

Abstract
Recent studies implicate hydrogen sulfide (H2S) oxidation as an important aspect of bacterial antibiotic resistance and sulfide homeostasis. The cst operon of the major human pathogen Staphylococcus aureus is induced by exogenous H2S stress and encodes enzymes involved in sulfide oxidation, including a group I flavoprotein disulfide oxidoreductase sulfide:quinone oxidoreductase (SQR). In this work, we show that S. aureus SQR catalyzes the two-electron oxidation of sodium sulfide (Na2S) into sulfane sulfur (S0) when provided flavin adenine dinucleotide and a water-soluble quinone acceptor. Cyanide, sulfite, and coenzyme A (CoA) are all capable of functioning as the S0 acceptor in vitro. This activity requires a C167–C344 disulfide bond in the resting enzyme, with the intermediacy of a C344 persulfide in the catalytic cycle, verified by mass spectrometry of sulfide-reacted SQR. Incubation of purified SQR and S. aureus CstB, a known FeII persulfide dioxygenase-sulfurtransferase also encoded by the cst operon, yields thiosulfate from sulfide, in a CoA-dependent manner, thus confirming the intermediacy of CoASSH as a product and substrate of SQR and CstB, respectively. Sulfur metabolite profiling of wild-type, Δsqr, and Δsqr::pSQR strains reveals a marked and specific elevation in endogenous levels of CoASSH and inorganic tetrasulfide in the Δsqr strain. We conclude that SQR impacts the cellular speciation of these reactive sulfur species but implicates other mechanisms not dependent on SQR in the formation of low-molecular weight thiol persulfides and inorganic polysulfides during misregulation of sulfide homeostasis.
Graphical abstract
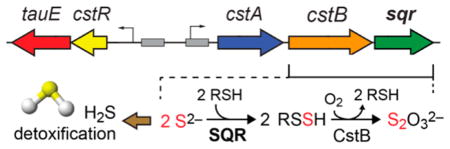
Hydrogen sulfide (H2S, HS−, or S2−) is a biologically important molecule with complex physiological functions, ranging from the well-known inhibitory effect on cellular respiration1 to the recently discovered beneficial aspect as a gasotransmitter or signaling molecule in mammalian systems.2 H2S passes through biological membranes freely and exists mainly as hydrosulfide anion (HS−) at physiological pH.3 HS− can be exported by a specialized efflux transporter, the only known instance of which is found in the human pathogen Clostridium difficile.4 In addition to exogenous sources, H2S is endogenously synthesized by three major enzymes of the transsulfuration and cysteine degradation pathways, cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfur transferase (3-MST), and this process has been reported to enhance bacterial resistance to antibiotic stress.5 Staphylococcus aureus encodes a CBS (cysM) and CSE (metB) but lacks 3-MST, while Escherichia coli encodes only 3-MST as a source of endogenous H2S.
The beneficial impact of endogenous H2S or H2S-derived per- and polysulfide species that are thought to be important for redox signaling6 and oxidative stress resistance,7,8 antibiotic resistance,9 and cysteine biosynthesis,10 for example, contrasts sharply with the well-known deleterious effects of sulfide as a potent inhibitor of heme-containing enzymes. These opposing functions of H2S suggest a requirement for endogenous sulfide homeostasis, although how this is accomplished in cells is not fully understood. The major human pathogen S. aureus encodes the cst (copper-sensing operon repressor-like sulfurtransferase) operon that is transiently and strongly induced by exogenous cellular sulfide stress,11 via transcriptional upregulation mediated by the per- and polysulfide-sensing repressor CstR.12 The cst-encoded proteins are thought to effect clearance of elevated LMW persulfides that derive from cellular sulfide stress.11 Genes encoded by the cst operon, including cstA, cstB, and sqr, have been shown to be necessary to mitigate the effects of cellular sulfide toxicity, added as exogenous NaHS or polysulfides to cells grown aerobically in a liquid culture.13 Both CstA and CstB have been biochemically characterized as a multidomain sulfurtransferase and non-heme FeII persulfide dioxygenase, respectively, and these findings support an H2S detoxification model in S. aureus.13,14 We emphasize, however, that the degree to which sulfide versus downstream sulfide oxidation products, e.g., per- and polysulfides, sulfite, and thiosulfate, are toxic species associated with sulfide misregulation remains an open question.6
The presumed initial step of H2S oxidation in S. aureus associated with the sulfide:quinone oxidoreductase (SQR) has not yet been characterized.13 Sulfide:quinone oxidoreductases represent a diverse family of group I pyridine nucleotide-dependent, FAD (flavin adenine dinucleotide)-containing flavoprotein disulfide reductases (FDRs) that have been structurally and biochemically characterized across all kingdoms of life.15 Group I FDRs include lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mycothione reductase. These FDRs employ NAD(P)H as a source of two electrons that are transferred to the flavin moeity, then to a non-flavin redox center, often a disulfide bond, and ultimately to an external e− acceptor.15 SQRs and related flavocytochrome c:sulfide dehydrogenases (FCSD) differ from other group I FDRs in that they do not bind NAD(P)H and utilize only sulfide (S2−) as both a substrate and a source of electrons, with sulfane sulfur (S0) thus formed and transferred to a nucleophilic acceptor. All SQRs contain two absolutely conserved cysteines that form a catalytic disulfide redox center, and a third more variable N-terminal Cys that in some cases has been shown to covalently bind the FAD cofactor. All three cysteine residues are retained in S. aureus SQR as Cys133, Cys167, and Cys344 (Figure S1).
SQRs have recently been structurally classified into six types (types I–VI), with FCSDs most closely related to the type II SQRs, which includes S. aureus SQR and the human mitochondrial enzyme.16 Type II SQRs lack both “sulfide-capping loops” that characterize the type I and type V SQRs exemplified by Aquifex aeolicus SQR17 and Acidianus ambivalens, respectively (Figure 1 and Figure S1).18 In each of these enzymes, the flavin is covalently attached to the enzyme by the 8-methylene group (C8M) of the isoalloxazine ring of FAD via either a thioether (A. ambivalens, C129) or perthioether (Aq. aeolicus, C124) linkage; in Allochromatium vinosum FCSD, a more N-terminal Cys (C42) forms the analogous C8M thioether linkage, with the catalytic disulfide positioned on the re face of the FAD (Figure 1).19 In contrast, this Cys is not covalently attached to the FAD in another type I enzyme, Acidithiobacillus ferrooxidans (Figure S1).20 In type II enzymes, the role of the N-terminal Cys is not yet clear, with a Tyr often found at this position (Figure S1);16 in the type II human SQR, the FAD is also noncovalently bound.21 Other features that distinguish different SQR types are a variable-length C-terminal amphipathic “tail” that dictates membrane association and/or assembly state, while eukaryotic SQRs harbor an N-terminal signal peptide that directs subcellular localization to mitochondria (Figure S1).21
Figure 1.

