targets have been reported and stored in databases [178-180] to facilitate retrieval of possible interactions of a query drug with proteins.
Proteomic data, without following a cohort over time, suffer from the same problem as genomic and transcriptomic data in the causal interpretation as I have mentioned before. In particular, from differential expression observed in many proteins, it is difficult to identify which is truly disease-causing. Different proteins change their abundance at different cell cycle phases. Without taking temporal and spatial heterogeneity of cells into consideration, comparison of protein profiles (or transcriptomic profiles) between matched patient/normal pairs will continue to pump out false positives that have little relevance to drug discovery. In animal models, it is possible to sample cells over different periods [176]. Performing single-cell characterization of transcriptomes and proteomes [181-183] over time to reconstruct a cell cycle profile of gene expression (i.e., reorder cell expression profiles characterized at phases 3, 1, 2, 2, 4 to phases 1, 2, 2, 3, 4) should yield much more informative results.
6. RIBOSOME PROFILING AND DRUG DISCOVERY
Protein abundance data have limitations because 1) low-concentration proteins, short peptides, or transient proteins often cannot be detected, 2) membrane proteins, which often serve as essential components in signal transduction, are difficult to isolate, separate and purify. Transcriptomic data once spawned the hope that proteomic data can be predicted from transcriptomic data, but differential translation efficiencies among mRNA [168, 184] and degradation efficiencies among proteins distort the relationship between mRNA abundance and protein abundance. However, ribosome profiling data, coupled with transcriptomic data, are expected to generate good predictions of protein production rate. Transcriptomic and ribosome profiling data provide information on mRNA abundance and translation efficiency, respectively. If genes A and B have mRNA abundance values NA and NB, respectively, from transcriptomic data, and their translation efficiency is RA and RB, respectively, from ribosome profiling data, then their relative protein production rate is NA*RA and NB*RB, respectively. Differences between such predicted protein abundance and observed protein abundance can be used to measure protein degradation rate. Such prediction should be facilitated by obtaining transcriptomic and proteomic data in the same experiment [185], ideally from a single cell [181-183].
Ribosome profiling data, traditionally from microarray [186-187], is now almost exclusively from deep sequencing of ribosome-protected fragments (RPF, ~30 nucleotides) of mRNA [188-190]. The two approaches, however, exhibit high concordance with data from the yeast [167]. The sequenced RPFs can be mapped to protein-coding genes to obtain the location of the ribosome on mRNA. Ribosomal density may be taken as a proxy of translation efficiency [167]. However, for an mRNA with poor codon usage, ribosomes may move slowly and become densely packed along the mRNA. For this reason, elongation efficiency needs to be controlled for, e.g., by regressing ribosome density on the index of translate elongation [168]. Ribosome profiling data are useful in characterizing regulatory motifs such as poly(A) tract that modulate translation efficiency [167], e.g., short poly(A) at 5’ UTR may facilitate the recruitment of translation initiation factors and enhance translation, but long poly(A) may bind to poly(A)-binding proteins and inhibit translation. Such regulatory motifs can serve easily identifiable drug targets that can be easily manipulated.
There are four major models of translation initiation cross-classified by two variables. The first is whether the translation machinery starts scanning for the start codon from the 5’ end of mRNA [191, 192] or from internal ribosome entry sites [169, 193-196]. The second is whether the small ribosomal subunit does the scanning for the start codon or a fully formed ribosome can also perform the scan. While there is little controversy now on the occurrence of internal ribosome entry, only recent ribosome profiling data have offered strong empirical support for fully formed ribosomes along 5’ UTR of mRNAs [197], suggesting that fully formed ribosomes may also scan for the start codon.
In contrast to eukaryotic internal ribosomal entry sites (IRESs) whose IRES activity decreases with the stability of secondary structure [198], many viral IRESs have strong secondary structure. Cricket paralysis virus (CrPV) has an IRES located at the intercistronic region that is capable of directly interacting with the ribosome via its complex secondary structure without any translation initiation factors [199, 202]. The hepatitis C virus (HCV) has an IRES that can mimic the translation initiation complex so that it does not need initiation factors essential for cap-dependent translation [203, 204]. The IRES mechanism of translation initiation allows viruses to carry on their translation while the host cap-dependent translation has been shut down, and viral IRESs, especially those with relatively rigid secondary and tertiary structure such as in HCV, have consequently been recognized as promising drug targets [205].
Translation regulation represents an important cellular mechanism capable of responding to extracellular environment. In the yeast Saccharomyces cerevisiae, a dozen or so genes are transcribed but not normally translated; they are translated when the surface nutrients have been depleted and their products enable yeast cells to burrow down into the culture medium to extract nutrients for growth [169]. Ribosome profiling data can reveal the translation status of these translation regulated messages, and consequently help us understand how organisms use translation regulation in response to environmental changes.
Ribosome profiling is the ultimate tools to discover new protein-coding genes many of which could be drug targets. That many protein-coding genes may remain unannotated is highlighted by the finding that even the extensively studied phage lambda may have unannotated protein-coding genes [206]. In human and mouse, ribosomes are frequently found on transcripts not annotated as coding sequences, with the consequent production of polypeptides [207]. Given that the majority of the human genome are in fact transcribed [115], many new protein-coding genes may be discovered by bioinformatic analysis of ribosome profiling data [208].
One fundamental problem in analyzing ribosome profiling data is with assigning RPFs to paralogous genes when an RPF matches multiple genes equally well. This problem is shared with transcriptomic and proteomic data where protein identification is typically done with peptide mass fingerprinting and a peptide fragment can match multiple proteins equally well [56, pp. 293-308]. Most programs offer two unsatisfactory options: 1) ignore sequence reads that match to multiple paralogous genes, and 2) allocate such reads equally among the matched paralogous genes. A recent program (MMR: Multiple Mapper Resolution) available at https://github.com/ratschlab/mmr intends to solve this problem but offers no methodological details. Because of the large number of duplicated genes in multicellular eukaryotes, inappropriate assignment of RPFs to paralogous genes will render all downstream analysis untrustworthy. I will outline the approach for assigning RPFs to three or more paralogous genes implemented in the computer program Tuxedo [154]. When a gene family has only two members, the assignment is relatively simple and will not be discussed here.
A phylogenetic tree is needed for proper allocation of sequence reads with three or more paralogous genes in a gene family. I illustrate the allocation principle with a gene family with three paralogous genes A, B, and C idealized into three segments in Fig. (4). The three genes shared one identical middle segment with 23 matched reads (that necessarily match equally well to all three paralogues). Genes B and C share an identical first segment to which 20 reads matched. Gene A has its first segment different from that of B and C and got four matched reads. The three genes also have a diverged third segment where paralogous gene A matched 3 reads, B matched 6 and C matched 12. Our task is then to allocate the 23 reads shared by all three and 20 reads shared by B and C to the three paralogues.
Fig. (4).
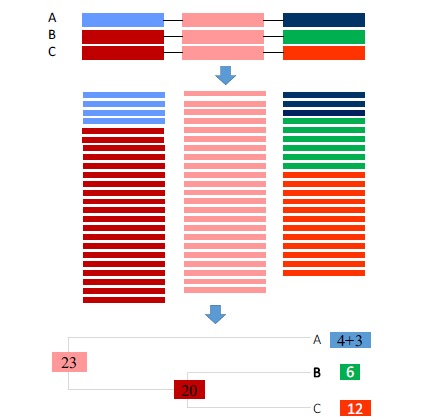
Allocation of shared reads in a gene family with three paralogous genes A, B and C with three idealized segments with a conserved identical middle segment, strongly homologous first segment that is identical in B and C, and a diverged third segment. Reads and the gene segment they match to are of the same color. (The color version of the figure is available in the electronic copy of the article).
TUXEDO uses a simple counting approach by applying the following:
| (3) |
Thus, we allocate the 23 equally matched reads to paralogous genes A, B and C according to PA, PB and PC, respectively. For the 20 reads that matched B and C equally well, we allocate 20*6/(6+12) to B and 20*12/(6+12) to C. This gives the estimated number of matches to each gene as:
| (4) |
7. STRUCTURAL BIOLOGY AND DRUG DISCOVERY
An ideal bioinformatic platform for drug discovery based on structural biology should allow one to 1) predict 3-D structure of a protein or RNA based on the cellular environment where it is translated or transcribed, 2) “BLAST” a known protein/RNA structure against databases of protein/RNA structures to retrieve all protein/RNA with similar structures to facilitate structure-function interpretations and assessment of functional redundancy of the query protein in the cell, and to understand structural convergence, e.g., nonhomologous proteins or RNAs with similar structures [209-210], 3) identify and retrieve all potential binding partners of a given query structure to facilitate the assessment of the query’s potential as a drug target or drug candidate, i.e., its efficiency and side effects as a consequence of physical interactions with other cellular components, 4) automatically identify proteins and RNA that can form a complex and assemble such complexes (e.g., ribosome and spliceosome) through structural modeling and simulation, 5) predict the function of protein/RNA with known structure, either alone or as a component in a complex, and 6) suggest new structures that can physically interact with the query to activate/deactivate the query protein/RNA function in the cell. Almost all of these can be done, although not perfectly, by databases and software tools compiled at http://www.click2drug.org/ .
When one has a protein of interest, the first is to check if its structure already exists in PDB [211-212]. If not, then one can use tools such as homology modeling to infer its structure based on one or more close homologues with known structure. Such tools include SWISS-MODEL [213] and TASSER [214] and its derivatives. Once the structure is refined, one can use UCSF Chimera [215] or PyMOL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC) to visualize the structure and use automated screening software such as SwissSimilarity [216] to identify potential drug candidate that can interact with the protein of interest. Such screening approach is greatly enhanced by metabolic and ligand databases such ChEMBL [217] and SuperSite [218]. Such analyses not only shed lights on identifying drug-target interactions, but also facilitate the identification of side effects of individual drugs, e.g., a drug that can bind to many biologically important enzymes in human is almost surely to have many side effects, but a drug that does the same in a pathogen would be quite desirable.
