

Abstract
In colorectal cancer (CRC), KRAS mutations are a strong negative predictor for treatment with the EGFR‐targeted antibodies cetuximab and panitumumab. Since it can be difficult to obtain appropriate tumor tissues for KRAS genotyping, alternative methods are required. Circulating tumor cells (CTCs) are believed to be representative of the tumor in real time. In this study we explored the capacity of a size‐based device for capturing CTCs coupled with a multiplex KRAS screening assay using droplet digital PCR (ddPCR). We showed that it is possible to detect a mutant ratio of 0.05% and less than one KRAS mutant cell per mL total blood with ddPCR compared to about 0.5% and 50–75 cells for TaqMeltPCR and HRM. Next, CTCs were isolated from the blood of 35 patients with CRC at various stage of the disease. KRAS genotyping was successful for 86% (30/35) of samples with a KRAS codon 12/13 mutant ratio of 57% (17/30). In contrast, only one patient was identified as KRAS mutant when size‐based isolation was combined with HRM or TaqMeltPCR. KRAS status was then determined for the 26 available formalin‐fixed paraffin‐embedded tumors using standard procedures. The concordance between the CTCs and the corresponding tumor tissues was 77% with a sensitivity of 83%. Taken together, the data presented here suggest that is feasible to detect KRAS mutations in CTCs from blood samples of CRC patients which are predictive for those found in the tumor. The minimal invasive nature of this procedure in combination with the high sensitivity of ddPCR might provide in the future an opportunity to monitor patients throughout the course of disease on multiple levels including early detection, prognosis, treatment and relapse as well as to obtain mechanistic insight with respect to tumor invasion and metastasis.
Keywords: Digital PCR, KRAS, Circulating tumor cells, Colorectal cancer, Liquid biopsy
Highlights
Combination of a size‐based CTC enrichment method and droplet digital PCR allows detection of less than one cells per mL blood.
This procedure is at least 10 fold more sensitive than TaqMelt PCR or High resolution melting methods.
The procedure provided high concordance for the KRAS status between circulating tumor cells (CTCs) and matched tumor tissues.
This assay allows prediction of KRAS tumor status based on CTCs.
1. Introduction
The recent development of next‐generation sequencing has allowed the identification of key genetic and epigenetic changes in human tumors. This has facilitated the development of a novel group of anticancer agents targeting oncogenic signaling pathways associated with specific mutations (Shen et al., 2015). In colorectal cancer (CRC), genetic alterations are frequent in genes encoding downstream effectors of the epidermal growth factor receptor (EGFR) pathway including KRAS, NRAS and BRAF as well as genes involved in pathway crosstalk like PTEN or PIK3CA (Therkildsen et al., 2014; Muzny DM et al., 2012). KRAS mutations are observed for 40–45% of CRC patients with the 7 most frequent mutations occurring in codons 12–13. These mutations have been identified as a strong negative predictive factor for the response to EGFR‐targeted antibodies including cetuximab and panitumumab. Therefore, the detection of KRAS status, and more recently, NRAS status has become mandatory before starting the treatment of metastatic CRC patients with EGFR‐directed antibodies. Previous clinical trials have demonstrated that patients with KRAS mutant tumors have marginal or no response to EGFR‐directed antibodies whereas up to 40% of patients with KRAS wild‐type (wt) tumors respond to the treatment (Amado et al., 2008; Lievre et al., 2006). However, the necessity to do KRAS genotyping of tumor tissues may be problematic in many clinical situations. For instance, for non‐metastatic CRC, most patients are rapidly operated after diagnosis. 25–30% of these patients will develop liver or lung metastasis within 5 years due to the growth of occult micrometastasis. In this case, the KRAS status in the primary tumor may no longer be available and serial biopsies of the metastasis is generally not feasible. Furthermore, the KRAS status of the primary tumor may not necessarily reflect the current KRAS status of the metastasis since tumors evolve during disease progression by both Darwinian selection and therapeutic pressure (Chisholm et al., 2015). The same argument holds for the 25% of patients harboring metastasis at diagnosis. Finally, following tumor removal it is advisable to determine the presence of residual disease and to monitor the efficiency or resistance to therapies (Misale et al., 2014; Bird et al., 2006). For all these reasons, alternative assays to KRAS testing of the primary tumor are warranted.
Liquid biopsies including circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) has become an exciting tool in oncology to investigate the dynamics of tumor development over time (Heitzer et al., 2015; Lianidou et al., 2015; Gingras et al., 2015; Ma et al., 2015). Besides ctDNA, one of the most promising applications would be molecular characterization of CTCs. CTCs are rare cells present in the blood and lymphatic vessels which have been released from the primary tumor and/or metastasis and which may have the potential to induce relapse or metastasis (Hardingham et al., 2015; Pantel and Speicher, 2015; Joosse et al., 2014). It is widely accepted that the number of CTCs is inversely correlated with the prognosis of the patients for multiple solid tumors including both metastatic and non‐metastatic CRC (Lim et al., 2014; Cohen et al., 2009; Cohen et al., 2008) suggesting that these cells are potentially harmful. At present, relatively little is known about their mutational status because molecular characterization of CTCs requires both a specific method to isolate the CTCs from the hematogenous cells (Alix‐Panabières and Pantel, 2014; Pantel and Alix‐Panabieres, 2013; Torino et al., 2013; Lianidou, 2014) as well as a molecular detection method with high sensitivity in order to be able to identify a few mutant copies against a strong background of wt DNA (Cai et al., 2015).
First‐generation Sanger sequencing is currently the gold standard for detection of mutant DNA, However, it is only reliable for detection of 10–20% mutant copies at the allelic level (Tsiatis et al., 2010). Since lower levels of mutant copies would be masked by a high background of wt DNA from contaminant leukocytes, Sanger sequencing would not be appropriate for molecular characterization of CTCs. Other PCR‐based approaches for detecting rare mutant copies have been developed, all with advantages and disadvantages in terms of cost, sensitivity and specificity. This includes pyrosequencing (Tsiatis et al., 2010), co‐amplification at lower denaturation temperature‐PCR (cold‐PCR) (Zuo et al., 2009), amplification refractory mutation system using a bifunctional self‐probing primer (Franklin et al., 2010), massively‐parallel sequencing (Peeters et al., 2013), high‐resolution melting (HRM) analysis (Simi et al., 2008; Mohamed Suhaimi et al., 2015) or allele specific blocker PCR (ASB‐PCR) (Mohamed Suhaimi et al., 2015). Recently, droplet digital PCR (ddPCR) (Hindson et al, 2011, 2013) has been developed to detect very low number of mutant copies. This system partitions the PCR products into approximately 20,000 droplets, each of which undergoes an individual reaction. Each individual droplet is defined on the basis of fluorescent amplitude as being either positive or negative. This technique has been successful for characterization of hotspot mutations in ctDNA (Hudecova, 2015; Bettegowda, 2014) including the detection of KRAS mutations (Thierry et al., 2014). Recent studies have explored the feasibility to detect KRAS mutant cells by ddPCR for the diagnosis of pancreatic cancer (Earl et al., 2014) or other type of mutations such as Estrogen‐receptor‐1 in metastatic breast cancer (Guttery, 2015) thereby opening new avenues for molecular characterization of CTCs.
