

Abstract
A pilot program was initiated using whole genome sequencing (WGS) to diagnose suspected genetic disorders in the Genetics Clinic at Children's Hospital of Wisconsin. Twenty-two patients underwent WGS between 2010 and 2013. Initially, we obtained a 14% (3/22) diagnosis rate over 2 years; with subsequent reanalysis, this increased to 36% (8/22). Disease causing variants were identified in SKIV2L, CECR1, DGKE, PYCR2, RYR1, PDGFRB, EFTUD2, and BCS1L. In 75% (6/8) of diagnosed cases, the diagnosis affected treatment and/or medical surveillance. Additionally, one case demonstrated a homozygous A18V variant in VLDLR that appears to be associated with a previously undescribed phenotype.
Keywords: genome, sequencing, diagnosis, clinic
Introduction
Research use of next-generation sequencing has successfully identified pathogenic variants in the genes responsible for numerous rare genetic disorders.1 One of the earliest uses of this technology in clinical practice was the successful diagnosis and treatment of a child with a rare immunologic disorder.2 This case showed that whole exome sequencing (WES) has a place in clinical care. The exome represents ∼1.5% of the genome and, in this case, exome sequencing provided a cost-effective approach for analysis of the patient's sample. WES was subsequently used in a research setting to establish disease etiology in a larger number of cases.3
Today, commercial and academic diagnostic laboratories offer WES and whole genome sequencing (WGS), together referred to as genomic or genome-wide testing, to clients across the United States and abroad. Genomic testing has been proven to be effective in the diagnosis of rare diseases4 in both children5 6 7 8 9 10 11 and adults.12 Recently, WES provided diagnostic and/or potentially actionable findings in 40% of pediatric cases with newly diagnosed solid tumors.13 Importantly, Miller14 has shown that testing can yield results rapidly. A WES approach is used more often than WGS for both gene discovery and clinical diagnostics due to its lower cost.
As genomic testing enters clinical practice broadly, many questions and proposals have emerged concerning next-generation sequencing's reliability, variant interpretation, integration of testing into current practice, impact of results on clinical management, test regulation, and test reimbursement.15 16 17 18 19 Nationally funded programs designed to deliver actionable genomic information into medical practice are underway.20
With these questions in mind, the Medical College of Wisconsin (MCW) joined with Children's Hospital of Wisconsin (CHW) in 2010 to create a unified genomic medicine program. This paper describes the development of that program, the findings from initial cases, and specific information describing how WGS can be integrated into clinical care.
Methods
Setting
Success with our initial case2 led to significant internal demand for genomic sequencing to be available as a part of routine clinical practice at CHW. In June 2010, a team with expertise in clinical genetics, laboratory genetics, pathology, and genomics met with senior leadership from MCW and CHW to establish a pilot program to provide genomic sequencing for clinical diagnostic purposes.
Case Referral
The pilot program sought to identify a molecular etiology in cases with a suspected Mendelian disorder where standard testing had failed to yield a diagnosis and where a diagnosis would enhance medical decision making. Because resources were limited, cost-per-case was high ($25,000 for sequencing alone at the outset), and insurance coverage was uncertain, a referral procedure and a multidisciplinary case review team to select cases were established. All specialties at CHW were encouraged to submit cases.
The review team met monthly. Referral required two physicians with expertise in the patient's disorder to determine that sequencing the patient's genome was clinically warranted and that other standard clinical assessments had been attempted and had failed to define the cause of the patient's disorder. A case summary of the patient's history (Supplement 1, available in the online version), family history (four-generation pedigree), physical examination, and laboratory evaluation was prepared for the review team.
Case Review Team and Case Selection
The case review team was led by the hospital's chief medical officer and included two ethicists, three physicians not directly involved with the case, a clinical geneticist with expertise in DNA testing, a genetic counselor, an informatics specialist, a genomics expert, a pathologist, and a CHW administrative leader. The informatics specialist was a PhD scientist with extensive experience in human genomic analysis and the developer of CarpeNovo, the software tool used to analyze all of the cases in this report. The genomics expert was a PhD scientist with extensive experience in genomics research. A case review process that accounted for the limitations of the technology was developed by the case review team with input from legal counsel, hospital administration, an internal ethics board, and by external ethics review.21
The team developed case selection requirements with the aim to integrate WGS into routine medical care (Table 1). The decision to recommend WGS was based on the information supplied by the referring physicians in a case summary (Supplement 1, available in the online version). This summary ranged from two to five pages.
Table 1. Case selection requirements for WGS.
| 1. Obtain a case summary from physicians with expertise in the patient's condition to help the laboratory correlate the patient's phenotype and family history with the genotype derived from sequencing. |
| 2. Confirm that the patient has had the current standard diagnostic testing used to evaluate that patient's phenotype to ensure that a cost-effective approach has been taken to diagnosis. |
| 3. Choose patients with an apparently undiagnosed monogenic genetic disorder ideally with a rare or distinctive phenotype. |
| 4. Focus on cases where a molecular diagnosis could help physicians/families with medical decision making/management such as treatment and family planning, a key to securing insurance preauthorization. |
| 5. Select cases with appropriate samples available to carry out initial genomic sequencing as well as follow-on testing including confirmatory functional assays in the patient and segregation analysis of variants in the parents and other family members. |
| 6. Choose cases where the cost of genomic sequencing appears to be less costly than testing many individual genes for the phenotype in question. |
Abbreviation: WGS, whole genome sequencing.
Note: The criteria that the review committee used to select cases for whole genome sequencing are summarized.
Following a review of each case, the case review team made one of the following decisions: (1) recommend WGS, (2) recommend additional testing and/or additional information prior to resubmission, (3) reserve for future consideration, and (4) WGS not recommended. As this was a clinical program, cases were selected without consideration of their research potential. Although the pilot program itself was clinical, some limited research aspects were also included. A study of patient outcomes and attitudes was approved by the Institutional Review Board at the MCW and patients (or their family members) provided written informed consent.
Genetic Counseling, Consent, and Sample Submission
Genetic counseling was provided by a genomics trained, board-certified genetic counselor prior to WGS. A note was entered into the medical record describing the counseling, and the consent form required by the laboratory was completed. Supplement 2, available in the online version lists the topics typically covered during counseling. Due to the nature of the test, counseling concerning incidental findings received special consideration. An incidental finding was defined as a variant identified in the patient that published literature identified as causing a Mendelian disorder unrelated to the patient's current phenotype. A form to indicate which types of incidental findings the family wished to have returned (Supplement 3, available in the online version) was completed and provided to the laboratory. This approach was based on the recognition of patient and parental rights as applied in other societal contexts and other areas of clinical medicine.22 Pharmacogenetic variants and risk alleles such as ApoE4 were not reported.
At the time of sample submission, the case summary (Supplement 1, available in the online version) was provided to the laboratory. In addition, a list of genes thought to be associated with the patient's phenotype was generated by the geneticist and clinician involved in the case (Supplement 4, available in the online version) and submitted to the laboratory. To reduce the chance of uncovering incidental findings, families were informed that if a diagnosis was established based on the gene list, further analysis of the genome would not be undertaken. The patient/family preference form (Supplement 3, available in the online version) was used by the laboratory to guide analysis.
