

Abstract
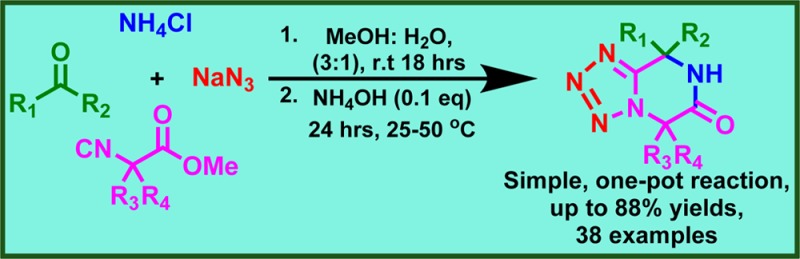
Ammonia in the tetrazole Ugi variation together with α-amino acid methyl ester-derived isocyanides provides tetrazolopiperidinones in good to high yields in one pot. The scope and limitations of this reaction were investigated by performing >70 reactions. The scaffold is useful to fill high-throughput screening decks and in structure-based drug design.
Keywords: tetrazolopiperidinone, isocyanide, multicomponent reaction, Ugi reaction, ammonia Ugi reaction, European Lead Factory
Introduction
Diketopiperazine (DKP) is a privileged scaffold present in many natural products as well as in screening libraries of synthetic compounds (Figure 1).1 While 1,5-disubstituted tetrazoles are bioisosteric to cis-amides,2 bicyclic tetrazolopiperidinones are bioisosteric to DKP, although one amide is morphed to the tetrazole with the consequence that one less hydrogen-bond donor and two more hydrogen-bond acceptors are present (Figure 2). The hydrogen-bond donor–acceptor count is a key molecular property influencing oral bioavailability, solubility, stability, and half-life time, just to mention a few.3
Figure 1.

DKP fungal natural product brevianamide F (cyan sticks) bound to prenyltransferase (gray sticks) (PDB ID 3O2K).2 Noteworthy is the fact that the DKP NH undergoes a hydrogen-bond-donor−π interaction (yellow dotted lines) to the Met backbone carbonyl. Hydrogen-bonding, van der Waals, dipolar, and π–π interactions are shown using red, yellow, cyan, and orange dotted lines, respectively. The stereopicture was rendered using PYMOL.3
Figure 2.
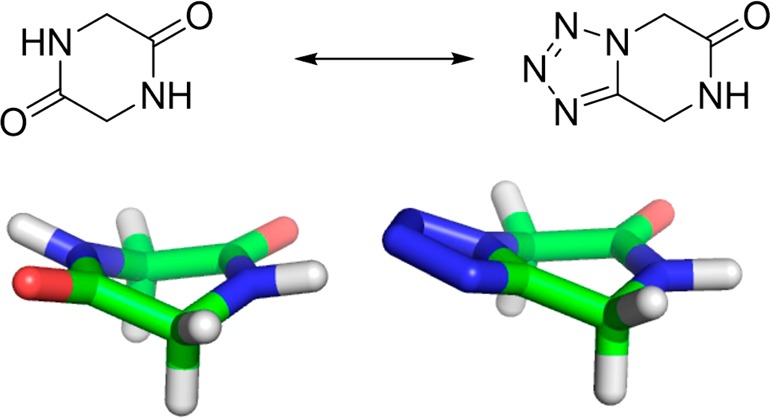
Bioisosterism of diketopiperazine and tetrazolopiperidinone.
Tetrazolopiperidinones are less prominent in the chemical literature compared with DKP and can be synthesized by multicomponent reaction (MCR) pathways (Scheme 1).4−10 MCR-based piperidinone syntheses are very versatile and have been recently reviewed.11 Previously we described the successful use of ammonia in the tetrazole Ugi reaction, an otherwise difficult and low-yielding starting material in other Ugi variations.12 Here we wish to describe the unprecedented, straightforward, and versatile synthesis of N-unsubstituted tetrazolopiperidinones by the Ugi reaction of TMS-N3, α-amino acid-derived isocyanoacetic acid methyl esters, oxo components, and ammonia.
Scheme 1. Examples of Previous DKP, Tetrazolylpipridine, and Tetrazolopiperidinone Syntheses and the Present Work.