To our knowledge the present study is the first to explore the potential of ddPCR to detect and characterize KRAS status in the CTCs of patients with colorectal cancer after isolation by size‐based technology. Our findings show that molecular characterization of CRCs using ddPCR is feasible with a good sensitivity and concordance between the KRAS phenotype in the CTCs and the corresponding tumor tissues.
2. Material and methods
2.1. Samples from colorectal cancer patients and healthy donors
A total of 35 patients with colorectal cancer (CRC) were included in this study. Patients enrolled in this study have been included in larger clinical studies (NCT01675999 and NCT 00958737) approved by the local ethical committee. All participants were informed by the surgeons (MK, GM and AP) and signed a specific consent form for this study. Ethylenediaminetetraacetic acid (EDTA)‐anticoagulated peripheral blood (6 mL) was obtained from all patients just before colorectal tumor resection. DNA was extracted from the enriched fraction of CTCs (see below) as well as from tumor tissues obtained after surgery. All samples from healthy donors (HD) were obtained anonymously from ten healthy volunteers (6 men and 4 women; mean age 42 ± 15 years) drawn at french national transfusion agency (Bichat‐Claude Bernard). According to the French law related to research of donated blood, general indications includes age <70 years without known chronic diseases, cancers, hematological disorders or viral infections and no transfusion or surgery during the last 4 months.
2.2. Determination of serum tumor markers
CEA (Carcinoma Embryonic Antigen) and CA19.9 were assayed in the serum of patients collected in an additional tube at the same time that the CTC isolation and cytological analysis. The immunoanalysis assay was performed on a KRYPTOR analyzer (B.R.A.H.M.S, Hennigsdorf, Germany) according to standard procedures.
2.3. Cell culture and spiking experiments
The human pancreatic adenocarcinoma PANC1 cell line and the human breast cancer cell line MCF‐7 were obtained from ATCC (ATCC, Manassas, USA). PANC‐1 were cultured with DMEM containing 10% foetal bovine serum and antibiotics while MCF7 were cultured with MEM and addition of 0.01 mg/mL human recombinant insulin, 10% foetal bovine serum and antibiotics. PANC1 is heterozygous for KRAS c.35G > A (G12D) and MCF7 harbor no mutations in KRAS gene. After trypsinization, PANC1 cells were picked one by one under an inverted microscope and transferred into 100 μL of nuclease‐free PBS1X before to be added into 3 or 6 mL of whole blood of HD. Then, blood was filtered for CTCs counting or KRAS status determination.
2.4. Cytological evaluation of CTCs
For cytological detection of CTCs, 3 mL blood was filtered according to the manufacturer's instructions using the ScreenCell Cyto kit (Screencell, Sarcelles, France) (Desitter et al., 2011). After completing the filtration, the filter was rinsed with 500 μL of phosphate‐buffered saline, dried on absorbent tissue, and stained with either Hematoxylin/eosin (H/E), Giemsa (G) or May‐Grunewald (MGG). The stained filters were analyzed by an experienced cytopathologist (JW) blinded to the histological diagnosis. Cells were categorized as “negative”, “suspicious”, or “malignant” based on cytomorphological features. Cells considered as “malignant” displayed an epithelial phenotype with enlarged nuclei, coarse chromatin, and irregular nuclear membranes. Cells classified as “suspicious”, also demonstrated an epithelial phenotype, but displayed bland nuclear features with round smooth nuclei and even chromatin.
2.5. Preparation of CTCs samples for detection of KRAS status
For molecular characterization of the CTCs, 6 mL blood was filtered using the ScreenCell MB™ (ScreenCell, Sarcelles, France) device according to the manufacturer's instructions (Desitter et al., 2011). DNA from cells captured on the filter was isolated using QIAampDNA Micro Kit (Qiagen®, Courtaboeuf, France). Next, Whole Genome Amplification (WGA) was performed using GenomePlex® Single Cell Whole Genome Amplification Kit (Sigma–Aldrich, Saint‐Quentin Fallavier, France). For each WGA reactions, DNA libraries were separated on a 1.5% agarose gel for visual confirmation of product amplification and quality. Then, they were diluted 1/50 before determination of KRAS status.
2.6. Detection of KRAS status in CRCs by droplet digital PCR (ddPCR), TaqMelt PCR, High Resolution Melting (HRM) or Sanger sequencing
For HRM, we used a home‐made assay performed in routine for clinical samples with LightCycler 480 (Roche Diagnostics, Meylan, France) using the following primers: for codon 12, F: GGCCTGCTGAAAATGACTGAA, R: GGTCCTGCACCAGTAATATGC and codon 13 F: CAGACTGTGTGTTTCTCCCTTCTCAGG, R: AGAAAGCCCTCCCCAGTCCTCA. For TaqMelt PCR, we used the Cobas® KRAS mutation kit with the COBAS z480 system (Roche Diagnostics, Meylan, France). At last, for ddPCR, we used the KRAS Screening Multiplex Kit able to detect the 7 most common mutations in KRAS genes with QX200 droplet digital PCR System (Biorad®, Marne‐la‐Coquette, France).
2.7. Detection of KRAS status in tumor tissues
DNA was extracted from several 10 μm sections of paraffin‐embedded tissues using Qiasymphony and QiampDNA minikit (Qiagen, Courtaboeuf, France) according to the manufacturer's instructions. The KRAS status was determined using the Trusight Tumor Panel on MiSeq System (Illumina Inc., San Diego, USA) with a detection limit of about 5%. DNA of lower quality was tested by HRM and Sanger sequencing.
2.8. Statistical analysis
Statistical evaluation was performed with Graph pad prism for categorical variables. Fisher exact test was performed in the instance of 2 and 3 variables.
3. Results
3.1. ddPCR allows detection of KRAS mutant cells from whole blood after size‐based enrichment with a higher sensitivity than Sanger sequencing, TaqMelt PCR and HRM methods
We first determined the capacity of droplet digital PCR to detect rare copies of mutant KRAS alleles in a KRAS wild‐type (wt) background with a multiplex KRAS assay able to detect 7 of the most frequent mutations (Figure 1A and B). For this purpose, we did whole genome amplification of DNA from PANC1 cells, bearing a heterozygous KRAS mutation (G12D) and of DNA from MCF‐7 cells, that have wt KRAS, and then spiked the DNA from the PANC1 cells into the DNA from the MCF‐7 cells. The results show that this assay is able to discriminate at least 5 copies of mutant allele for 100,000 wt copies (i.e. an abundance ratio of 0.05%) (Figure 1C and D). Additionally, we found that the performance of ddPCR is at least 10‐fold more sensitive than TaqMelt (using Cobas z480) and HRM (using LC480 analyzer) with a sensitivity limit for both techniques close to 0.5–1% (Supp. Data 1). Next, we compared the capacity of ddPCR, Sanger sequencing, TaqMelt and HRM to detect KRAS mutant cells in whole blood (Figure 2). For this purpose, we spiked 100, 75, 50, 10 or 5 cells into healthy donor blood and then used the size‐based ScreenCell technology to enrich the fraction of mutant cells from the leucocyte background. Next, DNA was extracted and amplified from the enriched cell fraction and then processed by the different molecular detection methods. For ddPCR, we defined a threshold after processing of three independent samples from healthy blood donor as “mean of the abundance ratio + 3 SD” thereby allowing us to fix the detection limit to 0.2% (Figure 3). The results showed that we were able to detect a true signal for blood samples spiked with 5 PANC1 cells (ie less than 1 KRAS mutant copy per mL blood). Moreover, we obtained a linear abundance ratio with samples spiked with a higher number of PANC1 cells. In contrast, Sanger sequencing showed a sensitivity close to 100 cells per 6 mL (ie about 16 copies per mL blood) (Figure 4A) while TaqMelt and HRM had a sensitivity close to 75 and 50 cells per 6 mL, respectively (ie about 12 copies or 8 copies per mL blood) (Figure 4B and C). A summary of these findings are presented in Table 1.