When testing failed to identify an etiology, routine yearly follow-up was offered, at which time, clinical information was updated and the genome reevaluated.
Genomic Sequencing and Analysis
All testing was performed in CLIA certified/College of American Pathologist accredited laboratories. Blood was drawn from each patient and both biological parents when possible. DNA was extracted using a Gentra Puregene Cell Kit (Qiagen, Germantown, MD, United States). Cases were sequenced on an Illumina HiSEq, 2000 or Illumina GAIIx system (Illumina Clinical Services Laboratory, San Diego, CA, United States). All variants identified as candidates for clinical reporting were evaluated by dideoxynucleotide sequencing using an ABI 3730 capillary sequencer (Advanced Genomics Laboratory, Children's Hospital of Wisconsin, Milwaukee, WI, United States). Parental DNA was used for variant segregation analysis.
Sequence analysis took place in three phases: primary, secondary, and tertiary.
Primary analysis: Primary analysis took place on the Illumina sequencer. Two × 100 base pair reads were produced using on-board cluster generation and paired-end sequencing.
Secondary analysis: The Illumina Clinical Services Laboratory analyzed the data using the CASAVA mapping, alignment, and variant calling software (versions 1.6–1.8 depending on the sample; for review of this step, see Worthey 2013).23 Sequence reads were quality selected (according to metrics in Supplement 5, available in the online version). Y chromosome trimming, creation of a consensus file, analysis of DNA sequencing, mapping to the NCBI Human reference genome (build 36 or 37), making use of variant data from dbSNP (versions 28–31), genotyping concordance, and finally variant calling were performed. The output was analyzed to ensure quality (an overall accuracy of > 99.9% and an accuracy at the poorest quality calls of 97%).
Tertiary analysis: Tertiary analysis to annotate identified variants was performed by an in-house developed tool (CarpeNovo). For a thorough review of the types of annotations incorporated, see Worthey 2013.23
In brief, CarpeNovo annotates each variant with the following: gene symbols and NCBI gene and transcript identifiers; genomic coordinates (hg18 or hg19 depending on when the sample was run); reference and patient's nucleotides; allele frequencies derived from dbSNP,24 1000 Genomes Project,25 Exome Variant Server,26 an in-house database of publicly available genomes called Valcrie, and the CarpeNovo in-house patients sample database; genomic and genic location information (intron, exon, UTR, intergenic, etc.); variant effect information (start site, stop site, splice site, nonsynonymous, deletion, etc.); amino acid and cDNA position and change; predicted effect on protein based on Polyphen,27 MutationAssessor,28 and SIFT29; protein length; nucleotide conservation score30; depth of coverage at the variant position; percentage of the reads supporting the variant call; presence of and disease terms for the specific variant in HGMD31 and/or OMIM32; presence of and disease terms for the gene in HGMD or OMIM; and whether the region containing the nucleotide maps to a unique location in the genome. Importantly, the software further annotates the variants based on their level of known or likely deleterious impact and identifies variants in genes associated previously with genetic diseases. All external databases and incorporated tools are tagged with version and date stamps.
Post-Tertiary Analysis and Interpretation
Following annotation, the CarpeNovo platform allows the user to filter and prioritize variants based on these annotations. In this pilot program, analysis of each case was performed by applying our custom prioritization schema, which takes into account the likely impact of the variant, the allelic frequency of the variant in reference datasets, and functional associations for the variant harboring gene (Supplement 6, available in the online version).
Since the patients in this pilot were presumed to have rare diseases, an allele frequency threshold of 1% or less was used to exclude common variants. Variant segregation among the patients' parents, cases reported in PubMed, and cases found in locus-specific databases were used to further include or exclude variants from consideration. For variants reported in the literature, the original publication and any supplemental publications were carefully reviewed to determine the quality and nature of the supporting evidence. Variant classification for reporting purposes followed standard guidelines.33
Evaluation of Coverage
Coverage of the genome was evaluated by GapMine, an analysis tool that evaluates sequence coverage at the genome, chromosome, gene, transcript, and individual exon level. For this analysis, we used a threshold of less than eightfold coverage (8×) to delineate low coverage. This threshold was not used as a strict cutoff for evaluation, but rather variants at positions less than 8× were considered potentially false positive calls and the apparent zygosity was deemed suspect. The chance that a variant allele will be missed is less than 1% when a nucleotide has greater than sevenfold coverage in a diallelic system based on a binomial probability. This depth of coverage is expected to yield an error rate comparable to the error rate that occurs in standard dideoxynucleotide sequencing for a single nucleotide variant.
Results
Committee Case Selection and Genetic Counseling
Of 57 cases submitted for review, 22 had WGS performed (Fig. 1). Table 2 briefly describes the case findings when testing occurred based on the case summaries supplied to the laboratory, WGS results, and impact of the diagnosis on the patient's care. Among the 22 patients, 10 had a DNA microarray performed prior to WGS employing either the Affymetrix Genome-Wide Human SNP Array 6.0 or a high-resolution oligonucleotide array CGH. Preliminary information concerning the 22 cases was previously reported.34
Fig. 1.
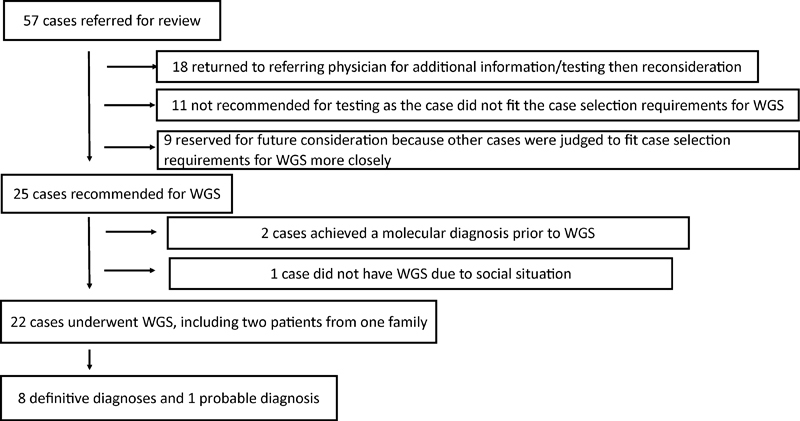
Decision tree for 57 cases. The case review team evaluated 57 cases submitted for whole genome sequencing. The disposition of the cases is shown.