Results and Discussion
Ammonia in the Ugi tetrazole reaction often leads to multiple reaction pathways.13−17 For example, the initially formed tetrazolomethylamine can react twice further in the Ugi reaction; ammonia as a strong base can induce aldol-type condensations with the oxo components; or ammonia can lead to amidation of the isocyanoacetic acid methyl esters. Not surprisingly, few successful examples of ammonia Ugi reactions are known.18−20 Thus, tritylamine as an ammonia surrogate was introduced in the Ugi MCR.21 In this work, we wanted to perform the one-pot synthesis of tetrazolopiperidinones C using a suitable ammonia source in the Ugi tetrazole reaction and basic reaction conditions for postcyclization. Therefore, it was not clear a priori whether the envisioned reaction could be performed successfully. Initially, we selected aqueous ammonia as the ammonia source and a base to promote the postcyclization reaction. Not surprisingly, a first reaction using aqueous ammonia in methanol at room temperature yielded only traces of the product (Table 1). Thus, we embarked on an extensive optimization exercise to increase the yield of the reaction by varying the azide and ammonia sources, temperature, time, solvent, and base.
Table 1. Optimization of the Reaction Conditions.

| entry | NH3 source | N3 source | solvent | base (equiv) | temp, time | yield (%) |
|---|---|---|---|---|---|---|
| 1 | NH4OH | TMSN3 | MeOH | – | RT, 8 h | traces |
| 2 | NH4OH | TMSN3 | MeOH | – | RT, 18 h + 60 °C, 18 h | 31 |
| 3 | NH4OH | TMSN3 | MeOH | – | 60 °C, 18 h | 48a |
| 4 | NH4OH | NaN3 | MeOH | – | RT, 18 h + 50 °C, 18 h | 35 |
| 5 | NH4OH | TMSN3 | MeOH | NaOMe (1.0) | RT, 18 h + RT, 18 h | 22 |
| 6 | NH4OH | NaN3 | MeOH | NaOMe (1.0) | RT, 18 h + 50 °C, 18 h | 16 |
| 7 | NH4OH | NaN3 | MeOH | Et3N (1.0) | RT, 18 h + 60 °C, 18 h | 9 |
| 8 | NH4Cl | NaN3 | MeOH/H2O (3:1) | Et3N (1.0) | RT, 18 h + RT, 18 h | 59 |
| 9 | NH4Cl | NaN3 | MeOH/H2O (3:1) | Et3N (1.0) | RT, 18 h + 60 °C, 18 h | 47 |
| 10 | NH4Cl | NaN3 | MeOH/H2O (3:1) | Et3N (1.0) | RT, 18 h + 40 °C, 18 h | 50 |
| 11 | NH4Cl | NaN3 | MeOH:H2O (3:1) | NaOMe (1.0) | RT, 18 h + 40 °C, 18 h | 27 |
| 12 | NH4Cl | NaN3 | MeOH/H2O (3:1) | NH4OH (1.0) | RT, 18 h + 40 °C, 18 h | 29 |
| 13 | NH4Cl | NaN3 | MeOH/H2O (3:1) | NaOMe (0.1) | RT, 18 h + 40 °C, 18 h | 55 |
| 14 | NH4Cl | NaN3 | MeOH/H2O (3:1) | NH4OH (0.1) | RT, 18 h +50 °C, 18 h | 67 |
The product was obtained as the
free tetrazole.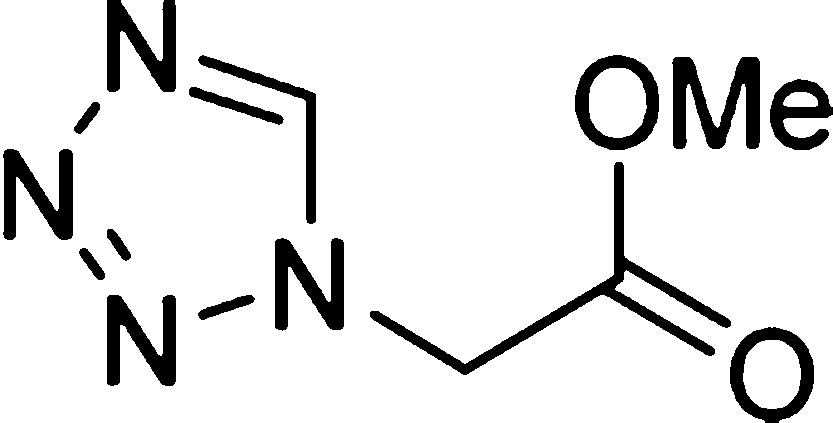
In order to optimize the reaction conditions, we first screened different ammonia sources. Ammonium hydroxide (1.2 equiv, 28–30 wt % solution of NH3 in water) as an ammonia source together with cyclohexanone (1 equiv) as an oxo component, methyl isocyanoacetate (1 equiv), and TMSN3 (1.2 equiv) in methanol (1 M) at room temperature for 18 h yielded trace amounts of the cyclized product (Table 1, entry 1). Furthermore, the same reaction mixture was heated at 60 °C for 18 h, and we observed a 31% yield of cyclized product (Table 1, entry 2). Unfortunately, when the