Figure 1.
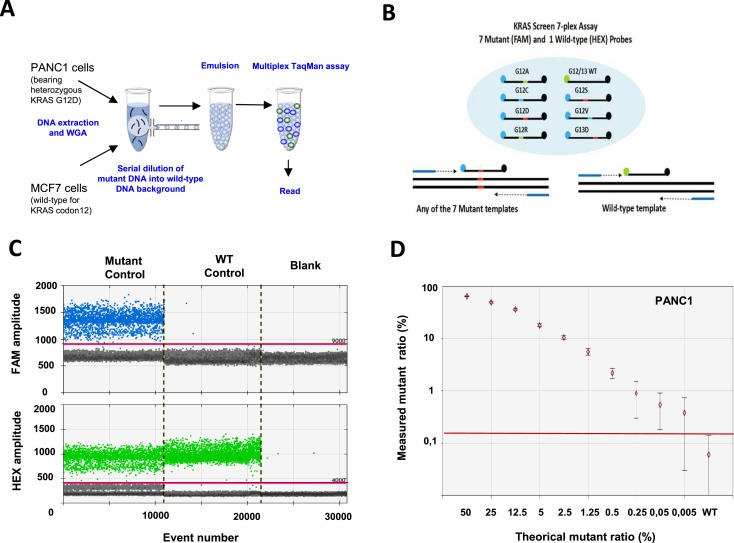
Sensitivity of the KRAS multiplex assay by droplet digital PCR. A. Protocol. DNA was extracted from 100 PANC1 cells bearing a heterozygous mutation c.35G > A in KRAS codon 12 (Gly12Asp) and 100 MCF7 cell lines (wild‐type for KRAS codon 12–13). After whole genome amplification, DNA from the two cell lines was mixed and droplet digital PCR was performed using a multiplex KRAS assay. B. KRAS screening assay. This TaqMan assay is designed to detect the seven most frequent mutations in codon 12 and codon 13 of KRAS. C. Experimental validation. 1D‐Dot plot. The blue histogram indicates the number of droplets considered as positive for mutant KRAS according to the fluorescence threshold. The green histogram corresponds to the number of wild‐type droplets. Mutant control, DNA containing a 1:1 mix of mutant and wildtype KRAS. WT control, DNA containing only KRAS wild‐type. Blank, water. D. Mutant ratio threshold determination (mutant copies/total copies in %) according to theoretical mutant ratio obtained by serial dilution. The red line indicates the detection threshold.
Figure 2.
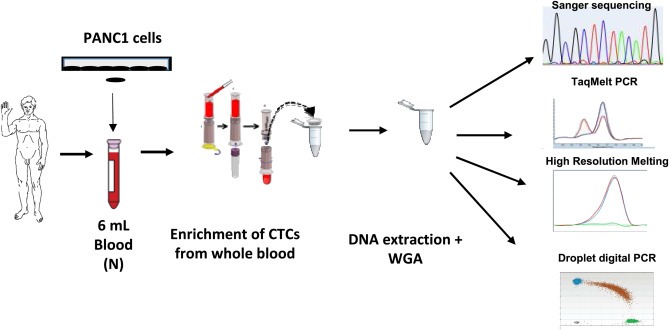
Validation of the protocol for KRAS testing. Whole blood was spiked with PANC1 cells harboring a KRAS mutation in codon 12–13 followed by size‐based enrichment and multiplex KRAS analysis using droplet digital PCR. N, “Normal” blood from healthy donors. WGA, Whole Genome Amplification.
Figure 3.
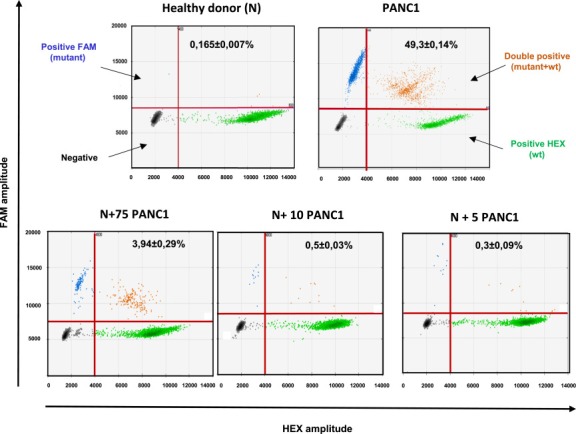
Detection threshold of KRAS from whole blood using droplet digital PCR. 2D‐Dot plot. Fluorescence results are plotted as two‐dimensional dot plots (similar to the depiction of flow cytometry data). The region of these plots can be sequentially separated based on the fluorescence intensity of each droplet. Grey dots correspond to empty droplets. Green dots correspond to droplets containing wild‐type copies of KRAS on codon 12–13. Blue dots correspond to droplets containing at least one codon 12–13 KRAS mutation. Orange dots correspond to droplets containing at least one KRAS wild type copy and one mutant copy. These droplets are considered for the analysis. Abundance ratio (KRAS mutant copies/total copies) is indicated for each dot plots. Each analysis was performed at least in triplicate. N, blood from “normal” healthy donors with no spiking of mutant KRAS cells. PANC1, blood from “normal” healthy donors spiked with PANC1 cells bearing the heterozygous c.35G > A mutation in KRAS codon 12 (Gly12Asp).
Figure 4.
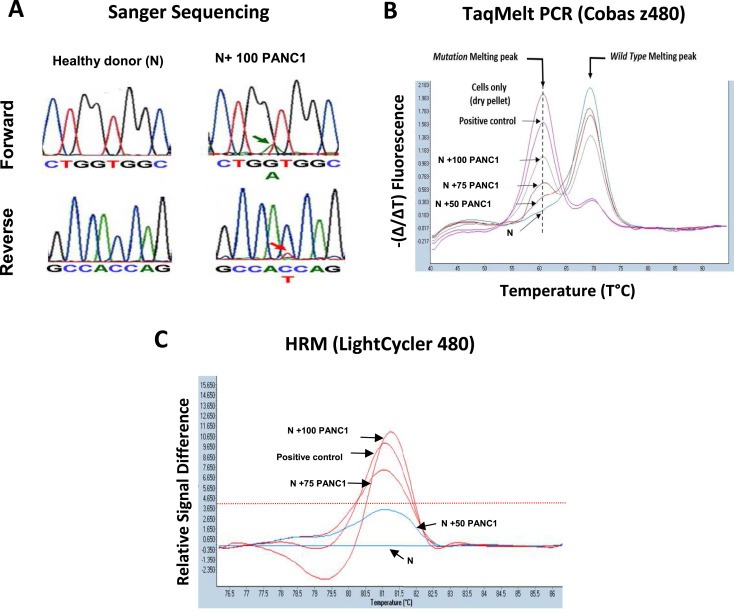
Comparison of the sensitivity for detection of KRAS mutant cells in whole blood. A. Sanger sequencing. Considering the weak signal, the detection limit is close to 100 cells. As control, no signal was detected in blood from healthy donors or in blood spiked with 50 PANC1 cells. B. TaqMeltPCR: The presence of a mutation melting peak at the correct annealing temperature indicate the presence of KRAS mutations. The sample of healthy donor blood spiked with 50 PANC1 cells is considered at the detection limit when applying validation guidelines. When the assay is performed in “CIVD mode” recommended for clinical applications, the detection limit is 100 cells. C, High Melting Resolution (HRM). The detection limit (red line) is between 75 and 50 cells.