Ribbon representation of the structure of flavocytochrome c:sulfide dehydrogenases (FCSD) from Al. vinosum19 (PDB entry 1fcd) (blue) bound to its electron acceptor (yellow) as a model for type II SQRs from S. aureus strain Newman (NWMN_0029) characterized here. The FAD cofactor and C161–C357 disulfide redox center are shown as sticks and spheres, respectively. The analogous Cys residues in S. aureus SQR are C167 and C344. One asterisk and two asterisks approximate insertion points of type V and type 1 SQR sulfide-capping loops (see Figure S1), respectively, not found in type II SQRs or in FCSDs.16 C42 in FCSD covalently binds the FAD from the si face via C8M, not conserved in type II SQRs; K129 marks the approximate position of C133 in the S. aureus enzyme, which is also not well conserved in type II SQRs.16
A plausible sulfide detoxification model in S. aureus is that the Cys167–Cys344 disulfide bridge in the resting SQR is attacked by HS− to form a persulfide on Cys344, which is ultimately transferred to a LMW thiol acceptor for further oxidation by the persulfide dioxygenase CstB (Figure 2).13 In a recent mechanistic proposal, the sulfide-derived electrons move through a charge transfer intermediate that is on-pathway to formation of a covalent FADH2 complex; this complex is subsequently attacked by an exogenous acceptor, e.g., sulfide, sulfite, CN−, or a reduced thiol,21,22 with the electrons ultimately transferred to a quinone acceptor (Figure 2).22
Figure 2.

Working mechanistic model for sulfide oxidation by S. aureus SQR based on a recent kinetic analysis of human SQR.22 (A) Sulfide (HS−) attacks the C167–C344 disulfide bridge in the resting enzyme (E·FAD) to create C344 persulfide and C167 thiolate. (B) One of these two thiolate species or the other forms a charge transfer (CT) complex with the oxidized flavin centered on carbon 4a to create the 4a adduct in panel C. (C) Attack by the exogenous sulfane sulfur (S0) acceptor, here shown as a LMW thiol persulfide (LMW-S−) on the C344 persulfide, leads to formation of the E·FADH2 complex in panel D, with release of the LMW persulfide and re-formation of the C167–C344 disulfide bridge. (D) The reduced E·FADH2 complex shown is then oxidized by a quinone species (CoQ) to regenerate the E·FAD enzyme and a reduced quinone (CoQH2). Note that there is no mechanistic role for the most N-terminal, only weakly conserved, C133 in this model.
While SQRs exist in all domains of life,23 the diverse ecological or physiological niches in which organisms exist make it likely that the physiological impact of an SQR may vary in a species-specific manner. For example, many α-proteobacteria utilize SQRs to obtain electrons from sulfide to incorporate into the respiratory chain for ATP synthesis, a function commonly found in facultative anaerobes.23 Other marine organisms that must survive in a sulfide-rich environment (up to ≈20 mM) rely on SQRs for reducing equivalents and mobilizable sulfur, and possibly to mediate sulfide detoxification.24 SQRs in eukaryotic mitochondrial H2S detoxification systems have also been extensively studied,21,25–27 where this activity is thought to be coupled to a persulfide dioxygenase (PDO), ETHE1, and a rhodanese (sulfurtransferase) to produce thiosulfate as a product of H2S detoxification.28 Interestingly, S. aureus harbors a mitochondrial-like, sulfide-inducible sulfide oxidation system encoded by the cst operon but whose physiological function is not well understood.12
In this work, we present the characterization of the S. aureus SQR placed in the context of previous studies of two other cst-encoded proteins, the multidomain sulfurtransferase CstA,14 and the FeII-dependent persulfide dioxygenase–rhodanese fusion, CstB.13 We show here that FAD is noncovalently bound to S. aureus SQR and the enzyme catalyzes the initial oxidation of sulfide utilizing the Cys167–Cys344 disulfide center with a number of water-soluble quinones capable of serving as electron acceptors. Cyanide, sulfite, and the abundant LMW thiol, coenzyme A (CoA), are all capable of functioning as in vitro S0 acceptors for this reaction. Both Cys167 and Cys344 are catalytically required, with the formation of the Cys344 persulfide intermediate detected by mass spectrometry on sulfide-reacted SQR (see Figure 2). We also show that SQR catalyzes the oxidation of sulfide to thiosulfate in a CoA-dependent, CstB-dependent reaction. Cellular profiling of inorganic and organic per- and polysulfides in wild-type versus Δsqr and complemented strains confirms that the sqr gene plays a physiological role in controlling sulfur speciation in this important microbial pathogen.
MATERIALS AND METHODS
Cloning and Purification of Recombinant SQR Proteins
Full-length wild-type SQR was cloned into the pHis parallel expression plasmid using NcoI and SpeI restriction sites. SQR mutants (C133S, C167S, and C344S) were generated by site-directed mutagenesis of the expression plasmid. All SQR expression plasmids were transformed into E. coli Rosetta, cultured in LB medium at 37 °C, induced with 1 mM isopropyl β-D-1-thiogalactopyranoside at an OD600 of 0.6–0.8, and expressed at 16 °C for 16 h. Cells were harvested by centrifugation and stored at −80 °C.
All full-length SQRs (wild-type, C167S, and C344S) were purified using a Ni-NTA affinity chromatography protocol followed by the removal of the N-terminal His6 tag. The cell pellet was resuspended in 25 mM Tris-HCl, 500 mM NaCl, 20 mM imidazole, 2 mM tris(2-carboxyethyl)phosphine (TCEP), and 10% glycerol (pH 8.0) and lysed by sonication. The cell lysate was clarified by centrifugation, and the supernatant was loaded onto a Ni-NTA column, which was pre-equilibrated with lysis buffer. The column was washed with lysis buffer containing 50 mM imidazole, followed by elution with lysis buffer containing 500 mM imidazole. His6-tagged SQRs were exchanged into lysis buffer and incubated with tobacco etch virus (TEV) protease at 4 °C overnight. The cleaved His6 tag was removed by reapplication of the TEV-incubated protein to the Ni-NTA column, and the cleaved SQRs were collected from the flow-through and the 50 mM imidazole wash fractions, followed by further purification via size exclusion chromatography (G200 16/60) in 25 mM Tris-HCl, 500 mM NaCl, 2 mM TCEP, and 10% glycerol (pH 8.0). Fractions containing SQRs with a purity of >95% as judged by overloaded sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis were pooled and stored at −80 °C until they were used with a final glycerol concentration of 20% (v/v) added.
Purified wild-type, C167S, and C344S SQRs were digested by several sequencing grade proteases (trypsin, Glu-C, Lys-C, chymotrypsin, and Asp-N) and confirmed as SQRs by matrix-assisted laser desorption ionization (MALDI) mass spectrometry (Figure S2). Previous proteomics analysis performed with S. aureus strain COL showed that SQR partitions between membrane and cytoplasmic fractions.29 Consistent with this finding, His6-tagged SQR when overexpressed in the E. coli Rosetta strain is present in both supernatant and pellet fractions in the cell lysate. Wild-type, C167S, and C344S SQRs utilized in this study were purified from the supernatant fraction from the cell lysate. The purified SQRs remain stable in 25 mM Tris-HCl, 500 mM NaCl, and 10% glycerol (pH 8.0) at room temperature for up to 48 h if kept at or below 50 μM (3.4 mg/mL). Addition of nonionic detergent Triton X-100 significantly increased the solubility of SQRs under these conditions, suggesting the interaction with detergents stabilizes SQRs in solution as previously reported for mitochondrial sulfide dehydrogenase (HMT2).30,31
Flavin Adenosine Dinucleotide (FAD) Content Analysis
SQR is predicted to be a FAD-dependent enzyme, and purified SQR is expected to display a bright yellow absorption indicating the potential FAD binding event. The FAD stoichiometry of SQRs was determined by UV–vis using an ε280SQR of 67270 M−1 cm−1 and an ε450FAD of 10500–11800 M−1 cm−1.32 Purified SQRs (~50 μM) were extensively exchanged in a 30 kDa centrifugal filter into degassed 25 mM Tris-HCl, 500 mM NaCl, and 10% glycerol (pH 8.0) to remove the reducing reagent; 400 μL of degassed 25 mM Tris-HCl, 500 mM NaCl, 10% glycerol, and 8 M urea (pH 8.0) was added to 100 μL of SQR, followed by concentrating denatured SQR to ≈100 μL. This process was repeated four more times with all the flow-through fractions collected and analyzed by UV–vis (290–600 nm), to compare the fractions with native SQR and denatured SQR.
