

Abstract
Background
The Anopheles hyrcanus group, which includes at least 25 species, is widely distributed in the Oriental and Palearctic regions. Some group members have been incriminated as vectors of malaria and other mosquito-borne diseases. It is difficult to identify Hyrcanus Group members by morphological features. Thus, molecular phylogeny has been proposed as an important complementary method to traditional morphological taxonomy.
Methods
Based on the GenBank database and our original study data, we used 466 mitochondrial DNA COI sequences belonging to 18 species to reconstruct the molecular phylogeny of the Hyrcanus Group across its worldwide geographic range.
Results
The results are as follows. 1) The average conspecific K2P divergence was 0.008 (range 0.002–0.017), whereas sequence divergence between congroup species averaged 0.064 (range 0.026–0.108). 2) The topology of COI tree of the Hyrcanus Group was generally consistent with classical morphological taxonomy in terms of species classification, but disagreed in subgroup division. In the COI tree, the group was divided into at least three main clusters. The first cluster contained An. nimpe; the second was composed of the Nigerrimus Subgroup and An. argyropus; and the third cluster was comprised of the Lesteri Subgroup and other unassociated species. 3) Phylogenetic analysis of COI indicated that ancient hybridizations probably occurred among the three closely related species, An. sinensis, An. belenrae, and An. kleini. 4) The results supported An. paraliae as a probable synonym of An. lesteri, and it was possible that An. pseudopictus and An. hyrcanus were the same species, as evident from their extremely low interspecific genetic divergence (0.020 and 0.007, respectively) and their phylogenetic positions.
Conclusions
In summary, we reconstructed the molecular phylogeny and analysed genetic divergence of the Hyrcanus Group using mitochondrial COI sequences. Our results suggest that in the future of malaria surveillance, we should not only pay much attention to those known vectors of malaria, but also their closely related species.
Electronic supplementary material
The online version of this article (doi:10.1186/s40249-017-0273-7) contains supplementary material, which is available to authorized users.
Keywords: Mosquito, DNA barcoding, Genetic distance, Malaria
Multilingual abstracts
Please see Additional file 1 for translations of the abstract into the five official working languages of the United Nations.
Backgrounds
According to the Action Plan of China Malaria Elimination (APCME), 2010–2020, most areas in China should have been malaria-free since 2015, except for the Yunnan Province. To solidify achievements and realize the goal of nationwide malaria elimination by 2020, the government must address concerns regarding the Plasmodium reservoirs, including surveillance of the remaining parasite reservoirs, and monitoring insecticide resistance in vector populations [1]. The primary malaria vectors in China are Anopheles sinensis Wiedemann, 1828, An. lesteri Baisas & Hu, 1936, An. dirus Peyton & Harrison, 1979, and An. minimus (Theobald, 1905) [2]. The distribution ranges of the former two are mainly in the elimination areas, whereas the latter two are a major threat in Yunnan Province [3]. All of them belong to the genus Anopheles. An. sinensis and An. lesteri are species in the Hyrcanus Group [4]. There are 25 recognized species in this group according to Harbach [4], and one provisionally designated member, An. hyrcanus spIR [5]. The group comprises several species that transmit not only malaria [6–9] and filariasis [10, 11], but also Japanese encephalitis virus [12–14]. Thus, it is important to devise an efficient and accurate method to identify members of the Hyrcanus Group [15], which is a prerequisite for malaria vector surveillance in practice [16, 17]. However, it is difficult to distinguish cryptic species in the Hyrcanus Group by morphological features [5, 18] because there of 1) the scarcity of trained morphologists in the field [19] and 2) the loss of taxonomic traits during daily surveillance activities, such as wing scales [15, 20].
Combined with morphological characteristics and molecular evidence [19, 21–23], the accuracy of mosquito identification has recently improved, both in fieldwork and scientific research. This was caused by 1) the rapid growth of molecular marker sequences in the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/), 2) the consistency between barcoding results and traditional morphology-based taxonomy [24] and 3) the ability to extract enough deoxyribonucleic acid (DNA) for species identification by molecular methods from any life stage of individual mosquito [15, 25]. The gene region being used as the standard barcode for almost all animal species is an average 650 base-pair region in the mitochondrial cytochrome c oxidase 1 gene [25–28].
Compared with ribosomal DNA (rDNA), mitochondrial DNA (mtDNA) COI is advantageous because 1) its evolutionary rate is 5–10 times faster than that of rDNA [29], making it especially suitable for classification at the species level [28, 30]; 2) mtDNA is haplotype, and thus, there is no cloning step before sequencing, which is usually required when using rDNA as a molecular marker [31]; and 3) it has multiple copies, which makes amplification much easier [26]. The differences between COI sequences increase in higher taxonomic categories [32]. The COI barcoding gap is usually 2% within species [28]. High divergence of intraspecific distance is probably caused by recent geographic isolation, suggesting the presence of cryptic species [28, 33]. Thus, more complete sampling of the geographic range, greater distance among sample locations, and more diversified the sampled individuals, the more accurate the genetic divergences within and among species [19, 27, 30, 34]. As such, it will be easier to distinguish cryptic species from studies based on large geographic range, especially cryptic species with short historical divergence [27, 35, 36]. The distribution of mosquitoes is based on the geographic distribution of animal species, that is zoogeography [37]. However, in previous research on molecular phylogenetic reconstruction for mosquitoes, most studies [21, 33, 38–41] have targeted local or regional species from a small administrative area. Thus, it is probable that most studies did not comprehensively sample different species because the barcoding gap is correlated with the geographic scale of sampling [34, 42] and the sample size of target species [43, 44]. The exponential growth of GenBank COI sequences, accumulated from around the world [45, 46] makes it possible to study DNA barcoding at a more comprehensive scale for target species.
To further clarify the relationships among the Anopheles hyrcanus group species, and stablize the group, we combined our newly collected COI sequences and other sequences deposited in GenBank across a worldwide geographic range and applied different phylogenetic analytical methods to address the molecular phylogeny of the Hyrcanus Group. This research will provide a valuable tool for large-scale vector identification in practice and the planning of the malaria surveillance program in practice.
Methods
DNA extraction
A total of 33 dry museum specimens from the Hyrcanus Group, belonging to five species, An. hyrcanus (Pallas, 1771), An. peditaeniatus (Leicester, 1908), An. sinensis, An. pullus Yamada, 1937, and An. liangshanensis Kang, Tan, Cao, Cheng, Yang & Huang, 1984, were randomly chosen for DNA extraction. All of them were collected less than 7 years. Species identification was accomplished with the national key [9]. Collection localities and other specimen information are available in Table 1. One leg was removed from each adult specimen, transferred to a dry Eppendorf tube, and ground to powder. Then it was incubated in lysis buffer overnight at 56 °C. Additional steps followed the manufacturer’s instructions for the Qiagen® DNA blood & tissue kit. Voucher specimens were stored in the Herbarium of National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention.