Table 2. Case summaries and WGS outcomes.
| Case number and case summary | Result | Medication | Anticipatory guidance | Medical surveillance | Reproductive consequences for parents |
|---|---|---|---|---|---|
| Case 1: 3-month-old female whose newborn T-cell receptor excision circles (TREC) assay identified a profound T cell lymphopenia. A majority of subsequent TREC analyses were 0. Additional testing found an interlukin-7 receptor signaling defect. | No diagnosis | ||||
| Case 2: 19-month-old male who presented with a seizure disorder at 2 to 3 weeks of age. The patient subsequently had poorly controlled seizures, severely delayed developmental, hypertonia, and profound symmetric growth failure. MRI showed diffuse supratentorial leukoencephalopathy with gray and white matter abnormalities and lactic acid peaks within the globus pallidus bilaterally. | No diagnosis | ||||
| Case 3: 3-year 7-month-old girl who presented in the first year of life with multiple infections. Testing found low T-cell numbers that were severely skewed toward the memory phenotype. The patient subsequently had failure to thrive, enteropathy, and recurrent infections. The patient's full brother also has a primary immune deficiency found on newborn T-cell receptor excision circles assay. | No diagnosis | ||||
| Case 4: 10-year 7-month-old girl who presented with motor delay at 1 year of age. The patient subsequently developed spastic diplegia, ataxia, and intellectual disability. At age 2 years, MRI found leukodystrophy. Cerebellar atrophy appeared at age 5 years 6 months. | No diagnosis | ||||
| Case 5: 4-year 9-month-old girl who was born at 30 weeks of gestation following a pregnancy complicated by oligohydramnios, intrauterine growth retardation, and maternal hypertension. The patient subsequently had recurrent infections, profound symmetric growth failure, and diarrhea. Additionally, the patient has left aural atresia, bilateral hearing loss, a small muscular VSD, and trichorrhexis nodosa. The patient was clinically diagnosed with trichohepatoenteric syndrome. | SKIV2Lc.3187C > T (p.R1063X); c.2203–1G > C (NM_006929.4) | Supported use of subcutaneous IgG | Disorder associated with severe diarrhea, protein losing enteropathy, immune dysfunction, abnormal neurocognitive development, short stature, hearing abnormality, hypothyroidism, liver dysfunction | Thyroid function testing, hepatic transaminase testing | One in four risk of recurrence |
| Case 6: 5-year 7-month-old boy who presented in the first 2 years of life with recurrent infections. He subsequently had recurrent fevers and hepatosplenomegaly. At 4 years 9 months he had an ischemic stroke of right thalamus with subsequent ischemic stroke of the midbrain and deep parenchymal hemorrhage involving the right frontal lobe. | CECR1arr 22q11.21(17695127–17723376)x1 (build 37); c.506G > A (R169Q) (NM_017424.2) | Supported use of subcutaneous IgG, etanercept, and anakinra. Research into use of recombinant ADA2 | Disorder associated with early-onset strokes, recurrent fevers, systemic vasculopathy, hepatosplenomegaly, immune disorder | Abdominal ultrasound for portal hypertension, hepatic transaminase testing | One in four risk of recurrence |
| Case 7: 4-year 6-month-old girl who presented with extreme irritability, hypotonia, and global developmental delays in the first few months of life. She subsequently had infantile spasms, cortical blindness, severe intellectual disability, spastic quadraparesis, sensorineural hearing loss, microcephaly, and dysphagia. | No diagnosis | ||||
| Case 8: 15-year 3-month-old boy who presented with developmental regression at 7 months of age. He subsequently had severe intellectual disability, seizure disorder, failure to thrive, and cortical blindness. | No diagnosis | ||||
| Case 9: 12-year 9-month-old girl who presented in infancy with tracheal compression and difficulty feeding due to a congenital plexiform neurofibroma of the right supraclavicular fossa and right neck requiring surgical debulking. The mass was followed by serial MRI throughout the patient's life with slow progression in the size of the plexiform neurofibroma. At age 12, a biopsy found a low-grade malignant peripheral nerve sheath tumor. | No diagnosis | ||||
| Case 10: 5-year 1-month-old girl who presented at 8 months of age with a dilated cardiomyopathy. She subsequently had recurrent hypoglycemia starting at 3 years 11 months. | No diagnosis | ||||
| Case 11: 2-year 6-month-old girl who presented at 5 months of age with hemolytic-uremic syndrome. She subsequently had several additional episodes. Two full brothers are also affected. The patient's parents are unaffected. The affected individuals were clinically diagnosed with atypical hemolytic-uremic syndrome. | DGKEc.966G > A (p.W322X); c.888 + 40A > G (NM_003647.2) | Supported decision not to use eculizumab or plasma therapy | Disorder associated with episodic hemolytic anemia, renal failure, and thrombocytopenia, onset before 1 year of age, fewer acute relapses with age, persistence of urinary findings of disease between relapses, and progression of renal insufficiency without relapses. If the disease requires renal transplant, then the underlying disorder will not affect the transplanted kidney. | Screen for/treat hypertension, urinalysis for hematuria and proteinuria | One in four risk of recurrence |
| Case 12: 9-year 7-month-old boy who presented at 9 months of age with failure to thrive, developmental delay, chorea, and hypertonia. He subsequently had dystonia, severe intellectual disability, spasticity, and continued feeding disorder. His brother has a very similar phenotype. | PYCR2c.784delCCT (p.261delSER); c.138 + 1G > T(NM_013328.3) | Disorder associated with microcephaly, severe psychomotor delay, failure to thrive, facial dysmorphology, ataxia, hyperkinetic movements, muscle atrophy, seizures, spasticity, and white matter hyperintensities with global brain atrophy. Intellectual and motor functions worsen over time with increased childhood mortality. | One in four risk of recurrence | ||
| Case 13: 25-year 11-month-old boy who presented at 16 years of age with severe rhabdomyolysis (CK of 313,000), myoglobinuria, pain, and fatigue. He subsequently had several other episodes of rhabdomyolysis that were less severe. Between episodes, he fatigues easily. | RYR1c.4178A > G (p.K1393R) (NM_000540.2) | Disorder associated with malignant hyperthermia susceptibility, rhabdomyolysis and/or exertional myalgia, development of muscle weakness over time. Choose career and activities that avoid excessive heat and intensive exertion. Risk associated with anesthesia (volatile anesthetics and muscle relaxants) and possibly statins. Identify and counsel other at-risk family members. | CK testing when symptomatic | One in two risk of recurrence | |
| Case 14: 2-year 4-month-old boy who presented at 18 months of age with episodes of weakness lasting minutes to hours. He subsequently had hypothyroidism and prolonged QT interval on ECG. | No diagnosis | ||||
| Case 15: 12-year 10-month-old girl who was born at 33.5 weeks of gestation following a pregnancy complicated by toxemia and decreased fetal movement. At 3 days of age, the patient had her first seizure. The patient subsequently had difficult to control seizures, failure to thrive, short stature, microcephaly, spasticity, scoliosis, cortical visual loss, severe intellectual disability, and cyclic vomiting. | No diagnosis | ||||