reaction was performed at 60 °C for 18 h without prestirring at room temperature, no product formation was observed; instead, the formation of the tetrazole according to Oliveri-Mandala and Alagna was observed as the major product, (Table 1, entry 3). In another attempt, we replaced TMSN3 with sodium azide, which did not improve the overall yield (Table 1, entry 4). Furthermore, we envisioned that the use of a base after the Ugi adduct formation could enhance the cyclization reaction of the free amino group with the methyl ester group to form the corresponding tetrazolopiperidinone C. We tested various bases such as triethylamine, sodium ethoxide, sodium hydroxide, and sodium hydride, with various solvents and temperatures (Table 1, entries 5–7). Unfortunately, these combinations failed to give good yields. Next, we decided to exchange ammonium hydroxide with ammonium chloride as an ammonia source. As per our previous experience with α-aminomethyltetrazole formation, we used a 3:1 methanol/water mixture as the solvent.8 Thus, we performed the reaction using ammonium chloride, cyclohexanone, methyl isocyanoacetate, and sodium azide in 3:1 methanol/water at room temperature for 18 h, followed by addition of triethylamine and stirring at room temperature for an additional 18 h. Surprisingly, we observed a 59% yield of cyclized product (Table 1, entry 8). Encouraged by these results, we tested the same reaction with various bases and temperatures (Table 1, entries 9–14). By using sodium methoxide as a base, we observed hydrolysis of the methyl ester of the Ugi adduct to form the corresponding acid as a major side product, and the yield of cyclized product was lowered to 27% (Table 1, entry 11). A similar observation was made when ammonium hydroxide was used with triethylamine (1.0 equiv) as the base. To avoid hydrolysis of the methyl ester of the Ugi adduct, we decided to use the base in catalytic amounts (Table 1, entries 13 and 14). Surprisingly, we observed that the addition of a catalytic amount (0.1 equiv) of ammonium hydroxide as a base at 50 °C for 18 h gave a good 67% yield of the cyclized product (Table 1, entry 14).
With these optimized reaction conditions in hand, we looked into the scope and limitations of the reaction, taking into account various functional groups (small, bulky, aliphatic, aromatic, heteroaromatic, cyclic, acyclic, and α,β-unsaturated) on the oxo components and the isocyanides derived from methyl esters of α-amino acids; in total, we performed more than 70 different reactions (see Table 2). We purposely included difficult-to-deactivate starting materials to provide a realistic scope and limitation study.
Table 2. Substrate Scope and Limitations in the One-Pot Tetrazolopiperidinone Reactiona.



Isolated yields are shown. Diastereomeric ratios (dr) were determined by SFC-MS and 1H NMR analyses.
The diastereomeric mixture was isolated in 9:1 dr.
The cis:trans ratio was 4:1.
The diastereomeric mixture was isolated in 1:1:0.1:0.1 dr.
The diastereomeric mixture was isolated in 4:3 dr.
The cis:trans ratio was 19:1.