Gel Filtration Chromatography
Purified SQR containing stoichiometric FAD was chromatographed on an analytical Superdex-200 Increase column by loading 100 μL at a concentration of 10 μM and eluting at a rate of 0.5 mL/min at 4 °C.
LC–MS/MS Analysis of Proteolytically Digested Peptides of SQR
In a 200 μL reaction mixture, 25 μM buffer-exchanged wild-type or C344S SQR or Na2S-reacted wild-type or C344S SQR was alkylated by addition of 200 μL of alkylating buffer containing 100 mM Tris-HCl, 200 mM iodoacetamide (IAM), and 8 M urea (pH 8.0) and incubated for 30 min in the dark. Following alkylation, the proteins were precipitated by addition of 100 μL of 100% trichloroacetic acid (TCA), with the supernatant removed by centrifugation at 13200 rpm for 15 min at 4 °C. The protein pellets were then washed with 500 μL of ice-cold acetone three times to remove TCA and dried in a SpeedVac concentrator. Dried protein pellets were resuspended in 20 μL of digestion buffer containing 100 mM NH4HCO3 and 2 M urea, mixed with 1 μL of 200 ng/μL sequencing grade endoproteinase trypsin and 1 μL of 40 ng/μL sequencing grade endoproteinase Asp-N, and digested overnight at 37 °C. The digestion was terminated by addition of 1 μL of 10% trifluoroacetic acid (TFA). Peptides were enriched from the digestion mixture with a C18 zip tip using a standard protocol, dried in a SpeedVac concentrator, and resuspended in 10 μL of 0.1% formic acid for LC–MS/MS analysis performed in the Laboratory for Biological Mass Spectrometry at Indiana University, using a Thermo Finnigan LTQ-Orbitrap XL mass spectrometer equipped with an Eksigent NanoLC Ultra 2D plus system. Briefly, 2 μg of peptide was loaded onto a self-packed C18 reversed phase trapping column (100 μm × 50 mm, 5 μm, 200 Å Magic C18AQ) for 6 min in 0.1% formic acid and further chromatographed on a self-packed C18 reversed phase analytical column (75 μm × 100 mm, 5 μm, 100 Å Magic C18AQ) using an acetonitrile-based gradient (solvent A, 0% acetonitrile and 0.1% formic acid; solvent B, 100% acetonitrile and 0.1% formic acid). The elution protocol was as follows: 3 to 7% B from 0 to 1 min (linear gradient), 7 to 40% B from 1 to 19 min (linear gradient), 40 to 50% B from 19 to 21 min (linear gradient), 50 to 90% B from 21 to 21.5 min (linear gradient), and 90% B from 21.5 to 25 min (isocratic) followed by re-equilibration to 3% B. Data were collected by the Xcalibur System and converted into Mascot Generic Format by the Proteomics Tool Suite. The extracted peak list was analyzed using Protein Prospector.
Sulfide:Quinone Oxidoreductase Activity
Sulfide:quinone oxidoreductase activity was measured using a UV–vis-based assay to detect the sulfide-dependent quinone reduction indicated by the decrease in absorption at a wavelength distinct for each water-soluble quinone. The SQR proteins (wild-type, C167S, and C344S) were exchanged into degassed 25 mM Tris-HCl, 500 mM NaCl, and 10% glycerol (pH 8.0) in an anaerobic chamber to remove the reducing reagent. The 400 μL reaction mixture contained 100 nM SQR, 200 μM duroquinone (DQ), decyl-ubiquinone (decyl-UQ), or 2,3-dimethyl-1,4-naphthoquinone (DMN), and 4 mM KCN and Na2S ranging from 10 to 640 μM, buffered by degassed 25 mM Tris-HCl and 200 mM NaCl (pH 8.0). These reactions, including the control groups that were added with BSA instead of the enzyme, were initiated via the addition of Na2S, and the mixtures were incubated for 4 min and assessed for the reduction of quinones by UV–vis spectrometry at various Na2S concentrations at 25 °C. The molar extinction coefficients are as follows: ε267DQ = 18200 M−1 cm−1, ε275decyl-UQ = 15000 M−1 cm−1, and ε280DMN = 15200 M−1 cm−1.31
To evaluate the catalytic efficiency of SQR over sulfide in the presence of different S0 acceptors, the same assay was used with DQ as the electron acceptor. Briefly, the 400 μL reaction mixture also contained 100 nM SQR, 200 μM DQ, 4 mM KCN or 4 mM sulfite or 100 μM CoA, and Na2S ranging from 10 to 640 μM, buffered by degassed 25 mM Tris-HCl and 200 mM NaCl (pH 8.0). These reactions, including the control groups that were added with BSA instead of enzyme, were also initiated with the addition of Na2S, and the mixtures were incubated for 4 min and assessed for the reduction of DQ by UV–vis spectrometry at various Na2S concentrations at 25 °C.
MALDI MS Analysis of Proteolytically Digested Peptides of SQR
The protein modification formed on SQR during the SQR-catalyzed sulfide oxidation was explored by MALDI mass spectrometry by analyzing the proteolytically digested peptides from sulfide-reacted SQR. In a 200 μL reaction mixture, 25 μM buffer-exchanged SQR was incubated with 500 μM sulfide substrate (Na2S) anaerobically for 1 h at 25 °C, buffered by degassed 25 mM MES and 100 mM NaCl (pH 6.0). The reactions were terminated by addition of 200 μL of alkylating buffer containing 100 mM Tris-HCl, 200 mM IAM, and 8 M urea (pH 8.0), and the mixtures were incubated for 30 min in the dark. Following alkylation, proteins were precipitated by addition of 100 μL of 100% TCA, with supernatant removed by centrifugation at 13200 rpm for 15 min at 4 °C. The protein pellets were then washed with 500 μL of ice-cold acetone three times to remove TCA and dried in a SpeedVac concentrator. Dried protein pellets were resuspended in 20 μL of digestion buffer containing 100 mM NH4HCO3 and 2 M urea, and 1 μL of 40 ng/μL sequencing grade endoproteinase Asp-N was added to each sample for overnight digestion at 37 °C. The digestion was terminated by addition of 1 μL of 10% TFA. Peptides were enriched from the digestion mixture with a C18 zip tip using the standard protocol and analyzed by MALDI MS. Data were collected and analyzed using flexImaging software.