| Case 16: 2-year 3-month-old girl whose newborn screening test found an elevated C3 acylcarnitine which responded to injectable hydorxocobalamin. Subsequently, hemolytic anemia, spherocytosis, abnormal osmotic fragility test, and abnormal band 3 fluorescein staining were found. She did not have a family history of hereditary spherocytosis. After a short trial off of hydroxocobalamin, her methylmalonic acid level increased; treatment was reinstituted. The patient developed a life-threatening anemia, absence seizures, and mild cognitive disability. | No diagnosis | ||||
| Case 17: 35-year 2-month-old African-American woman with recurrent myofibromas starting in infancy that continued to appear throughout lifetime. Some of the tumors were surgically removed, whereas others regressed spontaneously. The patient has had three children diagnosed with infantile myofibromatosis. One died in the first year of life. | PDGFRBc.1681C > T (p.R561C) (NM_002609.3) | Support use of protein kinase inhibitors such as imatinib | Disorder associated with solitary or multicentric myofibromas in the skin, muscle, bone, and viscera generally appearing early in life. Lesions tend to spontaneously regress and can recur throughout life. | MRI when symptomatic | One in two risk of recurrence |
| Case 18: 7-year 10-month-old boy who presented at birth with a tracheoesophageal fistula that was repaired. He subsequently had poor growth, cognitive disability, facial dysmorphology, microcephaly, epilepsy, and refractory cyclic vomiting. The patient was clinically diagnosed with Treacher Collins syndrome. | EFTUD2c.1608–1dupG (NM_004247.3) | Disorder associated with mandibular hypoplasia, malar hypoplasia, ear anomalies, preauricular tags, hearing loss, auditory canal stenosis, cleft palate, facial asymmetry, microcephaly, psychomotor delay, brain anomalies, epilepsy, esophageal atresia, congenital heart defects, hand anomalies, and renal anomalies. | Low risk of recurrence (de novo variant) | ||
| Case 19: 16-year 5-month-old girl who presented at 3 months of age with a seizure. She subsequently had profound cognitive disability, epilepsy, primary ovarian failure, osteoporosis, failure to thrive, feeding disorder, history of nephrolithiasis, myopia, and microcephaly. Her sister is case 20. | No diagnosis | ||||
| Case 20: 19-year 9-month-old girl with cognitive disability, autism, normal height, normal weight, epilepsy, and microcephaly. Her sister is case 19. | No diagnosis | ||||
| Case 21: 5-year 11-month-old boy who presented with an elevated tyrosine level on the newborn screening test and feeding difficulties. He subsequently had rickets, renal Fanconi syndrome, abnormal AST and ALT, and a positive ANA. A percutaneous liver biopsy at 3 years 5 months was consistent with autoimmune hepatitis. He developed cataracts while on systemic corticosteroid treatment. | BCS1Lc.166C > T (p.R56X); c.-147A > G (NM_004328.4) | Disorder associated with neonatal tubulopathy, hepatic failure, encephalopathy, pilli torti, deafness, failure to thrive, microcephaly, iron overload, cholestasis, and lactic acidosis | Bilirubin, hepatic transaminase, electrolyte, ferritin, and transferrin testing. Hearing screening. | One in four risk of recurrence | |
| Case 22: 13-year 9-month-old girl who presented with a short period of normal psychomotor development followed by neurological deterioration at 6 months of age. She subsequently had central nervous system calcifications, severe seizure disorder, acquired microcephaly, feeding disorder, progressive scoliosis, and severe cognitive disability. | No diagnosis |
Abbreviation: WGS, whole genome sequencing.
Note: A clinical summary for each whole genome sequencing case is shown. The information is based on details from the case summary supplied to the laboratory. In the cases where a diagnosis was established, the results and consequences of finding a diagnosis are described.
The current paper provides additional detail and diagnoses based on reanalysis. Genetic counseling at the outset of the program took 6 to 10 hours over the course of several face-to-face visits and follow-up phone conversations. With experience, we found that this could be accomplished with 2 hours of face-to-face time and follow-up phone time. Post-test results disclosure and counseling took longer than traditional results discussion when incidental findings were part of the discussion.
Confirmation of Variants Identified by WGS
Among 158 variants under consideration for reporting, 153 were confirmed with Sanger sequencing. Among the five variants that were not confirmed, four had a read depth of one or two reads. In case 4, WGS found a previously undescribed variant in VLDLR that appeared heterozygous. Dideoxynucleotide sequencing found that the patient was homozygous for the variant (data not shown). In case 12, WGS identified a single nucleotide substitution resulting in a leucine to valine amino acid change in NLGN4X. Dideoxynucleotide sequencing confirmed this variant but found a second nucleotide substitution two bases away. The two substitutions combined resulted in no amino acid change (data not shown).
Diagnoses Identified by WGS
Of the 22 cases (Table 2), we initially reached a diagnosis rate of 14% (3/22). With subsequent reanalysis, our diagnosis rate increased to 36% (8/22). In cases 5, 11, 12, and 17, subsequent research publications identified novel genes associated with the various phenotypes. In cases 11 and 17, newly published variants were identified. In cases 5 and 12, variants likely to be deleterious to the candidate gene, but not previously reported, were found. Case 6 was enrolled in a research project at the National Institutes of Health that led to a diagnosis.35
Variants of Uncertain Significance Identified by WGS Used in Research
An average of 3.6 variants of uncertain significance (VUS) (range 0–10) were identified per case. Cases without a diagnosis where VUS were found represent an opportunity for research. For example, in case 4, an apparent homozygous p.A18V VUS was identified in VLDLR. Pathogenic variants in this gene are associated with cerebellar hypoplasia and mental retardation.36 The patient's phenotype overlapped with this phenotype but with the addition of leukodystrophy. The amino acid change is located in the leader sequence of VLDLR, and protein modeling suggests that the change affects the leader sequence's function (data not shown). In case 14, two homozygous missense variants, p.V32A and p.R716Q, in the gene MYO18B are currently under investigation.
Use of Gene Lists to Limit Incidental Findings
The number of genes on the gene lists for each of the 22 cases ranged from 6 to 1074. The list of genes generally contained genes where no commercial testing was available and/or where the list was so long that commercial testing was not economical. In cases 13 and 21, variants in genes from the list associated with the patients' phenotypes were identified. While these results were pathogenic, it remained uncertain whether the variants explained the entire phenotype in the two cases. Therefore, the remainder of each genome was evaluated. No causal variants in other gene lists were identified.
Incidental Findings
Fig. 2 shows the incidental finding choices selected by the 21 patients and families (22 cases). Between zero and seven pathogenic incidental findings were identified in cases where incidental findings were requested. The number of incidental findings reported in a given case related, in part, to the family's requests concerning which type(s) of incidental findings they wished to have returned. Among the 41 incidental findings, 40 resulted in carrier status for a recessive disorder. One was associated with a dominant disorder (Supplement 7, available in the online version). Of note, among the 22 cases, no pathogenic incidental findings resulted in additional testing following disclosure.
Fig. 2.