Initially, we tested cyclohexanone (1b) with various isocyanides such as methyl isocyanoacetate and its derivatives (1a–6a). Reaction of 1b with isocyanide derivatives 2a, 4a, 5a, and 6a gave excellent yields (26c, 46c, 52c, and 66c, respectively). With these promising results in hand, we tested next the reactivity of methyl isocyanoacetate (1a) with various oxo components such as formaldehyde, p-chlorobenzaldehyde, 3-pentanone, and cyclic ketones such as basic N-benzyl- and neutral N-Cbz-4-piperidinone. Ketones gave generally good yields (2c–4c). In the case of aldehydes (Table 2, entries 5 and 6), however, multiple products were observed along with trace formation of product C, as confirmed by supercritical fluid chromatography–mass spectrometry (SFC-MS) analysis of the reaction mixtures. The initial Ugi tetrazole adducts using aldehydes as the oxo component together with ammonium chloride were more activated and resulted in an additional Ugi tetrazole reaction to form the bis(tetrazole) products instead of the cyclization with the methyl ester group of the Ugi adduct. In general, aldehydes gave bis(tetrazoles) as a major product with ammonia in the Ugi tetrzole reaction.8 Furthermore, we screened methyl 2-isocyano-2-methylpropanoate (2a) with different oxo components such as cyclic, acyclic, benzylic, unsaturated, alkyl, aryl, and heteroaryl oxo compounds (Table 2, entries 7–34). Acyclic ketones gave good yields (13c–16c), whereas α,β-unsaturated ketones gave only traces of the product (Table 2, entries 17–19). Cyclic ketones such as cyclohexanone, 4-phenylcyclohexanone, and N-Cbz- and N-ethoxycarbonyl-4-piperidinone gave excellent yields (26c–29c). α-Substituted cyclic ketones gave poor yields (24c, 8%; 25c, 14%), likely due to poor reaction in the Ugi adduct formation as a result of steric hindrance. Product 27c was isolated as a 9:1 mixture of diastereomers. Interestingly, all of the cyclic ketones gave spiro-tetrazolopiperidinone compounds. Benzylic ketones like acetophenone, benzophenone, and their derivatives failed to give any product; in a few cases, however, we observed trace formation of product, which was confirmed by SFC-MS analysis of the reaction mixture. Thus, potentially the product yields could be increased in these cases by prolonging the reaction time. Similar negative results were observed with cyclic benzylic ketones such as α-tetralone, chroman-4-one, and thiochroman-4-one. Similarly, 1,3-diketones such as acetylacetone and 1,3-cyclohexanedione exhibited the formation of multiple side products along with traces of the desired tetrazolopiperidinone C. Additionally, we tested paraformaldehyde, hydrocinnamaldehyde, furfural, phenylglyoxal, electron-rich 3,4,5-trimethoxybenzaldehyde, and electron-deficient 4-nitrobenzaldehyde with 2a. We always observed difficulties in purification due to formation of multiple side products along with poor yields of tetrazolopiperidinone derivatives C. Surprisingly, by using p-chlorobenzaldehyde we could isolate the required product 12c, but the yield was poor (22%). Moreover, different isocyanides (3a–6a) were employed and found to be good substrates to obtain tetrazolopiperidinones C. Methyl 2-isocyano-4-methyl-pentanoate (3a), for example, gave an excellent yield with acetone (36c, 75%), while hydroxyacetone gave a 65% yield of cyclized product (38c) with a diastereomeric ratio of 10:1. The bulky asymmetric methyl isopropyl ketone gave a moderate yield (37c, 46%) with a 9:1 diastereomeric ratio. Also, leucine-derived isocyanide 3a with cyclic ketones like cyclohexanone (41c, 80%), and N-benzylcyclohexanone (42c, 76%) give excellent to good yields. Methyl 2-isocyano-2-phenylacetate (4a) with acyclic and cyclic ketones gave good to excellent yields (44c–48c). Similarly, the reactions of methyl 2-isocyano-3-phenylpropanoate (5a) with cyclic ketones resulted in excellent yields (52c, 53c, 55c, 56c), except with N-methylpiperidinone (54c, 43%). The acyclic ketone acetone gave an excellent yield (50c, 79%), and the acyclic asymmetric ketone ethyl methyl ketone gave the product with a 4:3 diastereomeric ratio in good yield (51c, 65%). The tested aldehydes performed accordingly with other isocyanides (1a–6e); furthermore, the reaction of tryptophan-derived isocyanide 6a with cyclic ketones gave excellent yields (66c and 68c), except for N-methylpiperidinone, which gave a moderate yield (67c, 41%). Acyclic ketones like acetone (61c, 62%) and 3-pentanone (62c, 62%) gave moderate yields. Unfortunately, aldehydes such as hydrocinnamaldehyde and benzaldehyde and ketones like methyl vinyl ketone, acetophenone, benzophenone, α-tetralone, and chroman-4-one failed to produce pure cyclized product.
In general, acyclic and cyclic ketones, including those with tertiary amine substituents, are well-tolerated and give good to excellent yields irrespective of the isocyanide derivative. Aldehydes gave poor yields, as they formed hyperactive aminotetrazole derivatives as intermediates, which then react further with excess aldehyde present in the reaction mixture to give a second Ugi adduct to form bis(tetrazole) derivatives. α,β-Unsaturated ketones and benzylic ketones gave trace formation of product (as confirmed by SFC-MS analysis) irrespective of the isocyanide derivative used.