Oxidation of Sulfide into Thiosulfate by SQR and CstB Mixtures
The oxidation of sulfide to thiosulfate by SQR and CstB was assessed by a previously developed fluorescence-based HPLC assay to detect the production of thiosulfate.12 A typical 200 μL reaction mixture contained 2 μM SQR (wild-type, C167S, or C344S), 2 μM CstB, 400 μM CoA, 400 μM DQ, and Na2S ranging from 40 μM to 2.56 mM, buffered by air-saturated 25 mM MES and 100 mM NaBr (pH 6.0) at 25 °C. These reactions were initiated by the addition of enzymes or BSA (as a control); the mixtures were incubated for 2 min, and the reactions were terminated by addition of 1 μL of 5 M methanesulfonic acid (MA). Proteins were removed from the mixture by ultrafiltration, and 25 μL of the filtered solution was labeled in the dark with 75 μL of monobromobimane (mBBr) labeling buffer containing 25 mM Tris-HBr and 2 mM mBBr (pH 8.0) for 30 min at room temperature. The labeling reaction was terminated by addition of 100 μL of 16.4% methanol and 0.25% acetic acid (pH 3.9). Samples (20 μL) were injected in duplicate onto a Kinetex C18 reversed phase column (Phenomenex, P/No. 00F-4601-E0, 4.6 mm × 150 mm, 5 μm, 100 Å) outfitted with a Zorbax Eclipse Plus C18 guard column. Chromatographic analysis was conducted on a Waters 600 high-performance liquid chromatography system equipped with a Waters 717 plus autosampler, a Waters 474 scanning fluorescence detector (λex = 384 nm, and λem = 478 nm), and Empower chromatography software installed on a standard personal computer running Windows XP. A methanol-based gradient system was employed to analyze the samples at 25 °C [solvent A, 16.8% methanol and 0.25% acetic acid (pH 3.9); solvent B, 90% methanol and 0.25% acetic acid (pH 3.9)] with a flow rate of 1.2 mL/min and the following elution protocol: 0% B from 0 to 10 min (isocratic), 0 to 100% B from 10 to 12 min (linear gradient), and 100% B from 12 to 17 min (isocratic) followed by re-equilibration to 0% B. Quantitation of thiosulfate was permitted by running a series of authentic thiosulfate standards in separate chromatographic runs with an identical protocol. Other experiments (200 μL reaction mixtures) that included 10 μM SQR, 10 μM CstB, 400 μM CoA, 400 μM DQ, and 200 μM Na2S in 25 mM MES and 100 mM NaBr (pH 6.0) at 25 °C were used, with the reactions initiated, terminated, labeled, and analyzed as described above.
Cell Growth and Measurement of the Sulfide Concentration in the Growth Medium
sqr-related S. aureus Newman strains were inoculated from frozen glycerol stocks and grown in 10 mL of Tryptic Soy Broth (TSB) with 10 μg/mL chloramphenicol overnight, as all strains carry either the empty pOS1 vector (wild-type and Δsqr strain) or the wild-type sqr allele cloned into the pOS1 vector (Δsqr::pSQR).12 Overnight cultures were pelleted by centrifugation, resus-pended in an equal volume of a chemically defined Hussain–Hastings–White modified (HHWm) medium,33 and diluted to an OD600 of ≈0.008 into 15 mL of HHWm supplemented with 0.5 mM thiosulfate as the sole sulfur source and 10 μg/mL chloramphenicol, in the absence or presence of 0.2 mM NaHS. Cultures were grown at 37 °C while being shaken at 80 rpm in loosely capped 50 mL Falcon tubes. The sulfide concentration of the growth medium from two additional replicate cultures of wild-type, Δsqr, and Δsqr::pSQR strains, as well as a growth medium-only control to which 0.2 mM NaHS was added, was measured by withdrawing a 200 μL aliquot of growth medium every 2 h from 0 to 14 h, followed by centrifugation at 13000 rpm for 3 min. The supernatant was removed to a fresh tube and stored at −80 °C. Five microliters of the supernatant was then incubated with 95 μL of 20 mM Tris-HBr (pH 8.0), 50% acetonitrile, 1 mM monobromobimane (mBBr), and 2 μM N-acetyl-L-cysteine (NAC), an internal standard, at 60 °C for 1 h in the dark. The reaction was terminated with 300 μL of 10 mM methanesulfonic acid (MA), with 40 μL of each sample analyzed by liquid chromatography as previously described.13
Measurement of Cellular LMW Persulfides and Inorganic (Poly)sulfide Concentrations
Wild-type, Δsqr,12 and Δsqr::pSQR S. aureus strain Newman were grown in 10 mL of TSB medium supplemented with 10 μg/mL chloramphenicol overnight. Cells were pelleted and washed with phosphate-buffered saline (PBS), and cultures were initiated at an OD600 of ≈0.02 in HHWm medium supplemented with 0.5 mM thiosulfate and 10 μg/mL chloramphenicol; 0.2 mM NaHS was added to the cultures when the OD600 reached ≈0.2. All cultures were grown in loosely capped 50 mL Falcon tubes at 37 °C while being shaken at 200 rpm. Aliquots were removed 0 and 30 min following addition of NaHS, with 5 mL for LC–MS/MS sulfur metabolite detection and 1 mL for measurement of protein concentration, and harvested by centrifugation at 3000 rpm for 10 min, and the culture medium supernatant was discarded. Cell pellets were then washed with PBS, pelleted again by centrifugation at 13200 rpm for 5 min, and stored frozen at −80 °C until they were analyzed. Cell pellets were then thawed by resuspension in 100 μL of the mBBr labeling solution containing 20 mM Tris-HBr (pH 8.0), 50% acetonitrile, and 1 mM mBBr and frozen and thawed three times in liquid N2, in the dark in screw-capped Eppendorf tubes. Cellular debris and proteins were then pelleted by centrifugation at 13200 rpm for 5 min, and the supernatant was transferred to a fresh Eppendorf tube containing 100 μL of 15 mM MA to terminate the labeling reaction. Then samples were centrifuged at 13200 rpm for 5 min through a 0.2 μm centrifugal filter unit to remove particulates.
Quantitation of LMW persulfides was performed using a Waters Synapt G2S mass spectrometer running a Waters Acquity UPLC I-Class chromatography system. Samples (10 μL) were injected onto a YMC-Triart C18 column [50 mm × 2.0 mm (inner diameter); YMC]. Triplicate samples were typically analyzed using a methanol-based gradient [solvent A being 10% methanol and 0.25% acetic acid (pH 3.0) and solvent B being 90% methanol and 0.25% acetic acid (pH 3.0)] with the following elution protocol at 25 °C and a flow rate of 0.2 mL/min: 0% B from 0 to 3 min (isocratic), 0 to 25% B from 3 to 7 min (linear gradient), 25% B from 7 to 9 min (isocratic), 25 to 75% B from 9 to 12 min (linear gradient), 75 to 100% B from 12 to 14 min (linear gradient), 100% B from 14 to 14.5 min (isocratic), followed by re-equilibration to 0% B. Quantitation of LMW thiols and persulfides was permitted by spiking samples with a known concentration of authentic, mBBr-derivatized S34-LMW persulfide standards in each chromatography run. Analysis of peak areas was performed using MassLynx software.
RESULTS
Oligomerization State of Purified SQR
Chromatography of full-length wild-type recombinant S. aureus SQR on a preparative G200 gel filtration column reveals the presence of SQR both in the void volume and within the fractionation range of the column (data not shown). The material eluting within the fractionation range was reanalyzed by analytical gel filtration (Figure S3A), yielding a single peak with an estimated molecular weight of 32.6 kDa, somewhat smaller than that calculated for monomeric SQR (45.4 kDa). SDS–PAGE analysis revealed that denatured SQR runs at ≈45 kDa with no significant degradation products observed (Figure S3B). These data suggest that S. aureus SQR is prone to irreversible higher oligomerization states as also found for the type II human enzyme,22 likely attributed to the ability of SQR to partition between membrane and soluble fractions,29 with the soluble form predominantly monomeric under these solution conditions. A monomeric assembly state is consistent with that previously determined for the type V A. ambivalens SQR18 but contrasts with other SQRs that adopt higher oligomerization states, e.g., dimer for the human and Ac. ferrooxidans SQRs20,22 or hexamer for Aq. aeolicus SQR.17 Both monomeric SQRs lack the C-terminal extension found in other oligomeric type I and II enzymes (Figure S1). All purified S. aureus SQRs (wild-type, C167S, and C344S) exhibit an absorption spectrum consistent with stoichiometrically bound FAD cofactor as isolated, which is bound noncovalently to the enzyme (Figure S4). This finding is consistent with the type II human SQR21 but contrasts with that of type I and type V SQRs.17,18,20
An Intramolecular Disulfide Linkage in Purified SQR
Most group 1 FDRs harbor a redox active disulfide bond that packs against one of the faces of the isoalloxazine ring required for oxidation of sulfide.15 Analysis of proteolytically digested peptides from SQR by LC–MS/MS reveals that C167 is cross-linked to C344 through an intramolecular disulfide linkage (Figure 3), as previously observed for human SQR.21,22 A DTNB assay34 further reveals that ≈35% cysteine residues from purified SQR are reduced. Given that SQR has three cysteines (C133, C167, and C344), this is consistent with a C167–C344 disulfide bond, with C133 reduced in the resting enzyme.
