

Abstract
Despite the success of tyrosine kinase inhibitor (TKI) therapy in chronic myelogenous leukemia (CML), leukemic stem/progenitor cells remain detectable even in the state of deep molecular remission. Mechanisms that allow them to persist despite continued kinase inhibition remain unclear. We have previously shown that prolonged exposure to imatinib mesylate (IM) results in dysregulation of Akt/Erk 1/2 signaling, upregulation of miR-181a, enhanced adhesiveness, and resistance to high IM. To characterize the molecular basis and reversibility of those effects, we applied gene and protein expression analysis, quantitative phosphoproteomics, and direct miR-181a inhibition to our cellular model of CML cells subjected to prolonged exposure to IM. Those cells demonstrated upregulation of pluripotency markers (SOX2, SALL4) and adhesion receptors (CD44, VLA-4, CXCR4), as well as downregulation of Hippo signaling and upregulation of transcription coactivator YAP. Furthermore, inhibition of miR-181a using a microRNA sponge inhibitor resulted in decreased transcription of SOX2 and SALL4, decreased activation of YAP, and increased sensitivity to IM. Our findings indicate that long-term exposure to IM results in dysregulation of stem cell renewal–regulatory Hippo/YAP signaling, acquisition of expression of stem cell markers and that experimental interference with YAP activity may help to restore chemosensitivity to TKI.
Keywords: : chemoresistance, Hippo pathway, YAP, tyrosine kinase inhibitor, leukemic stem cells, imatinib mesylate
Introduction
In the last decade, the use of tyrosine kinase inhibitor (TKI)-based therapy for the treatment of BCR-ABL1-dependent chronic myelogenous leukemia (CML) redefined this disease from recalcitrant cancer to a manageable, chronic disorder with life expectancy approaching that of the general population [1]. In the randomized CML-Study IV, progression-free and overall survival rates, after 10 years of treatment with imatinib mesylate (IM), reached 82% and 84%, respectively [2]. However, IM treatment requires continuous administration of the drug, with a 76% cumulative incidence of adverse reactions at 8 years. Even though the rate of molecular remission (MR) reaches 59–92%, leukemic stem/progenitor cells (LS/PCs) are still detectable in the bone marrow of patients with deep MR defined as more than 4-log reduction of BCR-ABL1 mRNA from a standardized baseline [3–5]. It is, therefore, not surprising that attempts to stop TKI treatment in patients with MR consistently show relapse rates of about 60% within the first 6 months after TKI discontinuation [6,7]. These findings indicate that measurement of BCR-ABL1 transcript levels in the peripheral blood does not reflect eradication of the malignant clone, but also that the long-term effects of prolonged exposure to TKI at the molecular and stem cell levels are unknown. Since patients with CML are at present committed to life-long TKI therapy, understanding the long-term molecular effects of TKI treatment is of importance to the development of strategies to preserve and enhance chemosensitivity to these drugs.
Primitive cells that survive chemotherapy and contribute to tumor progression were first identified in human leukemia in 1960. CML stem cells were the first to be characterized as surviving in the presence of chemotherapeutics [8]. Detailed mechanistic studies of IM on stem cell survival have shown that BCR-ABL1-containing hematopoietic stem cells (HSCs) can survive in the absence of Bcr-Abl1 kinase activity [5,9]. These studies suggested that persistent LS/PCs, which are resistant to or unaffected by TKIs, may be responsible for relapse after TKI discontinuation [10]. This issue gains particular importance in the light of recent reports providing evidence that TKIs not only do not eliminate LS/PCs, but that prolonged TKI treatment may also paradoxically support their presence [11,12].
Signaling pathways that are essential for embryonic development and adult tissue homeostasis are often dysregulated in cancers. A population of rare cells termed cancer stem cells (CSCs) or cancer-initiating cells has been confirmed to exist in the majority of solid or hematological cancers. CSCs possess characteristics similar to normal stem cells, including the capacity for self-renewal and differentiation, which is thought to be responsible for sustained cancer growth and relapse [13,14]. Mounting evidence suggests that altered developmental signaling networks, such as those involving JAK/STAT, TGFβ, Wnt, Notch1, and Hedgehog, play an important role in maintaining CSCs and, thereby, the tumors themselves [15–17]. The Hippo pathway was recently identified as a mediator of developmental signals with a major role in organ homeostasis and stem-cell self-renewal [18]. It has also been found to have a role in carcinogenesis and CSCs biology [19,20]. More than 35 proteins have been identified in the human Hippo pathway. Upstream regulatory proteins control Yes-associated protein (YAP) and a transcriptional coactivator with PDZ-binding motif (TAZ or WW Domain-Containing Transcription Regulator 1 (WWTR1)) activity. This occurs through the regulation of the core kinase module, which consists of mammalian STE20-like protein kinase 1/2 (Mst1), Salvador homologue 1 (Sav1), Mob kinase activator 1A (Mob1A/B), and large tumor suppressor 1/2 (Lats1/2). Alternatively, upstream components, including tight/adherens junction proteins, members of the 14-3-3 family, scribble, or α catenin, can act directly on YAP and/or TAZ.
MicroRNAs are a class of small, noncoding RNAs about 22 nucleotides in length that are involved in the control of mRNA transcript stability [21]. Among 100 different miRNAs expressed in the hematopoietic system, the miR-181 family is the most highly conserved and the most preferentially expressed [22,23]. It includes six members encoded by three independent paralog precursors located on three separate chromosomes. The mature forms of miR-181a-1, miR-181a-2, miR-181b-1, and miR-181b-2 are identical. Together with miR-181c and miR-181d, they share the same 5′ seed sequence of 5′-AACAUUCA(A/U)(C/U)GCUGUCGGUG(A/G)GU-3′, indicating a high degree of functional redundancy between the members of the family. miR-181, which is strongly expressed in the thymus, is involved in T cell development, differentiation, and metabolic regulation [24–26]. It is also upregulated in mature B lymphocytes, suggesting a role in their differentiation [22]. Interestingly, miR-181 expression was also detected in lineage markers negative, undifferentiated progenitor cells [22], and in CD34+ cells, where it is involved in the inhibition of differentiation [27]. The miR-181 family also plays a significant role in the enhancement of OCT4, SOX2, and KLF4-driven reprogramming and promotion of pluripotency [28], consistent with its engagement in stem cell renewal and maintenance. These findings are consistent with our observations made using IM-resistant CML cells that were generated by prolonged exposure to IM. Microarray analysis of these cells showed significant upregulation of miR-181a that correlated with increased levels of NANOG and SOX2 [29]. However, the mechanism that links upregulation of miR-181a to increased expression of stem cell markers and IM resistance is unclear. In this study, we present evidence that long-term exposure to IM, while leading to a significant downregulation of Bcr-Abl1 protein level and its activity, results in inactivation of the Hippo pathway kinase module and upregulation of YAP, and that these changes correlate with increased expression of stem cell markers and acquired insensitivity to IM. Our observations are in agreement with a recent report showing increased levels of miR-181a in the plasma of CML patients who discontinued IM (STOP-IM group) [30]. In the present studies, we have observed that inhibition of miR-181a resulted in a decrease in YAP levels, an increase in its phosphorylation, and a modest but statistically significant increase in sensitivity to IM. Based on these data, we speculate that downregulation of the Hippo signaling kinase module and upregulation of YAP represents a new mechanism involved in maintenance of the LS/PC phenotype and leukemia recurrence after TKI discontinuation.
Materials and Methods
Cell lines and cell culture
The human chronic myeloid leukemia (CML) K562 cell line and its IM-resistant counterpart, clone K562-STI-R, were grown at 37°C, 5% CO2 in RPMI 1640 medium (HyClone) supplemented with 10% fetal bovine serum (Corning cellgro) and 2 mM GlutaMAX (Gibco). The K562-STI-R cell line was established by culture in incremental concentrations of IM (Cayman Chemicals). The adhesive phenotype of these cells was originally noted at around the 20th passage. All experiments were performed on cells maintained for 60–75 passages in the presence of 0.6 μM IM. Adherent cells were grown to achieve no more than 70% confluency. About 70–80% of K562-STI-R cells remain adherent at any given time [29]. Cells in a logarithmic growth phase were used for all experiments. For IM sensitivity experiments, cells were grown in 1, 10, 25, 50, or 100 μM IM. The number of apoptotic cells was evaluated by FACS using the Annexin V-FITC Early Apoptosis Detection Kit (#6592) from Cell Signaling Technology. Cell number was determined by counting in a hemocytometer using Trypan Blue dye exclusion.
Potassium EDTA-anticoagulated bone marrow cells were obtained from patients before initiation of IM therapy. CD34+ cell-enriched populations were isolated before cryopreservation using the MicroBeads-Based Kit from Miltenyi (>85% purity). Informed consent was obtained in accordance with the Declaration of Helsinki and procedures were approved by the Institutional Research Board of the Rhode Island Hospital. For suspension cultures: CD34+ cells were suspended in StemSpan Serum-Free Expansion Medium (SFEM II) supplemented with 10 ng/mL GM-CSF, 10 ng/mL G-CSF, 100 ng/mL SCF, 50 ng/mL LIF, 100 ng/mL MIP-1α, and 10 ng/mL hIL-6 (Stem Cell Technologies). A total of 5 × 104cells was then added to each well of a 24-well plate with or without 5 μM IM. Viable cell numbers were determined after 1 week of incubation by hemocytometer counts of Trypan Blue-excluding cells. For in vitro colony assays: 2 × 103 CD34+ cells were plated in quadruplicate in methylcellulose-based medium with recombinant cytokines SCF, IL-3, EPO, and GM-CSF (H4434; Stem Cell Technologies) in the presence or absence of 5 μM of IM. Colonies derived from burst-forming units erythroid (BFU-E), multilineage granulopoietic, erythroid, macrophage, and megakaryocytic colony-forming units (CFU-GEMM), granulocyte–macrophage colony-forming units (CFU-GM), and macrophage colony-forming units (CFU-M) were scored in situ after 14 days of incubation using an inverted microscope.
Immunophenotyping
Cell samples were analyzed by FACS using a LSRII flow cytometer (Becton Dickinson). Data were analyzed with FACSDiva software v8.0 and FlowJo v10.1. Cell viability was assessed by Trypan Blue dye exclusion. In no case did apoptosis for either the K562 or K562-STI-R cells exceed 5%. The percentage of CML CD34+ cells cultured for 1 week in the presence of 5 μM IM undergoing apoptosis was 39%. The following antibodies were used: human CD49d (9F10) APC, mouse/human CD44 (IM7) FITC, human NOTCH1 (MHN1-519) PE, human CD184 (12G5) PE, human CD34 (561) FITC (BioLegend), human CD133/1 (AC133) PE, human CD44 (DB105) APC, human CD24 (32D12) APC, and human CD34 (AC136) FITC (Miltenyi Biotec, Inc.). Isotype controls included mouse IgG1, κ (MOPC-21) APC, rat IgG2b, κ (RTK4530) FITC, mouse IgG1, κ (MOPC-21) PE, mouse IgG2a, κ (MOPC-173) PE, mouse IgG2a, κ (MOPC-173) FITC (BioLegend), and mouse IgG2a (S43.10) FITC (Miltenyi Biotec, Inc.). Unstained cells, isotype controls, and single-stained controls were used to determine gating.