Next, we investigated the structures of representative compounds in the solid state (Figure 3). We were able to grow crystals of seven products (14c, 16c, 28c, 29c, 46c, 61c, and 62c) suitable for single-crystal structure determination, which confirmed the tetrazolopiperidinone scaffold design. It is noteworthy that many different hydrogen-bonding motifs involving the tetrazolopiperidinone could be observed, underlining the structural diversity of these diketopiperazine bioisosteres.
Figure 3.

Diversity of 3D structures and crystal contacts observed in seven different tetrazolopiperidinones (nonpolar hydrogens have been omitted for clarity; key distances are given in Å). (A) 16c showing the coplanarity of the tetrazole and annulated piperidinone rings. (B) 14c exhibiting a hydrogen bond between the piperidinone NH and N-5 of a tetrazole moiety of an adjacent molecule. (C) Spiro-29c forming an unsymmetrical bifurcated hydrogen bond between the piperidinone NH and the N-3 and N-4 of a tetrazole moiety of an adjacent molecule. (D) Symmetrical dimer interaction involving both amide hydrogen-bond donors and acceptors in spiro-28c. (E) An even shorter dimeric interaction of spiro-46c similar to that in (D). (F, G) Similar hydrogen-bonding networks of Trp-substructure-containing 61c and 62c involving a piperidinone NH···N-5 tetrazole motif and a dimeric indole NH···piperidinone O motif.
Lipophilicity (expressed as log P) and molecular weight (MW) are key properties determining the usefulness of compound series for screening libraries. Ideal molecular weights are well below 400 Da and lipophilicities should be well below 4. Molecular weight, for example, is a major determinant of passive membrane diffusion and thus oral bioavailability. Lipophilicity on the other hand determines water solubility and also metabolic stability. Therefore, we calculated the theoretical log P values and molecular weights of N-alkyltetrazolopiperidinones reported earlier7 and our herein-reported molecules (Figure 4). We found that average molecular weight of the N-alkyltetrazolopiperidinones reported by Hulme was 366 and the average log P was 2.35. On the other hand, our N-unsubstituted tetrazolopiperidinones have a lower average molecular weight (293) and also a lower log P value (1.33) compared with the N-alkyltetrazolopiperidinones.
Figure 4.
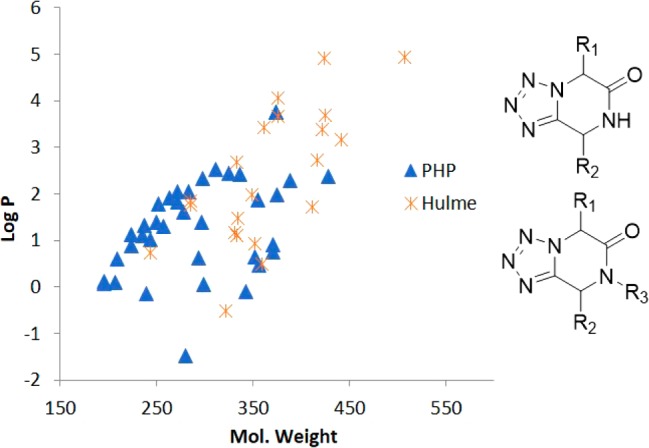
Plot of log P vs MW comparing tetrazolopiperidinone molecules synthesized by Hulme and co-workers7 (red) and our compounds synthesized and reported here (blue).
Conclusions
We have described a short, straightforward synthesis of the tetrazolopiperidinones. Although Hulme and co-workers7 described a similar reaction using primary amines, our method differs in several key respects: ammonia is a less investigated reagent in Ugi reactions, previously leading to a lot of side reactions and only recently leading to useful protocols;8 the herein-described scaffold is represented by a different pharmacophore model and includes one hydrogen-bond donor but five hydrogen-bond acceptors; and the average molecular weight and lipophilicity are much less than the corresponding N-substituted analogues. Thus, the current scaffold is suitable for compound enrichment of screening decks, such as the European Lead Factory.22−26
Acknowledgments
The work was financially supported by the NIH (2R01GM097082-05) and the Innovative Medicines Initiative (Grant Agreement 115489). Moreover, funding was also received from the European Union’s Horizon 2020 Research and Innovation Programme under MSC ITN “Accelerated Early Stage Drug Discovery” (AEGIS) (Grant Agreement 675555) and from the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract POIG.02.01.00-12-023/08).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombsci.7b00033.