Figure 3.

LC–MS/MS analysis reveals the formation of disulfide bond between C167 and C344 in the resting enzyme (see Figure 2). High-resolution ESI-MS/MS of trypsin and Asp-N-digested peptides from SQR in the (A) +2 charge state (inset) with the corresponding fragmentation pattern and (B) +4 charge state (inset) with the corresponding fragmentation pattern. y-ions and b-ions are shown for the C344-containing peptide, 339DGYTSCPIVTGYNR352 (red), and the C167-containing peptide, 167CGGAPM(Ox)K173 (cyan).
Sulfide Oxidation by SQR with Cyanide Served as the S0 Acceptor
We next used a standard sulfide:quinone oxidoreductase assay to measure the steady-state kinetics of S. aureus SQR first utilizing cyanide (CN−) as the S0 acceptor.21 Briefly, the initial rate of quinone reduction was measured as a function of sulfide concentration at a saturating CN− concentration (4 mM), in the presence of 100 nM wild-type SQR. A number of water-soluble quinones were chosen to serve as the electron acceptors as previously described for Bacillus stearothermophilus sulfide dehydrogenase.31 In the presence of DQ (Figure 4A) and decyl-UQ, S. aureus SQR exhibits Km values for sulfide of 22 ± 7 and 39 ± 6 μM, respectively (Table 1). With DMN as the electron acceptor, a lower sulfide Km was observed (Km = 642 ± 121 μM) (Table 1). These data reveal that S. aureus SQR is capable of utilizing a spectrum of water-soluble quinones as electron acceptors that vary widely in midpoint potentials (E1/2,DMN = −635 mV, E1/2,DQ = 5 mV, and E1/2,UQ = 45 mV),35,36 with DQ yielding the lowest Km for sulfide.
Figure 4.

Kinetic characterization of S. aureus SQR. (A) Wild-type (WT) SQR exhibits sulfide:quinone oxidoreductase activity with the sulfide substrate (Na2S), while C167S and C344S SQRs exhibit no activity. The duroquinone turnover is plotted as a function of Na2S concentration for WT SQR (○), C167S SQR (□), and C344S SQR (◇). The dashed lines through empty circles, empty squares, and empty diamonds are fits to the Michaelis–Menten equation (see Table 2). (B) SQR and CstB catalyze the production of thiosulfate from the sulfide substrate (Na2S), with no activity observed with the C167S and C344S SQRs. Thiosulfate turnover is plotted as a function of concentration of Na2S for WT SQR (○), C167S SQR (□), and C344S SQR (◇). The dashed lines through the empty circles, empty squares, and empty diamonds are fits to the Michaelis–Menten equation (see Table 2).
Table 1.
Sulfide:Quinone Oxidoreductase Activity of S. aureus SQR with Sulfide as the Substrate in the Presence of Different Quinones as the Electron Acceptora
| electron acceptor | Km (μM) | Vmax (μmol min−1 mg−1) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| duroquinone | 22 ± 7 | 0.41 ± 0.03 | 0.31 ± 0.02 | (1.41 ± 0.46) × 104 |
| decyl-ubiquinone | 39 ± 6 | 0.25 ± 0.01 | 0.19 ± 0.01 | (0.49 ± 0.08) × 104 |
| DMN | 642 ± 121 | 1.18 ± 0.13 | 0.90 ± 0.10 | (0.14 ± 0.03) × 104 |
Conditions: 40 nM SQR, 60 μM quinone electron acceptor, 4 mM KCN, 25 mM Tris-HCl, 0.2 M NaCl, pH 8.0, 25 °C.
Catalytic Role of the Cys167–Cys344 Pair for SQR during Sulfide Oxidation
We next measured the steady-state kinetics of sulfide oxidation using both FAD-bound C167S and C344S SQRs using DQ and CN− as the electron and S0 acceptors, respectively. As expected, the catalytic activities of both C167S and C344S mutant SQRs are abolished (Figure 4A), revealing the functional importance of the C167–C344 disulfide redox center in SQR activity (see Figure 2). Current models suggest that the Cys167–Cys344 pair is attacked by sulfide (HS−), with the formation of a C344 persulfide intermediate, with a covalent complex with the flavin cofactor mediated by Cys167 thiolate (Figure 2). Consistent with this, we find that in the absence of an electron acceptor quinone and an S0 acceptor, a large fraction of the Cys344-containing peptide shifts to a higher m/z by +32 Da following incubation of SQR with the sulfide substrate Na2S (Figure 5), a finding consistent with a C344 persulfide being the only observable modification for Na2S-reacted SQR. As expected, the ability to trap this persulfide is dependent on the C167–C344 disulfide bond because Na2S-reacted C167S SQR gives no persulfide product (Figure S5).
Figure 5.

MALDI MS analysis reveals a +32 Da mass shift for the Cys344-containing peptide from Na2S-reacted SQR in the absence of electron or S0 acceptors (see Figure 2). The unreacted SQR (top) and Na2S-reacted SQR (bottom) were alkylated with iodoacetamide, proteolytically digested with Asp-N, and analyzed by MALDI MS. The Cys344-containing peptide displays a mass of 2289.30 Da, and a portion displays a derivatized mass of 2321.28 Da after reaction with Na2S, indicating a mass shift of 31.98 Da, consistent with a persulfide moiety.
Reactivity of SQR for the Sulfide Oxidation Process with Different S0 Acceptors
S. aureus utilizes a bifunctional non-heme Fe(II) persulfide dioxygenase-sulfurtransferase, CstB, to oxidize LMW persulfides to thiosulfate,13 with both cstB and sqr shown to be critical in mitigating the effects of cellular sulfide toxicity in S. aureus.12 In this model, a LMW thiol delivers the S0 from SQR to CstB to connect these two activities. To investigate this, we used a cell-abundant LMW thiol, CoA, instead of CN−, as the S0 acceptor at 100 μM and measured the steady-state kinetics of SQR with sulfide. The Km values for sulfide are similar when CoA (32 ± 6 μM) or CN− (22 ± 7 μM) is used as the S0 acceptor, while the catalytic efficiencies differ by approximately 2-fold (Table 2). Sulfite was previously shown to be a physiologically important S0 acceptor for human mitochondrial SQR21,22 and thus was also assayed in the same way. Sulfite supports sulfide oxidation with a catalytic efficiency similar to that of CN− but with a reduced apparent sulfide Km of 249 ± 86 μM and a kcat/Km determined to be (1.4 ± 0.5) × 104 M−1 s−1 (Table 2). This may derive from competitive inhibition of initial sulfide binding by sulfite, which is capable of attacking the disulfide bond to form S-sulfocysteine, thus poisoning the enzyme.22
Table 2.