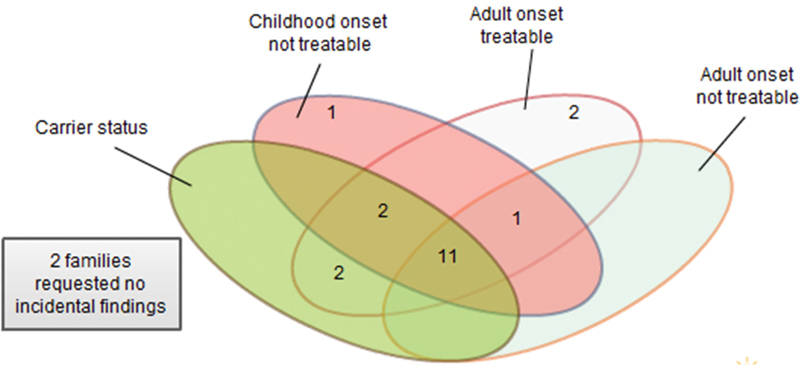
Choice of incidental findings. This Venn diagram summarizes the choice of incidental findings that each of the families selected.
Impact of Diagnosis on Patient Management
A definitive diagnosis was established in eight cases. In 75% (6/8) of these, the result had an impact on medical management or surveillance, with attendant economic consequences (Table 2).
In case 11, for example, treatment with eculizumab was considered prior to WGS. Such treatment is expensive (variable, but currently about $400,000 /year) and is associated with increased risk for sepsis due to encapsulated organisms.37 As the etiology involved DGKE, a gene that is not in the complement cascade, expert discussion following review of the WGS results established that eculizumab would be ineffective. Avoiding this therapy for one year in one of the affected siblings saved insurance carriers more money than the whole program cost. In addition, identifying a pathologic variant in DGKE supported the decision not to pursue any form of plasma therapy for atypical hemolytic uremic syndrome in this patient or her affected siblings in future disease relapses, which previously had been tried in an older affected sibling, but was thought likely to be ineffective.
In case 17, the identification of a variant in a tyrosine kinase (TK) gene, PDGFRB, immediately suggested a possible treatment with a TK inhibitor such as imatinib. In all cases where a diagnosis was established, the patients avoided further diagnostic testing, families received prognostic information concerning the disorder, and notably, parents were reassured that their actions during pregnancy did not “cause” their child's condition, a common parental concern. Further, genetic counseling and, as appropriate, testing were offered to the extended family. In the cases where the identified disorder was not amenable to therapeutic or management changes, establishment of a definitive diagnosis gave parents and providers confidence that the patient did not have a rare, currently treatable condition.
Discussion
Introduction of WGS into Clinical Practice
Introduction of WGS into clinical practice requires evidence that the test can be integrated into current clinical practice and that results are useful to physicians, patients, and families.
WGS established a diagnosis in 36% (8/22) of cases when all other reasonable testing had been exhausted and in 75% (6/8) of cases where a diagnosis was established, the result had an impact on management with attendant economic consequences (Table 2). WGS proved useful as a diagnostic tool even in patients who had had extensive diagnostic laboratory testing over many years. Consistent with other studies of clinical utility,10 11 WGS yielded results that were useful to physicians, patients, and families. Thus, we believe that use of genomic sequencing early in the diagnostic process for patients with suspected genetic disorders may reduce the cost of diagnosis by eliminating many individual tests and may improve outcome by reducing the time to diagnosis.
The improvement in the diagnosis rate from 14% (3/22) initially to 36% (8/22) with reanalysis is noteworthy. As new gene-phenotype connections appear in the literature, reanalysis of negative cases will increasingly yield a diagnosis. These diagnoses can result in specific patient management consideration.
OMIM catalogues 5,768 phenotypes for which the molecular basis is known among the 15,328 autosomal, X-linked, Y-linked, and mitochondrial mapped genes in the catalog.38 The fact that 62% of all known genes are not associated with a recognized phenotype explains, in part, the failure of genomic sequencing to yield a diagnosis in the majority of cases.
Our diagnosis rate is similar to other studies of WGS and WES. WES led to a molecular diagnosis in 25 to 29% of cases with a suspected genetic diagnosis.5 6 7 For patients with neurodevelopmental phenotypes, a WES diagnostic rate of 48% was seen.8 WGS led to a molecular diagnosis in ∼40% of cases with severe intellectual disability.9 In one study with adults, WES established a diagnosis in 18% of cases.12 When WES was employed as the first test in the evaluation of infants with suspected monogenic disease, a diagnosis was established in 57% of cases.10 A study using WGS as the initial test in a pediatric genetics clinic established a diagnosis in 34% of cases.11
WGS has an advantage over WES because it can detect variants that are outside protein coding regions and the immediately surrounding intronic regions covered by WES. Reports of pathogenic variants in deep intronic regions that result in hereditary disease are starting to appear.39 40 Review of the reported variants from our pilot cases showed that two of variants (case 11 and case 21 in Table 2) were in regions that are not well covered by WES.
Turnaround time is essential to the successful deployment of genomic sequencing in clinical practice. Toward the end of the pilot study (the last eight cases), a median time of 131 days was required from blood draw to final report. This is similar to the 15-week turnaround time reported for WES by Lazaridis.41 A commonly noted challenge to turnaround time in a high-throughput clinical setting is the time required for expert interpretation of identified variants. On average, WGS in our laboratory identified 3 to 4 million variants. Following prioritization, we found that ∼600 variants were prioritized for expert interpretation. By the end of this pilot program, ∼3 hours of clinical analyst time and 3 hours of director time were needed to select and interpret variants associated with the diagnosis under consideration and incidental findings if requested. Significant review is needed due to the presence of many incorrectly classified variants in the literature.18 42 Over time, with ongoing curation, the number of variants that have to be manually reviewed for each new case will drop, thus reducing the overall time required for analysis. Use of computer-assisted decision support should further reduce analysis time.
Our interpretation time differs significantly from other reports of WGS. To understand the discrepancy, it is important to consider the nature of the studies being compared. In their report, Dewey et al18 applied WGS to healthy individuals and evaluated all candidate inherited disease risk variants, certain coronary artery disease and diabetes genetic risk variants, and pharmacogenomic variants. The median number of variants that required manual curation was 108 per genome, and the median curation time was 54 minutes per variant. In contrast, data analysis in our study was limited to Mendelian disease, focused on rare variants in genes that might explain the patient's complex phenotype and when requested, incidental findings of published pathogenic variants in Mendelian disorders. These differences give rise to the significant differences in reporting times in the two studies.
Evidence is emerging that genomic sequencing can replace dideoxynucleotide sequencing and eliminate the time and expense related to confirmatory testing of WES and WGS results.43 44 45 The difference between the WGS result and the dideoxynucleotide sequencing results in case 4 and case 12 suggests that laboratories should proceed with caution.
Genomic sequencing raises a new challenge to the clinical molecular genetics laboratory. In cases where one variant is found and the disorder is recessive, then analysis of the rest of the gene in question must be undertaken as genomic sequencing may fail to detect a second variant, particularly in the case of insertions and deletions or variants larger than the detectable range for current next-generation sequencing methodologies. If a clinical laboratory offers dideoxynucleotide sequencing for the gene of interest, searching for the second variant is straightforward. However, there are many genes with no clinical laboratory testing available. Laboratories performing genomic sequencing should consider the need to provide dideoxynucleotide sequencing and insertion/deletion analysis of any gene on an “as needed” basis.