Table 1.
List of COI sequences of the Hyrcanus Group obtained from this study
| Species | Geographic localities | Genbank accession No. |
|---|---|---|
| An. hyrcanus | Xinjiang Prov., China | KT966851 |
| Xinjiang Prov., China | KT966852 | |
| Xinjiang Prov., China | KT966853 | |
| Xinjiang Prov., China | KU743222 | |
| Xinjiang Prov., China | KU743223 | |
| Xinjiang Prov., China | KU743224 | |
| Xinjiang Prov., China | KU743225 | |
| Xinjiang Prov., China | KU743226 | |
| Xinjiang Prov., China | KU743227 | |
| An. peditaeniatus | Yunnan Prov., China | KT966854 |
| Yunnan Prov., China | KT966855 | |
| Yunnan Prov., China | KT966856 | |
| Yunnan Prov., China | KT966857 | |
| An. sinensis | Yunnan Prov., China | KT966858 |
| Yunnan Prov., China | KT966859 | |
| Yunnan Prov., China | KT966860 | |
| Yunnan Prov., China | KT966861 | |
| Yunnan Prov., China | KT966862 | |
| Yunnan Prov., China | KT966863 | |
| Yunnan Prov., China | KT966864 | |
| Yunnan Prov., China | KT966865 | |
| Yunnan Prov., China | KT966866 | |
| Yunnan Prov., China | KT966867 | |
| Yunnan Prov., China | KT966868 | |
| Yunnan Prov., China | KT966869 | |
| An. pullus | Liaoning Prov., China | KT966870 |
| Liaoning Prov., China | KT966871 | |
| Liaoning Prov., China | KT966872 | |
| Liaoning Prov., China | KT966873 | |
| Liaoning Prov., China | KT966874 | |
| An. liangshanensis | Yunnan Prov., China | KU743228 |
| Yunnan Prov., China | KU743229 | |
| Yunnan Prov., China | KU743230 |
Sequence generation
Amplification of the COI region was performed with a universal primer pair. Universal primers LCO1490 (5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′, forward) and HCO2198 (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′) were used to amplify the COI sequences [47]. The amplified length was approximately 650 bp. The 25 μL reaction mixture contained 12.5 μL 2XTaq polymerase chain reaction (PCR) Master Mix (with dyes, DBI® Bioscience), 4 μL extracted DNA, and 6.5 μL ddH2O. The thermocycling profile consisted of one cycle of 2 min at 94 °C, five cycles of 30 s at 94 °C, 40 s at 45 °C, and 1 min at 72 °C, followed by 35 cycles of 30 s at 94 °C, 40 s at 51 °C, and 1 min at 72 °C, with a final extension at 72 °C (7 min). The PCR products were visualized on 1.2% 0.5XTBE agarose gels, then cleaned and sequenced by Shanghai Sangon (Shanghai, China).
Search for COI sequences of the Hyrcanus Group in GenBank
Based on the index of Harbach [4], there are 25 species in this group. We searched and downloaded COI sequences for the Hyrcanus Group members deposited in GenBank (Additional file 2) with the keywords “(species name) & COI.” We checked and trimmed odd sequences with the highest 5% intraspecific distances or the lowest 5% interspecific distances [42, 48, 49] to avoid sequences posted in GenBank that contained errors [50–52]. Although the interspecific distances of An. hyrcanus and An. pseudopictus Grassi, 1899; An. lesteri and An. paraliae Sandosham, 1959; An. sinensis, An. kleini Rueda, 2005 and An. belenrae Rueda, 2005 were less than 5%, the independent sequences for those species were used in present study because the taxonomic validity of An. pseudopictus, An. hyrcanus spIR, An. paraliae, and An. kleini are still controversial [5, 53, 54]. The information on locations for sample sequences and authors were also recorded (Additional file 2).
Tree building
The COI sequence dataset was combined with our original fragments and records retrieved from GenBank. ClustalW2 [55] was used to align sequences using the default settings, and we created a neighbour joining (NJ) tree with 1 000 bootstraps. Based on Akaike Information Criterion (AIC), the best-fit model for the alignment was determined using Modeltest 3.7 [56], in cooperation with PAUP*4.0b10 [57]. Consequently, the construction of the maximum likelihood (ML) and Bayesian likelihood completed under the TVM + I + G model. An. lindesayi Giles, 1900 and An. claviger (Meigen, 1804) were used as outgroup taxa based on a previous study [33]. The ML tree was performed by RAxML-HPC2 v7.4.4 [58, 59] on the CIPRES portal (www.phylo.org/) [60] with 1 000 bootstraps. The Bayesian tree was built with MrBayes v3.2.1 [61], run for 1 million generations, with the first 25% generations discarded as burn-in. The trees were visualized and edited in FigTree v1.4.2 [62].
Genetic diversity analysis
Pairwise distances within and between species were calculated using Kimura’s 2-parameter (K2P) distance model [63] in MEGA v5.10 [64]. DnaSP 5.10 [65] was applied to calculate the nucleotide diversity of COI sequences of each species, and we performed the neutrality tests for Fu’s Fs [66] and Tajima’s D value [67].
Results
There were 463 COI sequences of the Hyrcanus Group in GenBank belonging to 18 species. There were no COI records for An. chodukini Martini, 1929, An. engarensis Kanda & Oguma, 1978, An. hailarensis Xu & Luo, 1998, An. heiheensis Ma, 1981, An. hyrcanus spIR, An. sineroides Yamada, 1924, An. vietnamensis Nguyen, Tran & Nguyen, 1993, and An. pseudosinensis Baisas, 1935 in GenBank. The accession numbers of COI sequences, which we downloaded from GenBank, are available in Additional file 2, including the collection localities and author information. Because sequences submitted to GenBank came from labs worldwide, without further confirmation, we found some fragments that were distant from others in the same species (Additional file 2), but closer to sequences from their sister species, with peculiar phylogenetic positions in the pre-building phylogenetic tree (see Additional file 3). Thus, we removed them from further analyses.
Thirty-three newly collected sequences for five species (An. hyrcanus, An. peditaeniatus, An. sinensis, An. pullus, and An. liangshanensis) were included in this study. Amino acid translation showed that they were free of stop codons, indicating that none of them was rDNA sequences originating from mtDNA sequences. The GenBank accession numbers are listed in Table 1.