Sulfide:Quinone Oxidoreductase Activity of S. aureus SQR with Sulfide as the Substrate in the Presence of Different S0 Acceptorsa
| S0 acceptor | Km (μM) | Vmax (μmol min−1 mg−1) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| KCN | 22 ± 7 | 0.41 ± 0.03 | 0.31 ± 0.02 | (1.41 ± 0.46) × 104 |
| sulfite | 249 ± 86 | 4.51 ± 0.70 | 3.42 ± 0.53 | (1.37 ± 0.52) × 104 |
| CoAb | 32 ± 6 | 0.25 ± 0.01 | 0.19 ± 0.01 | (0.60 ± 0.12) × 104 |
Conditions: 40 nM SQR, 60 μM DQ, 4 mM S0 acceptor, 25 mM Tris-HCl, 0.2 M NaCl, pH 8.0, 25 °C.
The CoA concentration was 100 μM.
Oxidation of Sulfide by SQR/CstB Mixtures Leads to the Production of Thiosulfate
With SQR characterized as a type II SQR and CoA capable of functioning as an S0 acceptor, we next addressed if SQR might funnel the CoASSH product into the active site of CstB as a substrate for further oxidation.13 Activities analogous to those of SQR and CstB in mammalian mitochondrial H2S oxidation have been described but have not been assayed together.25 To test this, a previously described fluorescence-based sulfur metabolite profiling assay was used to quantify the production of thiosulfate,13 with sulfide substrate Na2S, in the presence of electron acceptor DQ and S0 acceptor CoA. In this mixing experiment, thiosulfate was indeed produced catalytically from oxidation of sulfide by stoichiometric SQR and CstB (Figure 4B). In contrast, if a C167S SQR/CstB or C344S SQR/CstB mixture is used, there is no detectable formation of thiosulfate (Figure 4B). However, when the initial rate of thiosulfate formation is plotted as a function sulfide concentration (Figure 4B), the activity of the complex is half-maximal at 2.1 ± 0.3 mM sulfide, far above the Km for sulfide for SQR alone, but similar to the Km for CstB-dependent CoASSH oxidation into thiosulfate, 1.1 ± 0.3 mM.13 The apparent kcat for the production of thiosulfate is 0.16 ± 0.01 s−1 and is comparable to the kcat for SQR with sulfide, 0.31 ± 0.02 s−1 (Table 1), but far lower than that of CstB with the CoASSH substrate, 19.3 ± 2.2 s−1.13 Thus, although the catalytic production of thiosulfate from sulfide is observed in vitro by mixing of the two enzymes, the turnover rate is limited by the substrate binding affinity of CstB and the turnover number for SQR. Thus, the catalytic “boost” one might expect to see from substrate “channeling” from SQR to CstB, for example, is not observed. In any case, control assays using high concentrations of enzyme revealed no significant thiosulfate production in the absence of CoA as an S0 acceptor, relative to robust production in the presence of CoA (Figure S6). We could find no compelling evidence that CstB and SQR strongly physically interact (data not shown), consistent with these kinetic findings.
LMW Persulfide and Inorganic (Poly)sulfides and Polysulfide Profiling of, WT, Δsqr, and Δsqr::pSQR Strains before and after Exogenous Sulfide Stress
Having biochemically characterized SQR, we next performed experiments to assess the physiological impact of an sqr deletion. The Δsqr strain exhibits a growth phenotype in the presence of exogenous 0.2 mM NaHS, evidenced by an extended lag phase in replicate experiments, which can be complemented by ectopic expression of the wild-type allele from an extrachromosomal plasmid to a degree similar to that of the wild-type strain (Figure 6B).12 This differential growth lag for the Δsqr versus the wild-type and SQR-complemented strains is not due to differential SQR-dependent depletion or volatilization of sulfide from the growth medium, as the rate of sulfide loss is independent of the strain being cultured (Figure 6A). We next measured the intracellular concentrations of both organic and inorganic reactive sulfur species (RSS) before (t = 0) and after (t = 30 min) exogenous sulfide (0.2 mM Na2S) stress to early mid log cells (OD600 ≈ 0.2) (Figure 7 and Figure S7). Consideration of simple substrate–product relationships results in the prediction that inorganic sulfide species would potentially accumulate in the Δsqr strain relative to the wild-type and complemented strains, while the LMW persulfide concentrations [BSSH, CoASSH, and CysSSH (Figure 7A–C)] would be lower in the Δsqr strain than in the wild-type strain.
Figure 6.

Sulfide stress induces an enhanced growth phenotype in a Δsqr strain that is complemented by SQR expression. (A) Total medium sulfide measured as a function of time after dissolution of 0.2 mM NaHS into sterile growth medium (HHWm and thiosulfate) (blue filled circles) compared to the culture medium obtained from WT::pOS1 (green empty circles), Δsqr::pOS1 (red empty left triangles), and Δsqr::SQR (red filled left triangles) cell growth. The dashed line is a linear fit to the average value determined at each time point, revealing only a small, strain-independent decrease in the total sulfide concentration over the time course of each growth curve. (B) Average growth curves for wild-type (WT), Δsqr, and plasmid-complemented Δsqr::SQR S. aureus Newman strains grown on HHWm/thiosulfate medium in the absence of NaHS (−NaHS, empty symbols) and 0.2 mM NaHS (+NaSH, filled symbols) added at time zero: WT (black circles), Δsqr (red left triangles), and Δsqr::pSQR (blue diamonds). Error bars from triplicate experiments are shown for the NaHS-stressed WT, Δsqr, and Δsqr::pSQR strains only, for the sake of clarity.
Figure 7.

Cellular LMW persulfide and inorganic sulfide profiling of wild-type (WT), Δsqr, and Δsqr::pSQR S. aureus Newman strains before (t = 0) and after (t = 30 min) addition of 0.2 mM Na2S to early log phase (OD600 = 0.2) cultures: (A) bacillithiol persulfide (BSSH) for WT (black bars), Δsqr (gray bars), and Δsqr::pSQR (white bars) strains, (B) CoASSH, and (C) cysteine persulfide (CysSSH). All cultures were grown in HHWm medium supplemented with 0.5 mM thiosulfate and 10 μg/mL chloramphenicol. WT and Δsqr::pSQR samples were analyzed as biological triplicates, and the Δsqr strain was analyzed as a biological replicate. Analogous data are shown for inorganic hydrodisulfide and tetrasulfide species (Figure S7).
What is actually observed differs dramatically from this prediction. A major observation is that significant reactive sulfur species (RSS) are found in unstressed cells (t = 0), with [BSSH] > [CoASSH] > [CysSSH] (Figure 7). Deletion of sqr gives rise to comparatively little change in BSSH (Figure 7A) and CysSSH (Figure 7C) concentrations. In contrast, the endogenous CoASSH concentration is ≈15-fold higher in the Δsqr strain, which is complemented by ectopic SQR expression (Figure 7C). Addition of exogenous sulfide, while increasing the concentrations of all RSSH ≈10-fold, tends to minimize the differences between these RSS among the three strains, with those of CoASSH and CysSSH increasing 2–4-fold, with SQR complementation slightly overcompensating. The one exception to this trend is BSSH, which is largely insensitive to sqr deletion before and after exogenous sulfide stress (Figure 7A). Parallel ≈50–100-fold increases in the hydrodisulfide and tetrasulfide concentrations are also observed upon sulfide stress, with some key differences (see Discussion) (Figure S7). These experiments show that deletion of SQR specifically impacts endogenous CoA persulfide levels, with comparatively smaller but measurable effects on other sulfur-containing species in cells.