Genomic sequencing used as a diagnostic tool will uncover incidental findings; this number will increase as our understanding of the genome improves. Some of these incidental findings will provide an opportunity to alter medical management. In this study, WGS uncovered 41 variants classified as incidental findings. None resulted in further investigation for disease in the patient. This is likely the result of limiting incidental findings to Mendelian disorders. In contrast, the study by Dewey et al evaluated all candidate inherited disease risk variants, certain coronary artery disease and diabetes genetic risk variants, and pharmacogenomic variants in healthy individuals and determined that the estimated cost of initial clinical follow-up is less than $1,000 per person.18
The prospect of discovering actionable incidental findings in the large number of individuals who are now undergoing genomic sequencing has started a lively debate concerning who should be involved in the decision to disclose incidental findings in patients who are adults and in patients who are children.22 46 47 48
When given a choice, patients and families made a range of decisions concerning return of incidental findings as noted in Fig. 2. In a cohort of adults undergoing WES, 6 of 38 did not consent to incidental finding reporting of one or more categories.49 Another study examined patient preferences regarding incidental findings discovered during tumor profiling; when offered a range of types of incidental findings that might be returned, there was substantial variability with regard to which types patients wished to receive.50 In an effort to identify diagnoses and limit incidental findings, we applied gene lists to all cases before extending analysis to all genes. This strategy yielded only two diagnoses in 22 cases. Uncertainty in these two cases prompted analysis of the remaining genes. This experience raises the question of whether the use of a gene list to limit incidental findings is valuable. Additionally, such a process might cause a laboratory to miss a diagnosis when a patient has two or more genetic diseases.5 11 The importance of providing the laboratory with extensive clinical information about the patient cannot be overemphasized. This arises from the nature of a genomic test. A molecular test for a recognized phenotype such as cardiomyopathy is very different from a genomic test for any known disease. In genomic testing, the prior probability of any given gene causing the patient's phenotype is low and therefore the laboratory must obtain detailed information about the patient's phenotype to carry out the analysis. Recent studies show that there are many rare variants that are population specific; not all of these are pathogenic.51 52
The VLDLR variant in case 4 is an example of the growing opportunity afforded by genomic sequencing to expand the number of genotype-phenotype associations. When a gene has not been associated with a patient's phenotype or when the gene has been associated with a different phenotype from that under consideration, this is called a “gene of uncertain significance” (GUS).53 There is now a database of undiagnosed cases where a GUS is identified that allows laboratories to exchange information about these cases.54 Additionally, it is important for clinical laboratories to work with research laboratories as the latter are in a position to pursue a GUS.
Genomic sequencing can also provide information about the other variants in a gene where pathogenic variants are found. Case 5 had two SKIV2L pathogenic variants in trans that together explained the patient's phenotype. There were seven additional variants identified by WGS. When a variant is observed in cis with a pathogenic variant in a recessive disorder, a clinical laboratory can use this information as evidence to help establish that the variant is benign. Among the five cases with recessive diseases, 114 potentially benign variants were identified. When a variant is observed in cis or trans with a pathogenic variant in a dominant disorder, a clinical laboratory can use this information as evidence to help establish that the variant is benign. Among the three cases with dominant diseases, 202 potentially benign variants were identified (Supplement 8, available in the online version). Contribution of this type of information along with pathogenic variants to publicly available databases, such as ClinVar,55 is useful to all genomic sequencing laboratories.
This study has some limitations. Case selection had several biases that may have dramatically affected the percentage of diagnoses achieved including a focus on cases with suspected rare inherited disease, cases with a distinctive phenotype, cases where extensive conventional genetic testing had failed to find a diagnosis, and cases where multiple additional expensive genetic tests might yield a diagnosis. Additionally, the number of cases evaluated in this study is small and therefore the percentage of diagnoses identified in this series may differ significantly when larger studies are undertaken. Due to the small sample size, our study is unable to provide information concerning the specificity of WGS for any given genetic disorder.
Development of a Genomic Medicine Clinic
Due to the subsequent availability of WES from several other clinical laboratories and the improvement in insurance coverage that occurred following the pilot program described above, we found that there was no longer a need for a case review team. The lessons learned from the case review process were used to initiate two genomic medicine clinics (GMCs).
Creating a GMC was a straightforward way to integrate WES and WGS into the current practice of medicine. Separate GMCs were started for children at CHW and for adults at Froedtert Hospital. These were staffed by clinical geneticists and genetic counselors and followed the guidelines developed by the team. While any physician could order WES or WGS, most physicians referred patients to the GMC to ensure that patients were properly counseled about genomic sequencing, that sequencing was undertaken when clinically appropriate, that abnormal results and incidental findings were properly explained, and that appropriate treatment and medical management were pursued. When genomic sequencing failed to find an answer, these patients continued to have regular follow-up in the GMC. For these visits, genomic data are rereviewed based on the updated literature, any new information about the patient, and application of updated sequencing and analysis capabilities. In this way, genomics is integrated into current genetics practice wherein undiagnosed cases are routinely reevaluated at regular intervals. As seen in this report, such reevaluation more than doubled our rate of diagnosis.
The number of cases at our institution and across the United States employing genomic sequencing is growing. Insurance companies generally require preauthorization of sequence-based testing. Therefore, in all cases where insurance coverage was requested, an insurance authorization letter was prepared (Supplement 9, available in the online version). The GMC's genetic counselors work with insurance authorization staff to prepare the letter. Over a 7-month period (from October 2013 to April 2014), 29 genomic sequencing cases were identified and submitted to payers for prior authorization. Three of the cases were denied. One of the three denials was appealed and later authorized. Twenty-seven cases were authorized and testing occurred.
As with all new medical technologies, some insurers cover or partially cover genomic testing while others consider it experimental and therefore, not a covered benefit. Clinical trials supporting clinical utility and cost-effectiveness are required to further convince insurance companies to expand coverage. Improved technology56 that reduces the price (currently $6,500–$9,500 for WGS57 58) will also help. Efforts to integrate genomic testing into routine reimbursement practices are underway.59 Reimbursement for this test is connected to the longstanding issues of adequate coverage of genetic counseling.60 Proper genetic counseling is essential for the effective deployment of genomic testing in clinical practice. Genomic testing now has specific CPT codes, which are needed in the insurance reimbursement process.
