

ABSTRACT
In the nosocomial opportunistic pathogen Acinetobacter baumannii, RecA-dependent mutagenesis, which causes antibiotic resistance acquisition, is linked to the DNA damage response (DDR). Notably, unlike the Escherichia coli paradigm, recA and DDR gene expression in A. baumannii is bimodal. Namely, there is phenotypic variation upon DNA damage, which may provide a bet-hedging strategy for survival. Thus, understanding recA gene regulation is key to elucidate the yet unknown DDR regulation in A. baumannii. Here, we identify a structured 5′ untranslated region (UTR) in the recA transcript which serves as a cis-regulatory element. We show that a predicted stem-loop structure in this 5′ UTR affects mRNA half-life and underlies bimodal gene expression and thus phenotypic variation in response to ciprofloxacin treatment. We furthermore show that the stem-loop structure of the recA 5′ UTR influences intracellular RecA protein levels and, in vivo, impairing the formation of the stem-loop structure of the recA 5′ UTR lowers cell survival of UV treatment and decreases rifampin resistance acquisition from DNA damage-induced mutagenesis. We hypothesize that the 5′ UTR allows for stable recA transcripts during stress, including antibiotic treatment, enabling cells to maintain suitable RecA levels for survival. This innovative strategy to regulate the DDR in A. baumannii may contribute to its success as a pathogen.
IMPORTANCE Acinetobacter baumannii is an opportunistic pathogen quickly gaining antibiotic resistances. Mutagenesis and antibiotic resistance acquisition are linked to the DNA damage response (DDR). However, how the DDR is regulated in A. baumannii remains unknown, since unlike most bacteria, A. baumannii does not follow the regulation of the Escherichia coli paradigm. In this study, we have started to uncover the mechanisms regulating the novel A. baumannii DDR. We have found that a cis-acting 5′ UTR regulates recA transcript stability, RecA protein levels, and DNA damage-induced phenotypic variation. Though 5′ UTRs are known to provide stability to transcripts in bacteria, this is the first example in which it regulates a bimodal DDR response through recA transcript stabilization, potentially enabling cells to have enough RecA for survival and genetic variability.
KEYWORDS: Acinetobacter, DNA damage, microbial genetics, RecA, gene expression, molecular biology
INTRODUCTION
Acinetobacter baumannii is a Gram-negative opportunistic pathogen that is a major problem in hospitals due to its ability to withstand desiccation (1), allowing it to ultimately reach immunocompromised individuals and cause diseases, including pneumonia and meningitis (2–4). Compounding this problem is the ability of A. baumannii to rapidly gain antibiotic resistances, such that there are now pan-resistant strains (5, 6). This may in part be due to A. baumannii's improved fitness during stress (7–10). However, fully understanding how A. baumannii rapidly acquires antibiotic resistances has been hindered by the limited knowledge of fundamental DNA repair processes and a limited genetic toolkit for the bacterium (11).
Notably, antibiotic resistance acquisition in A. baumannii has been linked to its DNA damage response (DDR) (12), which protects or repairs genetic material after DNA damage resulting from the environment, other bacteria, or antibiotics (13). One of the best-understood bacterial DDR systems is the conserved Escherichia coli paradigm (13–15). In this system, RecA, the cell's main recombinase, binds single-stranded DNA, the signal of DNA damage, to form the RecA nucleoprotein filament (RecA*) (16). Coprotease activity of RecA* promotes autocleavage and inactivation of LexA, the global DDR repressor, resulting in expression of >40 genes for DNA repair, including recA itself, and error-prone DNA polymerases (17–19). The lower fidelity of these polymerases leads to increased mutagenesis of native genes that when altered lead to antibiotic resistances; these genes include antibiotic targets and efflux machinery (12, 14, 15, 20, 21). Classic examples are mutations in the rpoB gene (conferring rifampin resistance [22]) and the gyrA gene (conferring nalidixic acid resistance [23]). Specifically, in A. baumannii, mutations in regulatory genes for the AdeABC efflux system lead to multidrug resistance (24).
In A. baumannii, the conserved E. coli DDR circuitry does not exist, in part because A. baumannii lacks a known functional LexA homologue and cognate binding boxes within DDR gene promoters (25, 26). Many opportunistic pathogens, including Legionella pneumophila, Streptococcus pneumoniae, and Helicobacter pylori (27–29), have noncanonical DNA damage responses (e.g., not mediated by a global LexA repressor).
In previous work we found that for A. baumannii ATCC 17978, the DDR is bimodal, unlike in E. coli (30). That is, in A. baumannii, when expression of a number of conserved DDR genes, including recA, is measured, we find DDRLow (low expression of DDR genes) and DDRHigh (high expression of DDR genes) phenotypic subpopulations. To our knowledge, this may be the first example of phenotypic variation in a DDR regulation. The DDRLow and DDRHigh cells have different survival capabilities upon DNA damage treatment, including the antibiotic ciprofloxacin (Cip), suggesting that this bimodal DDR expression may serve as a bet-hedging strategy conferring genomic plasticity and survival (31). We have shown that the DDR in A. baumannii is dependent on functional RecA and that activation of the DDR causes clinically relevant mutagenesis in the rpoB gene, resulting in rifampin resistance (12). Because of RecA's central role in the DDR and in antibiotic resistance acquisition, it is critical to understand both recA gene regulation and the DDR gene network in this bacterium.
In this study, we identified the recA promoter and showed that it has elements needed to respond to DNA damage. Importantly, we found that the recA transcript has a 5′ untranslated region (UTR), which we show contributes to both transcript stabilization and phenotypic variation. We demonstrated that a critical stem-loop structure in the 5′ UTR maintains mRNA half-life (t1/2). Furthermore, we found that the 5′ UTR is necessary for maintaining RecA levels in the cell and influences RecA-dependent processes in vivo, including survival of DNA damage and rifampin resistance acquisition. These data support a model of recA regulation in which the promoter is involved in DNA damage detection, while the 5′ UTR, specifically a predicted stem-loop structure, stabilizes and protects the recA transcript from degradation and maintains appropriate RecA levels. The 5′ UTR also affords bimodal DDR induction to support cell viability and mutagenesis. These findings expand our knowledge of bacterial DDRs beyond the conserved paradigm.
RESULTS
A. baumannii recA has a 99-nt 5′ UTR.
To begin dissecting the regulation of recA (A1S_1962) gene expression in A. baumannii ATCC 17978, we mapped its transcriptional start site (TSS). Using a modified standard primer extension assay experiment, we only found short products, indicating that there was extension very close to the reverse primer that stopped close to the recA starting methionine (see Fig. S1 in the supplemental material). Since this result did not account for the ribosomal binding site (RBS), we thought that there might be something interfering, e.g., a strong mRNA secondary structure, that did not permit full primer extension under our experimental conditions. To provide insight into the TSS, we examined published transcriptome sequencing (RNA-Seq) data for A. baummanii ATCC 17978 (32). The mRNA reads measured (from converted cDNA) were mapped onto the reference A. baummanii genome. Upstream of the recA coding sequence, we searched for the point that corresponds to the gene's 5′ boundary (the point where the number of mRNA reads mapped onto the DNA sequence starts to increase, shown in gray in Fig. 1A), which corresponds to the TSS (33). We predicted that the transcript began 95 to 100 nucleotides (nt) upstream of the recA start codon (Fig. 1A, red line). Consensus promoter −10 and −35 sequences (34) with the corresponding spacing indicated a possible +1 TSS 99 nt upstream of the recA start codon (Fig. 1C). To validate this prediction, total RNA was extracted from untreated A. baumannii cells and first-strand cDNA was synthesized. PCRs with nested forward primers (noted in Fig. 1A as arrows) were performed to determine the 5′ end of the cDNA generated from the recA mRNA transcript. This PCR amplification allowed for detection of products that we lacked the sensitivity to see in our extension experiments (Fig. S1). Products were obtained with primers F1 to F5, which are within the predicted transcript, while there was no product with primer F6, which is just outside the predicted TSS (Fig. 1B). These data supported the TSS predicted by RNA-Seq. Together, these results suggest that recA in A. baumannii contains an approximately 99-nt 5′ UTR (Fig. 1C).