Crystallographic data for 14c (CIF)
Crystallographic data for 16c (CIF)
Crystallographic data for 28c (CIF)
Crystallographic data for 29c (CIF)
Crystallographic data for 46c (CIF)
Crystallographic data for 61c (CIF)
Crystallographic data for 62c (CIF)
Experimental procedures, compound characterization data, NMR spectra, HRMS data, and crystal structure determinations (PDF)
Author Contributions
The article was written through contributions of P.P. and A.D. The crystallographic study was contributed by K.K. and J.K.-T.
The authors declare no competing financial interest.
Supplementary Material
References
- Borthwick A. D. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev. 2012, 112, 3641–3716. 10.1021/cr200398y. [DOI] [PubMed] [Google Scholar]
- Jost M.; Zocher G.; Tarcz S.; Matuschek M.; Xie X.; Li S.-M.; Stehle T. Structure–Function Analysis of an Enzymatic Prenyl Transfer Reaction Identifies a Reaction Chamber with Modifiable Specificity. J. Am. Chem. Soc. 2010, 132, 17849–17858. 10.1021/ja106817c. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Fuchs J. E.; Reutlinger M.; Stahl M.; Taylor N. R. Rationalizing Tight Ligand Binding through Cooperative Interaction Networks. J. Chem. Inf. Model. 2011, 51, 3180–3198. 10.1021/ci200319e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienaymé H.; Bouzid K. Synthesis of rigid hydrophobic tetrazoles using an Ugi multi-component heterocyclic condensation. Tetrahedron Lett. 1998, 39, 2735–2738. 10.1016/S0040-4039(98)00283-4. [DOI] [Google Scholar]
- Franckevicius V.; Longbottom D. A.; Turner R. M.; Ley S. V. 8,9,10,10a-Tetrahydro-6H-tetrazolo[1,5-a]pyrrolo[2,1-c]pyrazines: New Heterocyclic Frameworks Generated by an Ugi-Type Multicomponent Reaction. Synthesis 2006, 2006, 3215–3223. 10.1055/s-2006-950219. [DOI] [Google Scholar]
- Kalinski C.; Umkehrer M.; Gonnard S.; Jäger N.; Ross G.; Hiller W. A new and versatile Ugi/SNAr synthesis of fused 4,5-dihydrotetrazolo[1,5-a]quinoxalines. Tetrahedron Lett. 2006, 47, 2041–2044. 10.1016/j.tetlet.2006.01.027. [DOI] [Google Scholar]
- Nixey T.; Kelly M.; Hulme C. The one-pot solution phase preparation of fused tetrazole-ketopiperazines. Tetrahedron Lett. 2000, 41, 8729–8733. 10.1016/S0040-4039(00)01563-X. [DOI] [Google Scholar]
- Patil P.; Khoury K.; Herdtweck E.; Dömling A. MCR synthesis of a tetracyclic tetrazole scaffold. Bioorg. Med. Chem. 2015, 23, 2699–2715. 10.1016/j.bmc.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umkehrer M.; Kolb J.; Burdack C.; Ross G.; Hiller W. Synthesis of tetrazolopiperazine building blocks by a novel multi-component reaction. Tetrahedron Lett. 2004, 45, 6421–6424. 10.1016/j.tetlet.2004.06.133. [DOI] [Google Scholar]
- Zarganes-Tzitzikas T.; Patil P.; Khoury K.; Herdtweck E.; Dömling A. Concise Synthesis of Tetrazole–Ketopiperazines by Two Consecutive Ugi Reactions. Eur. J. Org. Chem. 2015, 2015, 51–55. 10.1002/ejoc.201403401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömling A.; Huang Y. Piperazine Scaffolds via Isocyanide-Based Multicomponent Reactions. Synthesis 2010, 2010, 2859–2883. 10.1055/s-0030-1257906. [DOI] [Google Scholar]
- Patil P.; de Haan M.; Kurpiewska K.; Kalinowska-Tłuścik J.; Dömling A. Versatile Protecting-Group Free Tetrazolomethane Amine Synthesis by Ugi Reaction. ACS Comb. Sci. 2016, 18, 170–175. 10.1021/acscombsci.5b00189. [DOI] [PubMed] [Google Scholar]