One may also use docking software such as SwissDock [219] to study physical interactions between protein and small molecules, or use SwissBioisostere [220] to design and refine ligands. Such structural studies improve the design of drugs against HIV-1 protease [221]. The protease is a homodimer each with 99 amino acids in each monomer, and an inhibitor typically needs to squeeze its way between the two monomers to disrupt the protease function [222-224].
Given one well documented protein-ligand interaction, it is natural to infer that other proteins with similar sequence or structure may also bind to the ligand. Such similarity-based approach [52, 225] is the conceptual foundation for the software SwissTargetPrediction [226].
It is important to keep in mind that a structure determined by X-ray crystallography or by NMR represents only a snapshot of structural dynamics, and that protein structure can change in response to different cellular environment. The software CHARMM [227] and its derivatives facilitate the characterization of such dynamic interactions of proteins with their binding partners. Such studies are facilitated by general databases of drug-target interactions [180] and special databases documenting protein interactions in cancer cells [179] or in membranes involving GPCR-ligand associations [178] or organism-specific databases such as that for Mycobacterium tuberculosis [228].
8. BIOINFORMATICS AND DRUG RESISTANCE
Bacterial resistance to penicillin became known soon after its discovery in 1928 and its regular medical applications in 1940 [229, 230]. Such resistance can also develop quickly in eukaryotic pathogens, e.g., in malaria parasite Plasmodium falciparum against the most effective anti-malaria drug artemisinin, soon after the large-scale application of artemisinin in Asian countries [231, 233]. Drug resistance often renders a costly developed drug useless, contributing to the high cost of drug development [1-2]. The rapid development of drug resistance in HIV-1 [234, 235] highlights the importance of understanding drug resistance.
Modern drug development against pathogens demands high specificity against the pathogen. If a drug is toxic to a specific bacterial pathogen, then drug-mediated selection will operate only in this particular bacterial pathogen population to favour individuals with drug resistance. However, if the drug is also toxic to 100 other non-pathogenic bacterial species, drug resistance may develop in all these species, often with subsequent transmission of drug resistance from a non-pathogenic species to a pathogenic one. Pathogenicity islands [41-43], i.e., distinct DNA segment in a large number of bacterial pathogens but not in their avirulent counterparts, serve as specific drug targets against pathogens, and bioinformaticians have created databases [236, 237] to facilitate the identification pathogenicity islands as drug targets.
Modern bioinformatic analysis and innovative experiments have shed light on how fast microbial pathogens can evolve drug resistance. In one experiment [238], error-prone PCR was used to introduce random mutations in Streptococcus pneumoniae genes. These mutated amplicons were then used to transform S. pneumoniae with some resulting colonies exhibiting resistance against antibiotic fusidic acid. DNA sequence analysis revealed a single mutation in the fusA gene accounting for the drug resistance. Many cases have been documented in HIV-1 protease in which a single mutation can significantly change the susceptibility of the protease to its inhibitors [239, 240]. Such studies allow us to estimate the proportion of mutations that confer drug resistance among all random mutations.
How rapid can bacterial and eukaryotic pathogens respond to drug resistance depends mainly on mutation rate, parasite population size and genetic diversity. Lack of genetic diversity implies that drug resistance need to arise de novo, in which case mutation rate becomes a major limiting factor in pathogens evolving drug resistance. Spontaneous mutation rate traditionally was measured in mutation accumulation experiments which are tedious and, for practical reasons, have been done mainly on viruses and a few rapidly replicating bacterial species [241, 242]. Dating the origin of pseudogenes and then comparing their divergence against their functional counterparts [243-246] allow for an estimation of spontaneous mutation rate (approximated by the neutral substitution rate) and mutation spectrum. High mutation rate and large population size increase the chance of parasites developing drug resistance.
For many years, it has been assumed that point mutations occur independent of each other, each being a separate mutation event. For this reason, two serine codons in the standard genetic code (UCU and AGU) are extremely unlikely to mutation into each other because they have to go through two nonsynonymous substitutions which are typically subject to strong purifying selection. However, bioinformatic research and modeling effort have revealed that multiple mutation events can happen in “clusters and showers” in a single generation not only in viruses and bacterial species [247-249], but also in eukaryotes [250, 251].
Genomics sequence analysis and phylogenetics have been frequently used to identify conserved sequence or structure that can guide the development of vaccine [252-254] and ligand designed as inhibitors against bacterial or viral pathogens because sequence and structural similarities often imply similarity in ligand binding. However, strongly conserved sites in a gene does not imply that mutations at these sites will necessarily cripple the gene function. Many amino acid sites in HIV-1 protease are invariant among subtypes of the M group suggesting that they are functionally important. However, drugs designed to inhibit HIV-1 protease quickly leads to mutations at these highly conserved sites, resulting in reduced susceptibility to protease inhibitors [239-240]. This adaptation to the drug-induced selection works in similar way as the development of antibiotics. In the absence of antibiotics, plasmids (regardless of whether they carrying antibiotic-resistant genes or not) in a bacterial species such as E. coli constitute a replication burden. They are consequently selected against and quickly lost in E. coli cultures. However, in the presence of antibiotics, the cost of a replication burden is more than offset by the benefit of antibiotic resistance, and the plasmids carrying the antibiotic-resistant genes will spread.
If we know the population size of the pathogen under the drug effect, the random mutation rate of the pathogen, the proportion of drug-resistant mutations among all random mutations, then it is possible to estimate the probability that a drug-resistant mutation will occur in the first generation after drug application, the probability of no such mutation until the second generation, or in general the probability of no such mutation until the Nth generation. One may also estimate the average number of generations for the first drug resistance mutation to occur. Such estimation is within the domain of population genetics.
9. BIOINFORMATIC SOFTWARE AND DATABASES
An extensive compilation of software, databases and web services directly related to drug discovery can be found at http://click2drug.org/ maintained by Swiss Institute of Bioinformatics. These are roughly grouped into 1) databases, 2) chemical structure representations, 3) molecular modeling and simulation, 4) homology modeling to infer the structure of a protein guided by a homologue of known structure, 5) binding site prediction, 6) docking, 7) screening for drug candidates, 8) drug target prediction, 9) ligand design, 10) binding free energy estimation, 11) QSAR, 12) ADME Toxicity. Many software packages are powerful and free, and supported by well-known institutions. These include databases such as ChEMBL [217] and SwissSidechain [255], software tools such as UCSF Chimera [215] which is not only a 3D visualization tool but also a platform for software developers interested in structural biology, SwissSimilarity for virtual screening [216], SwissBioisostere for ligand design [220], SwissTargetPrediction [226], SwissSideChain to facilitate experiments that expand the protein repertoire by introducing non-natural amino acids, and SwissDock [219] for docking drug candidates (small molecules) on proteins. Although some software are commercial, e.g., CHARMM [227] and PyMOL (Schrödinger), they typically have free versions for students and teachers.
Conclusion
Bioinformatics is a data-driven branch of science, with many of the algorithms and databases developed or adapted in response to new types of data. For this reason, bioinformatic tools often lag behind high-throughput data acquisition technologies. However, a large number of molecular biologists, computer scientists and mathematicians have dedicated their extensive effort to develop new and powerful software packages and databases to extend our views of nature, just as microscopes and telescopes extend our views to see patterns that we have never seen before. Taking a close look at this effort by pharmaceutical scientists may prove to be highly beneficial not only to pharmaceutical industry, but also to bioinformatics research community as well.
ACKNOWLEDGEMENTS
I thank reviewers and students in my lab for their comments and discussion.
CONFLICT OF INTEREST
The author is funded by the Discovery Grant from Natural Science and Engineering Research Council (NSERC, RGPIN/261252) of Canada, and has no conflict of interest in writing the paper.