FIG 1.

The A. baumannii recA transcript has a 5′ UTR of 99 nucleotides with a predicted stable stem-loop structure. (A) RNA-Seq alignment to the genome of A. baumannii ATCC 17978 upstream of the recA (A1S_1962) open reading frame (ORF) using CLC Genomics (Qiagen) (33). The density of mapped reads is shown in gray. The transcription start site (TSS) is predicted to be 99 nt upstream of the recA ORF start codon and is indicated with a red line. Arrows represent primers used to validate the predicted TSS. (B) RNA was extracted from untreated A. baumannii cells and converted to cDNA, which served as the template for PCR using the reverse primer R1 and a series of nested forward primers (F1 to F6), whose positions are indicated in panel A. The products obtained from primers F1 to F5 paired with R1 were separated by agarose gel electrophoresis and are annotated above their respective lane. No product was obtained when using primers F6 and R1 together. (C) Schematic of the region upstream of the recA ORF in A. baumannii. Conserved −35 and −10 promoter sites (34) are in bold. (D) Schematic of the predicted structure, base-pairing probability, and minimum free energy value for the native 5′ UTR using the RNAfold webserver (35). Indicated are a major stem-loop structure (i), an outer loop (ii), the start (AUG) codon, and +1 TSS. (E) A 347-nt product including the 99-nt 5′ UTR and its flanking regions was transcribed in vitro under the control of the T7 promoter. Products were separated by agarose gel electrophoresis. Lane 1, control fragment 2,200 nt in length (Thermo Fisher) heated for 10 min at 70°C prior to loading; lane 2, control fragment, unheated; lane 3, 347-nt recA fragment containing the 5′ UTR after heat treatment; lane 4, the same 347-nt fragment without heat treatment. The percentage of unstructured RNA per total RNA bands was quantified and is included below each lane.
The 5′ UTR of A. baumannii recA is predicted to form a stable stem-loop structure.
In silico modeling (35) showed a high probability for the 5′ UTR of A. baumannii recA to form a stable secondary structure (ΔG° = −21.55 kcal/mol [Fig. 1D]). Structural features included a major stem-loop from nucleotide U(+13) to A(+73) (Fig. 1D, loop i). Additionally, there was a loop out and shorter stem predicted near the start codon (Fig. 1D, loop ii). We then performed in vitro transcription of a section of the recA message containing the 5′ UTR, including the flanking native sequences to maintain sequence context, using a T7-dependent RNA polymerase in vitro transcription system with fluorescently labeled dUTPs incorporated for visualization. The products of T7 RNA polymerase were separated by gel electrophoresis either with or without heat treatment at 70°C prior to loading (Fig. 1E). Heat treatment was expected to denature most structures within the RNA transcript, leading to less structure and faster migration. In contrast, with no heat denaturation prior to loading, additional structure would be maintained and change the migration of the sample. In the control RNA provided by the kit (Thermo Fisher), we observed that 46% of the ∼2000-nt unheated sample migrated at a size similar to that of the heated sample (Fig. 1E, lane 2). In comparison, only 13% of the ∼350-nt unheated 5′ UTR-containing transcript migrated at a size similar to that of the heated sample (lane 4), indicating that despite the length of the transcript, the 5′ UTR had additional structure causing slower migration in the gel. These data provided further evidence that the 5′ UTR sequence is indeed structured in vitro.
Disruption of the predicted major stem-loop of the recA 5′ UTR lowers expression of a fluorescent reporter.
To investigate whether the structural features of the 5′ UTR have a regulatory role in cis on recA expression, a low-copy-number plasmid translational reporter was constructed in pNLAC1 (36), in which the recA promoter, 5′ UTR, and recA ribosomal start site were fused to the far-red fluorescent protein mKate2. The sequences flanking the recA gene coding sequence were included to maintain sequence context next to the 5′ UTR. Within the major stem-loop (Fig. 2Ai), we identified three consecutive cytosine-guanine pairings [C(+24), C(+25), and C(+26) pairing to G(+62), G(+61), and G(+60), respectively] that in silico modeling predicted were critical for maintaining the stem-loop structure. We called this native reporter PrecA-UTR(C::G)-mKate2 (Fig. 2Ai). We created a second plasmid in which the three cytosines C(+24), C(+25), and C(+26) were each changed to guanine [PrecA-UTR(G::G)-mKate2]. This change was predicted to disrupt the formation of the stem-loop structure in silico (Fig. 2Aii). The full sequences of all 5′ UTR variants are listed in Table S1 in the supplemental material. Using single-cell resolution fluorescence microscopy (Fig. 2Ai), we observed low levels of expression of A. baumannii cells with the native PrecA-UTR(C::G)-mKate2 plasmid in the absence of DNA damage treatment. We hypothesized that this represents basal expression. To quantify the expression, we measured the fluorescence intensity of >1,000 single cells using MicrobeJ (37) (Fig. 2B). Fluorescence microscopy was used instead of fluorescence-activated cell sorting due to higher sensitivity of fluorescent reporter detection. As expected, we observed induction of mKate2 upon Cip treatment (Fig. 2A and B), which inhibits DNA gyrase, leading to double-stranded breaks (38). This induction is similarly observed with UV radiation (Fig. S2). Remarkably, we found that in the absence of DNA damage treatment, A. baumannii cells with the PrecA-UTR(G::G)-mKate2 reporter (Fig. 2Aii) have lowered fluorescence [38% lower than native UTR(C::G) (Fig. 2A and B)], suggesting that impairing the formation of the native 5′ UTR structure decreased recA expression.
FIG 2.

Disrupting the predicted stem-loop structure of the recA 5′UTR decreases expression. (A) (i) Using the RNAfold webserver (35), the predicted native structure of the 5′ UTR of recA was found to contain the bases C(+24), C(+25), and C(+26) pairing to G(+62), G(+61), and G(+60), respectively [UTR(C::G)]. Changing each C(+24), C(+25), C(+26) to G [UTR(G::G)] results in the predicted structure (ii), which has a lower probability to be structured, while removing the outer loop (iii) [UTR(ΔOL)] results in partial loss of structure near the starting AUG. To test the functionality of the 5′ UTR and its predicted structure, mKate2 was fused to the recA promoter, UTR, and putative ribosomal binding site in a low-copy-number plasmid. Representative fluorescence microscopy images of mKate2 expression of the strains with each UTR variant without (−) or with treatment with 10× MIC of Cip (+) are shown. The white arrow points to a DDRHigh cell, while the gray arrow points to a DDRLow cell. The scale bar represents 10 μm. (B) Single-cell quantification of mKate2 expression of each reporter strain with (+) or without (−) Cip treatment using MicrobeJ (37). Each dot represents one cell. At least 1,000 cells for each strain were quantified for relative mKate2 fluorescence with the respective 5′ UTR variant. The x axis is in log10 scale. (C) RT-qPCR was performed in biological triplicate to determine steady-state expression of mKate2 mRNA from each of the plasmid reporters with the respective UTR derivatives in WT A. baumannii cells, standardized to 16S rRNA. Relative quantification of the mKate2 transcript is shown with the native UTR reporter set to be equal to 1. Error bars represent standard deviations. An unpaired two-tailed t test was used for statistical analysis compared to mKate2 expression of the native UTR(C::G). *, P < 0.05.
Removing the predicted outer loop in the 5′ UTR recA transcript elevates expression of the fluorescent reporter.
There is a predicted outer loop in the 5′ UTR structure (Fig. 1D). When this feature was removed, the predicted structure (Fig. 2Aiii) still maintained the recA 5′ UTR major stem-loop but lacked structure near the recA start codon. In the absence of DNA damage treatment, we found that A. baumannii cells with the plasmid lacking the outer loop, PrecA-UTR(ΔOL)-mKate2, were brighter than the native PrecA-UTR(C::G)-mKate2 reporter (Fig. 2Aiii versus Ai). Again, we observed induction after DNA damage treatment (Fig. 2Aiii).