Quantitative RT-PCR analysis
Total RNA from human K562 or K562-STI-R cells was purified using an RNeasy Mini Kit (Qiagen GmbH). Residual DNA was removed by on-column DNase I treatment (Qiagen GmbH). The iScript Advanced cDNA Synthesis Kit from Bio-Rad was used for reverse transcription according to the manufacturer's protocol. Relative levels of RNA were measured by quantitative real-time PCR using the C1000™ Thermal Cycler with CFX96™ Real-Time System (Bio-Rad) following the SsoAdvanced™ Universal SYBR® Green Supermix and Bio-Rad gene expression assay protocol. The following primers were designed for transcript quantification:
ABI1: For: 5′CTGGCACACTGTCGAGAACAA3′ and Rev: 5′CTTGGCGGTTTCTGAGTAGGA3′;
SALL4: 5′AGCACATCAACTCGGAGGAG3′ and Rev: 5′CATTCCCTGGGTGGTTCACTG′3′;
TDGF1: 5′TTTGAACTGGGATTAGTTGCCG3′ and Rev: 5′GGGGCCAAATGCTGTCATCT3′;
DNMT3B: For: 5′AGGGAAGACTCGATCCTCGTC3′ and Rev: 5′GTGTGTAGCTTAGCAGACTGG3′;
TAL1: For: 5′AGCCGGATGCCTTCCCTAT3′ and Rev: 5′GGGACCATCAGTAATCTCCATCT3′;
RUNX1: For: 5′CTGCCCATCGCTTTCAAGGT3′ and Rev: 5′GCCGAGTAGTTTTCATCATTGCC3′;
SOX2: For: 5′GCCGAGTGGAAACTTTTGTCG3′ and Rev: 5′GGCAGCGTGTACTTATCCTTCT3′;
NANOG: For: 5′TTTGTGGGCCTGAAGAAAACT3′ and Rev: 5′AGGGCTGTCCTGAATAAGCAG3′;
CITED2: For: 5′CCTAATGGGCGAGCACATACA3′ and Rev: 5′GGGGTAGGGGTGATGGTTGA3′;
POU5F1: For: 5′GACAGGGGGAGGGGAGGAGCTAGG3′ and Rev: 5′CTTCCCTCCAACCAGTTGCCCCAAAC3′;
AREG: For: 5′GTGGTGCTGTCGCTCTTGATA3′ and Rev: 5′CCCCAGAAAATGGTTCACGCT3′;
BIRC5: For: 5′AGGACCACCGCATCTCTACAT3′ and Rev: 5′AAGTCTGGCTCGTTCTCAGTG3′;
CTGF: For: 5′CAGCATGGACGTTCGTCTG3′ and Rev: 5′AACCACGGTTTGGTCCTTGG3′;
STAT3: For: 5′CAGCAGCTTGACACACGGTA3′ and Rev: 5′AAACACCAAAGTGGCATGTGA3′;
YAP1: For: 5′TAGCCCTGCGTAGCCAGTTA3′ and Rev: 5′TCATGCTTAGTCCACTGTCTGT3′;
WWTR1: For: 5′GATCCTGCCGGAGTCTTTCTT3′ and Rev: 5′CACGTCGTAGGACTGCTGG3′;
NOTCH1: For: 5′GAGGCGTGGCAGACTATGC3′ and Rev: 5′CTTGTACTCCGTCAGCGTGA3′';
CD44: For: 5′CTGCCGCTTTGCAGGTGTA3′ and Rev: 5′CATTGTGGGCAAGGTGCTATT3′;
S18: For: 5′GGGCGGAGATATGCTCATGTG3′ and Rev: 5′TCTGGGATCTTGTACTGTCGT3′;
ACTB: For: 5′CATGTACGTTGCTATCCAGGC3′ and Rev: 5′CTCCTTAATGTCACGCACGAT3′;
GAPDH: For: 5′TCATTGACCTCAACTACATGGTTT3′ and Rev: 5′GGCATGGACTGTGGTCATGAGTC3′.
All primers were purchased from IDT DNA. Results were normalized to the level of human ACTB, S18, or GAPDH. Human miR-181a, RNU19, and RNU6B were amplified using TaqMan Universal PCR Master Mix and miR-181a-specific and control TaqMan probes (Applied Biosystems by Life Technologies) [29]. Relative quantitation of gene expression was evaluated by CFX96 Real-Time System (Bio-Rad). Results were evaluated using Bio-Rad CFX manager.
Phosphopeptide enrichment and LC-MS/MS analysis
K562 or K452-STI-R cells in a logarithmic growth phase were harvested (five independently revived and grown cellular clones per cell line) and lysed (8 M urea, 1 mM sodium orthovanadate, 20 mM HEPES, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, pH 8.0, 20 min, 4°C), sonicated, and cleared by centrifugation (14,000 g, 15 min, 4°C). Protein concentration was measured (Pierce BCA Protein Assay; Thermo Fisher Scientific) and a total of 100 μg of protein per sample was subjected to trypsin digestion. The tryptic digested peptides were lyophilized and phosphopeptide enrichment was done using Titansphere Phos-TiO tips (GL Sciences) following the manufacturer's protocol with minor modifications. LC-MS/MS was performed on a fully automated proteomic technology platform [31]. Experiments were performed with an Agilent 1200 Series Quaternary HPLC system (Agilent Technologies) connected to a Q Exactive Plus Mass Spectrometer (Thermo Fisher Scientific).
Bioinformatics analysis
Peptide spectrum matching of MS/MS spectra of each file was searched against a human-specific database (UniProt; downloaded 2/1/2013) using MASCOT v. 2.4 (Matrix Science Ltd.). A concatenated database containing 144,156 “target” and “decoy” sequences was employed to estimate the false discovery rate (FDR). Msconvert from ProteoWizard (v. 3.0.5047), using default parameters and with the MS2Deisotope filter on, was employed to create peak lists for Mascot. Search parameters permitted variable modification of methionine oxidation (+15.9949 Da), and static modification of carbamidomethylation (+57.0215 Da) on cysteine. To identify the phosphoresidues' additional variable modification of phosphorylation (+79.9663 Da) on serine, threonine and tyrosine residues were included. Peptide assignments from the database search were filtered down to 1% FDR by a logistic spectral score, as previously described [32,33]. To validate the position of the phosphorylation sites, the Ascore algorithm [34] was applied and the reported phosphorylation site position reflected the top Ascore prediction.
Relative quantitation of the phosphopeptides
Relative quantitation of phosphopeptide abundance was performed through calculation of selected ion chromatogram (SIC) peak areas. Retention time alignment of individual replicate analyses was performed as previously described [35]. SIC peak areas were determined for every phosphopeptide that was identified by MS/MS. In the case of a missing MS/MS for a particular peptide in a particular replicate, the SIC peak area was calculated according to the peptide's isolated mass and retention time calculated from retention time alignment. A minimum SIC peak area equivalent to the typical spectral noise level of 1,000 was required of all data reported for label-free quantitation. Individual SIC peak areas were normalized to the peak area of the standard phosphopeptide DRVpYHPF that was added before phosphopeptide enrichment and reversed-phase elution into the mass spectrometer.
Proteomic result data analysis
Functional analysis of phosphoproteomics data was performed using MetaCore™ software suite. Volcano plot was calculated from the log 2 of the ratio of the fold change of each peptide peak area compared with the natural log of the q-value of each peptide.
Immunoblotting and immunofluorescence
Whole cell lysates were used in all experiments. Protein levels were equalized based on β-actin protein level. Western blotting was performed as described previously [29]. The following antibodies were used: rabbit polyclonal anti- Abi-1 #NB100-59845 (Novus Biologicals), mouse monoclonal anti-Abi-1 #D147-3 (MBL International Corporation), mouse monoclonal anti-SSH3BP1 [4E2] #ab11222 (Abcam), Mouse monoclonal anti-Abi1 (clone 2G8) unpublished (kind gift from L. Kotula, Upstate Medical University in Syracuse, NY), mouse monoclonal anti-c-Abl #OP20 (Calbiochem/Millipore), rabbit monoclonal phospho-anti-c-Abl (Tyr412) # 2865, rabbit polyclonal anti-Mst1 #3682, rabbit monoclonal anti-Sav1 #13301, rabbit monoclonal anti-Lats1 #3477, rabbit monoclonal anti-Mob1 #13730, rabbit monoclonal phospho-anti-YAP(S127) #1308, rabbit monoclonal phospho-anti-YAP(S397) #13619, mouse monoclonal anti-CrkL #3182, and rabbit polyclonal anti-phospho-CrkL (Tyr207) #3181 were from Cell Signaling Technology. Rabbit monoclonal anti-YAP/TAZ was from Abcam #ab52771. Mouse monoclonal anti-β-actin #A1978 was from Sigma-Aldrich. Peroxidase-conjugated AffiniPure goat anti-rabbit (H+L) and goat anti-mouse (H+L) were from Jackson ImmunoResearch Laboratories, Inc. Immunofluorescence was performed as described [36]; rabbit monoclonal anti-YAP/TAZ #ab52771 followed by goat anti-rabbit conjugated with AlexaFluor 555 were used, phalloidin conjugated with AlexaFluor 555 was used to visualize F-actin (Invitrogen). Confocal images were acquired with a Nikon C1si confocal (Nikon, Inc.) using diode lasers 402, 488, and 561. Serial optical sections were performed with EZ-C1 software (Nikon, Inc.). Z series sections were collected at 0.2 μm with a 60 × PlanApo objective and scan zoom of 2. Images were processed and projected in Elements software (Nikon, Inc.).
MicroRNA sponge design
To construct the miR-181a sponge, oligonucleotides were designed to contain two miR-181a binding sites. Each binding site was composed of a 22-nucleotide long sequence complementary to the sequence of miR-181a. Each binding site contained a central mismatch at positions 11–14, in which the nucleotide at position 14 was deleted. The two miR-181a binding sites were separated by a sequence of four nucleotides (AATT). Control sponges were designed to have a scrambled sequence at positions 3–8, the region that corresponds to the miR-181a seed sequence. Sense and antisense oligomers were synthesized, PAGE purified at a 100 nmol scale, and phosphorylated at the 5′ ends. The oligomers had overhangs compatible with the restriction endonuclease SanDI. Complementary oligomer pairs were mixed in a 1:1 ratio and annealed. T4 ligase was used for oligomer duplexes ligation into multimers. Empty pcDNA3-SanDI or pcDNA3-SanDI-GFP mammalian expression vectors (kind gift from Dr. Kluiver, University Medical Center, The Netherlands) were digested with SanDI restriction endonuclease and dephosphorylated with alkaline phosphatase (Fermentas). T4 Ligase (New England BioLabs, Inc.) was used to ligate the multimers into the pcDNA3-SanDI or pcDNA3-SanDI-GFP. The number of oligomers ligated into the expression vector was verified in a digestion reaction using EcoRI and BamHI restriction endonucleases. Vectors containing 8, 6, and 4 oligomer duplexes for control or miR-181a sponge (16-, 12-, 8-mers) were sequenced and tested for the effect on miR-181a. Vectors containing octamer control or octamer miR-181a sponge were selected for subsequent experiments.