Table 1.
Comparison of sensitivity of different methods to detect KRAS mutant cells and copy number in cells isolated from whole blood.
| Methods | Sensitivity | Sensitivity |
|---|---|---|
| Mutant cells/mL of blood | Mutant copies/mL of blood | |
| Sanger sequencing | 16 | 8 |
| HRM | 12 | 6 |
| Cobas KRAS kit (TaqMelt PCR) | 8 | 4 |
| ddPCR | <1 | <0.5 |
3.2. ddPCR can detect KRAS mutations in CTCs from CRC patients
Next, we wished to evaluate the detection of mutant KRAS in CTCs obtained from CRC patients. For this, we enrolled 35 CRC patients at any stage of the disease who were admitted to the digestive surgery department of the Pitié‐Salpêtrière University Hospital. The characteristics of this cohort are indicated in Table 2. First, we performed CTC analysis of blood samples obtained just before curative surgery. This analysis showed that 90% (26/29) of patients harbored at least one CTC per 3 mL of total blood and among then 7% (2/29) of patients had also cell clusters named circulating tumor microemboli or multicellular CTC clusters (Table 3 and Supp. Data 2). In contrast, the analysis of blood from ten healthy donors revealed no CTCs. We observed a trend of increasing numbers of CTCs according to disease stage ranging from in situ to metastatic disease. In contrast, no correlation was observed between the number of CTCs and serum concentrations of the tumor markers CEA (carcinoma embryonic antigen) and CA19.9 (Supp. data 2). Next, we performed the multiplex KRAS genotype assay by ddPCR. 86% (30/35) was performed successfully, since 5 samples could not be amplified by whole genome amplification. We found 57% of samples (17/30) positive for the presence of KRAS mutations on codons 12–13 (Table 3). Processing of the same samples by TaqMeltPCR revealed only one positive sample (patient #24) thereby demonstrating the clear superiority of ddPCR to detect very low amounts of mutant copies (Supp. data 3).
Table 2.
Patient characteristics. The stage of the disease was determined according to the classification of UICC (International Cancer Union).
| Sex | ||
| Male | 15 | 42.8% |
| Female | 20 | 57.1% |
| Total | 35 | |
| Age at the diagnosis (years) | 65 (41–94) | |
| Type | ||
| Colon | 28 | 80.0% |
| Right | 12 | 42.8% |
| Left | 14 | 50.0% |
| Transverse | 3 | 10.7% |
| Rectum | 7 | 20.0% |
| Stage (UICC) | ||
| is | 3 | 8.6% |
| I | 6 | 17.1% |
| II | 9 | 25.7% |
| III | 13 | 37.1% |
| IV | 4 | 11.5% |
| Neoadjuvant treatmenta | ||
| Yes | 6 | 17.1% |
| No | 29 | 82.8% |
Neoadjuvant treatment: 4 of the 6 patients had rectal cancer. Blood withdrawal for CTC analysis was done after neoadjuvant treatment.
Table 3.
Circulating tumor cells (CTCs), KRAS status of the tumor, KRAS status in the CTCs as determined by multiplex assay and serum tumor markers. Sex, F (Female) M (Male). Type, Col. Colon adenocarcinoma; Rec, Rectal adenocarcinoma. Stage, Tumor stage according to UICC.
| Patient | Sex | Tumor | CTCs/CTMs | Tumoral markers | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type | Stage | KRAS genotype | Cytology | CTCs | CTMs | Multiplex KRAS | ACE (μg/L) | CA199 (kU/L) | ||
| 3 | M | Col. | IV | WTa | + | 2 | 0 | − | 19.9 | 12.3 |
| 4 | F | Rec. | IIIb | WTa | + | 1 | 0 | − | 2.7 | 23.4 |
| 5 | M | Col. | IIIb | WTa | + | 7 | 0 | + | 1.8 | 13.1 |
| 6 | F | Col. | IIIb | c.35G > A (G12D)a | + | 15 | 0 | + | 1.9 | 15.8 |
| 7 | M | Col. | IIIc | WTa | + | 9 | 0 | − | 6.8 | <1.2 |
| 8 | M | Col. | IV | WTa | + | 25 | 30 | Failed | 1.4 | <1.2 |
| 9 | F | Col. | IIIb | WTa | + | 16 | 0 | − | 20.1 | 20.5 |
| 11 | M | Col. | I | c.38G > A (G13D)a | + | 2 | 0 | − | 2.4 | 15.6 |
| 12 | F | Col. | I | WTa | + | 2 | 0 | − | 7.6 | 17.3 |
| 13 | M | Col. | I | na | − | 0 | 0 | − | 1.1 | 7.1 |
| 14 | M | Col. | IIb | c.34G > A (G12S)b | na | na | na | + | 3.9 | <1.2 |
| 15 | F | Rec. | IIa | WTa | na | na | na | − | 0.4 | 32.5 |
| 17 | M | Col. | IIIb | Mutc | na | na | na | + | 0.6 | 8.9 |
| 18 | F | Col. | IIIb | WTc | na | na | na | − | 1.6 | 41.4 |
| 19 | M | Col. | is | c.38G > A (G13D)a | + | 2 | 1 | + | 1 | 12.7 |
| 20 | M | Rec. | I | WTc | na | na | na | − | 1.6 | 11 |
| 21 | F | Col. | IIa | c.35G > T (G12V)a | na | na | na | + | 11 | 5.7 |
| 22 | F | Col. | IIa | c.38G > A (G13D)a | + | 1 | 0 | + | 68.2 | 813.2 |
| 23 | M | Rec. | IIa | WTc | + | 10 | 0 | + | 1.8 | 18.1 |
| 24 | M | Col. | IIIb | c.38G > A (G13D)b | + | 1 | 0 | + | 1.8 | 42.8 |
| 26 | F | Col. | IV | c.35G > A (G12D)b | + | 2 | 0 | + | 26.5 | 4781 |
| 30 | M | Col. | is | na | + | 12 | 0 | + | 1.2 | 45.6 |
| 31 | F | Col. | IIa | WTb | + | 2 | 0 | + | 2.6 | 40.8 |
| 32 | F | Col. | IIIc | WTa | + | 1 | 0 | − | 1.3 | 12.5 |
| 33 | M | Rec. | I | na | − | 0 | 0 | + | 3.7 | 14.3 |
| 34 | F | Col. | IIIa | c.35G > A (G12D)a | + | 7 | 0 | + | 0.8 | 13 |
| 35 | F | Col. | IIa | WTa | + | 4 | 0 | Failed | 1.6 | 16 |
| 36 | M | Col. | IV | WTb | + | 3 | 0 | Failed | 36.8 | 990.6 |
| 37 | M | Col. | IIIb | c.38G > A (G13D)a | + | 3 | 0 | − | 1.6 | 38.3 |
| 38 | F | Rec. | IIIb | WTa | + | 1 | 0 | − | 0.3 | 6.4 |
| 39 | M | Col. | IIIa | WTa | + | 4 | 0 | + | 1.4 | 17 |
| 40 | M | Col. | IIa | WTa | + | 5 | 0 | Failed | 1.6 | 66.7 |
| 41 | F | Rec. | is | na | − | 0 | 0 | + | 0.5 | 3.5 |
| 42 | M | Col. | I | WTc | + | 10 | 0 | Failed | 2.4 | 5.6 |
| 43 | M | Col. | IIa | c.34G > T (G12C)a | + | 3 | 0 | + | 1.3 | 13.8 |
Tumor KRAS genotype as determined by a) tumor genotyping with MiSeq Illumina Tumor, b) HRM + Sanger sequencing or c) HRM only. Cytology, +, the number of circulating tumor cells was at least 1 CTCs per 3 mL whole blood. CTCs, number of circulating tumor cells per 3 mL whole blood. CTMs, number of circulating microemboli per 3 mL whole blood. The multiplex assay screen the 7 most frequent mutations in KRAS codons 12–13. The test is considered positive if the measured abundance ratio is >0.2% according to our validation specifications. Serum concentrations of the tumoral markers CEA and CA19.9 are indicated (normal CEA < 5 μg/L and CA19.9 37 kU/L). In Tumor KRAS genotype column a, b, c refers to the method used for genotyping Illumina, HRM + Sanger Sequencing or digital PCR, respectively. na: not available.