We then performed reverse transcriptase quantitative PCR (RT-qPCR) to determine if the recA 5′ UTR variants described above changed steady-state transcript levels. Using 16S rRNA as an endogenous control, we did not see a significant difference in transcript levels between the native reporter and the PrecA-UTR(ΔOL)-mKate2 reporter. This suggested that the increased expression observed (Fig. 2Aiii) might be due to increased translational efficiency, which may be caused by easier access to translational machinery. Interestingly, we found a significant (3-fold) decrease in mKate2 steady-state transcript levels in A. baumannii with PrecA-UTR(G::G)-mKate2 compared to the wild-type (WT) reporter [PrecA-UTR(C::G)-mKate2] (Fig. 2C).
The native recA 5′ UTR structure governs mRNA half-life and transcript stability.
To directly test the role of the 5′ UTR in mRNA stability, we measured the half-life of our reporter transcript with various UTR modifications (Fig. 3). To do this, cells with the appropriate reporter plasmids were grown to exponential phase and treated with rifampin to inhibit transcription. We then collected cells at consecutive times after rifampin treatment and determined the level of transcripts from these samples using RT-qPCR. By plotting the mKate2 transcript levels relative to the transcript levels at the start of rifampin treatment, we determined the rate of decay (k) of the mKate2 transcript in each strain and from it calculated the half-life of the native and variant UTR reporters (Fig. S3). The half-life of the mKate2 transcript from the native recA promoter and 5′ UTR is 4.5 min, while the half-life of the UTR(G::G) variant, which has lowered expression (Fig. 2Aii), is 35% lower (2.8 min) than that of the native UTR reporter construct (Fig. 3A). Remarkably, the UTR(ΔOL) half-life was not different from that of the native UTR (Fig. 3A; see also Fig. S3), which further supports the idea that the observed increase in mKate2 expression (Fig. 2A) is not due to transcript stability but may be due to a higher rate of translation. To test whether our results were based mostly on the 5′ UTR structure rather than the nucleotide sequence, we made the compensatory base-pairing variant of the native structure by taking the UTR(G::G) sequence and changing G(+62), G(+61), and G(+60) each to a cytosine, resulting in a 5′ UTR variant we called UTR(G::C) (Fig. 3B). This compensatory change resulted in a predicted structure similar to that of the native 5′ UTR (Fig. 3B). Indeed, the half-life of the respective mKate2 transcript was not significantly different from that of the native UTR (4.8 min [Fig. 3A]). This result indicated that the structure might be more important rather than the primary sequence for maintaining the stability of the recA transcript. Notably, we noticed that expression from cells with the compensatory stem-loop reporter was similar to that from cells with the native reporter. However, these cells had higher expression upon Cip treatment than the ones with the native reporter (Fig. S4). This suggests that while the secondary structure influences mRNA half-life, there may be additional levels of regulation for DNA damage induction, potentially through tertiary structure.
FIG 3.
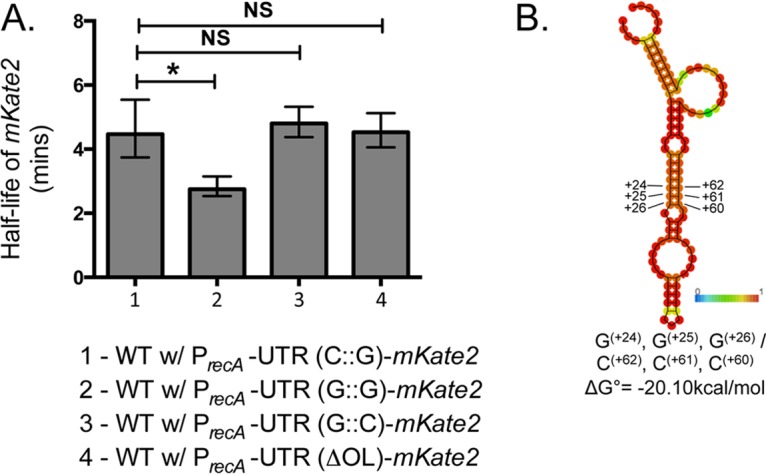
The native recA 5′ UTR structure governs mRNA half-life. (A) WT A. baumannii cells expressing mKate2 from plasmids containing different UTR variants were grown as indicated in Materials and Methods. Rifampin was added at different time points to inhibit further transcription. RT-qPCR was performed in triplicate to determine expression of mKate2 mRNA from samples collected at 2.5, 5, and 10 min after rifampin treatment standardized to 16S rRNA expression. The mean half-life of the transcripts from each plasmid (calculated as described in Materials and Methods and Fig. S3) is shown. Error bars represent standard deviations. Transcripts with the stem-loop [UTR(G::G)] have a significantly lower half-life than does the native UTR (bar 2). A UTR variant that has a compensatory mutation to rescue structure, PrecA-UTR(G::C)-mKate2 (B), has no significant change in half-life compared to that of the native or ΔOL UTR. An unpaired two-tailed t test was used for statistical analysis. *, P < 0.05. NS, not significant. (B) The predicted in silico structure of the compensatory UTR(G::C) has a structure similar to that of the native UTR based on the RNAfold webserver (35). This was constructed by changing G(+62), G(+61), and G(+60) of UTR(G::G) (Fig. 2A) each to C.
DNA damage response occurs at the promoter level.
Thus far, each strain with a 5′ UTR variant responded to DNA damage (Fig. 2A and B). To determine if the DNA damage response elements are within the UTR, we constructed a reporter with only the promoter and putative ribosomal binding site of recA fused to mKate2 (PrecA-NO UTR-mKate2). We found that the reporter responded, albeit weakly, to DNA damage (1.3-fold induction) (Fig. 4A). The level of induction that we observed in cells with the reporter containing the recA promoter without the 5′ UTR supports a role for the 5′ UTR in stabilizing transcripts from degradation but not in detecting DNA damage. Confirming that the observed levels of fluorescence in these cells were not fluctuations in background fluorescence, we observed that WT cells bearing an empty vector had, as expected, no increase in fluorescence in response to DNA damage above background (data not shown).
FIG 4.

The recA promoter senses DNA damage, while the 5′ UTR modulates the amplitude of recA expression in a nonuniform manner. Shown are representative fluorescence microscopy images of WT A. baumannii cells with (+) or without (−) Cip treatment with plasmid-borne mKate2 reporters. The reporter containing the recA promoter lacking the UTR, PrecA-NO UTR-mKate2, responds weakly to DNA damage treatment (A); the trpB promoter lacking the UTR, PtrpB-NO UTR-mKate2, has minimal expression (B); and the trpB promoter with the native recA UTR, PtrpB-UTR(C::G)-mKate2, displays bimodal mKate2 expression unresponsive to Cip treatment (C). Scale bars represent 10 μm. (D) Single-cell quantification of mKate2 expression (performed as for Fig. 2B) for each A. baumannii reporter strain, including that of the plasmid borne Escherichia coli PrecA-GFP used as a control for unimodal gene expression with (+) or without (−) Cip treatment. Native reporter (PrecA-UTR-mKate2) is included for comparison. The x axis is in log10 scale.
We then compared strains bearing an mKate2 reporter fused to the trpB promoter, a non-DDR gene, alone and with the native recA 5′ UTR (Fig. 4B and C, respectively). Cells bearing the reporter with the trpB promoter lacking the recA 5′ UTR (PtrpB-NO UTR-mKate2) showed very low mKate2 expression and did not respond to DNA damage (Fig. 4B). In contrast, cells with the trpB promoter fused to the recA 5′ UTR [PtrpB-UTR(C::G)-mKate2] had higher mKate2 expression (Fig. 4C). However, these cells did not show increased mKate2 fluorescence in response to DNA damage treatment (Fig. 4C, +cip). Fluorescence quantification is shown in Fig. 4D. Together, these data suggested that the recA promoter contributes to detecting DNA damage, while the recA 5′ UTR element can independently increase mKate2 expression, which we hypothesized was due to the ability of this structured feature to govern transcript stability.