Electroporation
Vectors used for electroporation were purified using the EndoFree Plasmid Maxi Kit (Qiagen GmbH). DNA purity and integrity were determined spectroscopically (OD260nm/OD280nm = 1.90–2.0), (OD260nm/OD230nm > 2.00). DNA electroporation was performed with Neon® Transfection System (Life Technology) according to optimized manufacturer's instruction. After electroporation, cells were seeded into 10-cm plates with 10 mL of prewarmed RPMI 1640 containing IM and incubated at 37°C in a humidified CO2 incubator for the next 24 h.
Statistical analyses
We performed statistical analyses using two-tailed unpaired Student's t-test. We considered values of P < 0.05 to be statistically significant (*P < 0.05, **P < 0.01, and ***P < 0.001). For phosphoproteomic analyses P-values were calculated from five replicates. To select phosphopeptides that show statistically significant change in abundance between K562-STI-R cells and K562 cell lines, two-tailed unpaired Student's t-test and q-values for multiple hypothesis tests were calculated based on the determined P-values using the R package QVALUE as previously described [37].
Results
Exposure to IM results in increased expression of stem cell markers and acquired insensitivity to IM
Recent reports suggest that TKI therapy not only fails to eliminate LS/PCs, but that it may also actually support their presence and contribute to upregulation of certain stem cell markers [11,12]. To determine the effect of IM on the genes that are associated with the stem cell and CSC phenotypes (SALL4, OCT4, SOX2, NANOG, CD44, CD133, and STAT3), we analyzed published microarray data that had examined gene expression in CML CD34+ cells isolated from the bone marrow at diagnosis and after IM initiation. Two data sets were analyzed: NCBI/GEO/GSE12211, includes data on six patients from whom samples were obtained before IM initiation, and six patients from whom samples were obtained 7 days after IM initiation [38]; NCBI/GEO/GSE33075, includes nine patients from whom samples were obtained before IM initiation and nine patients from whom samples were obtained 4 weeks after imatinib initiation [39]. In both studies, patients received a standard IM daily dose of 400 mg. Our analysis of these data (Fig. 1A) showed a significant increase in transcripts for NOTCH1, STAT3, and CD44 1 and 4 weeks following initiation of IM therapy. This suggested that both short and prolonged exposure to IM may result in upregulation of gene transcripts associated with the CSC phenotype.
FIG. 1.

NOTCH1, STAT3, and CD44 expression in hematopoietic progenitor cells obtained from CML patients. (A) Affymetrix Human Genome U219 Array results for NOTCH1, STAT3, CD44 expression were analyzed. Analyses were carried out on CD34+ cells obtained from the bone marrow of CML patients at diagnosis (n = 6, pre-IM) and 1 week after the initiation of therapy (n = 6, post-IM; top three panels), and at diagnosis (n = 9, pre-IM), and 4 weeks after initiation of therapy (n = 9, post-IM; lower three panels). The following probe sets were evaluated using the deposited GEO GSE12211 and GSE33075 databases (GPL571 and GPL570 platforms, respectively). The status of expression of probes 218902_at for NOTCH1, 208991_at for STAT3, and 204489_s_at for CD44 is presented as interquartile range and median of normalized, log-transformed raw data. P-values are denoted on the graphs. (B) Hematopoietic progenitor cells were obtained from CML patients (n = 3) before initiation of TKI therapy. NOTCH1, STAT3, and CD44 expression was determined on CML CD34+ cells cultured with or without 5 μM IM for 1 week. Normalized expression relative to ACTB expressed as fold change, is presented. Samples were cultured and run in triplicate **P < 0.01. CML, chronic myelogenous leukemia; IM, imatinib mesylate.
We also assessed the direct effect of IM on the expression of NOTCH1, STAT3, and CD44 in the CML CD34+ cells isolated from the bone marrow of CML patients prior initiation of IM therapy and cultured in vitro for 1 week in 5 μM IM in serum-free expansion medium supplemented with cytokines. The presence of 5 μM of IM for 1 week resulted in a five-fold decrease in CML CD34+ cell number (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/scd) and a five-fold decrease in the colony formation capacity of CML CD34+ cells (Supplementary Fig. S1B). Thirty-nine percent of surviving CML CD34+ cells were apoptotic. These observations are in line with previously described findings [5,40,41]. One week-long exposure to IM also resulted in increased expression of NOTCH1 (Fig. 1B), in agreement with findings presented on Fig. 1A. Notably, transcript levels of STAT3 remained unaffected, whereas mRNA levels of CD44 were significantly reduced (Fig. 1B). These data indicate that exposure to IM may directly affect expression of stem cell markers.
We extended these observations to our established cellular model of long-term exposure to IM. This model, the K562-STI-R cell line, was established by culturing K562 CML cells in concentrations of IM that were increased until selection resulted in cells that were viable in the presence of 10 μM IM. K562-STI-R cells were maintained in 0.6 μM IM for 60–75 passages; 70–80% of these cells persisted in being adherent. K562-STI-R cells display significant downregulation of Bcr-Abl1 protein levels and activity (Supplementary Fig. S2 and also [29]). We examined the K562-STI-R expression pattern of the pluripotency markers OCT4 (POU5F1), NANOG, SOX2, TDGF1, and DNMT3, and of the HSC markers SALL4, CITED2 (regulators of human hematopoiesis), and TAL1 (regulator of HSC quiescence). ACTB, S18, and GAPDH were used as controls. Results (Fig. 2A) showed upregulation of SOX2 and SALL4 in the K562-STI-R cells. NANOG, CITED2, DNMT3A, and TAL1 were downregulated (Supplementary Fig. S3A, B). Since both SOX2 and SALL4 are well-established regulators of cell fate during development whose upregulation is associated with maintenance of CSCs [42,43], we interpreted our data as indicating that long-term exposure to IM may promote the stem cell phenotype and acquisition of a CSC phenotype.
FIG. 2.


Stem cell markers and sensitivity to IM in K562 and IM-resistant K562-STI-R cells. (A) Expression of NANOG, POU5F1, SOX2, CITED2, SALL4, and TAL1 was determined in K562 or IM-resistant K562-STI-R cells. Data represent normalized expression relative to ACTB, S18, and GAPDH. Samples were run in triplicate. Representative data from three independent analyses are shown. ***P < 0.001 (B) Expression of CD45, CD44, CXCR4, VLA-4, CD24, NOTCH1, and CD34 was determined by FACS in K562 and K562-STI-R cells. 3D histograms and a bar graph summary of the quantitative results are presented. Representative data from three independent analyses are shown. ***P < 0.001 (C) Coexpression of CD44 and CD24 by FACS was determined for K562 and K562-STI-R cells. Analyses were performed in quadruplicate. Representative data are shown. (D) FACS analyses were used to determine the percentage of apoptotic cells in a population of K562 and K562-STI-R cells subjected to incremental concentrations of IM for 24 h. The percent Annexin V-FITC-positive cells was determined. Representative data from three independent analyses are shown *P < 0.05.
We then examined the expression of adhesion receptors that regulate interactions between HSCs and their microenvironment, and are associated with adhesion-mediated drug resistance and the CSC phenotype [44]. These include CD34, CXCR4 (C-X-C chemokine receptor type 4), VLA-4 (Very Late Antigen 4, CD49f), NOTCH1, CD44, CD24, and CD133. We found a significant upregulation of CD44 and VLA-4, a modest increase in expression of CXCR4 and NOTCH1, and a modest decrease in the expression of CD24 (Fig. 2B). The CD44hi/CD24low phenotype, present in the K562-STI-R cells (Fig. 2C), is associated with the CSC phenotype [45,46]. These results indicated that long-term exposure to IM induces upregulation of adhesion receptors that may contribute to an increase in adhesion.
Finally, we evaluated the sensitivity of K562-STI-R cells to incremental concentrations of IM. K562-STI-R cells that overexpressed genetic and surface markers associated with stem cell potential and the adhesive phenotype showed significant resistance to IM treatment. These cells were able to withstand very high concentrations of the drug, with an increase in apoptotic cells occurring only at IM concentrations in excess of 25 μM (Fig. 2D). These data indicate that cells exposed to long-term IM treatment display a high degree of resistance to IM and upregulate genetic (SOX2, SALL4) and surface (CD44, VLA-4, CXCR4) markers associated with the stem cell phenotype.
Long-term exposure to IM results in the inhibition of Hippo signaling kinase module and increased YAP activity
To investigate the signaling pathways involved in the phenotype associated with long-term exposure to IM, we performed label-free, quantitative phosphoproteomic analysis of K562 and K562-STI-R cells. For each cell type, a total of five biological replicates were analyzed using LC–MS/MS (experimental strategy presented in Supplementary Fig. S4). Using a 1% FDR, we quantified 4,807 unique phosphopeptides derived from 1,559 proteins. The complete list of the identified phosphopeptides subjected to quantitative analysis is presented in Supplementary Table S1. The pairwise analysis of replicate peak areas indicated a high degree of quantitative reproducibility between individual LC–MS runs used for label-free, intensity-based quantitation. Among all identified phosphopeptides, a total of 2,593 phosphopeptides representing 1,175 phosphoproteins showed a statistically significant difference between the two cell lines. Detection of significantly altered phosphopeptides was carried out by applying Student's t-test with a q-value correction for multiple comparisons. A volcano plot representing the relationship between fold change and q-value (Fig. 3A) illustrates that among 36.9% of identified phosphopeptides with an absolute fold change greater than 2, 88.3% of peptides showed lower abundance in the K562-STI-R cells, whereas 11.7% showed higher peak abundance. We performed a functional analysis of the significantly different phosphopeptides using MetaCore software. The top five identified pathways included cell cycle (chromosome condensation), cytoskeleton remodeling, Erk1/2 signaling, transport (AVP regulation of aquaporin 2), and development (negative regulation of Hippo pathway and positive regulation of YAP/TAZ function) (Fig. 3B). Based on its potential role in the K562-STI-R phenotype that we observed, we focused on the Hippo pathway.
FIG. 3.


Effect of long-term exposure to IM on the Hippo pathway and YAP. (A) Volcano plots showing fold change versus q-value of phosphopeptide peak areas were generated. Of the 4,807 unique phosphopeptides identified, 36.9% peptides (red in inset pie chart) showed a statistically significant (q < 0.05) difference and an absolute fold change greater than 2 comparing the K562-STI-R cells to control K562 cells. Within this group of significantly different peptides, 88.3% showed lower abundance in the K562-STI-R cells. (B) The five most highly scored pathways identified in K562-STI-R cells reflect the enrichment analysis of gene IDs corresponding to the proteins for which identified phosphopeptides showed statistically a significant (q < 0.05) fold change >2. (C) This schematic representation of Hippo/YAP cross-talk shows phosphosites that were significantly different in K562-STI-R cells versus control K562 cells. (D) Western blot analysis was performed on K562 and K562-STI-R whole cell lysates. Levels of Sav1, Lats1, Mob, and YAP along with phosphorylation of YAP on Ser127 and 397 were evaluated. Data from three independent, replicate cultures of each cell line representing three independent experiments are shown. (E) RT-PCR was performed to quantify AREG, BIRC5, and CTGF expression in K562 and IM-resistant K562-STI-R cells. Data are presented as normalized expression relative to ACTB. Samples were run in triplicate. Representative data from three independent analyses are shown *P < 0.05.