After combining COI records from GenBank with our original sequences and excluding suspicious fragments, 466 sequences of 18 Hyrcanus Group members were used for analyses of genetic diversity indices and reconstruct phylogenetic trees. The topology of the NJ tree, ML tree, and Bayesian tree were almost identical for the major lineages, although node confidence values were slightly different among the three (Fig. 1). Hence, only the NJ tree is presented here. The tree showed that the group could be divided into at least three main clusters. The first cluster was solely composed of An. nimpe Nguyen, Tran & Nguyen, 2000, which was coincidence with genetic distance analysis. An. nimpe exhibited extensive interspecific divergences (the minimum distance was 0.067) with other Hyrcanus Group members (Table 2, Fig. 2). The second cluster consisted of An. nigerrimus Giles, 1900, An. nitidus Harrison, Scanlon & Reid, 1973, An. pursati Laveran, 1902, and An. argyropus (Swellengrebel, 1914), and the third cluster included An. sinensis, and An. belenrae, An. kleini grouped; An. lesteri and An. paraliae grouped; An. crawfordi Reid, 1953; An. hyrcanus and An. pseudopictus grouped; An. liangshanensis; An. kweiyangensis Yao & Wu, 1944; An. peditaeniatus; An. sineroides; and An. pullus Yamada, 1937. Almost all node-linking sequences of individuals of the same species had a high bootstrap value. However, the relationships between hyrcanus/pseudopictus, lesteri/paraliae, and sinensis/belenrae/kleini were unclear. They exhibited very low pair-wise distance values (Table 2), and formed monoclades with high node confidence values (Fig. 1).
Fig. 1.
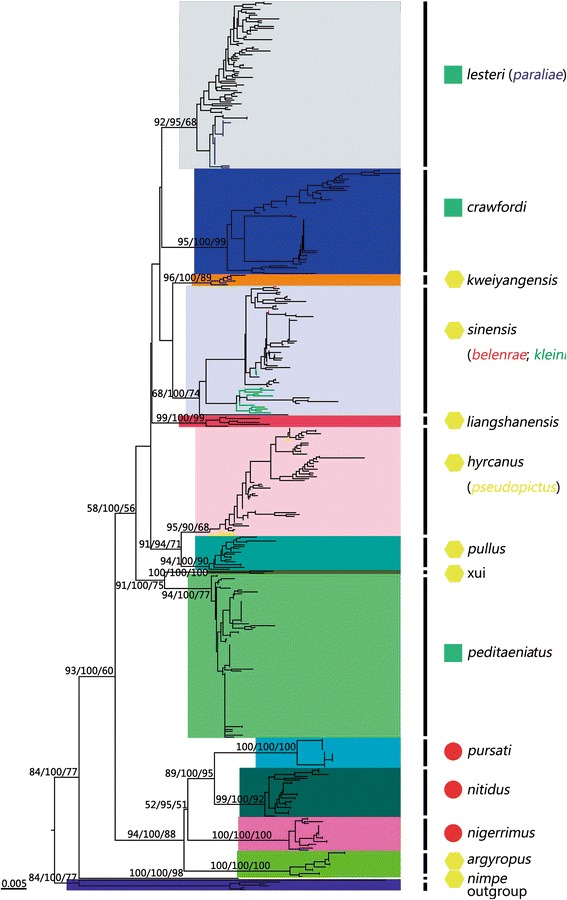
Neighbour joining tree based on COI sequences deposited in GenBank and our original data for the Hyrcanus Group. Bootstrap values (1 000 replicates, not shown for less than 50%) of Neighbour Joining, Bayesian, and Maximum likelihood analyses are shown above the main lineages, respectively. Lineage designation is indicated on the right. Branches representing COI sequences of An. belenrae, An. kleini, An. pseudopictus, and An. paraliae are indicated by red, green, yellow, and blue, respectively. The geometric shapes correspond to the different subgroups of the Hyrcanus Group, according to the classification of Harbach [4]. (green square) Lesteri Subgroup; (red circle) Nigerrimus Subgroup; (yellow hexagon) unclassified species. Bars represent 0.1 substitutions per site. An. lindesayi and An. claviger were used as outgroup taxa
Table 2.
Mean intra- and interspecific K2P distances of the COI gene in 18 Hyrcanus Group members
| Species | n | arg. | bel. | cra. | hyr. | kle. | kwe. | les. | lia. | nig. | nim. | nit. | par. | ped. | pse. | pul. | pur. | sin. | xui |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| arg. | 14 | 0.007 | |||||||||||||||||
| bel. | 2 | 0.070 | 0.008 | ||||||||||||||||
| cra. | 56 | 0.077 | 0.055 | 0.017 | |||||||||||||||
| hyr. | 55 | 0.078 | 0.051 | 0.058 | 0.008 | ||||||||||||||
| kle. | 13 | 0.075 | 0.024* | 0.058 | 0.055 | 0.014 | |||||||||||||
| kwe. | 6 | 0.073 | 0.039 | 0.056 | 0.040 | 0.045 | 0.008 | ||||||||||||
| les. | 63 | 0.087 | 0.057 | 0.055 | 0.048 | 0.044 | 0.045 | 0.011 | |||||||||||
| lia. | 6 | 0.065 | 0.049 | 0.043 | 0.046 | 0.056 | 0.040 | 0.050 | 0.006 | ||||||||||
| nig. | 18 | 0.062 | 0.067 | 0.069 | 0.062 | 0.072 | 0.066 | 0.068 | 0.068 | 0.005 | |||||||||
| nim. | 1 | 0.094 | 0.086 | 0.079 | 0.073 | 0.083 | 0.073 | 0.074 | 0.075 | 0.073 | n/c | ||||||||
| nit. | 26 | 0.065 | 0.081 | 0.094 | 0.086 | 0.085 | 0.072 | 0.077 | 0.082 | 0.052 | 0.094 | 0.007 | |||||||
| par. | 26 | 0.080 | 0.042 | 0.038 | 0.037 | 0.035 | 0.035 | 0.020* | 0.035 | 0.052 | 0.067 | 0.077 | 0.002 | ||||||
| ped. | 87 | 0.094 | 0.071 | 0.064 | 0.057 | 0.068 | 0.060 | 0.056 | 0.053 | 0.079 | 0.071 | 0.084 | 0.046 | 0.005 | |||||
| pse. | 3 | 0.076 | 0.046 | 0.055 | 0.007* | 0.053 | 0.037 | 0.045 | 0.044 | 0.059 | 0.070 | 0.083 | 0.034 | 0.055 | 0.005 | ||||
| pul. | 18 | 0.071 | 0.047 | 0.058 | 0.029 | 0.055 | 0.043 | 0.045 | 0.045 | 0.058 | 0.076 | 0.082 | 0.041 | 0.057 | 0.026 | 0.008 | |||
| pur. | 16 | 0.078 | 0.108 | 0.096 | 0.084 | 0.107 | 0.100 | 0.090 | 0.099 | 0.060 | 0.085 | 0.052 | 0.086 | 0.083 | 0.082 | 0.091 | 0.007 | ||
| sin. | 54 | 0.072 | 0.009* | 0.054 | 0.050 | 0.023* | 0.040 | 0.055 | 0.049 | 0.065 | 0.085 | 0.082 | 0.041 | 0.070 | 0.046 | 0.047 | 0.107 | 0.010 | |
| xui | 2 | 0.092 | 0.068 | 0.054 | 0.047 | 0.070 | 0.059 | 0.058 | 0.052 | 0.064 | 0.079 | 0.087 | 0.042 | 0.044 | 0.045 | 0.049 | 0.077 | 0.069 | 0.005 |
The numbers of intraspecific distances are shown in boldface for clarity. Numbers underlined indicate the highest intraspecific distance and the lowest interspecific distance. n = No. of sequences. The interspecific distances of hyrcanus/pseudopictus, lesteri/paraliae, sinensis/belenrae/kleini, were highlighted with an asterisk. arg. = An. argyropus; bel. = An. belenrae; cra. = An. crawfordi; hyr. = An. hyrcanus; kle. = An. kleini; kwe. = An. kweiyangensis; les. = An. lesteri; lia. = An. liangshanensis; nig. = An. nigerrimus; nim. = An. nimpe; nit. = An. nitidus; par. = An. paraliae; ped. = An. peditaeniatus; pul. = An. pullus; pur. = An. pursati; sin. = An. sinensis
Fig. 2.