The 5′ UTR endows nonuniform distribution of gene expression.
Using single-cell analyses, we visualized phenotypic variation of the WT reporter, observing DDRHigh and DDRLow cells upon treatment (Fig. 2A, white and gray arrows, respectively). To quantify the contribution of the promoter and the 5′ UTR of recA to the bimodality of the response, we compared the percent difference between the arithmetic and geometric mean of expression of the population (abbreviated as DAG) (Table 1). The arithmetic mean is strongly affected by extreme values, while the geometric mean takes into account the effects of a skewed population to better approximate the mean. For normal distributions, the percent difference between the mean values should be low, whereas for nonuniform (e.g., bimodal) distributions, these values would be higher (39). To set the DAG value limits for our experiments, we determined the DAG in a unimodally distributed system (30), in this case an E. coli strain with a PrecA-gfp plasmid reporter (Fig. 4D). The distribution of expression of the E. coli reporter was mostly normal, while the A. baumannii native reporter had characteristic clusters of expression. The DAG for the E. coli treated cells is 9.8% (Table 1). Thus, we chose a maximum cutoff of 15% for normally distributed expression. That is, any DAG value lower than 15% would mean that the cells are normally distributed and there is limited phenotypic variation.
TABLE 1.
Quantification of mKate2 expression of the different reporter strainsa
| Reporter strain | Ciprofloxacin treatment | AM | GM | % DAGb | Fold induction upon treatmentc |
|---|---|---|---|---|---|
| E. coli PrecA-gfp | − | 2.9 | 2.6 | 11.5 | 28.6 |
| + | 82.8 | 75.4 | 9.8 | ||
| A. baumannii | |||||
| PrecA-No UTR-mKate2 | − | 8.8 | 8.4 | 4.8 | 1.3 |
| + | 11 | 10.5 | 4.8 | ||
| PrecA-UTR(C::G)-mKate2 | − | 10.7 | 8.8 | 21.6 | 2.5 |
| + | 27.1 | 14.9 | 81.9 | ||
| PrecA-UTR(ΔOL)-mKate2 | − | 38.7 | 31.6 | 22.5 | 4.2 |
| + | 163.2 | 132.4 | 23.3 | ||
| PrecA-UTR(G::G)-mKate2 | − | 6.6 | 6.2 | 6.5 | 2.3 |
| + | 15.2 | 13.6 | 11.8 | ||
| PtrpB-No UTR-mKate2 | − | 6.3 | 6.3 | 0 | 1 |
| + | 6.5 | 6.5 | 0.62 | ||
| PtrpB-UTR(C::G)-mKate2 | − | 39.1 | 33.1 | 18 | 1.01 |
| + | 39.4 | 32.8 | 20.4 |
The fluorescence expression of >1,000 cells of each A. baumannii strain was quantified using the MicrobeJ plug-in for ImageJ (37). The arithmetic mean (AM) and geometric mean (GM) for each population were calculated.
The percent difference between the arithmetic mean and the geometric mean (DAG) was determined as 100 × (AM − GM)/AM.
Fold induction of mKate2 expression upon treatment with 10× MIC of Cip was determined as AMTreated/AMUntreated.
When we quantified the DAG for the native PrecA-UTR(C::G)-mKate2 reporter upon Cip treatment, we found that it has a nonuniform expression profile (DAG = 81.9%). Interestingly, the PrecA-UTR(G::G)-mKate2 reporter (which destabilizes the stem-loop structure) had a much more uniform distribution of expression (DAG = 11.8%). Furthermore, when mKate2 was expressed under the control of the recA promoter and no 5′ UTR, expression was normally distributed upon DNA damage (DAG = 4.8% [Table 1; Fig. 4A]). We next wanted to test whether the recA 5′ UTR could endow a non-DDR gene with phenotypic variation. When the 5′ UTR was fused to the trp promoter, mKate2 expression was nonuniform, in contrast to uniform expression without the 5′ UTR (DAG = 20.4% and 0.6%, respectively [Table 1; Fig. 4B and C]), indicating that the 5′ UTR may play a role in establishing the nonuniform expression of recA in A. baumannii.
The structured 5′ UTR maintains cellular RecA levels.
While mKate2 fluorescence experiments showed that protein levels are different with the variant UTRs (Fig. 2), we sought to determine the physiological consequences of altering the cis regulation by the 5′ UTR on recA in vivo. RecA is important for many cellular processes aside from its role in the DDR (40). Consistent with this, we observed that A. baumannii ΔrecA cells, which have no functional RecA protein, were elongated in late exponential phase (53% with a length of >2.5 μm [Fig. 5Aii]) compared to isogenic WT cells (<0.1% elongated [Fig. 5Ai]) in the absence of external DNA damage treatment. We used the elongated cell length phenotype of ΔrecA cells as an in vivo proxy for RecA intracellular levels. We complemented the phenotype using plasmids containing recA under the control of its native promoter with either the native or variant 5′ UTR and measured cell length. Indeed, native PrecA-UTR(C::G)-recA rescued the elongated ΔrecA cell morphology to WT levels: less than 1% (Fig. 5Aiii). However, when recA had the 5′ UTR predicted to form an impaired folding structure [PrecA-UTR(G::G)-recA], there was only a partial rescue of the cell length phenotype, with 24% of cells still remaining elongated, indicating that the 5′ UTR structure is critical for maintaining appropriate cellular RecA levels (Fig. 5Aiv). We observed that PrecA-UTR(G::C)-recA, the compensatory mutation that restores structure to the stem-loop (Fig. 3B), rescued the cell length phenotype of A. baumannii ΔrecA cells to less than 1% elongated (Fig. 5Av). This evidence suggested that the stem-loop structure of the recA UTR plays a role in regulating the intracellular levels of RecA in vivo.
FIG 5.

The recA 5′ UTR governs RecA intracellular concentration. (A) Plasmids used for these experiments were constructed with the recA gene under the control of its native promoter and UTR or with variant UTRs. Plasmids with the native regulatory region [PrecA-UTR(C::G)-recA], the mutated UTR(G::G) [PrecA-UTR(G::G)-recA], or the compensatory UTR(G::C) [PrecA-UTR(G::C)-recA] were introduced by transformation into A. baumannii ΔrecA cells. Untreated cells were imaged, and the percentage of cells equal to or longer than 2.5 μm was measured (shown underneath each representative phase-contrast image). The untreated WT A. baumannii cells have the typical coccobacillary shape of A. baumannii (i) and an undetectable number of elongated cells (<0.1%), while approximately 50% of the cells in the isogenic ΔrecA strain are elongated (ii). The elongation phenotype is rescued to the shorter coccobacilli by the plasmid-borne WT recA gene under the control of its native regulatory region, PrecA-UTR(C::G)-recA (iii), and only partially restored by PrecA-UTR(G::G)-recA (iv). A UTR variant that complements the stem-loop structure, PrecA-UTR(G::C)-recA, also rescued the cell length phenotype (v). Scale bar represents 10 μm. (B) Immunoblot to detect RecA was performed in each strain (lanes i to v) as described for panel A. Equal amounts of cell-free lysates were probed with polyclonal anti-RecA and monoclonal anti-RpoB antibodies (see Materials and Methods). RpoB was measured to account for differences in protein loading between samples. Relative RecA protein from each strain was quantified and standardized to ΔrecA cells with PrecA-UTR(C::G)-recA (lane iii) to compare the effects of altering the UTR structure (values shown below under each lane). RecA levels decrease upon disrupting formation of the UTR structure (lane iv). This value is rescued to native levels by the compensatory mutation in the UTR (lane v). **, undetectable. (C) UV survival (27 J/m2) of WT A. baumannii and isogenic ΔrecA strains with the plasmid-borne recA gene with native and variant 5′ UTRs. Only the strains with the UTRs that maintain a probable stem-loop structure survive UV treatment at WT levels. Error bars represent standard deviations. An unpaired two-tailed t test between indicated strains was used for statistical analysis. *, P < 0.05. (D) DNA damage-induced rifampin resistance acquisition of WT A. baumannii and ΔrecA cells with the plasmid-borne UTR-recA variants. Impairing formation of the major stem-loop results in an ∼10-fold-lower mutation frequency. Error bars represent standard deviations. An unpaired two-tailed t test between indicated strains was used for statistical analysis. *, P < 0.05.