The list of top five identified pathways-associated phosphopeptides that differed in abundance in the K562-STI-R cells relative to K562 cells is presented in Table 1. In Fig. 3C, we present a schematic outline of the Hippo cascade, with phosphosites identified in our phosphoproteomic analysis highlighted. Immunoblotting for components of the Hippo pathway (Fig. 3D) showed significantly decreased expression levels of Lats1, Sav1, and Mob in the IM-resistant cells. This was associated with an increase in YAP protein levels and a marked reduction in the stoichiometry of phosphorylation of YAP on Ser 127 and Ser 397. Evaluation of the expression levels of YAP targets showed downregulation of AREG and BIRC5 and upregulation of CTGF (Fig. 3E and Supplementary Fig. S5).
Table 1.
The List of Phosphopeptides Associated with Top Five Pathways Identified to be Dysregulated in the K562-STI-R Relative to K562 Cells
| Protein name | Peptide sequence (*phosphorylated, #methionine oxidation) | Phosphorylation site (p-site) | Accession no. for p-site | Ratio K562-STI-R/K562 | q-Values | Charge state | Gene symbol | HPRD accession |
|---|---|---|---|---|---|---|---|---|
| Cell cycle—chromosome condensation in prometaphase | ||||||||
| Histone H1x | K.AGGSAALS*PSKK.R | S31* | HPRD:04151_1 | 0.019244966 | 3.83359E-05 | 2 | H1FX | 04151_1 |
| Topoisomerase DNA II, beta | K.VVEAVNS*DS*DS*EFGIPK.K | S1517S1519S1521* | HPRD:00537_1 | 0.060399862 | 0.000632588 | 2 | TOP2B | 00537_1 |
| Topoisomerase DNA II, beta | K.VVEAVNS*DS*DSEFGIPKK.T | S1517S1519* | HPRD:00537_1 | 0.11168151 | 4.40595E-06 | 3 | TOP2B | 00537_1 |
| Inner centromere protein | R.AAAAAAAATMALAAPS*SPTPES*PTMLTK.K | S142S148* | HPRD:05103_1 | 0.134864416 | 0.000258977 | 3 | INCENP | 05103_2 |
| Topoisomerase DNA II, beta | K.YFAES*DEEEDDVDFAMFN.- | S1608 | HPRD:00537_1 | 0.157412811 | 0.040967043 | 2 | TOP2B | 00537_1 |
| Topoisomerase DNA II, beta | K.SEDDSAKFDS*NEEDSASVFSPSFGLK.Q | S1461* | HPRD:00537_1 | 0.168750517 | 0.005740955 | 3 | TOP2B | 00537_1 |
| Cyclin B2 | K.DLASPLIGRS*.- | S398 | HPRD:04131_1 | 0.170492751 | 0.000897379 | 2 | CCNB2 | 04131_1 |
| Inner centromere protein | R.IAQVS*PGPR.D | S263* | HPRD:05103_1 | 0.192521701 | 4.99393E-06 | 2 | INCENP | 05103_2 |
| Topoisomerase DNA II, beta | R.KAS*GS*ENEGDYNPGR.K | S1545S1547* | HPRD:00537_1 | 0.227852671 | 7.9002E-06 | 2 | TOP2B | 00537_1 |
| H3 Histone, family 3B | K.S*TELLIR.K | S58 | HPRD:03036_1 | 0.251507088 | 5.84008E-06 | 2 | H3F3B | 18825_1 |
| CDC2 | K.IGEGT*YGVVYK.G | T14* | HPRD:00302_1 | 0.254472604 | 0.000406451 | 3 | CDK1 | 00446_1 |
| Inner centromere protein | R.AAAAAAAATMALAAPSS*PTPES*PTMLTK.K | S143S148* | HPRD:05103_1 | 0.261545024 | 0.001363582 | 4 | INCENP | 05103_2 |
| KARP 1 binding protein | R.ST*GS*ATSLASQGER.R | T628S630* | HPRD:17172_1 | 0.323005541 | 0.000253714 | 2 | CEP170 | 17172_3 |
| Chromosome condensation protein G | K.TLHCEGTEINS*DDEQESKEVEETATAK.N | S674* | HPRD:12097_1 | 0.346120173 | 0.005823643 | 3 | NCAPG | 12097_1 |
| KARP 1 binding protein | R.S*DSEATISR.S | S1165 | HPRD:17172_1 | 0.35100536 | 3.89936E-05 | 2 | CEP170 | 17172_3 |
| KARP 1 binding protein | R.STGS*ATSLASQGER.R | S630* | HPRD:17172_1 | 0.36744914 | 0.000148779 | 2 | CEP170 | 17172_3 |
| Chromosome condensation protein G | K.LNLAQFLNEDLS*.- | S1015 | HPRD:12097_1 | 0.382107906 | 0.009815237 | 2 | NCAPG | 12097_1 |
| CDC2 | K.IGEGT*Y*GVVYKGR.H | T14Y15* | HPRD:00302_1 | 0.411471675 | 0.002256503 | 3 | CDK1 | 00302_2 |
| CND2 | R.LLAS*PSSR.S | S87 | HPRD:03820_1 | 0.442434391 | 4.82902E-05 | 2 | NCAPH | 03820_1 |
| Inner centromere protein | K.INPDNY*GMDLNS*DDST*DDEAHPR.K | Y818S824T828* | HPRD:05103_1 | 0.481723555 | 0.006483106 | 3 | INCENP | 05103_2 |
| Non-SMC condensin II complex, subunit H2 | K.ESRS*PQQSAALPR.R | S284* | HPRD:11783_1 | 0.677367739 | 0.003665133 | 3 | NCAPH2 | 11783_3 |
| Chromosome condensation protein G | R.CQT*AEADSES*DHEVPEPESEMK.M | T968S975* | HPRD:12097_1 | 0.679711752 | 0.020134351 | 3 | NCAPG | 12097_1 |
| Chromosome condensation protein G | R.CQTAEADS*ES*DHEVPEPESEMK.M | S973S975* | HPRD:12097_1 | 0.679877906 | 0.020178793 | 3 | NCAPG | 12097_1 |
| DNA topoisomerase II alpha | K.NENTEGS*PQEDGVELEGLKQR.L | S1247* | HPRD:00536_1 | 0.760612799 | 0.022737593 | 3 | TOP2A | 00536_1 |
| DNA topoisomerase II alpha | K.SVVS*DLEADDVK.G | S1377* | HPRD:00536_1 | 0.797929426 | 0.045978223 | 2 | TOP2A | 00536_1 |
| Chromosome condensation-related SMC-associated protein 1 | R.YQPLAS*TASDNDFVT*PEPR.R | S1330T1339* | HPRD:06472_1 | 2.062328833 | 0.011033288 | 3 | NCAPD2 | 06472_1 |
| Chromosome condensation-related SMC-associated protein 1 | R.YQPLAST*ASDNDFVT*PEPR.R | T1331T1339* | HPRD:06472_1 | 2.067195121 | 0.01091227 | 3 | NCAPD2 | 06472_1 |
| Chromosome condensation-related SMC-associated protein 1 | R.YQPLASTAS*DNDFVT*PEPR.R | S1333T1339* | HPRD:06472_1 | 2.06901788 | 0.010957473 | 3 | NCAPD2 | 06472_1 |
| Cytoskeleton remodeling | ||||||||
| Myosin heavy chain, nonmuscle type B | R.QLHLEGAS*LELS*DDDTESK.T | S1952S1956* | HPRD:01178_1 | 0.017049285 | 3.38772E-05 | 3 | MYH10 | 01178_1 |
| Myosin heavy chain, nonmuscle type B | R.GGPISFS*SSR.S | S1937 | HPRD:01178_1 | 0.047808617 | 2.16877E-05 | 2 | MYH10 | 01178_1 |
| Paxillin | R.FIHQQPQS*SSPVYGSSAK.T | S83* | HPRD:03937_1 | 0.074088561 | 0.000279604 | 3 | PXN | 03937_2 |
| Erythrocyte membrane protein band 4.1 | R.LFS*SFLK.R | S84 | HPRD:00558_1 | 0.104031741 | 1.76077E-05 | 2 | EPB41 | 00558_2 |
| Erythrocyte membrane protein band 4.1 | R.LFSS*FLK.R | S85 | HPRD:00558_1 | 0.104031741 | 1.76077E-05 | 2 | EPB41 | 00558_2 |
| MAP3K7 | R.S*IQDLTVTGTEPGQVS*SR.S | S439S454* | HPRD:04011_1 | 0.108429364 | 0.000178486 | 2 | MAP3K7 | 04011_4 |
| Myosin heavy chain, nonmuscle type B | R.QLHLEGASLELS*DDDTESK.T | S1956* | HPRD:01178_1 | 0.113452724 | 0.000402341 | 3 | MYH10 | 01178_1 |
| ERK2 | R.VADPDHDHTGFLT*EY*VATR.W | T185Y187* | HPRD:01496_1 | 0.128708662 | 7.37351E-05 | 3 | MAPK1 | 01496_2 |
| SHC (Src homology 2 domain containing) transforming protein 1 | R.ELFDDPSY*VNVQNLDK.A | Y427* | HPRD:02780_1 | 0.15029448 | 0.000762219 | 2 | SHC1 | 02780_2 |
| Williams Beuren syndrome chromosome region 1 | R.AYS*SFGGGRGSR.G | S13* | HPRD:11940_1 | 0.163809196 | 5.6494E-05 | 3 | EIF4H | 11940_2 |
| Zyxin | R.S*PGAPGPLTLK.E | S344* | HPRD:03592_1 | 0.193649965 | 2.9433E-05 | 2 | ZYX | 03592_2 |
| Erythrocyte membrane protein band 4.1 | K.IEVKEES*PQS*KAETELK.A | S188S191 | HPRD:00558_1 | 0.196565066 | 7.81528E-07 | 3 | EPB41 | 00558_2 |
| Filamin A, alpha | K.AFGPGLQGGS*AGSPAR.F | S1081* | HPRD:02060_1 | 0.225995795 | 4.14801E-07 | 2 | FLNA | 02060_1 |
| Myc proto-oncogene protein | M.PLNVS*FTNR.N | S6 | UNI:P01106 | 0.229391119 | 0.00011783 | 2 | MYC | |
| Erythrocyte membrane protein band 4.1 | R.LST*HSPFR.T | T710* | HPRD:00558_1 | 0.238296778 | 0.000122817 | 2 | EPB41 | 00558_4 |
| SOS1 | R.DGPPLLENAHS*S.- | S1332 | HPRD:01681_1 | 0.238884192 | 2.45255E-05 | 2 | SOS1 | 01681_1 |
| CRK | R.LLDQQNPDEDFS*.- | S304 | HPRD:01267_1 | 0.256892592 | 5.84008E-06 | 2 | CRK | 01267_1 |
| Filamin A, alpha | K.AFGPGLQGGSAGS*PAR.F | S1084* | HPRD:02060_1 | 0.320034539 | 0.002956223 | 2 | FLNA | 02060_1 |
| MAP3K7 | R.S*IQDLTVTGTEPGQVSSR.S | S439* | HPRD:04011_1 | 0.374568908 | 2.26197E-05 | 2 | MAP3K7 | 04011_4 |