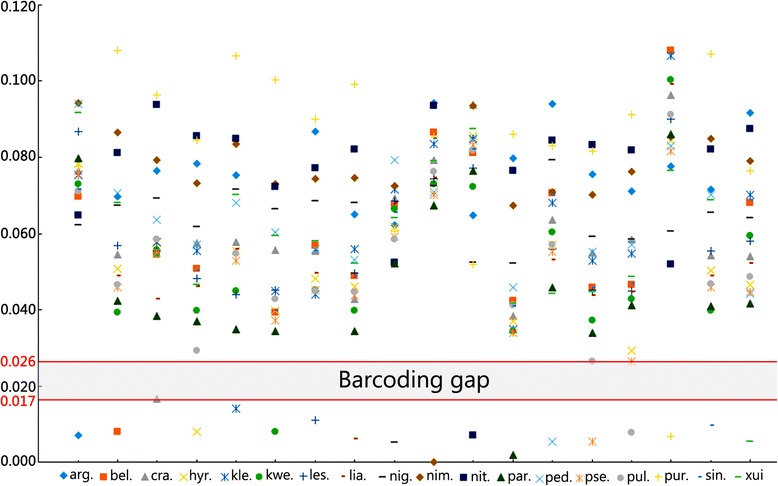
Plot of K2P distance of the 18 Hyrcanus Group members determined using NJ-K2P distances. Y-axis: genetic divergence; X-axis: Hyrcanus group members. arg. = An. argyropus; bel. = An. belenrae; cra. = An. crawfordi; hyr. = An. hyrcanus; kle. = An. kleini; kwe. = An. kweiyangensis; les. = An. lesteri; lia. = An. liangshanensis; nig. = An. nigerrimus; nim. = An. nimpe; nit. = An. nitidus; par. = An. paraliae; ped. = An. peditaeniatus; pul. = An. pullus; pur. = An. pursati; sin. = An. sinensis; xui = An. xui
Excluding these polytypic species above, based on the mtDNA COI sequence, the average K2P distances between and within the Hyrcanus Group species were 0.064 (range 0.027–0.108) and 0.008 (range 0.002–0.017), respectively (Table 2). On average, the differences between congroup species were 8-fold higher than the differences within species. The maximum K2P distance within species was in An. crawfordi (0.017), and the minimum K2P distance between the Hyrcanus Group members was 0.026 (Fig. 2).
It should be noted that An. belenrae and An. kleini had a genetic distance of 0.024 from each other, and 0.009 and 0.023, respectively, with An. sinensis. In addition, the distance between hyrcanus and pseudopictus was 0.007, and it was 0.020 between lesteri and paraliae.
The genetic diversity indices and the results of neutrality tests are showed in Table 3. Both Fu’s Fs and Tajima’s D values were significant in An. lesteri and An. pullus, suggesting past population expansion.
Table 3.
Genetic diversity indices and neutrality tests of the COI gene in 14 Hyrcanus Group members
| species | n | S | Pi | h | Hd | Fu’s Fs | Tajima’s D |
|---|---|---|---|---|---|---|---|
| An. argyropus | 14 | 13 | 0.00576 | 7 | 0.89 | −0.269 | −0.32403 |
| An. crawfordi | 56 | 37 | 0.01910 | 31 | 0.902 | −10.400** | 0.09433 |
| An. hyrcanus | 55 | 27 | 0.00880 | 25 | 0.925 | −13.586** | −1.25768 |
| An. kleini | 13 | 20 | 0.01230 | 11 | 0.962 | −3.224* | 0.35139 |
| An. kweiyangensis | 6 | 8 | 0.0054 | 6 | 1 | −3.07918* | −0.39875 |
| An. lesteri | 63 | 58 | 0.01026 | 57 | 0.997 | −74.707** | −1.74894* |
| An. liangshanensis | 6 | 10 | 0.00628 | 6 | 1.000 | −2.734 | −0.79480 |
| An. nigerrimus | 18 | 16 | 0.00631 | 10 | 0.882 | −1.923 | −0.61570 |
| An. nitidus | 26 | 24 | 0.00816 | 14 | 0.948 | −2.844* | −0.53529 |
| An. paraliae | 26 | 5 | 0.00150 | 5 | 0.591 | −0.832 | −0.70434 |
| An. peditaeniatus | 87 | 16 | 0.00533 | 19 | 0.837 | −9.149** | −1.08597 |
| An. pullus | 18 | 23 | 0.00707 | 16 | 0.987 | −11.407** | −1.70855* |
| An. pursati | 16 | 7 | 0.00540 | 3 | 0.692 | 5.031 | 2.40596 |
| An. sinensis | 54 | 29 | 0.00906 | 28 | 0.911 | −18.570** | −1.44523 |
n = number of sequences; S = number of polymorphic sites; pi = nucleotide diversity; h = number of haplotypes; Hd = haplotype diversity. The significance of Fu’s Fs or Tajima’s D values is indicated by asterisks (* P < 0.05; ** P < 0.01). Species represented by <3 specimens were excluded from the analyses
Discussion
The suspicious records for COI found in this study, as evidenced by their pairwise distances and phylogenetic positions, may have been caused by misidentification of specimens in previous studies. Misidentified COI sequences were detected in mosquitoes in a previous study [52]; however this was possibly caused by the presence of other cryptic species. Although it is possible that they were morphologically misidentified or cryptic species, we omitted them from the analyses of genetic diversity indices and tree building. The maximum intra-specific distance of Hyrcanus Group was 0.017. A same value was obtained for Anopheles by other labs [33, 38].