To directly measure the intracellular RecA protein concentration of these cells, we performed Western blot analysis (Fig. 5B) using polyclonal anti-RecA and monoclonal anti-RpoB antibodies raised against the E. coli proteins, as we have done before (12). First, we isolated cell extract from WT and ΔrecA cells to set up the assay (Fig. 5B, lanes i and ii, respectively). We detected the RecA protein in the WT and not in the ΔrecA cells, as expected. Cell extracts from ΔrecA cells containing the PrecA-UTR(G::G)-recA construct (Fig. 5B, lane iii) were used as a reference to which we standardized protein levels. We detected a decrease in RecA for cells with the PrecA-UTR(G::G)-recA construct (0.7 [Fig. 5B, lane iv]), which is consistent with the results using the in vivo proxy of cell length (Fig. 5Aiv). This evidence suggested that preventing the 5′ UTR from forming the native stem-loop structure, which, in turn, changes the mRNA half-life, has a downstream effect on RecA levels. Moreover, cells with the compensatory UTR structure had slightly increased RecA compared to the cells with the native UTR (1.1 [Fig. 5B, compare lane iii with lane v]).
Intriguingly, we observed that the relative amount of RecA in WT A. baumannii cells (0.2) was lower than in the ΔrecA cells bearing the PrecA-UTR(G::G)-recA variant (0.7). The WT cells in the absence of external DNA damage and with a chromosomal recA gene might express relatively constant levels of RecA in all cells in the population. In contrast, it is possible that a majority of the cells with UTR(G::G) have higher levels of RecA since the recA gene is plasmid borne, but a subset of these cells may have RecA below a certain threshold, explaining the in vivo elongated phenotype (Fig. 5Aiv). Altogether, this evidence showed that the 5′ UTR of the recA transcript plays a role in maintaining intracellular RecA levels.
The recA 5′ UTR confers cell survival to DNA damage and rifampin resistance acquisition.
It is known that RecA is necessary for A. baumannii to survive DNA damage treatment (40). To test the role of the recA 5′ UTR in cell survival of DNA damage, we treated cells bearing different UTR variants with UV and measured their percent survival. We found that ∼80% of WT A. baumannii cells survive the level of UV used, while we did not detect viable cells among A. baumannii ΔrecA cells (Fig. 5B). The ΔrecA UV lethality was rescued with a plasmid-borne copy of recA containing the native recA 5′ UTR (Fig. 5C, bar 3). Survival of the ΔrecA cells bearing a copy of recA with UTR(G::G) was significantly lower than that of the WT; less than 3% of cells survived UV treatment (Fig. 5C, bar 4). Notably, restoration of the structure of the 5′ UTR with UTR(G::C) resulted in UV survival similar to that of the WT (Fig. 5C, bar 5). As a control, we measured UV survival and determined cell length of A. baumannii ΔrecA cells with the PrecA-UTR(G::C)-mKate2 reporter plasmid and found no difference compared to A. baumannii ΔrecA cells (Fig. 5C; see also Fig. S5A). These data suggest that maintaining the recA 5′ UTR is critical for survival of DNA-damaging conditions.
Previously, we demonstrated that functional RecA is necessary for DNA damage-induced mutagenesis and subsequent rifampin resistance acquisition upon mutation of rpoB, which encodes the target of rifampin (12). To test the relevance of the recA 5′ UTR in acquisition of rifampin resistance upon DNA damage, we measured the frequency of UV-dependent rifampin resistance mutants (Rifr) in the ΔrecA strains with the native and 5′ UTR-recA variants. Rifampin is an antibiotic that is often used to treat multidrug-resistant A. baumannii infections (41). We irradiated cells with UV light as done previously (12) and determined the frequency of Rifr mutants. As expected, the ΔrecA strain showed no difference between spontaneous (uninduced) and UV-induced resistant mutants, demonstrating the dependency on functional RecA (Fig. 5D). For strains bearing a plasmid copy of recA, we found that the Rifr uninduced spontaneous mutation frequency was typically about 1 to 2% of the UV-induced mutation frequency. These values were subtracted from the values of the DNA damage-induced frequencies to provide the adjusted frequency of Rifr. We observed no significant difference in induced mutation frequency between WT and ΔrecA cells bearing an extrachromosomal copy of recA under the control of its native promoter and 5′ UTR (frequencies were both ∼1.5 × 10−8 [Fig. 5D, compare bars 1 and 3]). Remarkably, ΔrecA cells with the UTR(G::G)-recA variant had a significant (∼10-fold) decrease in Rifr mutant frequency compared to the native UTR(C::G) construct (Fig. 5D, compare bars 4 and 3). For the compensatory UTR(G::C)-recA, we did not find a significant difference compared to the native UTR(C::G).
We noted that the DNA damage-induced RecA-dependent processes were rescued to WT levels, but protein levels for untreated cells differed between the strains as we showed earlier (Fig. 5B). Western blot analysis on cells that were UV irradiated showed that WT and ΔrecA cells bearing the native UTR(C::G)-recA plasmids had similar relative protein levels (Fig. S5B). This finding was consistent with the similar UV survival and Rifr mutant frequency between these strains. Interestingly, ΔrecA cells with the compensatory UTR(G::C)-recA had increased relative levels of RecA protein compared to those with the native UTR. This was also consistent with the expression we observed for the mKate2 reporter upon DNA damage treatment (Fig. S4).
DISCUSSION
This work focused on understanding recA regulation in A. baumannii, as RecA is key for induction of the DDR, which directly impacts survival of DNA-damaging agents, mutagenesis, and virulence (12, 40). We found that the recA transcript in A. baumannii has a 5′ UTR cis-regulatory element (Fig. 1) whose secondary structure provides stability to the transcript (Fig. 3) and modulates the amplitude of recA expression in a nonuniform manner (Fig. 2 and 4; Table 1). Notably, impairing stem-loop structure formation of the 5′ UTR of the recA mRNA transcript by mutation (Fig. 2Aii and C and Fig. 3) caused abnormal cell elongation, indicating that there is not enough RecA to maintain homeostasis in a subset of cells (Fig. 5A), as it lowers endogenous RecA protein levels (Fig. 5B). Additionally, preventing formation of the stem-loop structure of the 5′ UTR leads to poor survival of DNA damage treatment and lowered frequency of DNA damage-induced mutagenesis (Fig. 5C and D). The 5′ UTR maintains and modulates the intracellular concentration of the recA transcript and ultimately of functional RecA. Under DNA-damaging conditions, transcription and other cellular processes may become stalled. However, if the recA transcript is stable through its own inherent 5′ UTR structure, RecA protein could still be made, allowing the cell to respond to stress via the DDR. Moreover, this may allow the cell to survive desiccation and acquire advantageous mutations in endogenous genes, such as those for antibiotic resistance. Additionally, we found that the promoter and not the 5′ UTR is involved in sensing DNA damage. The mechanism(s) underlying the induction of recA and other DDR genes upon DNA damage is beyond the scope of this work but remains a critical feature of A. baumannii biology to continue to investigate.
It has been shown that other bacterial genes employ 5′ UTRs to stabilize mRNA transcripts (42–44). One well-studied example is the 5′ UTR of ompA in E. coli, which contains a stem-loop that stabilizes the ompA transcript by preventing 5′ end-dependent degradation as well as increasing the life span of other RNA transcripts (42, 45, 46). However, this is the first time, to our knowledge, that this type of regulation has been seen in a DNA damage response. We have also shown that a gene central to the DNA damage response has a 5′ UTR with the ability to stabilize other transcripts and, moreover, independently transfer phenotypic variation (Fig. 4C). Thus, we show a role for a 5′ UTR cis-regulatory RNA element to tune the distribution of recA gene expression and afford phenotypic variation among the population.