| MAP2K2 | R.LNQPGT*PTR.T | T394* | HPRD:03164_1 | 0.40428212 | 2.40617E-05 | 2 | MAP2K2 | 03164_1 |
| MAP3K7 | R.SIQDLTVT*GTEPGQVSSR.S | T446* | HPRD:04011_1 | 0.459359346 | 0.000797977 | 3 | MAP3K7 | 04011_4 |
| MAP3K7 | R.SIQDLTVTGT*EPGQVSSR.S | T448* | HPRD:04011_1 | 0.46011863 | 0.000781265 | 3 | MAP3K7 | 04011_4 |
| MAP3K7 | R.SIQDLT*VTGTEPGQVSSR.S | T444* | HPRD:04011_1 | 0.462145995 | 0.000747788 | 3 | MAP3K7 | 04011_4 |
| EIF4G3 | R.S*PVPAQIAITVPK.T | S495* | HPRD:06802_1 | 0.524468385 | 0.037236161 | 2 | EIF4G3 | 06802_1 |
| Zyxin | K.LGHPEALSAGTGS*PQPPSFTYAQQR.E | S308* | HPRD:03592_1 | 0.537300804 | 0.016351316 | 3 | ZYX | 03592_2 |
| c-Myc | K.KFELLPT*PPLS*PSRR.S | T73S77* | HPRD:01818_1 | 0.58647132 | 0.019836997 | 3 | MYC | 01818_1 |
| Nucleolin | R.LELQGPRGS*PNAR.S | S563* | HPRD:01245_1 | 1.333398242 | 0.001838834 | 2 | NCL | 01245_1 |
| Nucleolin | K.KVVVS*PTKK.V | S67* | HPRD:01245_1 | 1.506374651 | 0.00264531 | 2 | NCL | 01245_1 |
| Nucleolin | K.KVVVSPT*KK.V | T69* | HPRD:01245_1 | 1.506374651 | 0.00264531 | 2 | NCL | 01245_1 |
| Nucleolin | K.GFGFVDFNS*EEDAK.A | S619* | HPRD:01245_1 | 1.601309703 | 0.014442235 | 2 | NCL | 01245_1 |
| Nucleolin | K.ALVAT*PGKK.G | T121* | HPRD:01245_1 | 1.796823121 | 0.024897338 | 2 | NCL | 01245_1 |
| Myosin heavy chain 9, nonmuscle | R.KGAGDGS*DEEVDGKADGAEAKPAE.- | S1943* | HPRD:01177_1 | 2.202907542 | 0.000407614 | 4 | MYH9 | 01177_1 |
| Myosin regulatory light chain MRLC2 | R.ATS*NVFAMFDQSQIQEFK.E | S20 | HPRD:10095_1 | 2.396215105 | 0.048226977 | 2 | MYL12A | 17619_2 |
| ERK 1/2 signaling pathway | ||||||||
| MAP3K7 | R.S*IQDLTVTGTEPGQVS*SR.S | S439S454* | HPRD:04011_1 | 0.108429364 | 0.000178486 | 2 | MAP3K7 | 04011_4 |
| Protein kinase C, beta 1 | R.HPPVLT*PPDQEVIR.N | T641* | HPRD:01499_1 | 0.123333002 | 1.75336E-05 | 3 | PRKCB | 01499_1 |
| ERK2 | R.VADPDHDHTGFLT*EY*VATR.W | T185Y187* | HPRD:01496_1 | 0.128708662 | 7.37351E-05 | 3 | MAPK1 | 01496_2 |
| SHC (Src homology 2 domain containing) transforming protein 1 | R.ELFDDPSY*VNVQNLDK.A | Y427* | HPRD:02780_1 | 0.15029448 | 0.000762219 | 2 | SHC1 | 02780_2 |
| SOS1 | R.DGPPLLENAHS*S.- | S1332 | HPRD:01681_1 | 0.238884192 | 2.45255E-05 | 2 | SOS1 | 01681_1 |
| MAP3K7 | R.S*IQDLTVTGTEPGQVSSR.S | S439* | HPRD:04011_1 | 0.374568908 | 2.26197E-05 | 2 | MAP3K7 | 04011_4 |
| MAP2K2 | R.LNQPGT*PTR.T | T394* | HPRD:03164_1 | 0.40428212 | 2.40617E-05 | 2 | MAP2K2 | 03164_1 |
| MAP2K2 | R.LNQPGT*PTR.T | T394* | HPRD:03164_1 | 0.40428212 | 2.40617E-05 | 2 | MAP2K2 | 03164_1 |
| MAP3K7 | R.SIQDLTVT*GTEPGQVSSR.S | T446* | HPRD:04011_1 | 0.459359346 | 0.000797977 | 3 | MAP3K7 | 04011_4 |
| MAP3K7 | R.SIQDLTVTGT*EPGQVSSR.S | T448* | HPRD:04011_1 | 0.46011863 | 0.000781265 | 3 | MAP3K7 | 04011_4 |
| MAP3K7 | R.SIQDLT*VTGTEPGQVSSR.S | T444* | HPRD:04011_1 | 0.462145995 | 0.000747788 | 3 | MAP3K7 | 04011_4 |
| MAP3K8 | R.NIGS*DFIPR.G | S141 | HPRD:01863_1 | 2.471643237 | 0.000126802 | 2 | MAP3K8 | 01863_1 |
| Protein kinase C delta | R.AS*TFCGTPDYIAPEILQGLK.Y | S506* | HPRD:01501_1 | 6.324500473 | 0.014223 | 3 | PRKCD | 01501_2 |
| Transport–the role of APV in regulation of aquaporin | ||||||||
| Protein kinase, cAMP dependent, regulatory type II, alpha | R.VADAKGDS*ES*EEDEDLEVPVPSR.F | S78S80* | HPRD:01484_1 | 0.058727412 | 2.57063E-05 | 2 | PRKAR2A | 01484_1 |
| Protein kinase, cAMP dependent, regulatory type II, alpha | K.MFGS*SVDLGNLGQ.- | S395 | HPRD:01484_1 | 0.08484677 | 0.001111705 | 2 | PRKAR2A | 01484_1 |
| Synaptosomal-associated protein, 23 kd | K.TTWGDGGENS*PCNVVSK.Q | S110 | HPRD:03962_1 | 0.141129472 | 0.000102945 | 2 | SNAP23 | 03962_1 |
| Protein kinase, cAMP dependent, regulatory, type II, beta | R.RAS*VCAEAYNPDEEEDDAESR.I | S114* | HPRD:01486_1 | 0.290252792 | 9.12088E-05 | 3 | PRKAR2B | 01486_1 |
| Protein kinase, cAMP dependent, regulatory, type I, alpha | R.TDSREDEIS*PPPPNPVVK.G | S83* | HPRD:01786_1 | 0.51476936 | 0.011064665 | 3 | PRKAR1A | 01786_3 |
| Guanine nucleotide-binding protein G(S), alpha subunit | R.ISTAS*GDGR.H | S995 | HPRD:00761_1 | 3.921542175 | 0.000408155 | 2 | GNAS | 00761_6 |
| Development–negative regulation of HIPPO and positive regulation of YAP/TAZ | ||||||||
| LIMD1 | R.SS*EGSLGGQNSGIGGR.S | S240 | HPRD:05177_1 | 0.038093765 | 0.000219693 | 2 | LIMD1 | 05177_1 |
| Rho guanine nucleotide exchange factor 2 | R.QSLLGS*R.R | S137 | HPRD:10458_1 | 0.087942015 | 0.020979399 | 2 | ARHGEF2 | 10458_2 |
| Rho guanine nucleotide exchange factor 2 | K.NNTALQSVS*LR.S | S109 | HPRD:10458_1 | 0.21884569 | 0.00040601 | 2 | ARHGEF2 | 10458_2 |
| Rho guanine nucleotide exchange factor 2 | R.S*LPAGDALYLSFNPPQPSR.G | S885* | HPRD:10458_1 | 0.22363664 | 0.003435839 | 3 | ARHGEF2 | 10458_2 |
| LATS1 | R.SNS*FNNPLGNR.A | S464* | HPRD:09147_1 | 0.251673373 | 0.000262497 | 2 | LATS1 | 09147_1 |
| LATS1 | K.QIT*TSPITVR.K | T611* | HPRD:09147_1 | 0.26611696 | 7.89302E-06 | 2 | LATS1 | 09147_1 |
| Ring finger protein 131 | K.SSAGDTEFVHQNS*QEIQR.S | S253 | HPRD:11436_1 | 0.266846173 | 0.000795098 | 3 | PJA2 | 11436_1 |
| Rho guanine nucleotide exchange factor 2 | R.EPALPLEPDSGGNT*SPGVTANGEAR.T | T694* | HPRD:10458_1 | 0.30272125 | 0.000207263 | 3 | ARHGEF2 | 10458_2 |
| Rho guanine nucleotide exchange factor 2 | R.EPALPLEPDS*GGNTSPGVTANGEAR.T | S690* | HPRD:10458_1 | 0.303583488 | 0.000208202 | 3 | ARHGEF2 | 10458_2 |
| Bromodomain adjacent to zinc finger domain, 1B | R.SDVQEES*EGSDT*DDNKDSAAFEDNEVQDEFLEK.L | S705T710* | HPRD:10416_1 | 0.328193897 | 0.002398843 | 4 | BAZ1B | 10416_3 |
| Bromodomain adjacent to zinc finger domain, 1B | R.SDVQEES*EGSDTDDNKDS*AAFEDNEVQDEFLEK.L | S705S716* | HPRD:10416_1 | 0.328193897 | 0.002398843 | 4 | BAZ1B | 10416_3 |
| BAZ2A | R.AGDPGEMPQS*PTGLGQPK.R | S1397* | HPRD:10417_1 | 0.374052449 | 0.005143065 | 2 | BAZ2A | 10417_1 |
| WW domain-containing transcription regulator protein 1 (Fragment) | R.KGPS*M#LGK.L | S13 | UNI:H7C4Z7 | 0.380989775 | 0.004328685 | 2 | WWTR1 | |
| ARHGEF11 | R.NSVLSDPGLDSPRT*S*PVIMAR.V | T294S295* | HPRD:09302_1 | 0.430525392 | 0.002227269 | 3 | ARHGEF11 | 09302_2 |
| Bromodomain adjacent to zinc finger domain 1A | R.KRQS*PEPSPVTLGR.R | S1531* | HPRD:12034_1 | 0.545102594 | 0.000565312 | 3 | BAZ1A | 12034_3 |
| Bromodomain adjacent to zinc finger domain, 1B | K.SLS*GSPLK.V | S347 | HPRD:10416_1 | 0.588509068 | 0.001764533 | 2 | BAZ1B | 10416_3 |
| Bromodomain adjacent to zinc finger domain, 1B | K.SLSGS*PLK.V | S349 | HPRD:10416_1 | 0.592427318 | 0.002032571 | 2 | BAZ1B | 10416_3 |
| LIM domain containing 2 | R.SKS*FSLR.A | S29 | HPRD:14407_1 | 24.16883591 | 1.33096E-05 | 2 | LIMD2 | 14407_1 |
The human CML CD34+ samples did not disclose changes in YAP or WWTR1 (TAZ) expression after 1 week of IM treatment (GEO/GSE12211) [38], however an increase in WWTR1(TAZ) transcript levels was present in CML CD34+ samples obtained after 4 weeks of IM administration (Fig. 4A) (GEO/GSE33075) [39]. In CML CD34+ cells cultured in the presence of 5 μm of IM in vitro we observed a two-fold increase in transcript levels of both YAP and WWTR1 after 1 week culture in the presence of the drug (Fig. 4B). We interpreted these data as indicating that exposure to IM results in inactivation of Hippo signaling and increased activation of YAP/TAZ.