REFERENCES
- 1.David E., Tramontin T., Zemmel R., Pharmaceutical R. D: the road to positive returns. Nat. Rev. Drug Discov. 2009;8:609–610. doi: 10.1038/nrd2948. [DOI] [PubMed] [Google Scholar]
- 2.Drews J., Ryser S. The role of innovation in drug development. Nat. Biotechnol. 1997;15:1318–1319. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- 3.Davies J., Davies D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boxall A.B., Rudd M.A., Brooks B.W., Caldwell D.J., Choi K., Hickmann S., Innes E., Ostapyk K., Staveley J.P., Verslycke T., Ankley G.T., Beazley K.F., Belanger S.E., Berninger J.P., Carriquiriborde P., Coors A., Deleo P.C., Dyer S.D., Ericson J.F., Gagne F., Giesy J.P., Gouin T., Hallstrom L., Karlsson M.V., Larsson D.G., Lazorchak J.M., Mastrocco F., McLaughlin A., McMaster M.E., Meyerhoff R.D., Moore R., Parrott J.L., Snape J.R., Murray-Smith R., Servos M.R., Sibley P.K., Straub J.O., Szabo N.D., Topp E., Tetreault G.R., Trudeau V.L., Van Der Kraak G. Pharmaceuticals and personal care products in the environment: what are the big questions? Environ. Health Perspect. 2012;120:1221–1219. doi: 10.1289/ehp.1104477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doolittle R.F., Hunkapiller M.W., Hood L.E., Devare S.G., Robbins K.C., Aaronson S.A., Antoniades H.N. Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science. 1983;221:275–277. doi: 10.1126/science.6304883. [DOI] [PubMed] [Google Scholar]
- 6.Waterfield M.D., Scrace G.T., Whittle N., Stroobant P., Johnsson A., Wasteson A., Westermark B., Heldin C.H., Huang J.S., Deuel T.F. Platelet-derived growth factor is structurally related to the putative transforming protein p28sis of simian sarcoma virus. Nature. 1983;304:35–39. doi: 10.1038/304035a0. [DOI] [PubMed] [Google Scholar]
- 7.Pietras K., Sjoblom T., Rubin K., Heldin C.H., Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–443. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 8.Bergsten E., Uutela M., Li X., Pietras K., Ostman A., Heldin C.H., Alitalo K., Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- 9.Ehnman M., Missiaglia E., Folestad E., Selfe J., Strell C., Thway K., Brodin B., Pietras K., Shipley J., Ostman A., Eriksson U. Distinct effects of ligand-induced PDGFRalpha and PDGFRbeta signaling in the human rhabdomyosarcoma tumor cell and stroma cell compartments. Cancer Res. 2013;73:2139–2149. doi: 10.1158/0008-5472.CAN-12-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibbs J.B. Mechanism-based target identification and drug discovery in cancer research. Science. 2000;287:1969–1973. doi: 10.1126/science.287.5460.1969. [DOI] [PubMed] [Google Scholar]
- 11.Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 12.Moffat J.G., Rudolph J., Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discov. 2014;13:588–602. doi: 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]
- 13.Ow G.S., Ivshina A.V., Fuentes G., Kuznetsov V.A. Identification of two poorly prognosed ovarian carcinoma subtypes associated with CHEK2 germ-line mutation and non-CHEK2 somatic mutation gene signatures. Cell Cycle. 2014;13:2262–2280. doi: 10.4161/cc.29271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song F., Amos C.I., Lee J.E., Lian C.G., Fang S., Liu H., MacGregor S., Iles M.M., Law M.H., Lindeman N.I., Montgomery G.W., Duffy D.L., Cust A.E., Jenkins M.A., Whiteman D.C., Kefford R.F., Giles G.G., Armstrong B.K., Aitken J.F., Hopper J.L., Brown K.M., Martin N.G., Mann G.J., Bishop D.T., Bishop J.A., Kraft P., Qureshi A.A., Kanetsky P.A., Hayward N.K., Hunter D.J., Wei Q., Han J. Identification of a melanoma susceptibility locus and somatic mutation in TET2. Carcinogenesis. 2014;35:2097–2101. doi: 10.1093/carcin/bgu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W., Tan A.Y., Blumenfeld J., Liu G., Michaeel A., Zhang T., Robinson B.D., Salvatore S.P., Kapur S., Donahue S., Bobb W.O., Rennert H. Papillary renal cell carcinoma with a somatic mutation in MET in a patient with autosomal dominant polycystic kidney disease. Cancer Genet. 2016;209:11–20. doi: 10.1016/j.cancergen.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Brucher B.L., Jamall I.S. Somatic Mutation Theory - Why it's Wrong for Most Cancers. Cell. Physiol. Biochem. 2016;38:1663–1680. doi: 10.1159/000443106. [DOI] [PubMed] [Google Scholar]
- 17.Garraway L.A., Lander E.S. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Ling S., Hu Z., Yang Z., Yang F., Li Y., Lin P., Chen K., Dong L., Cao L., Tao Y., Hao L., Chen Q., Gong Q., Wu D., Li W., Zhao W., Tian X., Hao C., Hungate E.A., Catenacci D.V., Hudson R.R., Li W.H., Lu X., Wu C.I. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA. 2015;112:E6496–E6505. doi: 10.1073/pnas.1519556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira B., Chin S.F., Rueda O.M., Vollan H.K., Provenzano E., Bardwell H.A., Pugh M., Jones L., Russell R., Sammut S.J., Tsui D.W., Liu B., Dawson S.J., Abraham J., Northen H., Peden J.F., Mukherjee A., Turashvili G., Green A.R., McKinney S., Oloumi A., Shah S., Rosenfeld N., Murphy L., Bentley D.R., Ellis I.O., Purushotham A., Pinder S.E., Borresen-Dale A.L., Earl H.M., Pharoah P.D., Ross M.T., Aparicio S., Caldas C. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baird A.L., Westwood S., Lovestone S. Blood-Based Proteomic Biomarkers of Alzheimer's Disease Pathology. Front. Neurol. 2015;6:236. doi: 10.3389/fneur.2015.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daily K., Patel V.R., Rigor P., Xie X., Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics. 2011;12:495. doi: 10.1186/1471-2105-12-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H.Y., Chien C.H., Jen K.H., Huang H.D. RegRNA: an integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 2006;34:W429-434. doi: 10.1093/nar/gkl333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie X., Rigor P., Baldi P. MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics. 2009;25:167–174. doi: 10.1093/bioinformatics/btn605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia X. Position Weight Matrix, Gibbs Sampler, and the Associated Significance Tests in Motif Characterization and Prediction. Scientifica, 2012, 2012: Article ID 917540, 15 pp. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rouchka E.C. A Brief Overview of Gibbs Sampling.; IBC Statistics Study Group. Washington University, Institute for Biomedical Computing; 1997. [Google Scholar]
- 26.Hua S., Sun Z. Support vector machine approach for protein sub- cellular localization prediction. Bioinformatics. 2001;17:721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- 27.Zien A., Ratsch G., Mika S., Scholkopf B., Lengauer T. Mul- ler, K.R. Engineering support vector machine kernels that recog- nize translation initiation sites. Bioinformatics. 2000;16:799–807. doi: 10.1093/bioinformatics/16.9.799. [DOI] [PubMed] [Google Scholar]
- 28.Kotokorpi P., Venteclef N., Ellis E., Gustafsson J.A., Mode A. The human ADFP gene is a direct liver-X-receptor (LXR) target gene and differentially regulated by synthetic LXR ligands. Mol. Pharmacol. 2010;77:79–86. doi: 10.1124/mol.109.059063. [DOI] [PubMed] [Google Scholar]
- 29.Xia X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013;30:1720–1728. doi: 10.1093/molbev/mst064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vlasschaert C., Xia X., Coulombe J., Gray D.A. Evolution of the highly networked deubiquitinating enzymes USP4, USP15, and USP11. BMC Evol. Biol. 2015;15:230. doi: 10.1186/s12862-015-0511-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krasemann E.W., Meier V., Korenke G.C., Hunneman D.H., Hanefeld F. Identification of mutations in the ALD-gene of 20 families with adrenoleukodystrophy/adrenomyeloneuropathy. Hum. Genet. 1996;97:194–197. doi: 10.1007/BF02265264. [DOI] [PubMed] [Google Scholar]
- 32.Morita M., Shimozawa N., Kashiwayama Y., Suzuki Y., Imanaka T. ABC subfamily D proteins and very long chain fatty acid metabolism as novel targets in adrenoleukodystrophy. Curr. Drug Targets. 2011;12:694–706. doi: 10.2174/138945011795378577. [DOI] [PubMed] [Google Scholar]
- 33.Pauling L., Itano H.A., Singer S.J., Wells I.C. Sickle cell anemia a molecular disease. Science. 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 34.Ingram V.M. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956;178:792–794. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 35.Ingram V.M. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- 36.Steinberg M.H., Rodgers G.P. Pathophysiology of sickle cell disease: role of cellular and genetic modifiers. Semin. Hematol. 2001;38:299–306. doi: 10.1016/s0037-1963(01)90023-x. [DOI] [PubMed] [Google Scholar]
- 37.Kutlar A. Sickle cell disease: a multigenic perspective of a single gene disorder. Hemoglobin. 2007;31:209–224. doi: 10.1080/03630260701290233. [DOI] [PubMed] [Google Scholar]
- 38.Kioussis D., Vanin E., deLange T., Flavell R.A., Grosveld F.G. Beta-globin gene inactivation by DNA translocation in gamma beta-thalassaemia. Nature. 1983;306:662–666. doi: 10.1038/306662a0. [DOI] [PubMed] [Google Scholar]