Upon DNA damage induction, we observed DDRLow and DDRHigh populations. It is possible that this was due to some unknown regulatory element acting on the recA 5′ UTR, which would result in the 5′ UTR adopting alternative conformations that may lead to differences in stability or translation of the transcript. Regulators of the 5′ UTR could be an RNA-binding protein or small RNA that would facilitate possible changes in the structure. However, we do not yet know if the 5′ UTR structure is indeed dynamic in this manner. Since expression of other DDR genes is RecA dependent (12), variations in recA expression could establish the bimodality of the system if there is a positive or double negative feedback loop (47). Thus, stochastic noise of RecA expression could also be a driver for establishing bimodality if there is a critical threshold level of RecA for cells to express the DDR. We hypothesize that because recA expression is critical to DDR gene expression, differences in cell-to-cell levels of RecA will lead to heterogeneous expression of DDR genes, including those that encode mutagenic error-prone polymerases and lead to antibiotic resistance acquisition (Fig. 5D). As mentioned previously, bimodal recA expression may serve as a bet-hedging strategy to increase fitness (30, 47, 48). It will be interesting to investigate the broader role for RNA-mediated cis regulation within the whole genome of A. baumannii, given the example of recA regulation that we have presented. This novel DDR strategy may contribute to the overall pathogenic success of A. baumannii and provide a target for developing new antimicrobials or treatments.
MATERIALS AND METHODS
Strains and culture conditions.
Strains and plasmids used are listed in Table S2 in the supplemental material. All cultures were routinely grown in rich LB medium and incubated at 37°C with shaking at 225 rpm for liquid cultures. Kanamycin (Kan; 35 μg/ml; Sigma-Aldrich, St. Louis, MO), tetracycline (Tet; 12 μg/ml; Sigma-Aldrich), rifampin (Rif; 100 μg/ml; Calbiotech, El Cajon, CA), and ciprofloxacin (Cip; A. baumannii MIC = 0.6 μg/ml, E. coli MIC = 0.01 μg/ml; Sigma-Aldrich) were added to the medium as indicated elsewhere in the text and in Table S2.
RNA-Seq analysis and PCR walking.
RNA-Seq data sets (SRP036862) published by Hare et al. (32) for A. baumannii ATCC 17978 were assembled using CLC Genomics (Qiagen, Hilden, Germany) and aligned to the genome of A. baumannii ATCC 17978 (NCBI accession no. NC_009085.1). Total RNA from untreated A. baumannii cells bearing different reporters was extracted using the Zymo Direct-zol RNA kit as directed (Zymo, Irvine, CA). The absence of genomic DNA was verified by carrying out a PCR with primers recAmidF and recAmidR (Table S3). RNA was converted to cDNA using the a high-capacity cDNA reverse transcription kit as suggested by the manufacturer (Applied Biosystems, Foster City, CA). Twenty cycles of PCR were performed using the cDNA as the template with primer R1 and each of the primers F1 to F6, respectively, using DreamTaq Mastermix (Thermo Fisher, Waltham, MA). PCR was performed on WT genomic DNA (gDNA) to test that primers and PCR conditions were appropriate. Products were separated by gel electrophoresis on a 2% agarose gel in 1× Tris-acetate-EDTA (TAE) buffer at 90 V.
In vitro transcription.
The A. baumannii recA 5′ UTR transcript was amplified by PCR using the PrecAF and recAmKate2R oligonucleotides (Table S3) and DreamTaq master mix (Thermo-Fisher). The resulting 347-bp product was ligated into the pGEM-TEasy vector (Promega, Madison, WI) using T4 DNA ligase (Promega). The insert was sequenced to verify that the sequence remained intact with a standard primer. In vitro transcription was performed using the TranscriptAid T7 high-yield kit (Thermo Fisher) as described by the manufacturer, with 5 μg of NcoI-linearized plasmid as the template, and a 2.5 mM concentration of fluorescently labeled dUTPs (ChromaTide Alexa Fluor 546–14-dUTP; Thermo Fisher) was added to the in vitro transcription mixes. Reaction mixtures were incubated for 2.5 h at 37°C. Samples were treated with DNase I (Qiagen), and each was split to have both an untreated control and a heat treatment (70°C for 10 min and chilled afterwards for 3 min on ice). The samples were separated on a 2% agarose gel in 1× TAE buffer at 90 V. The gel was visualized with a Cy3-555 laser on a Typhoon 8600 (GE, Fairfield, CT).
Ciprofloxacin treatment, UV survival, and rifampin resistance assay.
For Cip treatment, saturated liquid cultures were diluted 1:100 in LB and grown to exponential phase for 3 h. Cells were then treated with 10× MIC of Cip for 3 h and visualized with fluorescence microscopy. Multiple photos from independent experiments were used to quantify at least 1,000 cells per strain and per treatment using the MicrobeJ (37) plug-in for ImageJ (NIH, Bethesda, MD). UV survival was carried out using three independent saturated liquid cultures of each strain, which were diluted 10−5 in SMO (100 mM NaCl, 20 mM Tris-HCl [pH 7.5]). Fifty microliters of this dilution was evenly spread using sterile glass beads on LB agar plates with the appropriate antibiotic as indicated in the figure legends. Plates were irradiated in the dark under a UV germicidal lamp with 27 J/m2. Colonies on untreated and UV-treated plates were counted, and percent survival was standardized to untreated cells. For UV-induced mutagenesis and rifampin resistance acquisition, a modified version of an established protocol was followed (12). To assay strains with lowered UV survival, e.g., ΔrecA cells, 10 ml of culture was concentrated 5-fold prior to irradiation in the dark under a UV germicidal lamp at a dosage that resulted in approximately 15 to 20% survival for each strain, followed by 3 h of outgrowth. Rifampin resistance frequency was calculated by dividing the number of Rifr mutants by the total number of CFU. The frequency of spontaneous Rifr mutants from parallel untreated cultures was determined the same way. The adjusted DNA damage-induced rifampin resistance frequency was calculated by subtracting the spontaneous resistance from the UV-induced frequency. All assays were performed in triplicate, and an unpaired two-tailed t test was used for statistical analysis (P < 0.05).
Strain and plasmid construction.
To construct the PrecA-UTR(CG)-mKate2 plasmid reporter (pCC1 [Table S2]), the A. baumannii recA (A1S_1962) putative promoter region was amplified using primers PrecAF and recAmKate2R (Table S2). The mKate2 coding sequence was amplified using pYC251 (Table S2) as the template with the primers mKate2F and mKate2R (Table S3). The forward and reverse primers included 15-bp overhangs for isothermal assembly (49). The PCR amplicons were cleaned up using a QIAquick PCR purification kit (Qiagen), and plasmid pNLAC1 (Table S2) was digested with PstI (New England BioLabs [NEB], Ipswich, MA). After enzyme inactivation, the inserts and vector were assembled using Gibson assembly master mix (NEB). The plasmid was sequenced using the primers PNLACF and PNLACR (Table S3) to confirm correct assembly.
To create the PrecA-UTR(G::G)-mKate2 plasmid reporter (pCC2 [Table S2]), we site directed C(+22), C(+23), and C(+24) of the 5′ UTR plasmid with the reporter to G's. To do this, 100 ng of pCC1 [PrecA-UTR(G::G)-mKate2] was methylated with CpG methyltransferase (Thermo Fisher) and primers SD1F and SD1R (Table S3) were used following the protocol provided by the Gene Tailor site-directed mutagenesis system (Invitrogen, Carlsbad, CA). To create the PrecA-UTR(ΔOL)-mKate2 plasmid reporter (pCC3 [Table S2]), the same protocol was carried out using pCC1 as the template and primers SD2F and SD2R. The PrecA-UTR(C::G)-recA plasmid (pCC4 [Table S2]) was constructed using Gibson assembly as described above with the primers PrecAF and recAR (Table S3), which encompass the recA coding sequence, into PstI-digested pNLAC1. To generate the PrecA-UTR(G::G)-recA plasmid (pCC5 [Table S1]), site-directed mutagenesis was performed as described above using pCC4 as the template and primers SD1F and SD1R (Table S3). To generate the PrecA-UTR(G::C)-recA plasmid (pCC6 [Table S3]), PrecA-UTR(G::G)-recA was the template and primers SD3F and SD3R (Table S3) were used.