3.3. KRAS status observed in CTCs is significantly concordant with matched tumor tissues
In parallel, we compared the CTC results with the genotype detected in the corresponding tumor tissues. The concordance analysis was carried out for the 26 patients for whose CTC genotype and formalin‐fixed paraffin‐embedded (FFPE) tumors were available. 17 tumor samples were genotyped by MiSeq with the trusight illumina kit with a cut‐off fixed to 5% while the 9 additional samples were analyzed by HRM and sequencing due to a poorer quality of extracted DNA. For these 26 tumors, we found 12 (46%) with KRAS mutations in codon 12–13. Five tumors harbored mutation c38G > A (G13D), 3 tumors the mutation c35G > A (G12D), and one tumor each had the following mutations c34G > T (G12C), c34G > A (G12S), c.35G > T (G12V) while the exact mutation could not be determined for the last tumor. Comparing the KRAS genotype in the tumors with the presence of at least one mutation in the CTCs showed concordant results in 77% of cases (20/26) [Ki2 = 7.83, Pr = 0.019] (Table 4). Specifically, among 12 tumor samples with codon 12–13 KRAS mutations, we observed 10 positive samples for the 10 corresponding CTCs samples using the KRAS multiplex assay (Sensitivity∼83%) whereas 2 CTC samples revealed the wild‐type genotype, in contrast to the corresponding tumors (patients 11 and 37). Moreover, out of 14 tumor samples with wild‐type KRAS, 10 CTCs sample were negative (Specificity∼71.4%) while 4 were positive (patients 5, 23, 31 and 39). The 4 patients with CTC positive are stage II or III. Among these patients, only one patient with stage IIIb has relapsed after 4.1 months. The follow up period for the 3 others was 4.0, 19.3 and 20.6 months without relapse.
Table 4.
Concordance of KRAS genotype between CTCs and the matched tumor tissues.
| CTCs | WT | Mutant | Total |
|---|---|---|---|
| Tumor | |||
| WT | 10 | 4 | 14 |
| Mutant | 2 | 10 | 12 |
| Total | 12 | 14 | 26 |
4. Discussion
Increasing evidence suggests that characterization of CTCs from cancer patients may provide important information regarding early detection, prognosis, treatment and relapse as well as improved mechanistic insight with respect to tumor invasion and metastasis. However, so far, it has been difficult to establish CTCs as a biomarker for routine clinical use. There are several reasons for this. In particular, most of the existing procedures are based on identification of malignant cells by either cytological features or by antigen detection using immunofluorescence or immunochemistry. These methods require highly qualified cytologists and complex quality procedures. The arrival of the FDA‐approved CellSearch analyzer method has permitted reproductive detection of CTCs based on automatic numeration of cells with an epithelial phenotype thereby permitting the use of CTCs as a predictive factor in clinical trials (Andree KC et al., 2016). Nevertheless, there is growing evidence that detection of CTCs based on cytological or antigen detection does not reflect the total pool of disseminating cells and may even ignore highly clinically relevant subpopulations like the multicellular mesenchymal CTC clusters (Yang et al., 2015; Yu et al., 2013).
In the current study, we were able to detect at least one CTC in 90% of samples, including those from non‐metastatic CRCs, by using the ScreenCell size‐based device which is significantly higher than what has been reported by other technics, especially those using epithelial immunoselection. This is in agreement with the notion that the most widely used technics likely underestimate the real number of total CTCs by which supposedly are most likely to initiate new tumors or metastasis (Cao et al., 2014, 2014, 2015, 2013).
In contrast, we acknowledge that small CTCs, which are close to leucocytes in size, would be lost using this technic (Williams et al., 2012). Moreover, even if we are able to detect a higher number of tumor cells, it is at the expense of lower specificity due to leucocyte contamination. Consequently, if we wish to carry out molecular characterization of CTCs using this device, we need an extremely sensitive method in order to detect very small amounts of mutant DNA. We here propose a combination of techniques that improves our capacity to detect mutants of interest in CTCs based on multiplex KRAS ddPCR‐assay and which furthermore appear feasible to implement by a hospital platform.