- 39.Taramelli R., Kioussis D., Vanin E., Bartram K., Groffen J., Hurst J., Grosveld F.G. Gamma delta beta-thalassaemias 1 and 2 are the result of a 100 kbp deletion in the human beta-globin cluster. Nucleic Acids Res. 1986;14:7017–7029. doi: 10.1093/nar/14.17.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofer A., Steverding D., Chabes A., Brun R., Thelander L. Trypanosoma brucei CTP synthetase: a target for the treatment of African sleeping sickness. Proc. Natl. Acad. Sci. USA. 2001;98:6412–6416. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gal-Mor O., Finlay B.B. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell. Microbiol. 2006;8:1707–1719. doi: 10.1111/j.1462-5822.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- 42.Hacker J., Blum-Oehler G., Muhldorfer I., Tschape H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol. Microbiol. 1997;23:1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- 43.Hacker J., Kaper J.B. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 44.Bhatia B., Ponia S.S., Solanki A.K., Dixit A., Garg L.C. Identification of glutamate ABC-Transporter component in Clostridium perfringens as a putative drug target. Bioinformation. 2014;10:401–405. doi: 10.6026/97320630010401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cox S.S., van der Giezen M., Tarr S.J., Crompton M.R., Tovar J. Evidence from bioinformatics, expression and inhibition studies of phosphoinositide-3 kinase signalling in Giardia intestinalis. BMC Microbiol. 2006;6:45. doi: 10.1186/1471-2180-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fernandez-Pinar R., Lo Sciuto A., Rossi A., Ranucci S., Bragonzi A., Imperi F. In vitro and in vivo screening for novel essential cell-envelope proteins in Pseudomonas aeruginosa. Sci. Rep. 2015;5:17593. doi: 10.1038/srep17593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gruber T.D., Borrok M.J., Westler W.M., Forest K.T., Kiessling L.L. Ligand binding and substrate discrimination by UDP-galactopyranose mutase. J. Mol. Biol. 2009;391:327–340. doi: 10.1016/j.jmb.2009.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kincaid V.A., London N., Wangkanont K., Wesener D.A., Marcus S.A., Heroux A., Nedyalkova L., Talaat A.M., Forest K.T., Shoichet B.K., Kiessling L.L. Virtual Screening for UDP-Galactopyranose Mutase Ligands Identifies a New Class of Antimycobacterial Agents. ACS Chem. Biol. 2015;10:2209–2218. doi: 10.1021/acschembio.5b00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedersen L.L., Turco S.J. Galactofuranose metabolism: a potential target for antimicrobial chemotherapy. Cell. Mol. Life Sci. 2003;60:259–266. doi: 10.1007/s000180300021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beverley S.M., Owens K.L., Showalter M., Griffith C.L., Doering T.L., Jones V.C., McNeil M.R. Eukaryotic UDP-galactopyranose mutase (GLF gene) in microbial and metazoal pathogens. Eukaryot. Cell. 2005;4:1147–1154. doi: 10.1128/EC.4.6.1147-1154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wesener D.A., May J.F., Huffman E.M., Kiessling L.L. UDP-galactopyranose mutase in nematodes. Biochemistry (Mosc.) 2013;52:4391–4398. doi: 10.1021/bi400264d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding H., Takigawa I., Mamitsuka H., Zhu S. Similarity-based machine learning methods for predicting drug-target interactions: a brief review. Brief. Bioinform. 2014;15:734–747. doi: 10.1093/bib/bbt056. [DOI] [PubMed] [Google Scholar]
- 53.Bastianelli G., Bouillon A., Nguyen C., Crublet E., Petres S., Gorgette O., Le-Nguyen D., Barale J.C., Nilges M. Computational reverse-engineering of a spider-venom derived peptide active against Plasmodium falciparum SUB1. PLoS One. 2011;6:e21812. doi: 10.1371/journal.pone.0021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alderwick L.J., Seidel M., Sahm H., Besra G.S., Eggeling L. Identification of a novel arabinofuranosyltransferase (AftA) involved in cell wall arabinan biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2006;281:15653–15661. doi: 10.1074/jbc.M600045200. [DOI] [PubMed] [Google Scholar]
- 55.Jacob F., Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 56.Xia X. Bioinformatics and the cell: Modern computational approaches in genomics, proteomics and transcriptomics. New York: Springer US; 2007. p. 349. [Google Scholar]
- 57.Higgs P.G., Attwood T.K. Bioinformatics and molecular evolution. Malden: Blackwell; 2005. [Google Scholar]
- 58.Cardon L.R., Burge C., Clayton D.A., Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA. 1994;91:3799–3803. doi: 10.1073/pnas.91.9.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goto M., Washio T., Tomita M. Causal analysis of CpG suppression in the Mycoplasma genome. Microb. Comp. Genomics. 2000;5:51–58. doi: 10.1089/10906590050145267. [DOI] [PubMed] [Google Scholar]
- 60.Xia X. DNA methylation and mycoplasma genomes. J. Mol. Evol. 2003;57:S21–S28. doi: 10.1007/s00239-003-0003-6. [DOI] [PubMed] [Google Scholar]
- 61.Xia X., Phy P.A. Phylogenetic method with pairwise sequence alignment outperforms likelihood methods in phylogenetics involving highly diverged sequences. Mol. Phylogenet. Evol. 2016;102:331–343. doi: 10.1016/j.ympev.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 62.Korenke G.C., Fuchs S., Krasemann E., Doerr H.G., Wilichowski E., Hunneman D.H., Hanefeld F. Cerebral adrenoleukodystrophy (ALD) in only one of monozygotic twins with an identical ALD genotype. Ann. Neurol. 1996;40:254–257. doi: 10.1002/ana.410400221. [DOI] [PubMed] [Google Scholar]
- 63.Petronis A. The origin of schizophrenia: genetic thesis, epigenetic antithesis, and resolving synthesis. . Biol. Psychiatry. 2004;55:965–970. doi: 10.1016/j.biopsych.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 64.Petronis A. Epigenetics and twins: three variations on the theme. Trends Genet. 2006;22:347–350. doi: 10.1016/j.tig.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 65.Petronis A., Gottesman I.I., Kan P., Kennedy J.L., Basile V.S., Paterson A.D., Popendikyte V. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophr. Bull. 2003;29:169–178. doi: 10.1093/oxfordjournals.schbul.a006988. [DOI] [PubMed] [Google Scholar]
- 66.Zoghbi H.Y., Beaudet A.L. Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol. 2016;8:a019497. doi: 10.1101/cshperspect.a019497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang Y.H., Bressler J., Beaudet A.L. Epigenetics and human disease. Annu. Rev. Genomics Hum. Genet. 2004;5:479–510. doi: 10.1146/annurev.genom.5.061903.180014. [DOI] [PubMed] [Google Scholar]
- 68.Fatemi M., Hermann A., Pradhan S., Jeltsch A. The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J. Mol. Biol. 2001;309:1189–1199. doi: 10.1006/jmbi.2001.4709. [DOI] [PubMed] [Google Scholar]
- 69.Wade P.A., Wolffe A.P. ReCoGnizing methylated DNA. Nat. Struct. Biol. 2001;8:575–577. doi: 10.1038/89593. [DOI] [PubMed] [Google Scholar]
- 70.Insinga A., Minucci S., Pelicci P.G. Mechanisms of selective anticancer action of histone deacetylase inhibitors. Cell Cycle. 2005;4:741–743. doi: 10.4161/cc.4.6.1717. [DOI] [PubMed] [Google Scholar]
- 71.Insinga A., Monestiroli S., Ronzoni S., Gelmetti V., Marchesi F., Viale A., Altucci L., Nervi C., Minucci S., Pelicci P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 72.Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 73.Voelter-Mahlknecht S. Epigenetic associations in relation to cardiovascular prevention and therapeutics. Clin. Epigenetics. 2016;8:4. doi: 10.1186/s13148-016-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kungulovski G., Jeltsch A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016;32:101–113. doi: 10.1016/j.tig.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 75.Grigg G., Clark S. Sequencing 5-methylcytosine residues in genomic DNA. BioEssays. 1994;16:431–436. doi: 10.1002/bies.950160612. [DOI] [PubMed] [Google Scholar]
- 76.Grigg G.W. Sequencing 5-methylcytosine residues by the bisulphite method. DNA Seq. 1996;6:189–198. doi: 10.3109/10425179609008443. [DOI] [PubMed] [Google Scholar]
- 77.Robertson G., Hirst M., Bainbridge M., Bilenky M., Zhao Y., Zeng T., Euskirchen G., Bernier B., Varhol R., Delaney A., Thiessen N., Griffith O.L., He A., Marra M., Snyder M., Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 78.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., Sandstrom R., Bernstein B., Bender M.A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L.A., Lander E.S., Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muller H.J., Altenburg E. The Frequency of Translocations Produced by X-Rays in Drosophila. Genetics. 1930;15:283–311. doi: 10.1093/genetics/15.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pazin M.J., Hermann J.W., Kadonaga J.T. Promoter structure and transcriptional activation with chromatin templates assembled in vitro. A single Gal4-VP16 dimer binds to chromatin or to DNA with comparable affinity. J. Biol. Chem. 1998;273:34653–34660. doi: 10.1074/jbc.273.51.34653. [DOI] [PubMed] [Google Scholar]
- 81.Pazin M.J., Kamakaka R.T., Kadonaga J.T. ATP-dependent nucleosome reconfiguration and transcriptional activation from preassembled chromatin templates. Science. 1994;266:2007–2011. doi: 10.1126/science.7801129. [DOI] [PubMed] [Google Scholar]
- 82.Pazin M.J., Sheridan P.L., Cannon K., Cao Z., Keck J.G., Kadonaga J.T., Jones K.A. NF-kappa B-mediated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes Dev. 1996;10:37–49. doi: 10.1101/gad.10.1.37. [DOI] [PubMed] [Google Scholar]
- 83.Sheridan P.L., Sheline C.T., Cannon K., Voz M.L., Pazin M.J., Kadonaga J.T., Jones K.A. Activation of the HIV-1 enhancer by the LEF-1 HMG protein on nucleosome-assembled DNA in vitro. Genes Dev. 1995;9:2090–2104. doi: 10.1101/gad.9.17.2090. [DOI] [PubMed] [Google Scholar]
- 84.Ito T., Bulger M., Pazin M.J., Kobayashi R., Kadonaga J.T. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 85.Palstra R.J., Tolhuis B., Splinter E., Nijmeijer R., Grosveld F., de Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 2003;35:190–194. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- 86.Tolhuis B., Palstra R.J., Splinter E., Grosveld F., de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 87.Forrester W.C., Epner E., Driscoll M.C., Enver T., Brice M., Papayannopoulou T., Groudine M. A deletion of the human beta-globin locus activation region causes a major alteration in chromatin structure and replication across the entire beta-globin locus. Genes Dev. 1990;4:1637–1649. doi: 10.1101/gad.4.10.1637. [DOI] [PubMed] [Google Scholar]