FIG. 4.

The effect of IM exposure on expression of YAP1 and WWTR1 (TAZ) (A) Affymetrix Human Genome U219 Array data were used to examine YAP1 and WWTR1 (TAZ) transcript expression. CD34+ cells obtained from the bone marrow of CML patients at diagnosis (n = 6, pre-IM) and 1 week after the initiation of therapy (n = 6, post-IM), and at diagnosis (n = 9, pre-IM) and 4 weeks after the initiation of therapy (n = 9, post-IM). The following probe sets were evaluated in GEO GSE12211 and GSE33075 213342_at for YAP1 and 202134_s_at for WWTR (TAZ)1, expression of the probes is presented as interquartile range and median of normalized, log-transformed raw data. P-values are denoted on the graphs. (B) YAP and WWTR1 expression in CML CD34+ cells cultured in the presence of 5 μM IM was determined. CML CD34+ cells were obtained from the bone marrow of three CML patients prior initiation of TKI therapy. Normalized expression relative to ACTB expressed as fold change, is presented. Samples were cultured and run in triplicate. Representative data from three independent analyses are shown **P < 0.01.
Long-term exposure to IM results in increased levels of miR-181a in the plasma of CML patients
We have previously shown that K562 CML cells subjected to prolonged exposure to IM exhibit a decrease in Bcr-Abl1 protein and activity while maintaining high Akt/Erk 1/2 signaling, enhanced proliferation, and an adhesive phenotype ([29] and Supplementary Fig. S2). MicroRNA profiling of these cells indicated significant downregulation of miR-663b and -4726, and upregulation of miR-181a, -182, -183, -438a, -513a, -847–1587, -4537, 4539, and -4690 [29]. We focused our subsequent studies on miR-181a as miR-181 family is the most highly conserved and the most preferentially expressed in the hematopoietic system [22,23]. To explore the association of miR-181a upregulation with long-term exposure to IM, we analyzed a published microarray data set: GEO GSE75392; seven CML patients from STOP-IM group and seven healthy volunteers [30]. We found that miR-181a was significantly upregulated in plasma isolated from CML STOP-IM group patients (Fig. 5). These data, which correspond to our published findings, are consistent with an association of miR-181a overexpression with prolonged exposure to IM. We, therefore, pursued studies to investigate the functional significance of these changes.
FIG. 5.
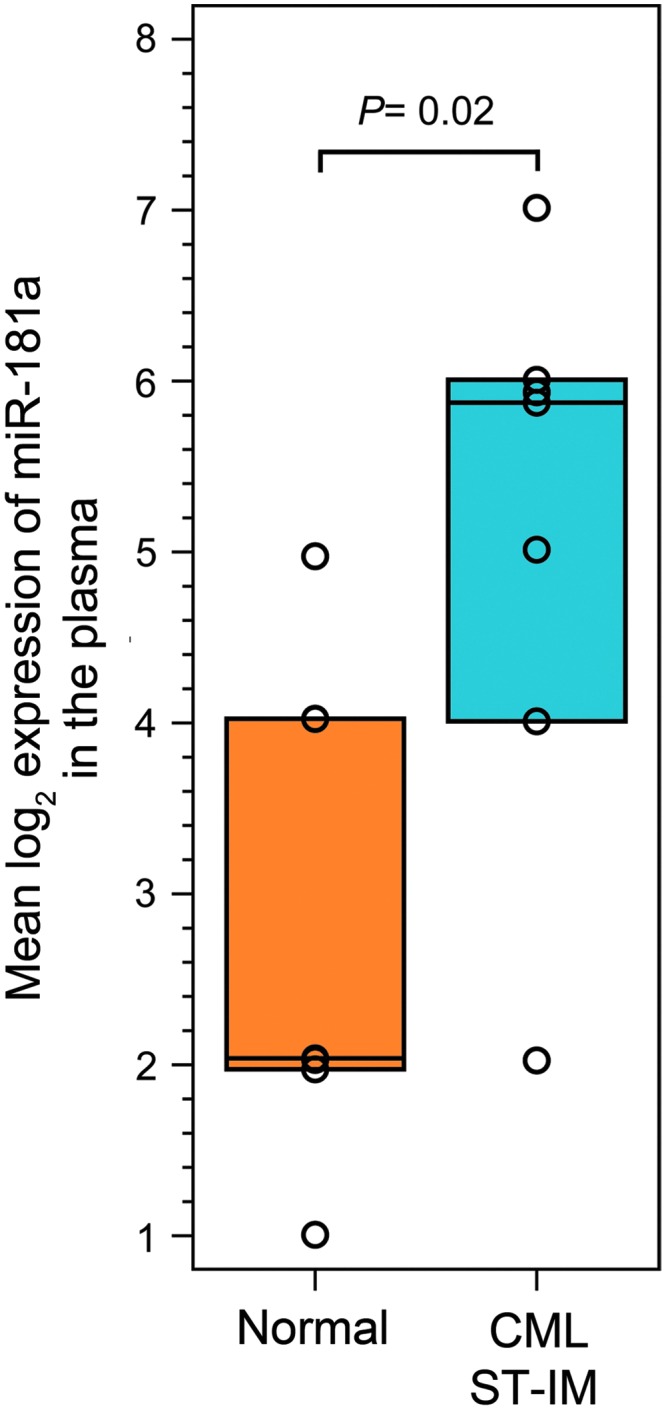
Effect of long-term exposure to IM on levels of miR-181a. Taqman Human MicroRNA A (Card v2.0) Array data were used to examine miR-181a expression in unselected CML patients from the STOP-IM group (n = 7) and 7 healthy volunteers (n = 7). The hsa-miR-181a-4373117 probe set data were extracted from the GEO GSE75392 (platform GPL13987) data set. The geometric mean of normalized, log-transformed raw data is presented. P-value is denoted on the graph.
Inhibition of miR-181a does not significantly affect proliferation or apoptosis
Upregulation of one of the micro RNAs from miR-181a family, miR-181c, was recently shown to inactivate the Hippo pathway, leading to hyperactivation of YAP/TAZ and chemoresistance in pancreatic cancer [47]. Interestingly, upregulation of the stem cell transcription factor SOX2 antagonizes the Hippo pathway leading to exaggerated YAP function [48]. The upregulation of miR-181a and SOX2 transcripts that we observed in K562-STI-R cells, and the positive regulation of YAP in these cells, indicated a mechanistic relationship between these elements. To elucidate that relationship, we designed a miR-181a sponge inhibitor according to the method established by Kluiver et al. [49] (Fig. 6A). Effectiveness of the sponge was evaluated by measuring the intracellular levels of miR-181a transcript by RT-PCR. The miR-181a sponge induced a 50% percent decrease in miR-181a transcript levels (Fig. 6B). As we had already shown that miR-181a targets the 3′ untranslated region of ABI1 (positions 131–137) [29], we quantified ABI1 transcript levels by RT-PCR. In K562-STI-R cells expressing the miR181a sponge, we observed a nearly 35% increase in ABI1 mRNA levels accompanied by an increase in Abi-1 protein levels (Supplementary Fig. S6A, B). To analyze the impact of miR-181a inhibition on cell proliferation and apoptosis, we evaluated the number of cells electroporated with a control sponge or with the miR-181a sponge, and the percentage of phosphatidylserine exposing early apoptotic cells at 24 h after electroporation. We noted no significant effect of miR-181a inhibition on proliferation or apoptosis (Fig. 6C, D). Electroporation efficiency in K562-STI-R cells was 50–55% as evaluated by FACS (Supplementary Fig. S7).
FIG. 6.

The effects of an miR-181a sponge inhibitor on IM-resistant K562-STI-R cells. (A) Schematic representation of the miR-181a sponge and the control sponge. The central mismatch at positions 11–14 (in which the nucleotide at position 14 was deleted) in each miRNA-binding site is shown in blue. The scrambled sequence for the control sponge is shown in red. Overhangs compatible with the restriction endonuclease SanDI are shown in underlined, bold font. (B) RT-PCR analyses of the levels of miR-181a in K562-STI-R cells electroporated with the control vector or that encoding the miR-181a sponge inhibitor. Expression relative to RU19 and RU6B is shown as fold difference. Triplicate analyses representative of data from three independent experiments are shown. ***P < 0.001 (C) FACS analyses of the percentage of apoptotic, Annexin V-FITC-positive K562-STI-R cells electroporated with the control vector or the miR-181a sponge inhibitor. Data represent four independent experiments. (D) K562-STI-R cells electroporated with the control vector or the miR-181a sponge inhibitor were cultured for 24 h. Cell count was determined. Data from three independent experiments are presented.
Inhibition of miR-181a results in decreased expression of stem cell markers SOX2 and SALL4 and negatively regulates YAP
To test the hypothesis that overexpression of miR-181a is associated with increased expression of the SOX2 and SALL4 stem cell markers, we evaluated the effect of miR-181a inhibition on the transcript levels of these genes using RT-PCR. We observed a negative effect of miR-181a inhibition on the expression of SOX2 and SALL4 (Fig. 7A), with transcript levels decreased by 20% and 30%, respectively, in K562-STI-R cells expressing the miR-181a sponge. Inhibition of miR-181a had no effect on the expression of adhesion receptors measured by FACS (Fig. 7B). Examination of the effect of miR-181a inhibition on the Hippo pathway showed positive effect on phosphorylation of YAP on Ser 127 and 397 and a decrease in the levels of YAP protein (Fig. 7C). Evaluation of the effect of miR-181a inhibition on cellular localization of YAP showed its preferential nuclear and perinuclear localization in K562-STI-R cells electroporated with control sponge, and increased cytoplasmic sequestration of YAP in K562-STI-R cells electroporated with miR-181a sponge (Fig. 7D). No actin stress fibers were observed in adherent K562-STIR cells and no significant effect of the miR-181a inhibition on cell shape and spreading was observed (Fig. 7E). Inhibition of miR-181a was associated with downregulation of the YAP target gene CTGF (Fig. 7F). We also evaluated the effect of miR-181a inhibition on responsiveness to IM treatment. K562-STI-R cells that expressed the miR-181a-inhibiting sponge showed modest, but significant increase in their sensitivity to IM treatment (Fig. 7G), indicating that inhibition of miR-181a may at least partially restore sensitivity to IM. In sum, these data suggest that inhibition of miR-181a can contribute to a decrease in the expression of stem cell markers and downregulation of YAP activity, consistent with a regulatory function of miR-181a in Hippo/YAP signaling and, ultimately, the adaptation of CML cells to tonic TKI exposure.