The Hyrcanus Group was monophyletic, as described by other authors [4, 68]. However, the subgroup division using COI was not the same as those based on morphologic characteristics [69, 70], nor as results obtained from nuclear marker phylogeny [5, 54, 68, 71]. The topology of COI tree obtained from this study was similar with the results described by Wijit et al. [72], with the same molecular marker, COI. Although the applied COI sequences in the former study did not contain sequences for An. nimpe, the NJ tree showed the other two main clusters. In the one comprised of An. nigerrimus, An. nitidus, An. pursati, and An. argyropus, the former three species were grouped into the Nigerrimus Subgroup. The remaining studied species were placed in the other cluster, including sinensis (unassociated species) and the Lesteri Subgroup (crawfordi, lesteri, paraliae, peditaeniatus). The trees from both studies indicated that the Lesteri Subgroup, as classified by morphological features, was not monophyletic.
Because of ITS2 sequence differences and the discrepancy of morphological identification, Rueda [16] distinguished and named two new species, An. belenrae and An. kleini, from An. sinensis. However, the pairwise differences of COI among the three species were below the lower threshold of the barcoding gap (Table 2, Fig. 2). In the phylogenetic tree (Fig. 1), the three species formed a monoclade with a high node confidence (NJ 68%; Bayes 100%; ML 74%). This suggested that the gene introgression at the mtDNA likely happened during species expansion [27, 73]. The hybridization experiments under laboratory conditions supported natural hybridization between An. sinensis and An. kleini [74]. It was inferred that the same situation probably happen between belenrae and sinensis in field.
Anopheles lesteri from Korea and An. paraliae from Thailand were suggested as conspecifics by Taai et al. [75], inferred from crossing experiments and molecular analyses. It is in agreement with the current study. The pairwise distance between An. lesteri and An. paraliae was 0.019, and the two species could not be distinguished in the phylogenetic tree (Fig. 1). An. lesteri is widely distributed across the Palaearctic and Oriental regions, north into the Primorsk region and Russia, and south to Philippines, Malaysia [9]. An. anthropophagus is endemic to China as evidenced by a synonym of An. lesteri [76, 77]. All COI sequences of An. paraliae in GenBank were obtained from specimens that were collected from Thailand. It is likely that An. lesteri, An. anthropophagus, and An. paraliae belong to a single species. The morphological differences among the three species were likely a result of recent geographic isolation.
The COI sequences of An. hyrcanus and An. pseudopictus were almost identical, with a distance of 0.008. An. pseudopictus clustered within the An. hyrcanus lineage in COI tree. Poncon et al. [53] demonstrated that the two species and their intermediate form were indistinguishable by nuclear markers. However, there was no further study on crossing experiments to support this supposition.
Two COI fragments of An. crawfordi (KF830735.1; KF830736.1) in GenBank collected from China (direct submission) were clustered with the lineage of An. xui Dong, Zhou, Dong & Mao, 2007 in the phylogenetic tree (Additional file 3). They were closer to sequences of An. xui than to those of their conspecifics. It implied that An. crawfordi might not exist in China, or quite possibly that the specimens were misidentified.
Because genes submitted to GenBank were without confirmation, there was probably some error sequences in the database [50, 78]. In this study, some sequences excluded in later phylogenetic analyses were related to authors (see Additional file 2) who submitted them to GenBank. Almost all COI sequences of the Hyrcanus Group submitted by some authors had peculiar phylogenetic positions; some even had considerable distances from other sequences from the same species. The validity of those data needs further research. It is possible that they could have been cryptic taxa.
Conclusions
The large data analysis showed that the COI barcoding gap (K2P distance) of the Hyrcanus Group species was 0.017 to 0.026. The average conspecific K2P divergence was 0.008 (range 0.002–0.017), whereas sequence divergence between congroup species averaged at 0.064 (range 0.026–0.108). The COI tree showed that the group could be divided into at least three main clusters. The first cluster contained An. nimpe; the second was composed of the Nigerrimus Subgroup and An. argyropus; the third cluster was comprised of the Lesteri Subgroup and other unassociated species. It was consistent with former phylogenetic analyses of the Hyrcanus Group with the same gene based on small sample sizes [72], but contradicted the morphological and rDNA ITS2-based classification when sorting out subgroups. In addition, phylogenetic analysis suggested that ancient hybridizations probably happen among the three species, An. sinensis, An. belenrae, and An. kleini. It supported that An. paraliae was synonymized with An. lesteri, whereas An. pseudopictus and An. hyrcanus may belong to a single species, as evidenced from extremely low interspecific genetic divergence (0.020 and 0.007, respectively), and their phylogenetic positions.
The neutrality tests indicated that several Hyrcanus Group members, An. lesteri, An. sinensis, An. hyrcanus, An. pullus, An. peditaeniatus, and An. pseudopictus, might have experienced population expansion or genetic hitchhiking. Almost all of these species are widespread and some of them have the capacity of malaria transmission.
An. sinensis, An. kleini and An. belenrae have been proved as suspected malaria vectors in South Korea [20, 79–81]. It can be inferred that closely related species may possess similar susceptibility to plasmodium infection. In addition, it has the possibility of natural hybridization happening among closely related species [27]. Therefore, we need to pay attention to An. paraliae, An. pseudopictus in future surveillance as well, since that their sister species has been incriminated as vectors of malaria [9, 82]. Integrated molecular phylogeny research combining both mtDNA and rDNA for the Anopheles hyrcanus group is underway in our lab.
Additional files
Multilingual abstracts in the five official working languages of the United Nations. (PDF 818 kb)
List of Anopheles hyrcanus group COI sequences deposited in GenBank and obtained from this study, with GenBank accession numbers, geographic localities, and corresponding authors. Suspicious sequences with high intraspecific genetic distance, and peculiar phylogenetic positions were annotated. (XLSX 33 kb)
Neighbour-joining phylogenetic tree of COI in the Hyrcanus Group showing suspicious sequences. Bootstrap values are shown above the main lineages. Lineage designation is indicated on the right. The geometric shape (red square) corresponds to suspicious sequences of the Hyrcanus Group in GenBank. (PDF 617 kb)
Acknowledgements
Not applicable.
Funding
This work was supported by the National Research and Development Plan of China (Grant No. 2016YFC1200500), and the National Special Science and Technology Project for Major Infectious Diseases of China (Grant No. 2012ZX10004219-009, 2012ZX10004-220, 2008ZX10004-011).
Availability of data and materials
All data generated or analysed during this study are included in this published article and Additional files 2 and 3.
Authors’ contributions
YF, WS and YZ designed the study; YF carried out laboratory experiments, performed phylogenetic analyses, and wrote the manuscript; WS and YZ revised the manuscript and gave approval of the version to be published. All the authors read and approved the final version of the manuscript.
Competing interests
The authors declared that they have no competing interests.
Consent for publication
Not applicable.