- 88.Deng W., Lee J., Wang H., Miller J., Reik A., Gregory P.D., Dean A., Blobel G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deng W., Rupon J.W., Krivega I., Breda L., Motta I., Jahn K.S., Reik A., Gregory P.D., Rivella S., Dean A., Blobel G.A. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hou C., Zhao H., Tanimoto K., Dean A. CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. USA. 2008;105:20398–20403. doi: 10.1073/pnas.0808506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma P., Xia X. Factors affecting splicing strength of yeast genes. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vlasschaert C., Xia X., Gray D.A. Selection preserves Ubiquitin Specific Protease 4 alternative exon skipping in therian mammals. Sci. Rep. 2016;6:20039. doi: 10.1038/srep20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bibikova M., Barnes B., Tsan C., Ho V., Klotzle B., Le J.M., Delano D., Zhang L., Schroth G.P., Gunderson K.L., Fan J.B., Shen R. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 94.Eckhardt F., Lewin J., Cortese R., Rakyan V.K., Attwood J., Burger M., Burton J., Cox T.V., Davies R., Down T.A., Haefliger C., Horton R., Howe K., Jackson D.K., Kunde J., Koenig C., Liddle J., Niblett D., Otto T., Pettett R., Seemann S., Thompson C., West T., Rogers J., Olek A., Berlin K., Beck S. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shoemaker R., Deng J., Wang W., Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20:883–889. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ohta T., Gray T.A., Rogan P.K., Buiting K., Gabriel J.M., Saitoh S., Muralidhar B., Bilienska B., Krajewska-Walasek M., Driscoll D.J., Horsthemke B., Butler M.G., Nicholls R.D. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am. J. Hum. Genet. 1999;64:397–413. doi: 10.1086/302233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen Q., Yan M., Cao Z., Li X., Zhang Y., Shi J., Feng G.H., Peng H., Zhang X., Qian J., Duan E., Zhai Q., Zhou Q. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351:397–400. doi: 10.1126/science.aad7977. [DOI] [PubMed] [Google Scholar]
- 98.Sharma U., Conine C.C., Shea J.M., Boskovic A., Derr A.G., Bing X.Y., Belleannee C., Kucukural A., Serra R.W., Sun F., Song L., Carone B.R., Ricci E.P., Li X.Z., Fauquier L., Moore M.J., Sullivan R., Mello C.C., Garber M., Rando O.J. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ingrosso D., Cimmino A., Perna A.F., Masella L., De Santo N.G., De Bonis M.L., Vacca M., D'Esposito M., D'Urso M. ; Galletti P., Zappia V. Folate treatment and unbalanced methyla- tion and changes of allelic expression induced by hyperhomocyste- inaemia in patients with uraemia. Lancet. 2003;361:1693–1699. doi: 10.1016/S0140-6736(03)13372-7. [DOI] [PubMed] [Google Scholar]
- 100.Ingrosso D., Perna A.F. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. . Cell, . 2009;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bigaud E., Corrales F.J. Methylthioadenosine (MTA) Regulates Liver Cells Proteome and Methylproteome: Implications in Liver Biology and Disease. Mol. Cell. Proteomics. 2016;15:1498–1510. doi: 10.1074/mcp.M115.055772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanehisa M. Molecular network analysis of diseases and drugs in KEGG. Methods Mol. Biol. 2013;939:263–275. doi: 10.1007/978-1-62703-107-3_17. [DOI] [PubMed] [Google Scholar]
- 103.Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D642. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tanabe M., Kanehisa M. Using the KEGG database resource. 2012. [DOI] [PubMed] [Google Scholar]
- 105.Jin P., Alisch R.S., Warren S.T. RNA and microRNAs in fragile X mental retardation. Nat. Cell Biol. 2004;6:1048–1053. doi: 10.1038/ncb1104-1048. [DOI] [PubMed] [Google Scholar]
- 106.Clark A.T. DNA methylation remodeling in vitro and in vivo. Curr. Opin. Genet. Dev. 2015;34:82–87. doi: 10.1016/j.gde.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bao J., Bedford M.T. Epigenetic regulation of the histone-to-protamine transition during spermiogenesis. Reproduction. 2016;151:R55–R70. doi: 10.1530/REP-15-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chu C., Qu K., Zhong F.L., Artandi S.E., Chang H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chu C., Quinn J., Chang H.Y. Chromatin isolation by RNA purification (ChIRP). J. Vis. Exp. 2012;61:3912. doi: 10.3791/3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X., Brugmann S.A., Goodnough L.H., Helms J.A., Farnham P.J., Segal E., Chang H.Y. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-Dinardo D., Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 112.Sendler E., Johnson G.D., Mao S., Goodrich R.J., Diamond M.P., Hauser R., Krawetz S.A. Stability, delivery and functions of human sperm RNAs at fertilization. Nucleic Acids Res. 2013;41:4104–4117. doi: 10.1093/nar/gkt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rodgers A.B., Morgan C.P., Leu N.A., Bale T.L. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc. Natl. Acad. Sci. USA. 2015;112:13699–13704. doi: 10.1073/pnas.1508347112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gapp K., Jawaid A., Sarkies P., Bohacek J., Pelczar P., Prados J., Farinelli L., Miska E., Mansuy I.M. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 2014;17:667–669. doi: 10.1038/nn.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Kuehn M.S., Taylor C.M., Neph S., Koch C.M., Asthana S., Malhotra A., Adzhubei I., Greenbaum J.A., Andrews R.M., Flicek P., Boyle P.J., Cao H., Carter N.P., Clelland G.K., Davis S., Day N., Dhami P., Dillon S.C., Dorschner M.O., Fiegler H., Giresi P.G., Goldy J., Hawrylycz M., Haydock A., Humbert R., James K.D., Johnson B.E., Johnson E.M., Frum T.T., Rosenzweig E.R., Karnani N., Lee K., Lefebvre G.C., Navas P.A., Neri F., Parker S.C., Sabo P.J., Sandstrom R., Shafer A., Vetrie D., Weaver M., Wilcox S., Yu M., Collins F.S., Dekker J., Lieb J.D., Tullius T.D., Crawford G.E., Sunyaev S., Noble W.S., Dunham I., Denoeud F., Reymond A., Kapranov P., Rozowsky J., Zheng D., Castelo R., Frankish A., Harrow J., Ghosh S., Sandelin A., Hofacker I.L., Baertsch R., Keefe D., Dike S., Cheng J., Hirsch H.A., Sekinger E.A., Lagarde J., Abril J.F., Shahab A., Flamm C., Fried C., Hackermuller J., Hertel J., Lindemeyer M., Missal K., Tanzer A., Washietl S., Korbel J., Emanuelsson O., Pedersen J.S., Holroyd N., Taylor R., Swarbreck D., Matthews N., Dickson M.C., Thomas D.J., Weirauch M.T., Gilbert J., Drenkow J., Bell I., Zhao X., Srinivasan K.G., Sung W.K., Ooi H.S., Chiu K.P., Foissac S., Alioto T., Brent M., Pachter L., Tress M.L., Valencia A., Choo S.W., Choo C.Y., Ucla C., Manzano C., Wyss C., Cheung E., Clark T.G., Brown J.B., Ganesh M., Patel S., Tammana H., Chrast J., Henrichsen C.N., Kai C., Kawai J., Nagalakshmi U., Wu J., Lian Z., Lian J., Newburger P., Zhang X., Bickel P., Mattick J.S., Carninci P., Hayashizaki Y., Weissman S., Hubbard T., Myers R.M., Rogers J., Stadler P.F., Lowe T.M., Wei C.L., Ruan Y., Struhl K., Gerstein M., Antonarakis S.E., Fu Y., Green E.D., Karaoz U., Siepel A., Taylor J., Liefer L.A., Wetterstrand K.A., Good P.J., Feingold E.A., Guyer M.S., Cooper G.M., Asimenos G., Dewey C.N., Hou M., Nikolaev S., Montoya-Burgos J.I., Loytynoja A., Whelan S., Pardi F., Massingham T., Huang H., Zhang N.R., Holmes I., Mullikin J.C., Ureta-Vidal A., Paten B., Seringhaus M., Church D., Rosenbloom K., Kent W.J., Stone E.A., Batzoglou S., Goldman N., Hardison R.C., Haussler D., Miller W., Sidow A., Trinklein N.D., Zhang Z.D., Barrera L., Stuart R., King D.C., Ameur A., Enroth S., Bieda M.C., Kim J., Bhinge A.A., Jiang N., Liu J., Yao F., Vega V.B., Lee C.W., Ng P., Shahab A., Yang A., Moqtaderi Z., Zhu Z., Xu X., Squazzo S., Oberley M.J., Inman D., Singer M.A., Richmond T.A., Munn K.J., Rada-Iglesias A., Wallerman O., Komorowski J., Fowler J.C., Couttet P., Bruce A.W., Dovey O.M., Ellis P.D., Langford C.F., Nix D.A., Euskirchen G., Hartman S., Urban A.E., Kraus P., Van Calcar S., Heintzman N., Kim T.H., Wang K., Qu C., Hon G., Luna R., Glass C.K., Rosenfeld M.G., Aldred S.F., Cooper S.J., Halees A., Lin J.M., Shulha H.P., Zhang X., Xu M., Haidar J.N., Yu Y., Ruan Y., Iyer V.R., Green R.D., Wadelius C., Farnham P.J., Ren B., Harte R.A., Hinrichs A.S., Trumbower H., Clawson H., Hillman-Jackson J., Zweig A.S., Smith K., Thakkapallayil A., Barber G., Kuhn R.M., Karolchik D., Armengol L., Bird C.P., de Bakker P.I., Kern A.D., Lopez-Bigas N., Martin J.D., Stranger B.E., Woodroffe A., Davydov E., Dimas A., Eyras E., Hallgrimsdottir I.B., Huppert J., Zody M.C., Abecasis G.R., Estivill X., Bouffard G.G., Guan X., Hansen N.F., Idol J.R., Maduro V.V., Maskeri B., McDowell J.C., Park M., Thomas P.J., Young A.C., Blakesley R.W., Muzny D.M., Sodergren E., Wheeler D.A., Worley K.C., Jiang H., Weinstock G.M., Gibbs R.A., Graves T., Fulton R., Mardis E.R., Wilson R.K., Clamp M., Cuff J., Gnerre S., Jaffe D.B., Chang J.L., Lindblad-Toh K., Lander E.S., Koriabine M., Nefedov M., Osoegawa K., Yoshinaga Y., Zhu B., de Jong P.J. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Murphy J., Mahony J., Ainsworth S., Nauta A., van Sinderen D. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013;79:7547–7555. doi: 10.1128/AEM.02229-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abdel-Hameed E.A., Ji H., Shata M.T. HIV-Induced Epigenetic Alterations in Host Cells. Adv. Exp. Med. Biol. 2016;879:27–38. doi: 10.1007/978-3-319-24738-0_2. [DOI] [PubMed] [Google Scholar]