FIG. 7.


Effect of miR-181a on SOX2 and SALL4 expression, YAP activity, and sensitivity to IM. (A) Analysis of expression of POU5F1, SOX2, CITED2, and SALL4 in K562-STI-R cells expressing the control sponge or the miR-181a sponge inhibitor. Normalized expression relative to ACTB, S18, and GAPDH is presented as fold difference. Triplicate analyses representing three independent experiments are shown. *P < 0.05 (B) Analyses of CD45, CD44, CXCR4, VLA-4, CD24, NOTCH1, and CD34 expression by FACS in K562-STI-R expressing the control sponge or the miR-181a sponge inhibitor. Representative data from three independent analyses are shown. (C) Western blot analysis was performed on the whole cell lysates prepared from K562-STI-R cells expressing the control sponge or the miR-181a sponge inhibitor. Levels of Lats1, Mob, and YAP along with phosphorylation of YAP on Ser127 and 397 were evaluated. Data from two independent electroporation reactions using control (ctrl sp: 1, 2) and miR-181a sponge (miR-181a sp: 1, 2) are presented. The results are representative of three independent experiments. Immunofluorescent analysis was performed to examine the cellular distribution of YAP (D) and F-actin (E). K562-STI-R cells were electroporated with GFP-tagged control sponge or miR-181a sponge. Anti-YAP followed by secondary antibody conjugated with AlexaFluor 555 and phalloidin conjugated with AlexaFluor 555 were used to visualize YAP and F-Actin, respectively (red), and DAPI (blue) was used to stain the nucleus. Confocal images were acquired with a Nikon C1si confocal. Z series sections were collected at 0.2 μm with a 60 × PlanApo objective. (F) RT-PCR analysis of AREG, BIRC5, and CTGF expression in K562-STI-R cells expressing the control sponge or the miR-181a sponge inhibitor. Normalized expression relative to ACTB and GAPDH is presented as fold difference. The triplicate analyses are representative of data from three independent experiments. *P < 0.05 (G) FACS analyses of the percentage of apoptotic cells in a population of K562-STI-R cells expressing control or miR-181a sponge inhibitor subjected to the incremental concentrations of IM. Cells 16 h after electroporation with vectors encoding the control sponge or the miR-181a sponge inhibitor were cultured for 24 h in the presence of IM. The percent of Annexin V-FITC-positive cells was determined. Representative data from three independent analyses are shown *P < 0.05.
Discussion
It is estimated that about 40% of patients who remain on IM for more than 5 years will achieve the status of undetectable minimal residual disease (UMRD), as defined by undetectable levels of BCR-ABL1 transcript in peripheral blood, and that 40% of those with UMRD will remain in MR upon TKI discontinuation [7]. However, TKI discontinuation in patients with 2-year UMRD resulted in a 53% rate of relapse within the first 4 months of stopping IM [7]. These and similar studies [6,10,50] demonstrate that TKI therapy is not able to eradicate CML-initiating cells, and that the measurement of BCR-ABL1 transcript levels in peripheral blood may not be the optimal criterion for treatment discontinuation. It is thought that rare LS/PCs that are insensitive to TKI therapy may be the cause of relapse [5,7]. At the same time, accumulating evidence points to a paradoxical involvement of TKI in induction of the stem cell phenotype and promotion of an adhesion-dependent phenotype [11,12,29].
We interpret our data as providing support for the emerging concept of therapy-dependent selection for cells, which, by upregulating certain clusters of markers typically associated with the stem cell phenotype and cell plasticity, are able to survive in the presence of the chemotherapeutic insult and retain disease-initiating capacity. Several reports that suggest chemotherapy-induced transition to a stem cell-like state [12,47,51–54] point to the role of multidrug resistance proteins, Wnt signaling, epithelial–mesenchymal (EMT) transition, and the Hippo/YAP/TAZ pathway in the acquisition of the chemoresistance-induced stem cell phenotype [47,54,55]. In this study we show that even short exposure to IM, both in vivo and in vitro may, within a population of CML CD34+ BM isolated cells, induce expression of genes such as NOTCH1, STAT3, or CD44. Since the population of CD34+ cells contains both CD38− and CD38+ fractions, a shift toward increased levels of stem cell markers may be associated with IM-induced reduction of the CD34+CD38+ population in favor of more primitive, IM-insensitive CD34+CD38− cells. Regardless of whether IM directly induces an increase in stem cell markers or spares the population of CML stem cells, its effect of protecting cells with a stem cell phenotype seems apparent.
In our cellular model of long-term exposure to IM, both genetic and surface markers that are typically associated with the stem cell-like phenotype (SOX2, SALL4, CD44, VLA-4, and CXCR4) are upregulated. With the goal of identifying a molecular mechanism that may be involved in the transition to a stem-like phenotype, we performed unlabeled quantitative phosphoproteomics. This approach identified the Hippo/YAP pathway as a dysregulated pathway induced by long-term IM exposure. Comparing our data to the published Hippo pathway interaction networks determined by affinity purification mass spectrometry [56,57], we were able to confirm dysregulation of 9 out of 52 Hippo core interactors and 146 out of 715 Hippo pathway interactors in K562-STI-R cells (Supplementary Table S2). Having seen high levels of expression of YAP and TAZ, one would expect YAP/TAZ target genes to be significantly upregulated (eg, CTGF, AREG, and BIRC5 [58–60]). The fact that we only observed increased transcript levels of CTGF may be associated with the mechanism by which YAP and TAZ regulate expression. Both translocate to the nucleus and bind to sequence-specific transcription factors TEAD1–TEAD4 and other transcription factors, such as SMAD, RUNX, TP73, TBX5, and PAX, thus enabling the transcription of target genes. That is, YAP/TAZ activity depends on the presence of other transcription factors. These other factors may be absent or inactive in IM-resistant cells.
Although we observed upregulation of NOTCH1 in CML CD34+ cells cultured in the presence of IM in vitro, we did not see the same trends in STAT3 or CD44 transcript levels. These markers are clearly upregulated in CML CD34+ cells isolated from the bone marrow after systemic treatment with the drug. This discrepancy may indicate involvement of the microenvironment or the bone marrow stroma in regulation of the expression of these stem cell markers.
The Hippo pathway was first defined in Drosophila melanogaster. It is crucial for tissue homeostasis, cell proliferation, development, and stem cell renewal [61]. It has recently been recognized to have a role in cancer and chemoresistance [55,62]. Although the core Hippo pathway has been characterized, the regulatory mechanisms that affect this signaling cascade are poorly understood. Recent reports point to a role of miR-181 in the negative regulation of Hippo signaling resulting in hyperactivity of YAP and accrual of chemoresistance [47].
The role of miR-181 in malignant hematopoiesis remains unclear. In acute myeloid leukemia (AML), upregulation of miR-181a was associated with a higher complete remission rate and better survival [63,64]. In other studies, inhibition of miR-181 family members reversed a differentiation block [65]. In chronic lymphocytic leukemia (CLL), a decrease in miR-181a expression is implicated in disease pathogenesis [66]. miR-181a was also shown to be downregulated in del17p-CLL, an ultra high-risk subtype of CLL, and upregulated in a subset of trisomy 12-associated CLL [67]. Conflicting results have been reported regarding the role of miR-181a in CML. Some reports show low levels of miR-181a in CML cells [68,69], whereas other reports show increased levels of miR-181a in CML blast crises [70]. Taken together, these studies support a role for miR-181 in leukemogenesis. Inconsistencies in its reported expression levels may be a result of the genetic and cytogenetic complexity of these malignancies as well as variation in cell sources (peripheral blood vs. bone marrow) and cell types.
Given the significant miR-181a upregulation in plasma of IM-exposed CML patients and in K562-STI-R cells, and the significant increase in levels of YAP, we reasoned that this microRNA may be involved in the regulation of Hippo pathway activity. Indeed, direct and specific inhibition of miR-181a resulted in downregulation of YAP levels along with decreases in SOX2 and SALL4 transcripts. Furthermore, miR-181a inhibition was associated with an increase in responsiveness to IM. However, it should be noted that the observed effect of miR-181a inhibition, while significant, remains relatively modest. This may reflect a high degree of functional redundancy between the members of the family, particularly miR-181a, -c, and -d.
In summary, our studies provide evidence that TKI treatment, while highly efficient in eradicating BCR-ABL1-positive progenitor cells, may be paradoxically involved in protecting a population of rare leukemic stem cells, and promoting or maintaining their stem cell potential. Our findings are consistent with a mechanism that involves downregulation of Hippo signaling and increased activity of the transcriptional coeffector YAP. Our observations indicate a role for miR-181a, and identify miR-181a and components of the Hippo/YAP pathway as potential targets to enhance the therapeutic effectiveness of TKIs in leukemia.