Ethical approval and consent to participate
No specific permits were required for this study. The study did not involve endangered or protected species. Therefore, the local ethics committee deemed that approval was unnecessary.
Abbreviations
- AIC
Akaike information criterion
- COI
Cytochrome c oxidase subunit I
- DNA
Deoxyribonucleic acid
- K2P distance
Kimura 2-parameter distance
- ML
Maximum likelihood
- mtDNA
Mitochondrial DNA
- NJ
Neighbour joining
- PCR
Polymerase chain reaction
- rDNA
Ribosomal DNA
- TBE
Tris/Borate/EDTA
Contributor Information
Yuan Fang, Email: cindy.yuanfang07@gmail.com.
Wen-Qi Shi, Email: shiwq998@163.com.
Yi Zhang, Email: zhang1972003@163.com.
References
- 1.Cotter C, Sturrock HJ, Hsiang MS, Liu J, Phillips AA, Hwang J, et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet. 2013;382(9895):900–911. doi: 10.1016/S0140-6736(13)60310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Z. The malaria situation in the People’s Republic of China. Bull World Health Organ. 1981;59(6):931–936. [PMC free article] [PubMed] [Google Scholar]
- 3.Sinka ME. Global distribution of the dominant vector species of malaria. In: Manguin S, editor. Anopheles mosquitoes: New insights into malaria vectors. Croatia: InTech; 2013. pp. 109–143. [Google Scholar]
- 4.Harbach RE. The phylogeny and classification of Anopheles. In: Manguin S, editor. Anopheles mosquitoes: New insights into malaria vectors. Croatia: InTech; 2013. pp. 3–55. [Google Scholar]
- 5.Djadid ND, Jazayeri H, Gholizadeh S, Rad Sh P, Zakeri S. First record of a new member of Anopheles Hyrcanus Group from Iran: molecular identification, diagnosis, phylogeny, status of kdr resistance and Plasmodium infection. J Med Entomol. 2009;46(5):1084–1093. doi: 10.1603/033.046.0515. [DOI] [PubMed] [Google Scholar]
- 6.Ho C, Chou TC, Chen TH, Hsueh AT. The Anopheles hyrcanus group and its relation to malaria in east China. Chin Med J. 1962;81:71–78. [PubMed] [Google Scholar]
- 7.Joshi D, Kim JY, Choochote W, Park MH, Min GS. Preliminary vivax malaria vector competence for three members of the Anopheles hyrcanus group in the Republic of Korea. J Am Mosq Control Assoc. 2011;27(3):312–314. doi: 10.2987/10-6086.1. [DOI] [PubMed] [Google Scholar]
- 8.Votypka J, Seblova V, Radrova J. Spread of the West Nile virus vector Culex modestus and the potential malaria vector Anopheles hyrcanus in central Europe. J Vector Ecol. 2008;33(2):269–277. doi: 10.3376/1081-1710-33.2.269. [DOI] [PubMed] [Google Scholar]
- 9.Lu BL, Xu JJ, Dong XS. Fauna Sinica, Insecta, vol.9. Diptera: Culicidae II. 1. Beijing: Science Press; 1997. [Google Scholar]
- 10.Sen P. Anopheles hyrcanus as a vector of filariasis in West Bengal. Nature. 1956;178:749–750. doi: 10.1038/178749a0. [DOI] [PubMed] [Google Scholar]
- 11.Feng LC. Anopheles hyrcanus var. sinensis Wied., transmittor of Wuchereria (Filaria) bancrofti in Woosung District, Shanghai, China. AM J Epidemoil. 1931;14(2):502–514. doi: 10.1093/oxfordjournals.aje.a117786. [DOI] [Google Scholar]
- 12.Rosen L. The natural history of Japanese encephalitis virus. Annu Rev Microbiol. 1986;40:395–414. doi: 10.1146/annurev.mi.40.100186.002143. [DOI] [PubMed] [Google Scholar]
- 13.Huang CH. Studies of Japanese encephalitis in China. Adv Virus Res. 1982;27:71–101. doi: 10.1016/S0065-3527(08)60433-9. [DOI] [PubMed] [Google Scholar]
- 14.Kanojia PC, Shetty PS, Geevarghese G. A long-term study on vector abundance & seasonal prevalence in relation to the occurrence of Japanese encephalitis in Gorakhpur district, Uttar Pradesh. Indian J Med Res. 2003;117:104–110. [PubMed] [Google Scholar]
- 15.Garros C, Dujardin J-P. Genetic and phenetic approaches to Anopheles systematics. In: Manguin S, editor. Anopheles mosquitoes: New insights into malaria vectors. Croatia: InTech; 2013. pp. 81–105. [Google Scholar]
- 16.Rueda LM. Two new species of Anopheles (Anopheles) Hyrcanus Group (Diptera: Culicidae) from the Republic of South Korea. Zootaxa. 2005;941:1–26. doi: 10.11646/zootaxa.941.1.1. [DOI] [Google Scholar]
- 17.Chen B, Butlin RK, Pedro PM, Wang XZ, Harbach RE. Molecular variation, systematics and distribution of the Anopheles fluviatilis complex in southern Asia. Med Vet Entomol. 2006;20(1):33–43. doi: 10.1111/j.1365-2915.2006.00604.x. [DOI] [PubMed] [Google Scholar]
- 18.Collins FH, Paskewitz SM. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol Biol. 1996;5(1):1–9. doi: 10.1111/j.1365-2583.1996.tb00034.x. [DOI] [PubMed] [Google Scholar]
- 19.Paredes-Esquivel C, Harbach RE, Townson H. Molecular taxonomy of members of the Anopheles hyrcanus group from Thailand and Indonesia. Med Vet Entomol. 2011;25(3):348–352. doi: 10.1111/j.1365-2915.2010.00937.x. [DOI] [PubMed] [Google Scholar]
- 20.Rueda LM, Brown TL, Kim H-C, Klein TA, Thongkukiatkul A, Sherwood V. Description and comparison of morphological structures of the eggs of Anopheles hyrcanus group and related species (Diptera: Culicidae) from the Republic of Korea. Zootaxa. 2009;2268:23–40. [Google Scholar]
- 21.Chan A, Chiang LP, Hapuarachchi HC, Tan CH, Pang SC, Lee R, et al. DNA barcoding: complementing morphological identification of mosquito species in Singapore. Parasit Vectors. 2014;7:569. doi: 10.1186/s13071-014-0569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen B, Harbach RE, Butlin RK. Molecular and morphological studies on the Anopheles minimus group of mosquitoes in southern China: taxonomic review, distribution and malaria vector status. Med Vet Entomol. 2002;16(3):253–265. doi: 10.1046/j.1365-2915.2002.00373.x. [DOI] [PubMed] [Google Scholar]
- 23.Linton YM, Pecor JE, Porter CH, Mitchell LB, Garzon-Moreno A, Foley DH, et al. Mosquitoes of eastern Amazonian Ecuador: biodiversity, bionomics and barcodes. Mem Inst Oswaldo Cruz. 2013;108(Suppl 1):100–109. doi: 10.1590/0074-0276130440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt S, Schmid-Egger C, Moriniere J, Haszprunar G, Hebert PD. DNA barcoding largely supports 250 years of classical taxonomy: identifications for Central European bees (Hymenoptera, Apoidea partim) Mol Ecol Resour. 2015;15(4):985–1000. doi: 10.1111/1755-0998.12363. [DOI] [PubMed] [Google Scholar]