To construct the reporter plasmid with the trpB promoter instead of the recA promoter (pCC7 [Table S3]), the oligonucleotide Ptrp-UTR(C::G)-mKate2 was synthesized using Integrated DNA Technologies (IDT) (Table S2) and assembled into PstI-digested pNLAC1 using Gibson assembly as described above. To construct plasmid reporters with the trpB or recA promoter lacking the 5′ UTR (pCC8 and pCC9, respectively [Table S2]), first a promoterless mKate2 plasmid (pCC8) was constructed. The mKate2 coding sequence with the putative ribosomal binding site (RBS) of A. baumannii recA was amplified from a fluorescent reporter that does not contain the recA coding sequence, using primers MCSmKate2F (with restriction sites for PstI, SacI, and XbaI [Table S3]) and MCSmKate2R (with a restriction site for PstI [Table S3]). The PCR amplicon and pNLAC1 were digested with PstI (NEB) and ligated using T4 ligase (Promega). Primers PrecAF2 and PrecAR (Table S3) were used to amplify the recA promoter (excluding the UTR), and primers PtrpBF and PtrpBR (Table S3) were used to amplify the promoter of trpB (A1S_1692). The forward and reverse primers included restriction sites for ScaI and XbaI, respectively. The PCR amplicons were cleaned up as before (QIAquick PCR purification kit; Qiagen), and both the amplicons and pCC8 were digested with XbaI and SacI. Both digests were separated using a 1% agarose gel and extracted using a QIAquick gel purification kit (Qiagen). The insert and vector were then ligated using T4 ligase (Promega) at 4°C for 18 h. All plasmids were confirmed with sequencing using primers PNLACF and PNLACR (Table S2) to confirm correct assembly after transformation into competent E. coli and selection for Tetr. All plasmids were introduced into A. baumannii cells by electroporation (30). Transformants were selected for on LB agar supplemented with Tet. All plasmids introduced into A. baumannii were confirmed by PCR using oligonucleotides PNLACF and PNLACR. Cycling conditions are available upon request.
Quantitative real-time PCR.
Total RNA from untreated A. baumannii cells bearing different reporters was extracted using the Zymo Direct-zol RNA kit as directed (Zymo). The absence of genomic DNA was verified by carrying out a PCR with the same oligonucleotide sets described below for qPCR. The total RNA concentration was measured with a NanoDrop 2000 (Thermo Scientific). RNA was converted to cDNA using a high-capacity cDNA reverse transcription kit as suggested by the manufacturer (Applied Biosystems). qPCR was performed using primers 16SRNAF and 16SRNA and primers mKate2qPCRF and mKate2qPCRR (Table S3) on serially diluted cDNA by following the protocol for Fast SYBR green master mix (Applied Biosystems) with a StepOnePlus real-time PCR system (Applied Biosystems). The comparative threshold cycle (ΔΔCT) was calculated for each strain in biological and technical triplicates. The relative expression was standardized using the endogenous 16S control with A. baumannii PrecA-UTR(CG)-mKate2 as the reference sample. An unpaired two-tailed t test was used for statistical analysis (P < 0.05).
Determination of mRNA half-life.
Saturated cultures were diluted 1:100 in fresh LB medium. After 3 h of growth to reach exponential phase, rifampin was added to the cultures at a final concentration of 250 μg/ml. At 2.5, 5, and 10 min posttreatment, 0.5-ml samples of each culture were collected and resuspended in bacterial RNAprotect reagent according to the manufacturer (Qiagen). Total RNA extraction and qPCR were performed as described above for each sample. The comparative ΔΔCT of mKate2 expression was calculated for each strain in biological and technical triplicates relative to 16S rRNA expression. The relative expression of mKate2 in these strains was transformed using the natural logarithm, and these values were plotted as a function of time to generate a linear decay curve. From this, we found the slope of each regression line of best fit, which corresponds to the half-coefficient k (see Fig. S3B). Half-life was then calculated using the formula t1/2 = ln (2)/k.
Microscopy and analysis.
One milliliter of cells was spun in a microcentrifuge at 14,000 rpm for 1 min and resuspended in 100 μl of LB. For microscopy, a 1-μl drop of these cells was placed on a pad with 1% agarose in water and covered with a coverslip. Background fluorescence was determined using wild-type cells with no reporter. Imaging across strains was done at the same exposure settings using a Leica MicroStation5000 with a Leica DM3000G camera (Leica, Wetzlar, Germany). Single-cell fluorescence of >1,000 cells was quantified using the MicrobeJ (37). Cell length was measured manually for 300 individual cells with ImageJ (NIH) by determining the distance between the two poles.
Western blot analysis.
Saturated cultures were diluted as described above and grown to an optical density (OD) of ∼0.6 to 0.8. For UV-treated cells, 27 J/m2 UV treatment was performed as described above for UV-induced Rifr. Cells were then collected by centrifugation. Western blot analysis was performed as previously described using the same antibodies (12) but detected with Radiance Plus chemiluminescent substrate (Azure Biosystems, Dublin, CA), followed by imaging on a ChemiDox XRS+ system and Image Lab software (Bio-Rad, Hercules, CA). Densitometry analysis of the bands was performed using ImageJ (NIH). Boxes were drawn around each band, and the average pixel intensity was measured. The same box was used to measure background pixel intensity of a region with no band, and this average intensity was subtracted from the average intensity of the band. These values were then standardized to one of the samples to determine a relative ratio of RecA expression.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Malamy, L. Sonenshein, and the members of the Godoy and Chai labs for critical discussions as well as the insightful reviewers that made this work better. We are indebted to T. Russo for the generous gift of the pNLAC1 plasmid and G. Bou for the generous gift of the A. baumannii ΔrecA mutant.
An NIH grant (GM088230) to V.G.G. and a startup grant from Northeastern University to Y.C. supported this work. C.C. was partly supported by a Northeastern University grant for continuing graduate research, and B.H. was funded by the DAAD program (ISAP 57166474).
C.C. and V.G.G. initiated the research. C.C., K.G., Y.C., and V.G.G. designed the experiments. C.C., K.G., and V.G.G. performed experiments. B.H. performed the in vitro transcription. C.C. and K.G. analyzed the data. C.C., K.G., and V.G.G. wrote the manuscript.
We declare that we have no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00799-16.