Supplementary Material
Acknowledgments
The authors thank Mandy Pereira, Mark Dooner, Elaine Pappa, Michael DelTatto, Joan Boylan, Aleksander F. Sikorski, Loren Fast, and Peter Quesenberry for helpful suggestions and technical assistance. This work was supported by a Brown Biomed Division DEANS Award grant (PMD), University of Medicine Foundation Integration Funding (PMD), Rhode Island Foundation grant (PMD), a pilot grant from the Lifespan COBRE Center for Cancer Research Development (1P30GM110759-01A) (PMD), Savit Foundation (PMD), and R01HL121796 (DT). AJO is supported by the American Society of Hematology Research Scholar award.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Bower H, Bjorkholm M, Dickman PW, Hoglund M, Lambert PC. and Andersson TM. (2016). Life Expectancy of Patients With Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J Clin Oncol 34:2851–2857 [DOI] [PubMed] [Google Scholar]
- 2.Rousselot P, Charbonnier A, Cony-Makhoul P, Agape P, Nicolini FE, Varet B, Gardembas M, Etienne G, Rea D, et al. (2014). Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J Clin Oncol 32:424–430 [DOI] [PubMed] [Google Scholar]
- 3.Chomel JC, Bonnet ML, Sorel N, Bertrand A, Meunier MC, Fichelson S, Melkus M, Bennaceur-Griscelli A, Guilhot F. and Turhan AG. (2011). Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 118:3657–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomel JC, Bonnet ML, Sorel N, Sloma I, Bennaceur-Griscelli A, Rea D, Legros L, Marfaing-Koka A, Bourhis JH, et al. (2016). Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 7:35293–35301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW. and Druker BJ. (2011). Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest 121:396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, et al. (2010). Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 11:1029–1035 [DOI] [PubMed] [Google Scholar]
- 7.Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, Dang P, Goyne JM, Slader C, et al. (2013). Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood 122:515–522 [DOI] [PubMed] [Google Scholar]
- 8.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L. and Holyoake TL. (2002). Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 99:319–325 [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Chen Y, Douglas L. and Li S. (2009). beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 23:109–116 [DOI] [PubMed] [Google Scholar]
- 10.Saußele S, Richter J, Hochhaus A, Mahon FX. (2016). The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 30:1638–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charaf L, Mahon FX, Lamrissi-Garcia I, Moranvillier I, Beliveau F, Cardinaud B, Dabernat S, de Verneuil H, Moreau-Gaudry F. and Bedel A. (2017). Effect of tyrosine kinase inhibitors on stemness in normal and chronic myeloid leukemia cells. Leukemia 31:65–74 [DOI] [PubMed] [Google Scholar]
- 12.Puissant A, Dufies M, Fenouille N, Ben Sahra I, Jacquel A, Robert G, Cluzeau T, Deckert M, Tichet M, et al. (2012). Imatinib triggers mesenchymal-like conversion of CML cells associated with increased aggressiveness. J Mol Cell Biol 4:207–220 [DOI] [PubMed] [Google Scholar]
- 13.Beck B. and Blanpain C. (2013). Unravelling cancer stem cell potential. Nat Rev Cancer 13:727–738 [DOI] [PubMed] [Google Scholar]
- 14.Greaves M. and Maley CC. (2012). Clonal evolution in cancer. Nature 481:306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX. and Ivy SP. (2015). Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 12:445–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H, Lee H, Herrmann A, Buettner R. and Jove R. (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14:736–746 [DOI] [PubMed] [Google Scholar]
- 17.Blank U. and Karlsson S. (2015). TGF-beta signaling in the control of hematopoietic stem cells. Blood 125:3542–3550 [DOI] [PubMed] [Google Scholar]
- 18.Varelas X. and Wrana JL. (2012). Coordinating developmental signaling: novel roles for the Hippo pathway. Trends Cell Biol 22:88–96 [DOI] [PubMed] [Google Scholar]
- 19.Harvey KF, Zhang X. and Thomas DM. (2013). The Hippo pathway and human cancer. Nat Rev Cancer 13:246–257 [DOI] [PubMed] [Google Scholar]
- 20.Piccolo S, Dupont S. and Cordenonsi M. (2014). The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94:1287–1312 [DOI] [PubMed] [Google Scholar]
- 21.Ha M. and Kim VN. (2014). Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524 [DOI] [PubMed] [Google Scholar]
- 22.Chen CZ, Li L, Lodish HF. and Bartel DP. (2004). MicroRNAs modulate hematopoietic lineage differentiation. Science 303:83–86 [DOI] [PubMed] [Google Scholar]
- 23.O'Connell RM. and Baltimore D. (2012). MicroRNAs and hematopoietic cell development. Curr Top Dev Biol 99:145–174 [DOI] [PubMed] [Google Scholar]
- 24.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, et al. (2007). miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 129:147–161 [DOI] [PubMed] [Google Scholar]
- 25.Fu G, Rybakin V, Brzostek J, Paster W, Acuto O. and Gascoigne NR. (2014). Fine-tuning T cell receptor signaling to control T cell development. Trends Immunol 35:311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams A, Henao-Mejia J, Harman CC. and Flavell RA. (2013). miR-181 and metabolic regulation in the immune system. Cold Spring Harb Symp Quant Biol 78:223–230 [DOI] [PubMed] [Google Scholar]
- 27.Georgantas RW, 3rd, Hildreth R, Morisot S, Alder J, Liu CG, Heimfeld S, Calin GA, Croce CM. and Civin CI. (2007). CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A 104:2750–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Judson RL, Greve TS, Parchem RJ. and Blelloch R. (2013). MicroRNA-based discovery of barriers to dedifferentiation of fibroblasts to pluripotent stem cells. Nat Struct Mol Biol 20:1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chorzalska A, Salloum I, Shafqat H, Khan S, Marjon P, Treaba D, Schorl C, Morgan J, Bryke CR, et al. (2014). Low expression of Abelson interactor-1 is linked to acquired drug resistance in Bcr-Abl-induced leukemia. Leukemia 28:2165–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohyashiki K, Umezu T, Katagiri S, Kobayashi C, Azuma K, Tauchi T, Okabe S, Fukuoka Y. and Ohyashiki JH. (2016). Downregulation of Plasma miR-215 in Chronic Myeloid Leukemia Patients with Successful Discontinuation of Imatinib. Int J Mol Sci 17:570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu K. and Salomon AR. (2010). HTAPP: high-throughput autonomous proteomic pipeline. Proteomics 10:2113–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elias JE. and Gygi SP. (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4:207–214 [DOI] [PubMed] [Google Scholar]
- 33.Yu K, Sabelli A, DeKeukelaere L, Park R, Sindi S, Gatsonis CA. and Salomon A. (2009). Integrated platform for manual and high-throughput statistical validation of tandem mass spectra. Proteomics 9:3115–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beausoleil SA, Villen J, Gerber SA, Rush J. and Gygi SP. (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24:1285–1292 [DOI] [PubMed] [Google Scholar]
- 35.Demirkan G, Yu K, Boylan JM, Salomon AR. and Gruppuso PA. (2011). Phosphoproteomic profiling of in vivo signaling in liver by the mammalian target of rapamycin complex 1 (mTORC1). PLoS One 6:e21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dubielecka PM, Ladwein KI, Xiong X, Migeotte I, Chorzalska A, Anderson KV, Sawicki JA, Rottner K, Stradal TE. and Kotula L. (2011). Essential role for Abi1 in embryonic survival and WAVE2 complex integrity. Proc Natl Acad Sci U S A 108:7022–7027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Storey JD. and Tibshirani R. (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruennert D, Czibere A, Bruns I, Kronenwett R, Gattermann N, Haas R. and Neumann F. (2009). Early in vivo changes of the transcriptome in Philadelphia chromosome-positive CD34+ cells from patients with chronic myelogenous leukaemia following imatinib therapy. Leukemia 23:983–985 [DOI] [PubMed] [Google Scholar]
- 39.Benito R, Lumbreras E, Abaigar M, Gutierrez NC, Delgado M, Robledo C, Garcia JL, Rodriguez-Vicente AE, Canizo MC. and Rivas JM. (2012). Imatinib therapy of chronic myeloid leukemia restores the expression levels of key genes for DNA damage and cell-cycle progression. Pharmacogenet Genomics 22:381–388 [DOI] [PubMed] [Google Scholar]
- 40.Jiang X, Forrest D, Nicolini F, Turhan A, Guilhot J, Yip C, Holyoake T, Jorgensen H, Lambie K, et al. (2010). Properties of CD34+ CML stem/progenitor cells that correlate with different clinical responses to imatinib mesylate. Blood 116:2112–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holtz MS, Forman SJ. and Bhatia R. (2005). Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia 19:1034–1041 [DOI] [PubMed] [Google Scholar]
- 42.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Tam WL, Tong GQ, Wu Q, Chan HY, Soh BS, Lou Y, Yang J, Ma Y, et al. (2006). Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol 8:1114–1123 [DOI] [PubMed] [Google Scholar]
- 44.Li L. and Bhatia R. (2011). Stem cell quiescence. Clin Cancer Res 17:4936–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ. and Clarke MF. (2003). Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100:3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS, Allan AL. and Chambers AF. (2011). Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res 9:834–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen M, Wang M, Xu S, Guo X. and Jiang J. (2015). Upregulation of miR-181c contributes to chemoresistance in pancreatic cancer by inactivating the Hippo signaling pathway. Oncotarget 6:44466–44479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu-Roy U, Bayin NS, Rattanakorn K, Han E, Placantonakis DG, Mansukhani A. and Basilico C. (2015). Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat Commun 6:6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kluiver J, Slezak-Prochazka I, Smigielska-Czepiel K, Halsema N, Kroesen BJ. and van den Berg A. (2012). Generation of miRNA sponge constructs. Methods 58:113–117 [DOI] [PubMed] [Google Scholar]
- 50.O'Hare T, Zabriskie MS, Eiring AM. and Deininger MW. (2012). Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer 12:513–526 [DOI] [PubMed] [Google Scholar]
- 51.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, 2nd, Samuel S, Kim MP, Lim SJ. and Ellis LM. (2009). Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res 69:1951–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polyak K. and Weinberg RA. (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265–273 [DOI] [PubMed] [Google Scholar]
- 53.Singh A. and Settleman J. (2010). EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29:4741–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Touil Y, Igoudjil W, Corvaisier M, Dessein AF, Vandomme J, Monte D, Stechly L, Skrypek N, Langlois C, et al. (2014). Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res 20:837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zanconato F, Cordenonsi M. and Piccolo S. (2016). YAP/TAZ at the Roots of Cancer. Cancer Cell 29:783–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, et al. (2013). Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal 6:rs15. [DOI] [PubMed] [Google Scholar]
- 57.Wang W, Li X, Huang J, Feng L, Dolinta KG. and Chen J. (2014). Defining the protein-protein interaction network of the human hippo pathway. Mol Cell Proteomics 13:119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moroishi T, Hansen CG. and Guan KL. (2015). The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, Brugge JS, Dyson NJ. and Haber DA. (2009). YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol 11:1444–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu H, Jiang D, Chi F. and Zhao B. (2012). The Hippo pathway regulates stem cell proliferation, self-renewal, and differentiation. Protein Cell 3:291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu FX. and Guan KL. (2013). The Hippo pathway: regulators and regulations. Genes Dev 27:355–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Y. and Yang X. (2015). The Hippo pathway in chemotherapeutic drug resistance. Int J Cancer 137:2767–2773 [DOI] [PubMed] [Google Scholar]
- 63.Schwind S, Maharry K, Radmacher MD, Mrozek K, Holland KB, Margeson D, Whitman SP, Hickey C, Becker H, et al. (2010). Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28:5257–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hickey CJ, Schwind S, Radomska HS, Dorrance AM, Santhanam R, Mishra A, Wu YZ, Alachkar H, Maharry K, et al. (2013). Lenalidomide-mediated enhanced translation of C/EBPalpha-p30 protein up-regulates expression of the antileukemic microRNA-181a in acute myeloid leukemia. Blood 121:159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su R, Lin HS, Zhang XH, Yin XL, Ning HM, Liu B, Zhai PF, Gong JN, Shen C, et al. (2015). MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene 34:3226–3239 [DOI] [PubMed] [Google Scholar]
- 66.Marton S, Garcia MR, Robello C, Persson H, Trajtenberg F, Pritsch O, Rovira C, Naya H, Dighiero G. and Cayota A. (2008). Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 22:330–338 [DOI] [PubMed] [Google Scholar]
- 67.Visone R, Rassenti LZ, Veronese A, Taccioli C, Costinean S, Aguda BD, Volinia S, Ferracin M, Palatini J, et al. (2009). Karyotype-specific microRNA signature in chronic lymphocytic leukemia. Blood 114:3872–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, et al. (2005). MicroRNA expression profiles classify human cancers. Nature 435:834–838 [DOI] [PubMed] [Google Scholar]
- 69.Wang G, Zhao R, Zhao X, Chen XI, Wang D, Jin Y, Liu XI, Zhao CI, Zhu Y, et al. (2015). MicroRNA-181a enhances the chemotherapeutic sensitivity of chronic myeloid leukemia to imatinib. Oncol Lett 10:2835–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Machova Polakova K, Lopotova T, Klamova H, Burda P, Trneny M, Stopka T. and Moravcova J. (2011). Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer 10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.