- 25.Monaghan MT, Balke M, Gregory TR, Vogler AP. DNA-based species delineation in tropical beetles using mitochondrial and nuclear markers. Philos Trans R Soc Lond B Biol Sci. 2005;360(1462):1925–1933. doi: 10.1098/rstb.2005.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruiz-Lopez F, Wilkerson RC, Conn JE, McKeon SN, Levin DM, Quinones ML, et al. DNA barcoding reveals both known and novel taxa in the Albitarsis Group (Anopheles: Nyssorhynchus) of Neotropical malaria vectors. Parasit Vectors. 2012;5:44. doi: 10.1186/1756-3305-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebert PD, Stoeckle MY, Zemlak TS, Francis CM. Identification of Birds through DNA Barcodes. PLoS Biol. 2004;2(10) doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hebert PD, Ratnasingham S, DeWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270(1):S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castro JA, Picornell A, Ramon M. Mitochondrial DNA: a tool for populational genetics studies. Int Microbiol. 1998;1(4):327–332. [PubMed] [Google Scholar]
- 30.Beebe NW, van den Hurk AF, Chapman HF, Frances SP, Williams CR, Cooper RD. Development and evaluation of a species diagnostic polymerase chain reaction-restriction fragment-length polymorphism procedure for cryptic members of the Culex sitiens (Diptera: Culicidae) subgroup in Australia and the southwest Pacific. J Med Entomol. 2002;39(2):362–369. doi: 10.1603/0022-2585-39.2.362. [DOI] [PubMed] [Google Scholar]
- 31.Hemmerter S, Slapeta J, Beebe NW. Resolving genetic diversity in Australasian Culex mosquitoes: incongruence between the mitochondrial cytochrome c oxidase I and nuclear acetylcholine esterase 2. Mol Phylogenet Evol. 2009;50(2):317–325. doi: 10.1016/j.ympev.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Cywinska A, Hunter FF, Hebert PD. Identifying Canadian mosquito species through DNA barcodes. Med Vet Entomol. 2006;20(4):413–424. doi: 10.1111/j.1365-2915.2006.00653.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang G, Li C, Guo X, Xing D, Dong Y, Wang Z, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS ONE. 2012;7(10) doi: 10.1371/journal.pone.0047051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, Balke M, et al. The effect of geographical scale of sampling on DNA barcoding. Syst Biol. 2012;61(5):851–869. doi: 10.1093/sysbio/sys037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hebert PD, Penton EH, Burns JM, Janzen DH, Hallwachs W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci U S A. 2004;101(41):14812–14817. doi: 10.1073/pnas.0406166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mutanen M, Kaila L, Tabell J. Wide-ranging barcoding aids discovery of one-third increase of species richness in presumably well-investigated moths. Sci Rep. 2013;3:2901. doi: 10.1038/srep02901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holt BG, Lessard JP, Borregaard MK, Fritz SA, Araujo MB, Dimitrov D, et al. An update of Wallace’s zoogeographic regions of the world. Science. 2013;339(6115):74–78. doi: 10.1126/science.1228282. [DOI] [PubMed] [Google Scholar]
- 38.Foster PG, Bergo ES, Bourke BP, Oliveira TM, Nagaki SS, Sant’Ana DC, et al. Phylogenetic analysis and DNA-based species confirmation in Anopheles (Nyssorhynchus) PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0054063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashfaq M, Hebert PD, Mirza JH, Khan AM, Zafar Y, Mirza MS. Analyzing mosquito (Diptera: culicidae) diversity in Pakistan by DNA barcoding. PLoS ONE. 2014;9(5) doi: 10.1371/journal.pone.0097268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karimian F, Oshaghi MA, Sedaghat MM, Waterhouse RM, Vatandoost H, Hanafi-Bojd AA, et al. Phylogenetic analysis of the oriental-Palearctic-Afrotropical members of Anopheles (Culicidae: Diptera) based on nuclear rDNA and mitochondrial DNA characteristics. Jpn J Infect Dis. 2014;67(5):361–367. doi: 10.7883/yoken.67.361. [DOI] [PubMed] [Google Scholar]
- 41.Blouin MS, Yowell CA, Courtney CH, Dame JB. Substitution bias, rapid saturation, and the use of mtDNA for nematode systematics. Mol Biol Evol. 1998;15(12):1719–1727. doi: 10.1093/oxfordjournals.molbev.a025898. [DOI] [PubMed] [Google Scholar]
- 42.Candek K, Kuntner M. DNA barcoding gap: reliable species identification over morphological and geographical scales. Mol Ecol Resour. 2015;15(2):268–277. doi: 10.1111/1755-0998.12304. [DOI] [PubMed] [Google Scholar]
- 43.Meyer CP, Paulay G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005;3(12) doi: 10.1371/journal.pbio.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang AB, He LJ, Crozier RH, Muster C, Zhu CD. Estimating sample sizes for DNA barcoding. Mol Phylogenet Evol. 2010;54(3):1035–1039. doi: 10.1016/j.ympev.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 45.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2014;42:D32–D37. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2015;43:D30–D35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3(5):294–299. [PubMed] [Google Scholar]
- 48.Meier R, Shiyang K, Vaidya G, Ng PK. DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst Biol. 2006;55(5):715–728. doi: 10.1080/10635150600969864. [DOI] [PubMed] [Google Scholar]
- 49.Meier R, Zhang G, Ali F. The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst Biol. 2008;57(5):809–813. doi: 10.1080/10635150802406343. [DOI] [PubMed] [Google Scholar]
- 50.Harris DJ. Can you bank on GenBank? Trends in Ecology and Evolution. 2003;18(7):317–319. doi: 10.1016/S0169-5347(03)00150-2. [DOI] [Google Scholar]
- 51.Fietz K, Graves JA, Olsen MT. Control control control: a reassessment and comparison of GenBank and chromatogram mtDNA sequence variation in Baltic grey seals (Halichoerus grypus) PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0072853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gunay F, Alten B, Simsek F, Aldemir A, Linton YM. Barcoding Turkish Culex mosquitoes to facilitate arbovirus vector incrimination studies reveals hidden diversity and new potential vectors. Acta Trop. 2014;143:112–120. doi: 10.1016/j.actatropica.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Poncon N, Toty C, Kengne P, Alten B, Fontenille D. Molecular evidence for similarity between Anopheles hyrcanus (Diptera: Culicidae) and Anopheles pseudopictus (Diptera: Culicidae), sympatric potential vectors of malaria in France. J Med Entomol. 2008;45(3):576–580. doi: 10.1093/jmedent/45.3.576. [DOI] [PubMed] [Google Scholar]