Collectively, our data show that it is feasible to use the KRAS mutant multiplex screening assay exploiting ddPCR technologies. The sensitivity observed in the studies presented here (about 0.05%) is clearly superior to what is observed for other technics in current use. The association of size‐based capture of CTCs with ddPCR allowed us to detect mutant DNA in CTCs in 57% of samples (17 out of 30) with a concordance of 83% compared to the corresponding tumor tissues. This represents a clear improvement compared to other recent studies. For example, the KRAS status was determined in CTCs isolated by CellSearch by different methods including COLD‐PCR (Transgenomic™), real‐time PCR (Entrogen™) or Nested‐ASB (allele specific blocker) PCR (Mostert et al., 2013). The results showed that Nested‐ASB had the best sensitivity (about 0.2%) and allowed the detection of KRAS mutation in 5 (12%) out of 43 CRC patients; unfortunately, a correlation with the corresponding tumor tissues was not reported. Interestingly, the same study reported that it was possible to detect mutant DNA even if the number of total CTCs measured either by cytological or immunoselection method was low or undetectable, which is in agreement with the results presented here. These findings are in favor of characterizing CTCs with a molecular approach even if only part of the CTCs harbors the mutation of interest. In another recent study, the IBN microsieve size based filtration unit (Lim et al., 2012) was coupled to molecular KRAS mutation detection using HRM (High Resolution method) and ASPCR (Allele specific PCR) methodology (Mohamed Suhaimi et al., 2015). This study reported sensitivity for both technics estimated at 1.25%. They also found a number of patients with KRAS mutant CTCs of 32% (14/44) for HRM and 23% (10/44) with ASPCR as well as a significant concordance of KRAS status for matched tumor tissues. Similar findings have been reported in other studies using more or less complex methodologies. (Lyberopoulou et al., 2015; Buim et al., 2015; Raimondi et al., 2014; Gutierrez et al., 2013). Therefore, ddPCR provides increased sensitivity and can overcome the inherent problem of the ScreenCell method with retention of a leucocyte subfraction. Furthermore, it has been reported that multiplex picodroplet digital PCR could be used to detect KRAS mutations in circulating DNA from plasma of colorectal cancers (Taly et al., 2013). Collectively, this study and our demonstrates that multiplex ddPCR allows for screening of multiple mutations simultaneously with sensitivity sufficient to detect mutations by minimal invasive blood sampling. Future research will be needed to compare molecular analysis of circulating tumor DNA or CTCs as the more pertinent marker for prognosis, theranostic or treatment efficiency.
With regard to the discordance detected between the genotype of the CTCs and the genotype of the tumor, the explanation may, at least in part, be biological. Human tumors are genetically instable and therefore prone to evolve during disease progression due to both to Darwinian selection and therapeutic pressure (Chisholm et al., 2015). Several studies have reported a discordance between the primary tumor and the metastasis with respect to KRAS status (Mostert et al., 2013). Furthermore, single cell analysis provided evidence for genetic heterogeneity of CTCs in agreement with the known heterogeneity of their tissue of origin (Gasch et al., 2013). Moreover, in the cases where the tumor was wild‐type and the CTCs mutant, we also have to consider the method used for the genotyping of the tumor. Generally, methods in routine use do not allow the detection of low levels of mutant cells and/or low mutant copy numbers. Furthermore, considering the heterogeneity of solid tumors, we cannot exclude that the tumor section that was selected for analysis may not have been representative of the total tumor population. A particularly pertinent question would be to establish which mutations are overrepresented in CTCs able to initiate new tumors compared to the tumor of origin. Detailed analysis with highly sensitive methods such as ddPCR or assimilated should be able to provide answers to these fundamental questions (Laurent‐Puig et al., 2015).
In conclusion, the data presented here suggest that it is feasible to detect KRAS mutations in CTCs from blood samples of colorectal cancer patients that are predictive for those present in the tumor. The minimal invasive nature of this procedure in combination with the high sensitivity of ddPCR might provide in the future an opportunity to monitor patients throughout the course of disease on multiple levels including early detection, prognosis, treatment and relapse as well as to provide improved mechanistic insight with respect to tumor invasion and metastasis. Although the current study was focused on KRAS mutations, this strategy should also allow us to monitor other clinically relevant mutations, or combinations of mutations, for improved patient care.
Declaration of interests
This work was supported in part by ScreenCell SA.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
The authors thank Martial Saumier (Biorad) for expert advice on ddPCR as well as for his contribution to the data analysis. We also thank Dr. Georges Uzan (UMR‐S1197, Villejuif, France) for proofreading of the manuscript and Dr. Juliette Nectoux (Department of Genetics, Assistance‐Publique Hôpitaux de Paris, Cochin hospital, Paris, France) for helpful discussion about ddPCR and the access to facilities.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.05.009.
Denis Jérôme Alexandre, Patroni Alexia, Guillerm Erell, Pépin Dominique, Benali-Furet Naoual, Wechsler Janine, Manceau Gilles, Bernard Maguy, Coulet Florence, Larsen Annette K., Karoui Mehdi, Lacorte Jean-Marc, (2016), Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery, Molecular Oncology 10, doi: 10.1016/j.molonc.2016.05.009.
References
- Alix-Panabieres, C. , Pantel, K. , 2014. Technologies for detection of circulating tumor cells: facts and vision. Lab Chip 14, 57–62. [DOI] [PubMed] [Google Scholar]
- Amado, R.G. , Wolf, M. , Peeters, M. , Van Cutsem, E. , Siena, S. , Freeman, D.J. , Juan, T. , Sikorski, R. , Suggs, S. , Radinsky, R. , Patterson, S.D. , Chang, D.D. , 2008. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 1626–1634. [DOI] [PubMed] [Google Scholar]