- Pick R.; Bauer M.; Kazmaier U.; Hebach C. Ammonia in Ugi Reactions - Four-Component versus Six-Component Couplings. Synlett 2005, 757–760. 10.1055/s-2005-863722. [DOI] [Google Scholar]
- Barthelon A.; El Kaim L.; Gizzi M.; Grimaud L. Ammonia in Ugi-Smiles and Ugi Couplings. Synlett 2010, 2010, 2784–2788. 10.1055/s-0030-1259004. [DOI] [Google Scholar]
- Kazmaier U.; Hebach C. Peptide Syntheses via Ugi Reactions with Ammonia. Synlett 2003, 11, 1591–1594. 10.1055/s-2003-40987. [DOI] [Google Scholar]
- Just G.; Chung B. Y.; Grözinger K. An attempted synthesis of an oxacepham derivative using isonitriles as β-lactam forming agents. On the stability of dihydrooxazines and the inadequacy of ammonia as the fourth component in the Ugi reaction. Can. J. Chem. 1977, 55, 274–283. 10.1139/v77-042. [DOI] [Google Scholar]
- Banfi L.; Basso A.; Damonte G.; De Pellegrini F.; Galatini A.; Guanti G.; Monfardini I.; Riva R.; Scapolla C. Synthesis and biological evaluation of new conformationally biased integrin ligands based on a tetrahydroazoninone scaffold. Bioorg. Med. Chem. Lett. 2007, 17, 1341–1345. 10.1016/j.bmcl.2006.11.085. [DOI] [PubMed] [Google Scholar]
- Gulevich A. V.; Balenkova E. S.; Nenajdenko V. G. The First Example of a Diastereoselective Thio-Ugi Reaction: A New Synthetic Approach to Chiral Imidazole Derivatives. J. Org. Chem. 2007, 72, 7878–7885. 10.1021/jo071030o. [DOI] [PubMed] [Google Scholar]
- Seganish W. M.; Bercovici A.; Ho G. D.; Loozen H. J. J.; Timmers C. M.; Tulshian D. A synthesis of dihydroimidazo[5,1- a]isoquinolines using a sequential Ugi–Bischler–Napieralski reaction sequence. Tetrahedron Lett. 2012, 53, 903–905. 10.1016/j.tetlet.2011.12.050. [DOI] [Google Scholar]
- Thompson M. J.; Chen B. Ugi Reactions with Ammonia Offer Rapid Access to a Wide Range of 5-Aminothiazole and Oxazole Derivatives. J. Org. Chem. 2009, 74, 7084–7093. 10.1021/jo9014529. [DOI] [PubMed] [Google Scholar]
- Zhao T.; Boltjes A.; Herdtweck E.; Dömling A. Tritylamine as an Ammonia Surrogate in the Ugi Tetrazole Synthesis. Org. Lett. 2013, 15, 639–641. 10.1021/ol303348m. [DOI] [PubMed] [Google Scholar]
- Mullard A. Nat. Rev. Drug Discovery 2013, 12, 173–175. 10.1038/nrd3956. [DOI] [PubMed] [Google Scholar]
- Besnard J.; Jones P. S.; Hopkins A. L.; Pannifer A. D. The Joint European Compound Library: boosting precompetitive research. Drug Discovery Today 2015, 20, 181–186. 10.1016/j.drudis.2014.08.014. [DOI] [PubMed] [Google Scholar]
- Nelson A.; Roche D. Innovative approaches to the design and synthesis of small molecule libraries. Bioorg. Med. Chem. 2015, 23, 2613. 10.1016/j.bmc.2015.02.046. [DOI] [PubMed] [Google Scholar]
- Paillard G.; Cochrane P.; Jones P. S.; van Hoorn W. P.; Caracoti A.; van Vlijmen H.; Pannifer A. D. The ELF honest data broker: informatics enabling public-private collaboration in a precompetitive arena. Drug Discovery Today 2016, 21, 97–102. 10.1016/j.drudis.2015.11.005. [DOI] [PubMed] [Google Scholar]
- Karawajczyk A.; Orrling K. M.; de Vlieger J. S. B.; Rijnders T.; Tzalis D. The European Lead Factory: A Blueprint for Public–Private Partnerships in Early Drug Discovery. Front. Med. 2017, 3 (75), 1–7. 10.3389/fmed.2016.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.