- 118.Bierne H., Hamon M., Cossart P. Epigenetics and bacterial infections. Cold Spring Harb. Perspect. Med. 2012;2:a010272. doi: 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Arbibe L., Sansonetti P.J. Epigenetic regulation of host response to LPS: causing tolerance while avoiding Toll errancy. Cell Host Microbe. 2007;1:244–246. doi: 10.1016/j.chom.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 120.Berger M.F., Levin J.Z., Vijayendran K., Sivachenko A., Adiconis X., Maguire J., Johnson L.A., Robinson J., Verhaak R.G., Sougnez C., Onofrio R.C., Ziaugra L., Cibulskis K., Laine E., Barretina J., Winckler W., Fisher D.E., Getz G., Meyerson M., Jaffe D.B., Gabriel S.B., Lander E.S., Dummer R., Gnirke A., Nusbaum C., Garraway L.A. Integrative analysis of the melanoma transcriptome. Genome Res. 2010;20:413–427. doi: 10.1101/gr.103697.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arvaniti E., Moulos P., Vakrakou A., Chatziantoniou C., Chadjichristos C., Kavvadas P., Charonis A., Politis P.K. Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci. Rep. 2016;6:26235. doi: 10.1038/srep26235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bell D., Bell A.H., Bondaruk J., Hanna E.Y., Weber R.S. In-depth characterization of the salivary adenoid cystic carcinoma transcriptome with emphasis on dominant cell type. Cancer. 2016;122:1513–1522. doi: 10.1002/cncr.29959. [DOI] [PubMed] [Google Scholar]
- 123.Furukawa R., Hachiya T., Ohmomo H., Shiwa Y., Ono K., Suzuki S., Satoh M., Hitomi J., Sobue K., Shimizu A. Intraindividual dynamics of transcriptome and genome-wide stability of DNA methylation. Sci. Rep. 2016;6:26424. doi: 10.1038/srep26424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Haustead D.J., Stevenson A., Saxena V., Marriage F., Firth M., Silla R., Martin L., Adcroft K.F., Rea S., Day P.J., Melton P., Wood F.M., Fear M.W. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-kappaB. Sci. Rep. 2016;6:26846. doi: 10.1038/srep26846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mlera L., Lam J., Offerdahl D.K., Martens C., Sturdevant D., Turner C.V., Porcella S.F., Bloom M.E. Transcriptome Analysis Reveals a Signature Profile for Tick-Borne Flavivirus Persistence in HEK 293T Cells. MBio. 2016:7. doi: 10.1128/mBio.00314-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Eder J., Sedrani R., Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discov. 2014;13:577–587. doi: 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- 127.Miller L.H., Su X. Artemisinin: discovery from the Chinese herbal garden. Cell. 2011;146:855–858. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Swinney D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013;93:299–301. doi: 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- 129.Swinney D.C., Anthony J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 130.Swinney Z.T., Haubrich B.A., Xia S., Ramesha C., Gomez S.R., Guyett P., Mensa-Wilmot K., Swinney D.C. A Four-Point Screening Method for Assessing Molecular Mechanism of Action (MMOA) Identifies Tideglusib as a Time-Dependent Inhibitor of Trypanosoma brucei GSK3beta. PLoS Negl. Trop. Dis. 2016;10:e0004506. doi: 10.1371/journal.pntd.0004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ulferts R., de Boer S.M., van der Linden L., Bauer L., Lyoo H.R., Mate M.J., Lichiere J., Canard B., Lelieveld D., Omta W., Egan D., Coutard B., van Kuppeveld F.J. Screening of a Library of FDA-Approved Drugs Identifies Several Enterovirus Replication Inhibitors That Target Viral Protein 2C. Antimicrob. Agents Chemother. 2016;60:2627–2638. doi: 10.1128/AAC.02182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Snell T.W., Johnston R.K., Srinivasan B., Zhou H., Gao M., Skolnick J. Repurposing FDA-approved drugs for anti-aging therapies. Biogerontology. 2016;17(5-6):907–920. doi: 10.1007/s10522-016-9660-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ozsvari B., Lamb R., Lisanti M.P. Repurposing of FDA-approved drugs against cancer - focus on metastasis. Aging (Albany, N.Y.) 2016;8:567–568. doi: 10.18632/aging.100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Singh V.K., Chang H.H., Kuo C.C., Shiao H.Y., Hsieh H.P., Coumar M.S. Drug repurposing for chronic myeloid leukemia: in silico and in vitro investigation of DrugBank database for allosteric Bcr-Abl inhibitors. J. Biomol. Struct. Dyn. 2016:1–16. doi: 10.1080/07391102.2016.1196462. [DOI] [PubMed] [Google Scholar]
- 135.Wishart D.S. Introduction to Cheminformatics. Curr. Protoc. Bioinformatics. 2016;53:1411–1421. doi: 10.1002/0471250953.bi1401s53. [DOI] [PubMed] [Google Scholar]
- 136.Wishart D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016;15(7):473–484. doi: 10.1038/nrd.2016.32. [DOI] [PubMed] [Google Scholar]
- 137.Xia J., Psychogios N., Young N., Wishart D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652-660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Swinney D.C. The role of binding kinetics in therapeutically useful drug action. Curr. Opin. Drug Discov. Devel. 2009;12:31–39. [PubMed] [Google Scholar]
- 139.Muller P.Y., Milton M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012;11:751–761. doi: 10.1038/nrd3801. [DOI] [PubMed] [Google Scholar]
- 140.Gabrielsson J., Green A.R. Quantitative pharmacology or pharmacokinetic pharmacodynamic integration should be a vital component in integrative pharmacology. J. Pharmacol. Exp. Ther. 2009;331:767–774. doi: 10.1124/jpet.109.157172. [DOI] [PubMed] [Google Scholar]
- 141.Holford N.H., Sheiner L.B. Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin. Pharmacokinet. 1981;6:429–453. doi: 10.2165/00003088-198106060-00002. [DOI] [PubMed] [Google Scholar]
- 142.Holford N.H., Sheiner L.B. Pharmacokinetic and pharmacodynamic modeling in vivo. Crit. Rev. Bioeng. 1981;5:273–322. [PubMed] [Google Scholar]
- 143.Xia X., Xie Z. AMADA: Analysis of microarray data. Bioinformatics. 2001;17:569–570. doi: 10.1093/bioinformatics/17.6.569. [DOI] [PubMed] [Google Scholar]
- 144.Gentleman R., Carey V., Huber W., Irizarry R., Dudoit S. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer-Verlag New York; 2005. p. 473. [Google Scholar]
- 145.Deng Q., Ramskold D., Reinius B., Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- 146.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Langmead B., Hansen K.D., Leek J.T. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11:R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roberts A., Schaeffer L., Pachter L. Updating RNA-Seq analyses after re-annotation. Bioinformatics. 2013;29:1631–1637. doi: 10.1093/bioinformatics/btt197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Roberts A., Trapnell C., Donaghey J., Rinn J.L., Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xia X. ARSDA: A comprehensive software package for analyzing RNA-Seq data., XiaLab http://dambe.bio.uottawa.ca/ARSDA/ ARSDA.aspx : Ottawa, 2016.
- 155.Poulos M.G., Batra R., Charizanis K., Swanson M.S. Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol. 2011;3:a000778. doi: 10.1101/cshperspect.a000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Alam S., Suzuki H., Tsukahara T. Alternative splicing regulation of APP exon 7 by RBFox proteins. Neurochem. Int. 2014;78:7–17. doi: 10.1016/j.neuint.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 157.Gaul G., Dutly F., Frei K., Foguet M., Lubbert H. APP RNA splicing is not affected by differentiation of neurons and glia in culture. FEBS Lett. 1992;307:329–332. doi: 10.1016/0014-5793(92)80706-m. [DOI] [PubMed] [Google Scholar]
- 158.Oltersdorf T., Fritz L.C., Schenk D.B., Lieberburg I., Johnson-Wood K.L., Beattie E.C., Ward P.J., Blacher R.W., Dovey H.F., Sinha S. The secreted form of the Alzheimer's amyloid precursor protein with the Kunitz domain is protease nexin-II. Nature. 1989;341:144–147. doi: 10.1038/341144a0. [DOI] [PubMed] [Google Scholar]
- 159.Rockenstein E.M., McConlogue L., Tan H., Power M., Masliah E., Mucke L. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer's disease. J. Biol. Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 160.Tsukahara T., Kunika N., Momoi T., Arahata K. Regulation of alternative splicing in the amyloid precursor protein (APP) mRNA during neuronal and glial differentiation of P19 embryonal carcinoma cells. Brain Res. 1995;679:178–183. doi: 10.1016/0006-8993(95)00216-d. [DOI] [PubMed] [Google Scholar]
- 161.Jin Y., Suzuki H., Maegawa S., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22:905–912. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A., Kiezun A., Hammerman P.S., McKenna A., Drier Y., Zou L., Ramos A.H., Pugh T.J., Stransky N., Helman E., Kim J., Sougnez C., Ambrogio L., Nickerson E., Shefler E., Cortes M.L., Auclair D., Saksena G., Voet D., Noble M., DiCara D., Lin P., Lichtenstein L., Heiman D.I., Fennell T., Imielinski M., Hernandez B., Hodis E., Baca S., Dulak A.M., Lohr J., Landau D.A., Wu C.J., Melendez-Zajgla J., Hidalgo-Miranda A., Koren A., McCarroll S.A., Mora J., Lee R.S., Crompton B., Onofrio R., Parkin M., Winckler W., Ardlie K., Gabriel S.B., Roberts C.W., Biegel J.A., Stegmaier K., Bass A.J., Garraway L.A., Meyerson M., Golub T.R., Gordenin D.A., Sunyaev S., Lander E.S., Getz G. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dosanjh A., Robison E., Mondala T., Head S.R., Salomon D.R., Kurian S.M. Genomic meta-analysis of growth factor and integrin pathways in chronic kidney transplant injury. BMC Genomics. 2013;14:275. doi: 10.1186/1471-2164-14-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mahanthappa N. Translating RNA interference into therapies for human disease. Pharmacogenomics. 2005;6:879–883. doi: 10.2217/14622416.6.8.879. [DOI] [PubMed] [Google Scholar]
- 165.Tiang J.M., Butcher N.J., Cullinane C., Humbert P.O., Minchin R.F. RNAi-mediated knock-down of arylamine N-acetyltransferase-1 expression induces E-cadherin up-regulation and cell-cell contact growth inhibition. PLoS One. 2011;6:e17031. doi: 10.1371/journal.pone.0017031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vangamudi B., Paul T.A., Shah P.K., Kost-Alimova M., Nottebaum L., Shi X., Zhan Y., Leo E., Mahadeshwar H.S., Protopopov A., Futreal A., Tieu T.N., Peoples M., Heffernan T.P., Marszalek J.R., Toniatti C., Petrocchi A., Verhelle D., Owen D.R., Draetta G., Jones P., Palmer W.S., Sharma S., Andersen J.N. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015;75:3865–3878. doi: 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xia X., MacKay V., Yao X., Wu J., Miura F., Ito T., Morris D.R. Translation Initiation: A Regulatory Role for Poly(A) Tracts in Front of the AUG Codon in Saccharomyces cerevisiae. Genetics. 2011;189:469–478. doi: 10.1534/genetics.111.132068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xia X. A Major Controversy in Codon-Anticodon Adaptation Resolved by a New Codon Usage Index. Genetics. 2015;199:573–579. doi: 10.1534/genetics.114.172106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gilbert W.V., Zhou K., Butler T.K., Doudna J.A. Cap-independent translation is required for starvation-induced differentiation in yeast. Science. 2007;317:1224–1227. doi: 10.1126/science.1144467. [DOI] [PubMed] [Google Scholar]