- Andree, K.C. , van Dalum, G. , Terstappen, L.W. , 2016. Challenges in circulating tumor cell detection by the CellSearch system. Mol. Oncol. 10, (3) 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere, G. , Fici, P. , Gallerani, G. , Fabbri, F. , Zoli, W. , Rigaud, M. , 2014. Circulating tumor cells and epithelial, mesenchymal and stemness markers: characterization of cell subpopulations. Ann. Transl Med. 2, 109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere, G. , Tartary, M. , Rigaud, M. , 2014. Epithelial mesenchymal transition: a new insight into the detection of circulating tumor cells. ISRN Oncol. 2012, 382010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda, C. , Sausen, M. , Leary, R.J. , Kinde, I. , Wang, Y. , Agrawal, N. , Bartlett, B.R. , Wang, H. , Luber, B. , Alani, R.M. , Antonarakis, E.S. , Azad, N.S. , Bardelli, A. , Brem, H. , Cameron, J.L. , Lee, C.C. , Fecher, L.A. , Gallia, G.L. , Gibbs, P. , Le, D. , Giuntoli, R.L. , Goggins, M. , Hogarty, M.D. , Holdhoff, M. , Hong, S.M. , Jiao, Y. , Juhl, H.H. , Kim, J.J. , Siravegna, G. , Laheru, D.A. , Lauricella, C. , Lim, M. , Lipson, E.J. , Marie, S.K. , Netto, G.J. , Oliner, K.S. , Olivi, A. , Olsson, L. , Riggins, G.J. , Sartore-Bianchi, A. , Schmidt, K. , Shih l, M. , Oba-Shinjo, S.M. , Siena, S. , Theodorescu, D. , Tie, J. , Harkins, T.T. , Veronese, S. , Wang, T.L. , Weingart, J.D. , Wolfgang, C.L. , Wood, L.D. , Xing, D. , Hruban, R.H. , Wu, J. , Allen, P.J. , Schmidt, C.M. , Choti, M.A. , Velculescu, V.E. , Kinzler, K.W. , Vogelstein, B. , Papadopoulos, N. , Diaz, L.A. , 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, N.C. , Mangnall, D. , Majeed, A.W. , 2006. Biology of colorectal liver metastases: a review. J. Surg. Oncol. 94, 68–80. [DOI] [PubMed] [Google Scholar]
- Buim, M.E. , Fanelli, M.F. , Souza, V.S. , Romero, J. , Abdallah, E.A. , Mello, C.A. , Alves, V. , Ocea, L.M. , Mingues, N.B. , Barbosa, P.N. , Tyng, C.J. , Chojniak, R. , Chinen, L.T. , 2015. Detection of KRAS mutations in circulating tumor cells from patients with metastatic colorectal cancer. Cancer Biol. Ther. 16, 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, X. , Janku, F. , Zhan, Q. , Fan, J.B. , 2015. Accessing genetic information with liquid biopsies. Trends Genet. 31, 564–575. [DOI] [PubMed] [Google Scholar]
- Cao, H. , Xu, E. , Liu, H. , Wan, L. , Lai, M. , 2015. Epithelial-mesenchymal transition in colorectal cancer metastasis: a system review. Pathol. Res. Pract. 211, 557–569. [DOI] [PubMed] [Google Scholar]
- Chisholm, R.H. , Lorenzi, T. , Lorz, A. , Larsen, A.K. , de Almeida, L.N. , Escargueil, A. , Clairambault, J. , 2015. Emergence of drug tolerance in cancer cell populations: an evolutionary outcome of selection, nongenetic instability, and stress-induced adaptation. Cancer Res. 75, 930–939. [DOI] [PubMed] [Google Scholar]
- Cohen, S.J. , Punt, C.J. , Iannotti, N. , Saidman, B.H. , Sabbath, K.D. , Gabrail, N.Y. , Picus, J. , Morse, M. , Mitchell, E. , Miller, M.C. , Doyle, G.V. , Tissing, H. , Terstappen, L.W. , Meropol, N.J. , 2008. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 3213–3221. [DOI] [PubMed] [Google Scholar]
- Cohen, S.J. , Punt, C.J. , Iannotti, N. , Saidman, B.H. , Sabbath, K.D. , Gabrail, N.Y. , Picus, J. , Morse, M.A. , Mitchell, E. , Miller, M.C. , Doyle, G.V. , Tissing, H. , Terstappen, L.W. , Meropol, N.J. , 2009. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann. Oncol. 20, 1223–1229. [DOI] [PubMed] [Google Scholar]
- Desitter, I. , Guerrouahen, B.S. , Benali-Furet, N. , Wechsler, J. , Janne, P.A. , Kuang, Y. , Yanagita, M. , Wang, L. , Berkowitz, J.A. , Distel, R.J. , Cayre, Y.E. , 2011. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer Res. 31, 427–441. [PubMed] [Google Scholar]
- Earl, J. , Garcia-Nieto, S. , Martinez-Avila, J.C. , Montans, J. , Sanjuanbenito, A. , Rodriguez-Garrote, M. , Lisa, E. , Mendia, E. , Lobo, E. , Malats, N. , Carrato, A. , Guillen-Ponce, C. , 2014. Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin, W.A. , Haney, J. , Sugita, M. , Bemis, L. , Jimeno, A. , Messersmith, W.A. , 2010. KRAS mutation: comparison of testing methods and tissue sampling techniques in colon cancer. J. Mol. Diagn. 12, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch, C. , Bauernhofer, T. , Pichler, M. , Langer-Freitag, S. , Reeh, M. , Seifert, A.M. , Mauermann, O. , Izbicki, J.R. , Pantel, K. , Riethdorf, S. , 2013. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem. 59, 252–260. [DOI] [PubMed] [Google Scholar]
- Gingras, I. , Salgado, R. , Ignatiadis, M. , 2015. Liquid biopsy: will it be the ‘magic tool’ for monitoring response of solid tumors to anticancer therapies?. Curr. Opin. Oncol. 27, 560–567. [DOI] [PubMed] [Google Scholar]
- Guttery, D.S. , Page, K. , Hills, A. , Woodley, L. , Marchese, S.D. , Rghebi, B. , Hastings, R.K. , Luo, J. , Pringle, J.H. , Stebbing, J. , Coombes, R.C. , Ali, S. , Shaw, J.A. , 2015. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin Chem. 61, 974–982. pancreatic cancer. BMC Cancer 15, 797 [DOI] [PubMed] [Google Scholar]
- Gutierrez, C. , Rodriguez, J. , Patino-Garcia, A. , Garcia-Foncillas, J. , Salgado, J. , 2013. Mutational status analysis of peripheral blood isolated circulating tumor cells in metastatic colorectal patients. Oncol Lett. 6, 1343–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham, J.E. , Grover, P. , Winter, M. , Hewett, P.J. , Price, T.J. , Thierry, B. , 2015. Detection and clinical significance of circulating tumor cells in colorectal cancer-20 years of progress. Mol. Med. 21, (Suppl. 1) S25–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer, E. , Ulz, P. , Geigl, J.B. , 2015. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 61, 112–123. [DOI] [PubMed] [Google Scholar]
- Hindson, B.J. , Ness, K.D. , Masquelier, D.A. , Belgrader, P. , Heredia, N.J. , Makarewicz, A.J. , Bright, I.J. , Lucero, M.Y. , Hiddessen, A.L. , Legler, T.C. , Kitano, T.K. , Hodel, M.R. , Petersen, J.F. , Wyatt, P.W. , Steenblock, E.R. , Shah, P.H. , Bousse, L.J. , Troup, C.B. , Mellen, J.C. , Wittmann, D.K. , Erndt, N.G. , Cauley, T.H. , Koehler, R.T. , So, A.P. , Dube, S. , Rose, K.A. , Montesclaros, L. , Wang, S. , Stumbo, D.P. , Hodges, S.P. , Romine, S. , Milanovich, F.P. , White, H.E. , Regan, J.F. , Karlin-Neumann, G.A. , Hindson, C.M. , Saxonov, S. , Colston, B.W. , 2011. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson, C.M. , Chevillet, J.R. , Briggs, H.A. , Gallichotte, E.N. , Ruf, I.K. , Hindson, B.J. , Vessella, R.L. , Tewari, M. , 2013. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 10, 1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecova, I. , 2015. Digital PCR analysis of circulating nucleic acids. Clin. Biochem. 48, 948–956. [DOI] [PubMed] [Google Scholar]
- Joosse, S.A. , Gorges, T.M. , Pantel, K. , 2014. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent-Puig, P. , Pekin, D. , Normand, C. , Kotsopoulos, S.K. , Nizard, P. , Perez-Toralla, K. , Rowell, R. , Olson, J. , Srinivasan, P. , Le Corre, D. , Hor, T. , El Harrak, Z. , Li, X. , Link, D.R. , Bouche, O. , Emile, J.F. , Landi, B. , Boige, V. , Hutchison, J.B. , Taly, V. , 2015. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 21, 1087–1097. [DOI] [PubMed] [Google Scholar]