Physician Scope of Practice
Because WGS and WES are relatively new and complex tests, some institutions may elect to limit test ordering to physicians with expertise in genomics. Hospitals routinely define a “scope of practice” for providers when expensive or high-risk tests and treatments are involved in an effort to control cost and reduce patient risk exposure. At CHW, geneticists have been involved in all requests for WES and WGS to date. However, as the price of sequencing drops and the number of clinically important genes and variants increases, it will be essential for all physicians to be able to appropriately order genomic testing and use relevant genomic data effectively. Physicians must also be prepared to counsel about and then handle incidental findings. As more physicians start to order genomic testing at CHW, the electronic medical record system may require the physician ordering WES or WGS to attest that the patient/family has had genetic counseling and that insurance preauthorization was obtained. Broad efforts to educate physicians about genomic medicine are needed.61
Challenges to Increased Use of WGS in the Clinic
Extracting relevant aspects of the patient's phenotype from the medical record to interpret genomic data are a significant challenge. Just as a tertiary analysis tool such as CarpeNovo has been developed to present the laboratory director with an efficient interface to help decide which variants should be reported, a similar interface will be needed to assemble and present clinical information to the laboratory director from the medical record using a standardized vocabulary of phenotypic abnormalities found in human disease such as the Human Phenotype Ontology.62 Efforts in this direction are underway.63 64
The capacity for storage and transfer of data must keep pace as medicine increasingly utilizes genomic testing. The data from a single genome (raw image data, BAM files, and VCF files) requires approximately one terabyte of storage. As a result, the MCW genomics laboratory uses more storage capacity than the entire combined storage needs of the two hospitals, CHW and Froedtert Hospital. Transfer of a single patient's genome to another institution via the Internet can take days; therefore, the data are often copied to a hard drive and sent via overnight courier when this is required. It can be argued that as sequencing accuracy improves and the cost of sequencing decreases, it will be safer and cheaper to resequence a patient rather than storing and reanalyzing old data.
At present, only the final analytic report is provided by the laboratory to the physician and stored in the medical record. The laboratory retains the genomic data. Increased bandwidth for data transfer and HIPPA compliant “cloud” storage will be required if patients and physicians expect to place the genomic data in the medical record. This would be similar to MRI images that are stored separately from the electronic medical record but are linked to it. Such a system would facilitate reevaluation of the genome when initial testing fails to find the answer or when a new clinical question arises. The physician would order such testing allowing different laboratories to access the clinical record and associated genomic data to facilitate reevaluation.
In order for the promise of genomic medicine to be fully realized, a new specialist will be needed, the “genomicist.” These will be physicians with training in the use of genomic data in clinical practice. At present, these individuals are geneticists and pathologists. Genomic sequencing can be divided into two steps. First, the laboratory generates a list of variants for a patient's genome. Next, the relevant variants are connected to the patient's condition using tertiary analysis software. The latter step would be accomplished by the genomicist. With time, each medical specialty may develop their own genomicists to evaluate patients for genetic disorders connected to their specialty. No pathologist or geneticist is likely to have as much knowledge of medical specialties such as cardiology compared with board-certified specialists in the respective area. This has revenue implications for each specialty in medicine. Current CPT coding already permits the laboratory to bill the technical component and the physician to bill the professional (interpretive) component.
Conclusion
Application of WGS in a rare disease setting dramatically increased our ability to make diagnoses even in difficult cases where other methods had failed. We found, over time, that the rate of diagnosis increased without the need to produce additional sequence data. This has a significant advantage over approaches that limit testing and analysis to the current scientific knowledge available at the time of testing. In summary, we believe that WGS is an appropriate and valuable molecular diagnostic methodology for use in clinical practice today.
Acknowledgments
The authors thank the patients and families for participating in our genomics program and Adriana Stout for assistance in the preparation of this manuscript.
Supplementary Material
References
- 1.Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59(1):5–15. doi: 10.1038/jhg.2013.114. [DOI] [PubMed] [Google Scholar]
- 2.Worthey E A, Mayer A N, Syverson G D. et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 3.Gahl W A, Boerkoel C F, Boehm M. The NIH Undiagnosed Diseases Program: bonding scientists and clinicians. Dis Model Mech. 2012;5(1):3–5. doi: 10.1242/dmm.009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gahl W A, Mulvihill J J, Toro C. et al. The NIH Undiagnosed Diseases Program and Network: applications to modern medicine. Mol Genet Metab. 2016;117(4):393–400. doi: 10.1016/j.ymgme.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Muzny D M, Xia F. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Etterer K, Juusola J, Cho M T. et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 7.Sawyer S L, Hartley T, Dyment D A. et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89(3):275–284. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nolan D, Carlson M. Whole exome sequencing in pediatric neurology patients: clinical implications and estimated cost analysis. J Child Neurol. 2016;31(7):887–894. doi: 10.1177/0883073815627880. [DOI] [PubMed] [Google Scholar]
- 9.Gilissen C Hehir-Kwa J Y Thung D T et al. Genome sequencing identifies major causes of severe intellectual disability Nature 2014511(7509):344–347. [DOI] [PubMed] [Google Scholar]
- 10.Stark Z, Tan T Y, Chong B. et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;2016:1–7. doi: 10.1038/gim.2016.1. [DOI] [PubMed] [Google Scholar]
- 11.Stavropoulos D J Merico D Jobling R et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine npj Genomic Med 2016115012. Doi: 10.1038/npjgenmed.2015.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Posey J E, Rosenfeld J A, James R A. et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med. 2016;18(7):678–685. doi: 10.1038/gim.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsons D W, Roy A, Yang Y. et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;1200(5):1–9. doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller N A Farrow E G Gibson M et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases Genome Med 201571100. Doi: 10.1186/s13073-015-0221-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowdin S, Gilbert A, Bedoukian E. et al. Recommendations for the integration of genomics into clinical practice. Genet Med. 2016;18(11):1075–1084. doi: 10.1038/gim.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delaney S K, Hultner M L, Jacob H J. et al. Toward clinical genomics in everyday medicine: perspectives and recommendations. Expert Rev Mol Diagn. 2016;16(5):521–532. doi: 10.1586/14737159.2016.1146593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fogel B L, Satya-Murti S, Cohen B H. Clinical exome sequencing in neurologic disease. Neurol Clin Pract. 2016;6(2):164–176. doi: 10.1212/CPJ.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dewey F E, Grove M E, Pan C. et al. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311(10):1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lapin V, Mighion L C, da Silva C P, Cuperus Y, Bean L JH, Hegde M R. Regulating whole exome sequencing as a diagnostic test. Hum Genet. 2016;135(6):655–673. doi: 10.1007/s00439-016-1677-3. [DOI] [PubMed] [Google Scholar]