DISCUSSION
In this work, we present a functional characterization of a bacterial type II SQR from a major human pathogen. The sqr gene is encoded by the cst operon and can be induced by exogenous sulfide stress, consistent with a role in sulfide and RSS homeostasis.12 S. aureus SQR is monomeric, harbors a noncovalently bound flavin cofactor, and generally exhibits properties consistent with human and other bacterial SQRs previously characterized. We provide direct evidence of a persulfide intermediate on the C-terminal Cys of the disulfide redox center (C344) and show that when SQR is mixed with the non-heme FeII persulfide dioxygenase CstB, thiosulfate is synthesized in a CoA-requiring process, thus invoking the intermediacy of CoASSH as a product and substrate for SQR and CstB, respectively. However, we could obtain no strong kinetic or physical support for the idea that SQR and CstB functionally interact. CoASSH is readily detected in early log phase and unstressed S. aureus cells, and deletion of sqr specifically increases endogenous CoASSH concentrations in these cells.
The repressor of the cst operon, CstR, senses both inorganic (poly)sulfide species as well as organic persulfides of LMW thiols,12 which accumulate in cells following exogenous sulfide stress (H. Peng, unpublished observations). However, these RSS are also present at significant concentrations in unstressed cells (Figure 7), with endogenous concentrations readily genetically manipulated in ΔcstR and ΔcysM/ΔmetB strains, the latter of which encodes two enzymes of the transsulfuration pathway and significant sources of endogenous H2S in S. aureus.5 These endogenous concentrations of BSSH, CoASSH, and CysSSH are in the range of 10–60 μM, with inorganic polysulfide species in unstressed cells present in the range of 70 μM (Figure 7 and Figure S7), assuming a cell diameter of 0.6 μm. These endogenous concentrations of sulfide are on the order of the SQR Km for sulfide (Tables 1 and 2), thus suggesting that any residual expression of the cst operon under housekeeping conditions will tend to maintain intracellular sulfide, and by extension other LMW thiol persulfides, in this concentration range. In contrast, these concentrations of organic persulfides are far below the measured Km for CstB,13 suggesting that this enzyme may not be strongly relevant to organic persulfide clearance unless the cell experiences massive sulfide influx or dysregulation.
Our RSS profiling experiments suggest that SQR, perhaps in conjunction with other cst-encoded enzymes, may be primarily tasked with ensuring that endogenous levels of CoA persulfide (at ≈1% of the CoA pool, H. Peng, unpublished observations) are not permitted to accumulate under unstressed or ambient growth conditions. Deletion of the sqr gene has no effect on the persulfide concentrations of the two other major thiols in S. aureus, BSSH and CysSSH, while CoASSH levels rise ≈15–20-fold. Inorganic tetrasulfide (S4) levels also spike dramatically relative to hydrodisulfide levels, which fall, suggesting a significant change in the inorganic sulfur specification in the cell as well (Figure S7). These findings are not easily rationalized on the basis of simple SQR substrate–product relationships, with the possible exception of hydrodisulfide, which human SQR can synthesize when sulfide functions as both the substrate and the S0 acceptor (see Figure 2).21 In unstressed cells, the level of hydrodisulfide is lower in the Δsqr strain, but this effect it not clearly complemented.
One possibility is that the loss of SQR leads to a sulfane sulfur “scrambling” via trans-persulfidation or other related processes6 among inorganic, organic, and proteome-derived S-sulfhydration (H. Peng, unpublished observations), creating a dynamic and interconvertible pool of bioactive sulfur that differentially impacts CoA persulfidation. CoA, unlike other major thiols BSH and Cys, is a metabolically important cofactor and precursor to the high-energy thioester, acetyl-CoA; extensive persulfidation of CoA would potentially disrupt the TCA cycle and other key energy-yielding processes, e.g., fatty acid metabolism, that depend on CoA availability. It is interesting to note that treatment of S. aureus MRSA and MSSA strains with nonlethal doses of antibiotics, e.g., oxacillin, results in an upregulation of peptidoglycan biosynthesis and, curiously, enzymes encoded by coaBC and coaE that lead to CoA from pantothenate.37 Because endogenously synthesized sulfide protects S. aureus from the effects of general antibiotic stress,5 it seems possible that SQR and other cst-encoded enzymes may function to reduce the impact of collateral metabolic damage mediated by extensive CoA persulfidation. If true, this would provide a rationale for the finding that a duplicated core cst operon appears genetically linked to methicillin resistance determinants in many MRSA strains.13 Studies are underway to further elucidate the physiological impact of the cst operon on oxidative and nitrosative stress resistance8,38 and antibiotic susceptibility.5
Supplementary Material
Acknowledgments
Funding
The authors gratefully acknowledge support by the National Institutes of Health (R01 GM097225 and R35 GM118157 to D.P.G.).
This work has been submitted by J.S. to the Graduate School of Indiana University in partial fulfillment of the requirements for the Ph.D. in Biochemistry. The authors thank Mr. Aaron Lee and Drs. Katie Edmonds and Julia Martin for many helpful discussions about this work.
ABBREVIATIONS
- Cst
CsoR-like sulfurtransferase
- DQ
duroquinone
- decyl-UQ
decyl-ubiquinone
- DMN
2,3-dimethyl-1,4-naphthoquinone
- HHWm
Hussain–Hastings–White modified chemically defined medium
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- LMW
low-molecular weight
- PDB
Protein Data Bank
- RSS
reactive sulfur species
- SQR, sulfide
quinone oxidoreductase
- S0
sulfane sulfur
- TCEP
tris(2-carboxyethyl)-phosphine
- TS
thiosulfate
Footnotes
Author Contributions
J.S. and D.P.G. designed the research. J.S. and H.P. performed the research. Y.Z. performed LC–MS/MS analysis. J.S. and D.P.G. wrote the manuscript.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.6b00714.