- 170.Wang M., Weiss M., Simonovic M., Haertinger G., Schrimpf S.P., Hengartner M.O., von Mering C. PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics. 2012;11:492–500. doi: 10.1074/mcp.O111.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Prabhakaran R., Chithambaram S., Xia X. E. coli and Staphylococcus phages: Effect of translation initiation efficiency on differential codon adaptation mediated by virulent and temperate lifestyles. J. Gen. Virol. 2015 doi: 10.1099/vir.0.000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chithambaram S., Prabhakaran R., Xia X. Differential Codon Adaptation between dsDNA and ssDNA Phages in Escherichia coli. Mol. Biol. Evol. 2014;31:1606–1617. doi: 10.1093/molbev/msu087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chithambaram S., Prabhakaran R., Xia X. The Effect of Mutation and Selection on Codon Adaptation in Escherichia coli Bacteriophage. Genetics. 2014;197:301–315. doi: 10.1534/genetics.114.162842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ujcikova H., Vosahlikova M., Roubalova L., Svoboda P. Proteomic analysis of protein composition of rat forebrain cortex exposed to morphine for 10days; comparison with animals exposed to morphine and subsequently nurtured for 20days in the absence of this drug. J. Proteomics. 2016;145:11–23. doi: 10.1016/j.jprot.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 175.Franklin J.L., Mirzaei M., Wearne T.A., Homewood J., Goodchild A.K., Haynes P.A., Cornish J.L. Quantitative Proteomic Analysis of the Orbital Frontal Cortex in Rats Following Extended Exposure to Caffeine Reveals Extensive Changes to Protein Expression: Implications for Neurological Disease. J. Proteome Res. 2016;15:1455–1471. doi: 10.1021/acs.jproteome.5b01043. [DOI] [PubMed] [Google Scholar]
- 176.Bitsika V., Duveau V., Simon-Areces J., Mullen W., Roucard C., Makridakis M., Mermelekas G., Savvopoulos P., Depaulis A., Vlahou A. High-Throughput LC-MS/MS Proteomic Analysis of a Mouse Model of Mesiotemporal Lobe Epilepsy Predicts Microglial Activation Underlying Disease Development. J. Proteome Res. 2016;15:1546–1562. doi: 10.1021/acs.jproteome.6b00003. [DOI] [PubMed] [Google Scholar]
- 177.Garg N.J., Soman K.V., Zago M.P., Koo S.J., Spratt H., Stafford S., Blell Z.N., Gupta S., Nunez Burgos J., Barrientos N., Brasier A.R., Wiktorowicz J.E. Changes in Proteome Profile of Peripheral Blood Mononuclear Cells in Chronic Chagas Disease. PLoS Negl. Trop. Dis. 2016;10:e0004490. doi: 10.1371/journal.pntd.0004490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chan W.K., Zhang H., Yang J., Brender J.R., Hur J., Ozgur A., Zhang Y. GLASS: a comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics. 2015;31:3035–3042. doi: 10.1093/bioinformatics/btv302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gohlke B.O., Nickel J., Otto R., Dunkel M., Preissner R. CancerResource-updated database of cancer-relevant proteins, mutations and interacting drugs. Nucleic Acids Res. 2016;44:D932–D937. doi: 10.1093/nar/gkv1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hecker N., Ahmed J., von Eichborn J., Dunkel M., Macha K., Eckert A., Gilson M.K., Bourne P.E., Preissner R. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012;40:D1113–D1117. doi: 10.1093/nar/gkr912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Heath J.R., Ribas A., Mischel P.S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 2016;15:204–216. doi: 10.1038/nrd.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Saadatpour A., Lai S., Guo G., Yuan G.C. Single-Cell Analysis in Cancer Genomics. Trends Genet. 2015;31:576–586. doi: 10.1016/j.tig.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wu J., Tzanakakis E.S. Deconstructing stem cell population heterogeneity: single-cell analysis and modeling approaches. Biotechnol. Adv. 2013;31:1047–1062. doi: 10.1016/j.biotechadv.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kudla G., Murray A.W., Tollervey D., Plotkin J.B. Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Smircich P., Eastman G., Bispo S., Duhagon M.A., Guerra-Slompo E.P., Garat B., Goldenberg S., Munroe D.J., Dallagiovanna B., Holetz F., Sotelo-Silveira J.R. Ribosome profiling reveals translation control as a key mechanism generating differential gene expression in Trypanosoma cruzi. BMC Genomics. 2015;16:443. doi: 10.1186/s12864-015-1563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Arava Y., Wang Y., Storey J.D., Liu C.L., Brown P.O., Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.MacKay V.L., Li X., Flory M.R., Turcott E., Law G.L., Serikawa K.A., Xu X.L., Lee H., Goodlett D.R., Aebersold R., Zhao L.P., Morris D.R. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics: response of yeast to mating pheromone. Mol. Cell. Proteomics. 2004;3:478–489. doi: 10.1074/mcp.M300129-MCP200. [DOI] [PubMed] [Google Scholar]
- 188.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kozak M. Evaluation of the “scanning model” for initiation of protein synthesis in eucaryotes. Cell. 1980;22:7–8. doi: 10.1016/0092-8674(80)90148-8. [DOI] [PubMed] [Google Scholar]
- 192.Jackson R.J., Hellen C.U., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Doudna J.A., Sarnow P. Translation Initiation by Viral Internal Ribosome Entry Sites. In: Mathews M.B., Sonenberg N., Hershey J., editors. Translational Control in Biology and Medicine. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2007. pp. 129–154. [Google Scholar]
- 194.Sonenberg N., Meerovitch K. Translation of poliovirus mRNA. Enzyme. 1990;44:278–291. doi: 10.1159/000468765. [DOI] [PubMed] [Google Scholar]
- 195.Yu Y., Sweeney T.R., Kafasla P., Jackson R.J., Pestova T.V., Hellen C.U. The mechanism of translation initiation on Aichivirus RNA mediated by a novel type of picornavirus IRES. EMBO J. 2011;30:4423–4436. doi: 10.1038/emboj.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Elroy-Stein O., Merrick W. Translation Initiation via Cellular Internal Ribosome Entry Sites. In: Mathews M.B., Sonenberg N., Hershey J., editors. Transl. Control Biol. Med. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2007. pp. 155–172. [Google Scholar]
- 197.Ingolia N.T., Brar G.A., Stern-Ginossar N., Harris M.S., Talhouarne G.J., Jackson S.E., Wills M.R., Weissman J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Reports. 2014;8:1365–1379. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Xia X., Holcik M. Strong Eukaryotic IRESs Have Weak Secondary Structure. PLoS One. 2009;4:e4136. doi: 10.1371/journal.pone.0004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jan E., Sarnow P. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J. Mol. Biol. 2002;324:889–902. doi: 10.1016/s0022-2836(02)01099-9. [DOI] [PubMed] [Google Scholar]
- 200.Jan E., Thompson S.R., Wilson J.E., Pestova T.V., Hellen C.U., Sarnow P. Initiator Met-tRNA-independent translation mediated by an internal ribosome entry site element in cricket paralysis virus-like insect viruses. Cold Spring Harb. Symp. Quant. Biol. 2001;66:285–292. doi: 10.1101/sqb.2001.66.285. [DOI] [PubMed] [Google Scholar]
- 201.Pestova T.V., Lomakin I.B., Hellen C.U. Position of the CrPV IRES on the 40S subunit and factor dependence of IRES/80S ribosome assembly. EMBO Rep. 2004;5:906–913. doi: 10.1038/sj.embor.7400240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schuler M., Connell S.R., Lescoute A., Giesebrecht J., Dabrowski M., Schroeer B., Mielke T., Penczek P.A., Westhof E., Spahn C.M. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol. 2006;13:1092–1096. doi: 10.1038/nsmb1177. [DOI] [PubMed] [Google Scholar]
- 203.Pestova T.V., Shatsky I.N., Fletcher S.P., Jackson R.J., Hellen C.U. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Boehringer D., Thermann R., Ostareck-Lederer A., Lewis J.D., Stark H. Structure of the Hepatitis C Virus IRES Bound to the Human 80S Ribosome: Remodeling of the HCV IRES. Structure. 2005;13:1695. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 205.Komar A.A., Hatzoglou M. Internal ribosome entry sites in cellular mRNAs: mystery of their existence. J. Biol. Chem. 2005;280:23425–23428. doi: 10.1074/jbc.R400041200. [DOI] [PubMed] [Google Scholar]