- Lianidou, E.S. , 2014. Molecular characterization of circulating tumor cells: Holy Grail for personalized cancer treatment?. Clin. Chem. 60, 1249–1251. [DOI] [PubMed] [Google Scholar]
- Lianidou, E.S. , Markou, A. , Strati, A. , 2015. The role of CTCs as tumor biomarkers. Adv. Exp. Med. Biol. 867, 341–367. [DOI] [PubMed] [Google Scholar]
- Lievre, A. , Bachet, J.B. , Le Corre, D. , Boige, V. , Landi, B. , Emile, J.F. , Cote, J.F. , Tomasic, G. , Penna, C. , Ducreux, M. , Rougier, P. , Penault-Llorca, F. , Laurent-Puig, P. , 2006. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 66, 3992–3995. [DOI] [PubMed] [Google Scholar]
- Lim, L.S. , Hu, M. , Huang, M.C. , Cheong, W.C. , Gan, A.T. , Looi, X.L. , Leong, S.M. , Koay, E.S. , Li, M.H. , 2012. Microsieve lab-chip device for rapid enumeration and fluorescence in situ hybridization of circulating tumor cells. Lab Chip 12, 4388–4396. [DOI] [PubMed] [Google Scholar]
- Lim, S.H. , Becker, T.M. , Chua, W. , Caixeiro, N.J. , Ng, W.L. , Kienzle, N. , Tognela, A. , Lumba, S. , Rasko, J.E. , de Souza, P. , Spring, K.J. , 2014. Circulating tumour cells and circulating free nucleic acid as prognostic and predictive biomarkers in colorectal cancer. Cancer Lett. 346, 24–33. [DOI] [PubMed] [Google Scholar]
- Lyberopoulou, A. , Aravantinos, G. , Efstathopoulos, E.P. , Nikiteas, N. , Bouziotis, P. , Isaakidou, A. , Papalois, A. , Marinos, E. , Gazouli, M. , 2015. Mutational analysis of circulating tumor cells from colorectal cancer patients and correlation with primary tumor tissue. PLoS One 10, e0123902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, M. , Zhu, H. , Zhang, C. , Sun, X. , Gao, X. , Chen, G. , 2015. “Liquid biopsy”-ctDNA detection with great potential and challenges. Ann. Transl. Med. 3, 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale, S. , Di Nicolantonio, F. , Sartore-Bianchi, A. , Siena, S. , Bardelli, A. , 2014. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 4, 1269–1280. [DOI] [PubMed] [Google Scholar]
- Mohamed Suhaimi, N.A. , Foong, Y.M. , Lee, D.Y. , Phyo, W.M. , Cima, I. , Lee, E.X. , Goh, W.L. , Lim, W.Y. , Chia, K.S. , Kong, S.L. , Gong, M. , Lim, B. , Hillmer, A.M. , Koh, P.K. , Ying, J.Y. , Tan, M.H. , 2015. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol. Oncol. 9, 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostert, B. , Jiang, Y. , Sieuwerts, A.M. , Wang, H. , Bolt-de Vries, J. , Biermann, K. , Kraan, J. , Lalmahomed, Z. , van Galen, A. , de Weerd, V. , van der Spoel, P. , Ramirez-Moreno, R. , Verhoef, C. , Ijzermans, J.N. , Wang, Y. , Gratama, J.W. , Foekens, J.A. , Sleijfer, S. , Martens, J.W. , 2013. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int. J. Cancer 133, 130–141. [DOI] [PubMed] [Google Scholar]
- Muzny, D.M. , Bainbridge, M.N. , Chang, K. , Dinh, H.H. , Drummond, J.A. , Fowler, G. , Kovar, C.L. , Lewis, L.R. , 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel, K. , Alix-Panabieres, C. , 2013. Real-time liquid biopsy in cancer patients: fact or fiction?. Cancer Res. 73, 6384–6388. [DOI] [PubMed] [Google Scholar]
- Pantel, K. , Speicher, M.R. , 2015. The biology of circulating tumor cells. Oncogene 35, (10) 1216–1224. [DOI] [PubMed] [Google Scholar]
- Peeters, M. , Douillard, J.Y. , Van Cutsem, E. , Siena, S. , Zhang, K. , Williams, R. , Wiezorek, J. , 2013. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 31, 759–765. [DOI] [PubMed] [Google Scholar]
- Raimondi, C. , Nicolazzo, C. , Gradilone, A. , Giannini, G. , De Falco, E. , Chimenti, I. , Varriale, E. , Hauch, S. , Plappert, L. , Cortesi, E. , Gazzaniga, P. , 2014. Circulating tumor cells: exploring intratumor heterogeneity of colorectal cancer. Cancer Biol. Ther. 15, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, T. , Pajaro-Van de Stadt, S.H. , Yeat, N.C. , Lin, J.C. , 2015. Clinical applications of next generation sequencing in cancer: from panels, to exomes, to genomes. Front. Genet. 6, 215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simi, L. , Pratesi, N. , Vignoli, M. , Sestini, R. , Cianchi, F. , Valanzano, R. , Nobili, S. , Mini, E. , Pazzagli, M. , Orlando, C. , 2008. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am. J. Clin. Pathol. 130, 247–253. [DOI] [PubMed] [Google Scholar]
- Taly, V. , Pekin, D. , Benhaim, L. , Kotsopoulos, S.K. , Le Corre, D. , Li, X. , Atochin, I. , Link, D.R. , Griffiths, A.D. , Pallier, K. , Blons, H. , Bouche, O. , Landi, B. , Hutchison, J.B. , Laurent-Puig, P. , 2013. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 59, (12) 1722–1731. 10.1373/clinchem.2013.206359 [DOI] [PubMed] [Google Scholar]
- Therkildsen, C. , Bergmann, T.K. , Henrichsen-Schnack, T. , Ladelund, S. , Nilbert, M. , 2014. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 53, 852–864. [DOI] [PubMed] [Google Scholar]
- Thierry, A.R. , Mouliere, F. , El Messaoudi, S. , Mollevi, C. , Lopez-Crapez, E. , Rolet, F. , Gillet, B. , Gongora, C. , Dechelotte, P. , Robert, B. , Del Rio, M. , Lamy, P.J. , Bibeau, F. , Nouaille, M. , Loriot, V. , Jarrousse, A.S. , Molina, F. , Mathonnet, M. , Pezet, D. , Ychou, M. , 2014. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 20, 430–435. [DOI] [PubMed] [Google Scholar]
- Torino, F. , Bonmassar, E. , Bonmassar, L. , De Vecchis, L. , Barnabei, A. , Zuppi, C. , Capoluongo, E. , Aquino, A. , 2013. Circulating tumor cells in colorectal cancer patients. Cancer Treat. Rev. 39, 759–772. [DOI] [PubMed] [Google Scholar]
- Tsiatis, A.C. , Norris-Kirby, A. , Rich, R.G. , Hafez, M.J. , Gocke, C.D. , Eshleman, J.R. , Murphy, K.M. , 2010. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J. Mol. Diagn. 12, 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, A. , Balic, M. , Datar, R. , Cote, R. , 2012. Size-based enrichment technologies for CTC detection and characterization. Recent Results Cancer Res. 195, 87–95. [DOI] [PubMed] [Google Scholar]
- Yang, M.H. , Imrali, A. , Heeschen, C. , 2015. Circulating cancer stem cells: the importance to select. Chin J. Cancer Res. 27, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, M. , Bardia, A. , Wittner, B.S. , Stott, S.L. , Smas, M.E. , Ting, D.T. , Isakoff, S.J. , Ciciliano, J.C. , Wells, M.N. , Shah, A.M. , Concannon, K.F. , Donaldson, M.C. , Sequist, L.V. , Brachtel, E. , Sgroi, D. , Baselga, J. , Ramaswamy, S. , Toner, M. , Haber, D.A. , Maheswaran, S. , 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, Z. , Chen, S.S. , Chandra, P.K. , Galbincea, J.M. , Soape, M. , Doan, S. , Barkoh, B.A. , Koeppen, H. , Medeiros, L.J. , Luthra, R. , 2009. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod. Pathol. 22, 1023–1031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data