Supplemental Figures S1–S7 and supplemental references (PDF)
References
- 1.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 2.Kolluru GK, Shen X, Bir SC, Kevil CG. Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide. 2013;35:5–20. doi: 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathai JC, Missner A, Kugler P, Saparov SM, Zeidel ML, Lee JK, Pohl P. No facilitator required for membrane transport of hydrogen sulfide. Proc Natl Acad Sci U S A. 2009;106:16633–16638. doi: 10.1073/pnas.0902952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czyzewski BK, Wang DN. Identification and characterization of a bacterial hydrosulphide ion channel. Nature. 2012;483:494–497. doi: 10.1038/nature10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shatalin K, Shatalina E, Mironov A, Nudler E. H2S: A universal defense against antibiotics in bacteria. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- 6.Mishanina TV, Libiad M, Banerjee R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat Chem Biol. 2015;11:457–464. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin VS, Lippert AR, Chang CJ. Cell-trappable fluorescent probes for endogenous hydrogen sulfide signaling and imaging H2O2-dependent H2S production. Proc Natl Acad Sci U S A. 2013;110:7131–7135. doi: 10.1073/pnas.1302193110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luhachack L, Nudler E. Bacterial gasotransmitters: an innate defense against antibiotics. Curr Opin Microbiol. 2014;21:13–17. doi: 10.1016/j.mib.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 10.Soutourina O, Poupel O, Coppee JY, Danchin A, Msadek T, Martin-Verstraete I. CymR, the master regulator of cysteine metabolism in Staphylococcus aureus, controls host sulphur source utilization and plays a role in biofilm formation. Mol Microbiol. 2009;73:194–211. doi: 10.1111/j.1365-2958.2009.06760.x. [DOI] [PubMed] [Google Scholar]
- 11.Grossoehme N, Kehl-Fie TE, Ma Z, Adams KW, Cowart DM, Scott RA, Skaar EP, Giedroc DP. Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J Biol Chem. 2011;286:13522–13531. doi: 10.1074/jbc.M111.220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luebke JL, Shen J, Bruce KE, Kehl-Fie TE, Peng H, Skaar EP, Giedroc DP. The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol Microbiol. 2014;94:1343–1360. doi: 10.1111/mmi.12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen J, Keithly ME, Armstrong RN, Higgins KA, Edmonds KA, Giedroc DP. Staphylococcus aureus CstB Is a novel multidomain persulfide dioxygenase-sulfurtransferase involved in hydrogen sulfide detoxification. Biochemistry. 2015;54:4542–4554. doi: 10.1021/acs.biochem.5b00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins KA, Peng H, Luebke JL, Chang FM, Giedroc DP. Conformational analysis and chemical reactivity of the multidomain sulfurtransferase. Biochemistry. 2015;54:2385–2398. doi: 10.1021/acs.biochem.5b00056. [DOI] [PubMed] [Google Scholar]
- 15.Argyrou A, Blanchard JS. Flavoprotein disulfide reductases: advances in chemistry and function. Prog Nucleic Acid Res Mol Biol. 2004;78:89–142. doi: 10.1016/S0079-6603(04)78003-4. [DOI] [PubMed] [Google Scholar]
- 16.Marcia M, Ermler U, Peng G, Michel H. A new structure-based classification of sulfide:quinone oxidoreductases. Proteins: Struct, Funct, Genet. 2010;78:1073–1083. doi: 10.1002/prot.22665. [DOI] [PubMed] [Google Scholar]
- 17.Marcia M, Ermler U, Peng G, Michel H. The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc Natl Acad Sci U S A. 2009;106:9625–9630. doi: 10.1073/pnas.0904165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brito JA, Sousa FL, Stelter M, Bandeiras TM, Vonrhein C, Teixeira M, Pereira MM, Archer M. Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry. 2009;48:5613–5622. doi: 10.1021/bi9003827. [DOI] [PubMed] [Google Scholar]
- 19.Chen ZW, Koh M, Van Driessche G, Van Beeumen JJ, Bartsch RG, Meyer TE, Cusanovich MA, Mathews FS. The structure of flavocytochrome c sulfide dehydrogenase from a purple phototrophic bacterium. Science. 1994;266:430–432. doi: 10.1126/science.7939681. [DOI] [PubMed] [Google Scholar]
- 20.Cherney MM, Zhang Y, Solomonson M, Weiner JH, James MN. Crystal structure of sulfide:quinone oxidor-eductase from Acidithiobacillus ferrooxidans: insights into sulfidotrophic respiration and detoxification. J Mol Biol. 2010;398:292–305. doi: 10.1016/j.jmb.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 21.Jackson MR, Melideo SL, Jorns MS. Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry. 2012;51:6804–6815. doi: 10.1021/bi300778t. [DOI] [PubMed] [Google Scholar]
- 22.Mishanina TV, Yadav PK, Ballou DP, Banerjee R. Transient kinetic analysis of hydrogen sulfide oxidation catalyzed by human sulfide quinone oxidoreductase. J Biol Chem. 2015;290:25072–25080. doi: 10.1074/jbc.M115.682369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theissen U, Hoffmeister M, Grieshaber M, Martin W. Single eubacterial origin of eukaryotic sulfide:quinone oxidoreductase, a mitochondrial enzyme conserved from the early evolution of eukaryotes during anoxic and sulfidic times. Mol Biol Evol. 2003;20:1564–1574. doi: 10.1093/molbev/msg174. [DOI] [PubMed] [Google Scholar]
- 24.Grieshaber MK, Volkel S. Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu Rev Physiol. 1998;60:33–53. doi: 10.1146/annurev.physiol.60.1.33. [DOI] [PubMed] [Google Scholar]
- 25.Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 26.Ma YB, Zhang ZF, Shao MY, Kang KH, Shi XL, Dong YP, Li JL. Response of sulfide:quinone oxidoreductase to sulfide exposure in the echiuran worm Urechis unicinctus. Mar Biotechnol. 2012;14:245–251. doi: 10.1007/s10126-011-9408-1. [DOI] [PubMed] [Google Scholar]
- 27.Vande Weghe JG, Ow DW. A fission yeast gene for mitochondrial sulfide oxidation. J Biol Chem. 1999;274:13250–13257. doi: 10.1074/jbc.274.19.13250. [DOI] [PubMed] [Google Scholar]
- 28.Jackson MR, Melideo SL, Jorns MS. Role of human sulfide: quinone oxidoreductase in H2S metabolism. Methods Enzymol. 2015;554:255–270. doi: 10.1016/bs.mie.2014.11.037. [DOI] [PubMed] [Google Scholar]
- 29.Becher D, Hempel K, Sievers S, Zuhlke D, Pane-Farre J, Otto A, Fuchs S, Albrecht D, Bernhardt J, Engelmann S, Volker U, van Dijl JM, Hecker M. A proteomic view of an important human pathogen–towards the quantification of the entire Staphylococcus aureus proteome. PLoS One. 2009;4:e8176. doi: 10.1371/journal.pone.0008176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibata H, Kobayashi S. Characterization of a HMT2-like enzyme for sulfide oxidation from Pseudomonas putida. Can J Microbiol. 2006;52:724–730. doi: 10.1139/w06-022. [DOI] [PubMed] [Google Scholar]
- 31.Shibata H, Suzuki K, Kobayashi S. Menaquinone reduction by an HMT2-like sulfide dehydrogenase from Bacillus stearothermophilus. Can J Microbiol. 2007;53:1091–1100. doi: 10.1139/W07-077. [DOI] [PubMed] [Google Scholar]
- 32.Aliverti A, Curti B, Vanoni MA. Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol Biol. 1999;131:9–23. doi: 10.1385/1-59259-266-X:9. [DOI] [PubMed] [Google Scholar]
- 33.Toledo-Arana A, Merino N, Vergara-Irigaray M, Debarbouille M, Penades JR, Lasa I. Staphylococcus aureus develops an alternative, ica-independent biofilm in the absence of the arlRS two-component system. J Bacteriol. 2005;187:5318–5329. doi: 10.1128/JB.187.15.5318-5329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 35.Nasiri HR, Panisch R, Madej MG, Bats JW, Lancaster CR, Schwalbe H. The correlation of cathodic peak potentials of vitamin K(3) derivatives and their calculated electron affinities. The role of hydrogen bonding and conformational changes. Biochim Biophys Acta, Bioenerg. 2009;1787:601–608. doi: 10.1016/j.bbabio.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 36.Denke E, Merbitz-Zahradnik T, Hatzfeld OM, Snyder CH, Link TA, Trumpower BL. Alteration of the midpoint potential and catalytic activity of the Rieske iron-sulfur protein by changes of amino acids forming hydrogen bonds to the iron-sulfur cluster. J Biol Chem. 1998;273:9085–9093. doi: 10.1074/jbc.273.15.9085. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Hu Y, Pai PJ, Chen D, Lam H. Label-free quantitative proteomics analysis of antibiotic response in Staphylococcus aureus to oxacillin. J Proteome Res. 2014;13:1223–1233. doi: 10.1021/pr400669d. [DOI] [PubMed] [Google Scholar]
- 38.Filipovic MR, Eberhardt M, Prokopovic V, Mijuskovic A, Orescanin-Dusic Z, Reeh P, Ivanovic-Burmazovic I. Beyond H2S and NO interplay: hydrogen sulfide and nitroprusside react directly to give nitroxyl (HNO). A new pharmacological source of HNO. J Med Chem. 2013;56:1499–1508. doi: 10.1021/jm3012036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.