- 206.Liu X., Jiang H., Gu Z., Roberts J.W. High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc. Natl. Acad. Sci. USA. 2013;110:11928–11933. doi: 10.1073/pnas.1309739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yoon J.H., De S., Srikantan S., Abdelmohsen K., Grammatikakis I., Kim J., Kim K.M., Noh J.H., White E.J., Martindale J.L., Yang X., Kang M.J., Wood W.H., III, Noren Hooten N., Evans M.K., Becker K.G., Tripathi V., Prasanth K.V., Wilson G.M., Tuschl T., Ingolia N.T., Hafner M., Gorospe M. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat. Commun. 2014;5:5248. doi: 10.1038/ncomms6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Popa A., Lebrigand K., Barbry P., Waldmann R. Pateamine A-sensitive ribosome profiling reveals the scope of translation in mouse embryonic stem cells. BMC Genomics. 2016;17:52. doi: 10.1186/s12864-016-2384-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dykeman E.C., Stockley P.G., Twarock R. Packaging signals in two single-stranded RNA viruses imply a conserved assembly mechanism and geometry of the packaged genome. J. Mol. Biol. 2013;425:3235–3249. doi: 10.1016/j.jmb.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 210.Naveed H., Hameed U.S., Harrus D., Bourguet W., Arold S.T., Gao X. An integrated structure- and system-based framework to identify new targets of metabolites and known drugs. Bioinformatics. 2015;31:3922–3929. doi: 10.1093/bioinformatics/btv477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rose P.W., Prlic A., Bi C., Bluhm W.F., Christie C.H., Dutta S., Green R.K., Goodsell D.S., Westbrook J.D., Woo J., Young J., Zardecki C., Berman H.M., Bourne P.E., Burley S.K. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43:D345–D356. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Westbrook J., Feng Z., Jain S., Bhat T.N., Thanki N., Ravichandran V., Gilliland G.L., Bluhm W., Weissig H., Greer D.S., Bourne P.E., Berman H.M. The Protein Data Bank: unifying the archive. Nucleic Acids Res. 2002;30:245–248. doi: 10.1093/nar/30.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252-258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang Y., Arakaki A.K., Skolnick J. TASSER: an automated method for the prediction of protein tertiary structures in CASP6. Proteins. 2005;61(Suppl. 7):91–98. doi: 10.1002/prot.20724. [DOI] [PubMed] [Google Scholar]
- 215.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 216.Zoete V., Daina A., Bovigny C., Michielin O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016;56:1399–1404. doi: 10.1021/acs.jcim.6b00174. [DOI] [PubMed] [Google Scholar]
- 217.Gaulton A., Bellis L.J., Bento A.P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J.P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bauer R.A., Gunther S., Jansen D., Heeger C., Thaben P.F., Preissner R. SuperSite: dictionary of metabolite and drug binding sites in proteins. Nucleic Acids Res. 2009;37:D195–D200. doi: 10.1093/nar/gkn618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Grosdidier A., Zoete V., Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270-277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wirth M., Zoete V., Michielin O., Sauer W.H. SwissBioisostere: a database of molecular replacements for ligand design. Nucleic Acids Res. 2013;41:D1137–D1143. doi: 10.1093/nar/gks1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Heal J.W., Jimenez-Roldan J.E., Wells S.A., Freedman R.B., Romer R.A. Inhibition of HIV-1 protease: the rigidity perspective. Bioinformatics. 2012;28:350–357. doi: 10.1093/bioinformatics/btr683. [DOI] [PubMed] [Google Scholar]
- 222.Broglia R., Levy Y., Tiana G. HIV-1 protease folding and the design of drugs which do not create resistance. Curr. Opin. Struct. Biol. 2008;18:60–66. doi: 10.1016/j.sbi.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 223.Wlodawer A., Erickson J.W. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 224.Wlodawer A., Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- 225.Ekins S., de Siqueira-Neto J.L., McCall L.I., Sarker M., Yadav M., Ponder E.L., Kallel E.A., Kellar D., Chen S., Arkin M., Bunin B.A., McKerrow J.H., Talcott C. Machine Learning Models and Pathway Genome Data Base for Trypanosoma cruzi Drug Discovery. PLoS Negl. Trop. Dis. 2015;9:e0003878. doi: 10.1371/journal.pntd.0003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gfeller D., Grosdidier A., Wirth M., Daina A., Michielin O., Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014;42:W32-38. doi: 10.1093/nar/gku293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Brooks B.R., Brooks C.L., III, Mackerell A.D., Jr, Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A.R., Feig M., Fischer S., Gao J., Hodoscek M.Im, W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R.W., Post C.B., Pu J.Z., Schaefer M., Tidor B., Venable R.M., Woodcock H.L., Wu X., Yang W., York D.M., Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kinnings S.L., Xie L., Fung K.H., Jackson R.M., Bourne P.E. The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLOS Comput. Biol. 2010;6:e1000976. doi: 10.1371/journal.pcbi.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Abraham E.P., Chain E. An enzyme from bacteria able to destroy penicillin. Rev. Infect. Dis. 1940;10:677–678. [PubMed] [Google Scholar]
- 230.Abraham E.P., Chain E., Fletcher C.M., Florey H.W., Gardner A.D., Heatley N.G., Jennings M.A. Further observations on penicillin. Lancet. 1941;238:177–189. [PubMed] [Google Scholar]
- 231.Noedl H., Se Y., Schaecher K., Smith B.L., Socheat D., Fukuda M.M. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 232.Noedl H., Se Y., Sriwichai S., Schaecher K., Teja-Isavadharm P., Smith B., Rutvisuttinunt W., Bethell D., Surasri S., Fukuda M.M., Socheat D., Chan Thap L. Artemisinin resistance in Cambodia: a clinical trial designed to address an emerging problem in Southeast Asia. Clin. Infect. Dis. 2010;51:e82–e89. doi: 10.1086/657120. [DOI] [PubMed] [Google Scholar]
- 233.Noedl H., Socheat D., Satimai W. Artemisinin-resistant malaria in Asia. N. Engl. J. Med. 2009;361:540–541. doi: 10.1056/NEJMc0900231. [DOI] [PubMed] [Google Scholar]
- 234.Smyth R.P., Davenport M.P., Mak J. The origin of genetic diversity in HIV-1. Virus Res. 2012;169:415–429. doi: 10.1016/j.virusres.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 235.Smyth R.P., Schlub T.E., Grimm A.J., Waugh C., Ellenberg P., Chopra A., Mallal S., Cromer D., Mak J., Davenport M.P. Identifying recombination hot spots in the HIV-1 genome. J. Virol. 2014;88:2891–2902. doi: 10.1128/JVI.03014-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pundhir S., Vijayvargiya H., Kumar A. PredictBias: a server for the identification of genomic and pathogenicity islands in prokaryotes. In Silico Biol. 2008;8:223–234. [PubMed] [Google Scholar]
- 237.Yoon S.H., Park Y.K., Kim J.F. PAIDB v2.0: exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 2015;43:D624–D630. doi: 10.1093/nar/gku985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Belanger A.E., Lai A., Brackman M.A., LeBlanc D.J. PCR-based ordered genomic libraries: a new approach to drug target identification for Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2002;46:2507–2512. doi: 10.1128/AAC.46.8.2507-2512.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Rhee S.Y., Taylor J., Fessel W.J., Kaufman D., Towner W., Troia P., Ruane P., Hellinger J., Shirvani V., Zolopa A., Shafer R.W. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob. Agents Chemother. 2010;54:4253–42561. doi: 10.1128/AAC.00574-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Young T.P., Parkin N.T., Stawiski E., Pilot-Matias T., Trinh R., Kempf D.J., Norton M. Prevalence, mutation patterns, and effects on protease inhibitor susceptibility of the L76V mutation in HIV-1 protease. Antimicrob. Agents Chemother. 2010;54:4903–4906. doi: 10.1128/AAC.00906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Drake J.W. Studies on the induction of mutations in bacteriophage t4 by ultraviolet irradiation and by proflavin. J. Cell. Physiol. 1964;64(Suppl. 1):19–31. [PubMed] [Google Scholar]
- 242.Drake J.W. Spontaneous mutations accumulating in bacteriophage T4 in the complete absence of DNA replication. Proc. Natl. Acad. Sci. USA. 1966;55:738–743. doi: 10.1073/pnas.55.4.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chang B.H., Li W.H. Estimating the intensity of male-driven evolution in rodents by using X-linked and Y-linked Ube 1 genes and pseudogenes. J. Mol. Evol. 1995;40:70–77. doi: 10.1007/BF00166597. [DOI] [PubMed] [Google Scholar]
- 244.Li W.H., Gojobori T., Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292:237–239. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- 245.Li W.H., Wu C.I., Luo C.C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- 246.Wu C.I., Li W.H., Shen J.J., Scarpulla R.C., Limbach K.J., Wu R. Evolution of cytochrome c genes and pseudogenes. J. Mol. Evol. 1986;23:61–75. doi: 10.1007/BF02100999. [DOI] [PubMed] [Google Scholar]
- 247.Drake J.W. Too many mutants with multiple mutations. Crit. Rev. Biochem. Mol. Biol. 2007;42:247–258. doi: 10.1080/10409230701495631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Drake J.W. Mutations in clusters and showers. Proc. Natl. Acad. Sci. USA. 2007;104:8203–8204. doi: 10.1073/pnas.0703089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Drake J.W., Bebenek A., Kissling G.E., Peddada S. Clusters of mutations from transient hypermutability. Proc. Natl. Acad. Sci. USA. 2005;102:12849–12854. doi: 10.1073/pnas.0503009102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Schrider D.R., Hourmozdi J.N., Hahn M.W. Pervasive multinucleotide mutational events in eukaryotes. Curr. Biol. 2011;21:1051–1054. doi: 10.1016/j.cub.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Averof M., Rokas A., Wolfe K.H., Sharp P.M. Evidence for a high frequency of simultaneous double-nucleotide substitutions. Science. 2000;287:1283–1286. doi: 10.1126/science.287.5456.1283. [DOI] [PubMed] [Google Scholar]
- 252.Manocheewa S., Mittler J.E., Samudrala R., Mullins J.I. Composite sequence-structure stability models as screening tools for identifying vulnerable targets for HIV drug and vaccine development. Viruses. 2015;7:5718–5735. doi: 10.3390/v7112901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.O'Connell R.J., Kim J.H., Excler J.L. The HIV-1 gp120 V1V2 loop: structure, function and importance for vaccine development. Expert Rev. Vaccines. 2014;13:1489–1500. doi: 10.1586/14760584.2014.951335. [DOI] [PubMed] [Google Scholar]
- 254.Anisimova M. Darwin and Fisher meet at biotech: on the potential of computational molecular evolution in industry. BMC Evol. Biol. 2015;15:76. doi: 10.1186/s12862-015-0352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gfeller D., Michielin O., Zoete V. SwissSidechain: a molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2013;41:D327–D332. doi: 10.1093/nar/gks991. [DOI] [PMC free article] [PubMed] [Google Scholar]