REFERENCES
- 1.Jawad A, Seifert H, Snelling AM, Heritage J, Hawkey PM. 1998. Survival of Acinetobacter baumannii on dry surfaces: comparison of outbreak and sporadic isolates. J Clin Microbiol 36:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fournier PE, Richet H. 2006. The epidemiology and control of Acinetobacter baumannii in health care facilities. Clin Infect Dis 42:692–699. doi: 10.1086/500202. [DOI] [PubMed] [Google Scholar]
- 3.Dijkshoorn L, Nemec A, Harald S. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
- 4.Maragakis LL, Perl TM. 2008. Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clin Infect Dis 46:1254–1263. doi: 10.1086/529198. [DOI] [PubMed] [Google Scholar]
- 5.Valencia R, Arroyo LA, Conde M, Aldana JM, Torres M, Fernández-Cuenca F, Garnacho-Montero J, Cisneros JM, Ortíz C, Pachón J, Aznar J. 2009. Nosocomial outbreak of infection with pan-drug-resistant Acinetobacter baumannii in a tertiary care university hospital. Infect Control Hosp Epidemiol 30:257–263. doi: 10.1086/595977. [DOI] [PubMed] [Google Scholar]
- 6.Custovic A, Smajlovic J, Tihic N, Hadzic S, Ahmetagic S, Hadzagic H. 2014. Epidemiological monitoring of nosocomial infections caused by Acinetobacter baumannii. Med Arch 68:402–406. doi: 10.5455/medarh.2014.68.402-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mugnier PD, Poirel L, Nordmann P. 2009. Functional analysis of insertion sequence ISAba1, responsible for genomic plasticity of Acinetobacter baumannii. J Bacteriol 191:2414–2418. doi: 10.1128/JB.01258-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F, Zhu Y, Yi Y, Lu N, Zhu B, Hu Y. 2014. Comparative genomic analysis of Acinetobacter baumannii clinical isolates reveals extensive genomic variation and diverse antibiotic resistance determinants. BMC Genomics 15:1163. doi: 10.1186/1471-2164-15-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geisinger E, Isberg RR. 2015. Antibiotic modulation of capsular exopolysaccharide and virulence in Acinetobacter baumannii. PLoS Pathog 11:1–27. doi: 10.1371/journal.ppat.1004691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas I. 2015. Genetic tools for manipulating Acinetobacter baumannii genome: an overview. J Med Microbiol 64:657–669. doi: 10.1099/jmm.0.000081. [DOI] [PubMed] [Google Scholar]
- 12.Norton MD, Spilkia AJ, Godoy VG. 2013. Antibiotic resistance acquired through a DNA damage-inducible response in Acinetobacter baumannii. J Bacteriol 195:1335–1345. doi: 10.1128/JB.02176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedberg E, Walker GC, Siede W, Wood RD, Schultz R. 2006. DNA repair and mutagenesis, 2nd ed. ASM Press, Washington, DC. [Google Scholar]
- 14.Cirz RT, O'Neill BM, Hammond JA, Head SR, Romesberg FE. 2006. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J Bacteriol 188:7101–7110. doi: 10.1128/JB.00807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirz RT, Jones MB, Gingles NA, Minogue TD, Jarrahi B, Peterson SN, Romesberg FE. 2007. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J Bacteriol 189:531–539. doi: 10.1128/JB.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, Yang H, Pavletich NP. 2008. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453:489–494. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 17.Little JW, Mount DW. 1982. The SOS regulatory system of Escherichia coli. Cell 29:11–22. [DOI] [PubMed] [Google Scholar]
- 18.Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- 20.Cirz RT, Romesberg FE. 2007. Controlling mutation: intervening in evolution as a therapeutic strategy. Crit Rev Biochem Mol Biol 42:341–354. doi: 10.1080/10409230701597741. [DOI] [PubMed] [Google Scholar]
- 21.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. 2015. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 22.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida H, Bogaki M, Nakamura M, Nakamura S. 1990. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob Agents Chemother 34:1271–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon E-J, Courvalin P, Grillot-Courvalin C. 2013. RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: major role for AdeABC overexpression and AdeRS mutations. Antimicrob Agents Chemother 57:2989–2995. doi: 10.1128/AAC.02556-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horii T, Ogawa T, Ogawa H. 1980. Organization of the recA gene in Escherichia coli. Proc Natl Acad Sci U S A 77:313–317. doi: 10.1073/pnas.77.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson A, Brzoska AJ, Turner KM, Withers R, Harry EJ, Lewis PJ, Dixon NE. 2010. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol Mol Biol Rev 74:273–297. doi: 10.1128/MMBR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charpentier X, Kay E, Schneider D, Shuman HA. 2011. Antibiotics and UV radiation induce competence for natural transformation in Legionella pneumophila. J Bacteriol 193:1114–1121. doi: 10.1128/JB.01146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varhimo E, Savijoki K, Jalava J, Kuipers OP, Varmanen P. 2007. Identification of a novel streptococcal gene cassette mediating SOS mutagenesis in Streptococcus uberis. J Bacteriol 189:5210–5222. doi: 10.1128/JB.00473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorer MS, Sessler TH, Salama NR. 2011. Recombination and DNA repair in Helicobacter pylori. Annu Rev Microbiol 65:329–348. doi: 10.1146/annurev-micro-090110-102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macguire AE, Ching MC, Diamond BH, Kazakov A, Novichkov P, Godoy VG. 2014. Activation of phenotypic subpopulations in response to ciprofloxacin treatment in Acinetobacter baumannii. Mol Microbiol 92:138–152. doi: 10.1111/mmi.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veening J-W, Smits WK, Kuipers OP. 2008. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol 62:193–210. doi: 10.1146/annurev.micro.62.081307.163002. [DOI] [PubMed] [Google Scholar]
- 32.Hare JM, Ferrell JC, Witkowski TA, Grice AN. 2014. Prophage induction and differential RecA and UmuDAb transcriptome regulation in the DNA damage responses of Acinetobacter baumannii and Acinetobacter baylyi. PLoS One 9:e93861. doi: 10.1371/journal.pone.0093861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lisser S, Margalit H. 1993. Compilation of E. coli mRNA promoter sequences. Nucleic Acids Res 21:1507–1516. doi: 10.1093/nar/21.7.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. 2008. The Vienna RNA websuite. Nucleic Acids Res 36:70–74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luke NR, Sauberan SL, Russo TA, Beanan JM, Olson R, Loehfelm TW, Cox AD, Michael FS, Vinogradov EV, Campagnari AA. 2010. Identification and characterization of a glycosyltransferase involved in Acinetobacter baumannii lipopolysaccharide core biosynthesis. Infect Immun 78:2017–2023. doi: 10.1128/IAI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ducret A, Quardokus E, Brun Y. 2016. MicrobeJ, a high throughput tool for quantitative bacterial cell detection and analysis. Nat Microbiol 1:1607. doi: 10.1038/nmicrobiol.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CR, Malik M, Snyder M, Drlica K. 1996. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol 258:627–637. doi: 10.1006/jmbi.1996.0274. [DOI] [PubMed] [Google Scholar]
- 39.Issa MM, Nejem RM, Van Staden RIS, Aboul-Enein HY. 2015. New approach application of data transformation in mean centering of ratio spectra method. Spectrochim Acta A Mol Biomol Spectrosc 142:204–209. doi: 10.1016/j.saa.2015.01.064. [DOI] [PubMed] [Google Scholar]
- 40.Aranda J, Bardina C, Beceiro A, Rumbo S, Cabral MP, Barbé J, Bou G. 2011. Acinetobacter baumannii RecA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J Bacteriol 193:3740–3747. doi: 10.1128/JB.00389-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giannouli M, Di Popolo A, Durante-Mangoni E, Bernardo M, Cuccurullo S, Amato G, Tripodi MF, Triassi M, Utili R, Zarrilli R. 2012. Molecular epidemiology and mechanisms of rifampicin resistance in Acinetobacter baumannii isolates from Italy. Int J Antimicrob Agents 39:58–63. doi: 10.1016/j.ijantimicag.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 42.Emory SA, Bouvet P, Belasco JG. 1992. A 5′-terminal stem-loop structure can stabilize mRNA in Escherichia coli. Genes Dev 6:135–148. doi: 10.1101/gad.6.1.135. [DOI] [PubMed] [Google Scholar]
- 43.Unniraman S, Chatterji M, Nagaraja V. 2002. A hairpin near the 5′ end stabilises the DNA gyrase mRNA in Mycobacterium smegmatis. Nucleic Acids Res 30:5376–5381. doi: 10.1093/nar/gkf697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amilon KR, Letley DP, Winter JA, Robinson K, Atherton JC. 2015. Expression of the Helicobacter pylori virulence factor vacuolating cytotoxin A (vacA) is influenced by a potential stem-loop structure in the 5′ untranslated region of the transcript. Mol Microbiol 98:831–846. doi: 10.1111/mmi.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emory SA, Belasco JG. 1990. The ompA 5′ untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J Bacteriol 172:4472–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deana A, Celesnik H, Belasco JG. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- 47.Smits WK, Kuipers OP, Veening J-W. 2006. Phenotypic variation in bacteria: the role of feedback regulation. Nat Rev Microbiol 4:259–271. doi: 10.1038/nrmicro1381. [DOI] [PubMed] [Google Scholar]
- 48.Kussell E, Leibler S. 2005. Phenotypic diversity, population growth, and information in fluctuating environments. Science 309:2075–2078. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- 49.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.