- 54.Hwang UW. Revisited ITS2 phylogeny of Anopheles (Anopheles) Hyrcanus group mosquitoes: reexamination of unidentified and misidentified ITS2 sequences. Parasitol Res. 2007;101(4):885–894. doi: 10.1007/s00436-007-0553-4. [DOI] [PubMed] [Google Scholar]
- 55.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 56.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 57.Wilgenbusch JC, Swofford D. Inferring evolutionary trees with PAUP*. Curr Protoc Bioinformatics. 2003;6:6.4. doi: 10.1002/0471250953.bi0604s00. [DOI] [PubMed] [Google Scholar]
- 58.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 59.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008;57(5):758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 60.Miller MA, Pfeiffer W, Schwartz T. Proceedings of the Gateway Computing Environments Workshop (GCE) New Orleans: Louisiana; 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees; pp. 1–8. [Google Scholar]
- 61.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morariu VI, Srinivasan BV, Raykar VC, Duraiswami R, Davis LS. Automatic online tuning for fast Gaussian summation. Adv Neural Inf Proces Syst. 2008;21:1113–1120. [Google Scholar]
- 63.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 64.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 66.Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma Y, Xu J. The Hyrcanus group of Anopheles (Anopheles) in China (Diptera: Culicidae): species discrimination and phylogenetic relationships inferred by ribosomal DNA internal transcribed spacer 2 sequences. J Med Entomol. 2005;42(4):610–619. [PubMed] [Google Scholar]
- 69.Reid JA. The Anopheles hyrcanus group in south-east Asia (Diptera: Culicidae) B Entomol Res. 1953;44:5–76. doi: 10.1017/S0007485300022938. [DOI] [Google Scholar]
- 70.Harrison BA. A new interpretation of affinities within the Anopheles hyrcanus complex of southeast Asia. Mosq Syst. 1972;4:73–83. [Google Scholar]
- 71.Gao Q, Beebe NW, Cooper RD. Molecular identification of the malaria vectors Anopheles anthropophagus and Anopheles sinensis (Diptera: Culicidae) in central China using polymerase chain reaction and appraisal of their position within the Hyrcanus group. J Med Entomol. 2004;41(1):5–11. doi: 10.1603/0022-2585-41.1.5. [DOI] [PubMed] [Google Scholar]
- 72.Wijit A, Saeung A, Baimai V, Otsuka Y, Thongsahuan S, Taai K, et al. DNA barcoding for the identification of eight species members of the Thai Hyrcanus Group and investigation of their stenogamous behavior. C R Biol. 2013;336(9):449–456. doi: 10.1016/j.crvi.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 73.Petit RJ, Excoffier L. Gene flow and species delimitation. Trends Ecol Evol. 2009;24(7):386–393. doi: 10.1016/j.tree.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 74.Choochote W, Min GS, Intapan PM, Tantrawatpan C, Saeung A, Lulitanond V. Evidence to support natural hybridization between Anopheles sinensis and Anopheles kleini (Diptera: Culicidae): possibly a significant mechanism for gene introgression in sympatric populations. Parasit Vectors. 2014;7:36. doi: 10.1186/1756-3305-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taai K, Baimai V, Saeung A, Thongsahuan S, Min GS, Otsuka Y, et al. Genetic compatibility between Anopheles lesteri from Korea and Anopheles paraliae from Thailand. Mem Inst Oswaldo Cruz. 2013;108(3):312–320. doi: 10.1590/S0074-02762013000300009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilkerson RC, Li C, Rueda LM, Kim H-C, Klein TA, Song G-H, et al. Molecular confirmation of Anopheles (Anopheles) lesteri from the Republic of South Korea and its genetic identity with An. (Ano.) anthropophagus from China (Diptera: Culicidae) Zootaxa. 2003;378:1–14. doi: 10.11646/zootaxa.378.1.1. [DOI] [Google Scholar]
- 77.Hwang UW, Tang LH, Kobayashi M, Yong TS, Ree HI. Molecular evidence supports that Anopheles anthropophagus from China and Anopheles lesteri from Japan are the same species. J Am Mosq Control Assoc. 2006;22(2):324–326. doi: 10.2987/8756-971X(2006)22[324:MESTAA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 78.Bidartondo MI. Preserving accuracy in GenBank. Science. 2008;319(5870):1616. doi: 10.1126/science.319.5870.1616a. [DOI] [PubMed] [Google Scholar]
- 79.Rueda LM, Kim HC, Klein TA, Pecor JE, Li C, Sithiprasasna R, et al. Distribution and larval habitat characteristics of Anopheles Hyrcanus group and related mosquito species (Diptera: Culicidae) in South Korea. J Vector Ecol. 2006;31(1):198–205. doi: 10.3376/1081-1710(2006)31[198:DALHCO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 80.Rueda LM, Li C, Kim HC, Klein TA, Foley DH, Wilkerson RC. Anopheles belenrae, a potential vector of Plasmodium vivax in the Republic of Korea. J Am Mosq Control Assoc. 2010;26(4):430–432. doi: 10.2987/10-6057.1. [DOI] [PubMed] [Google Scholar]
- 81.Lee WJ, Klein TA, Kim HC, Choi YM, Yoon SH, Chang KS, et al. Anopheles kleini, Anopheles pullus, and Anopheles sinensis: potential vectors of Plasmodium vivax in the Republic of Korea. J Med Entomol. 2007;44(6):1086–1090. doi: 10.1603/0022-2585(2007)44[1086:akapaa]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 82.Poncon N, Toty C, L’Ambert G, Le Goff G, Brengues C, Schaffner F, et al. Biology and dynamics of potential malaria vectors in Southern France. Malar J. 2007;6(1):18. doi: 10.1186/1475-2875-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multilingual abstracts in the five official working languages of the United Nations. (PDF 818 kb)
List of Anopheles hyrcanus group COI sequences deposited in GenBank and obtained from this study, with GenBank accession numbers, geographic localities, and corresponding authors. Suspicious sequences with high intraspecific genetic distance, and peculiar phylogenetic positions were annotated. (XLSX 33 kb)
Neighbour-joining phylogenetic tree of COI in the Hyrcanus Group showing suspicious sequences. Bootstrap values are shown above the main lineages. Lineage designation is indicated on the right. The geometric shape (red square) corresponds to suspicious sequences of the Hyrcanus Group in GenBank. (PDF 617 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and Additional files 2 and 3.