- 20.Weitzel K W Alexander M Bernhardt B A et al. The IGNITE network: a model for genomic medicine implementation and research BMC Med Genomics 2016911. Doi: 10.1186/s12920-015-0162-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bick D, Dimmock D. Whole exome and whole genome sequencing. Curr Opin Pediatr. 2011;23(6):594–600. doi: 10.1097/MOP.0b013e32834b20ec. [DOI] [PubMed] [Google Scholar]
- 22.Strong K A, Derse A R, Dimmock D P. et al. In the absence of evidentiary harm, existing societal norms regarding parental authority should prevail. Am J Bioeth. 2014;14(3):24–26. doi: 10.1080/15265161.2013.879959. [DOI] [PubMed] [Google Scholar]
- 23.Worthey E A Analysis and annotation of whole-genome or whole-exome sequencing-derived variants for clinical diagnosis Curr Protoc Hum Genet 2013797924. Doi: 10.1002/0471142905.hg0924s79 [DOI] [PubMed] [Google Scholar]
- 24.Sherry S T, Ward M H, Kholodov M. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auton A Brooks L D Durbin R M et al. A global reference for human genetic variation. 1000 Genomes Project Consortium Nature 2015526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Exome Variant Server http://evs.gs.washington.edu/EVS/. Accessed January 1, 2016
- 27.Adzhubei I A, Schmidt S, Peshkin L. et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reva B Antipin Y Sander C Predicting the functional impact of protein mutations: application to cancer genomics Nucleic Acids Res 20113917e118. Doi: 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P, Henikoff S, Ng P C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 30.Hubisz M J, Pollard K S, Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform. 2011;12(1):41–51. doi: 10.1093/bib/bbq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenson P, Mort M, Ball E, Shaw K, Phillips A, Cooper D. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genet. 2014;133(1):1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.OMIM Nathans Institute of Genetic Medicine Johns Hopkins University; http://www.omim.org/. Accessed January 1, 2016
- 33.Richards C S, Bale S, Bellissimo D B. et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10(4):294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 34.Jacob H J Abrams K Bick D P et al. Genomics in clinical practice: lessons from the front lines Sci Transl Med 20135194194cm5. Doi: 10.1126/scitranslmed.3006468 [DOI] [PubMed] [Google Scholar]
- 35.Zhou Q, Yang D, Ombrello A K. et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–920. doi: 10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kruer M C, Jepperson T N, Weimer J M. et al. Mutations in VLDLR associated with ataxia with secondary vitamin E deficiency. Mov Disord. 2013;28(13):1904–1905. doi: 10.1002/mds.25573. [DOI] [PubMed] [Google Scholar]
- 37.Eculizumab . Formulary Journal http://formularyjournal.modernmedicine.com/formulary-journal/news/clinical/clinical-pharmacology/eculizumab. Published 2007. Accessed January 1, 2016
- 38.Gene Map Statistics OMIM http://www.omim.org/statistics/geneMap. Published 2016. Accessed July 15, 2016
- 39.Gillis E, Kempers M, Salemink S. et al. An FBN1 deep intronic mutation in a familial case of Marfan syndrome: an explanation for genetically unsolved cases? Hum Mutat. 2014;35(5):571–574. doi: 10.1002/humu.22540. [DOI] [PubMed] [Google Scholar]
- 40.Qian Y, Johnson J A, Connor J A. et al. The 253-kb inversion and deep intronic mutations in UNC13D are present in North American patients with familial hemophagocytic lymphohistiocytosis 3. Pediatr Blood Cancer. 2014;61(6):1034–1040. doi: 10.1002/pbc.24955. [DOI] [PubMed] [Google Scholar]
- 41.Lazaridis K N, McAllister T M, Babovic-Vuksanovic D. et al. Implementing individualized medicine into the medical practice. Am J Med Genet C Semin Med Genet. 2014;166C(1):15–23. doi: 10.1002/ajmg.c.31387. [DOI] [PubMed] [Google Scholar]
- 42.Dorschner M O, Amendola L M, Turner E H. et al. Actionable, pathogenic incidental findings in 1,000 participants' exomes. Am J Hum Genet. 2013;93(4):631–640. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sikkema-Raddatz B, Johansson L F, de Boer E N. et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat. 2013;34(7):1035–1042. doi: 10.1002/humu.22332. [DOI] [PubMed] [Google Scholar]
- 44.Strom S P, Lee H, Das K. et al. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med. 2014;16(7):510–515. doi: 10.1038/gim.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baudhuin L M, Lagerstedt S A, Klee E W, Fadra N, Oglesbee D, Ferber M J. Confirming variants in next-generation sequencing panel testing by sanger sequencing. J Mol Diagn. 2015;17(4):456–461. doi: 10.1016/j.jmoldx.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Green R C, Berg J S, Grody W W. et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burke W, Antommaria A H, Bennett R. et al. Recommendations for returning genomic incidental findings? We need to talk! Genet Med. 2013;15(11):854–859. doi: 10.1038/gim.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borry P Evers-Kiebooms G Cornel M C Clarke A Dierickx K; Public and Professional Policy Committee (PPPC) of the European Society of Human Genetics (ESHG). Genetic testing in asymptomatic minors: background considerations towards ESHG Recommendations Eur J Hum Genet 2009176711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shahmirzadi L, Chao E C, Palmaer E, Parra M C, Tang S, Gonzalez K DF. Patient decisions for disclosure of secondary findings among the first 200 individuals undergoing clinical diagnostic exome sequencing. Genet Med. 2014;16(5):395–399. doi: 10.1038/gim.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yushak M L, Han G, Bouberhan S. et al. Patient preferences regarding incidental genomic findings discovered during tumor profiling. Cancer. 2016;122(10):1588–1597. doi: 10.1002/cncr.29951. [DOI] [PubMed] [Google Scholar]
- 51.Nelson M R Wegmann D Ehm M G et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people Science 2012337(6090):100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keinan A Clark A G Recent explosive human population growth has resulted in an excess of rare genetic variants Science 2012336(6082):740–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richards S, Aziz N, Bale S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Philippakis A A, Azzariti D R, Beltran S. et al. The Matchmaker exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36(10):915–921. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Landrum M J, Lee J M, Benson M. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Dijk E L, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30(9):418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 57.Clinical Services Lab - FAQS https://clinicallab.org/faqs/. Accessed August 4, 2016
- 58.Illumina TruGenome Clinical Sequencing Services http://www.illumina.com/clinical/illumina_clinical_laboratory/trugenome-clinical-sequencing-services.html. Accessed August 4, 2016
- 59.NHGRI Genomic Medicine Centers Meeting IV. physician education in genomics https://www.genome.gov/27552294/. Accessed January 1, 2016
- 60.Gustafson S L, Pfeiffer G, Eng C. A large health system's approach to utilization of the genetic counselor CPT® 96040 code. Genet Med. 2011;13(12):1011–1014. doi: 10.1097/GIM.0b013e3182296344. [DOI] [PubMed] [Google Scholar]
- 61.Dougherty M J, Wicklund C, Johansen Taber K A. Challenges and opportunities for genomics education: insights from an Institute of Medicine Roundtable Activity. J Contin Educ Health Prof. 2016;36(1):82–85. doi: 10.1097/CEH.0000000000000019. [DOI] [PubMed] [Google Scholar]
- 62.Köhler S Doelken S C Mungall C J et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data Nucleic Acids Res 201442(database issue): D966–D974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang L Zhang C Watkins J Jin Y McNutt M Yin Y SoftPanel: a website for grouping diseases and related disorders for generation of customized panels BMC Bioinformatics 2016171153. Doi: 10.1186/s12859-016-0998-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smedley D Robinson P N Phenotype-driven strategies for exome prioritization of human Mendelian disease genes Genome Med 20157181. Doi: 10.1186/s13073-015-0199-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.