

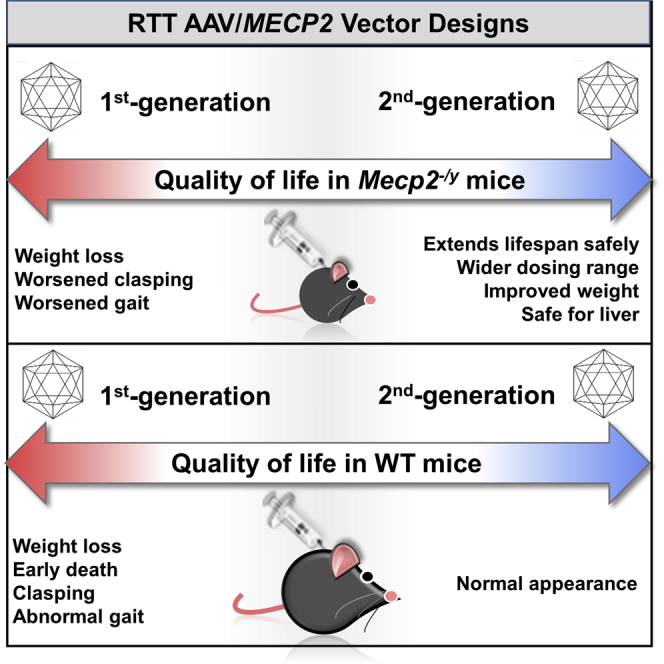
Abstract
Intravenous administration of adeno-associated virus serotype 9 (AAV9)/hMECP2 has been shown to extend the lifespan of Mecp2−/y mice, but this delivery route induces liver toxicity in wild-type (WT) mice. To reduce peripheral transgene expression, we explored the safety and efficacy of AAV9/hMECP2 injected into the cisterna magna (ICM). AAV9/hMECP2 (1 × 1012 viral genomes [vg]; ICM) extended Mecp2−/y survival but aggravated hindlimb clasping and abnormal gait phenotypes. In WT mice, 1 × 1012 vg of AAV9/hMECP2 induced clasping and abnormal gait. A lower dose mitigated these adverse phenotypes but failed to extend survival of Mecp2−/y mice. Thus, ICM delivery of this vector is impractical as a treatment for Rett syndrome (RTT). To improve the safety of MeCP2 gene therapy, the gene expression cassette was modified to include more endogenous regulatory elements believed to modulate MeCP2 expression in vivo. In Mecp2−/y mice, ICM injection of the modified vector extended lifespan and was well tolerated by the liver but did not rescue RTT behavioral phenotypes. In WT mice, these same doses of the modified vector had no adverse effects on survival or neurological phenotypes. In summary, we identified limitations of the original vector and demonstrated that an improved vector design extends Mecp2−/y survival, without apparent toxicity.
Keywords: Rett syndrome, MeCP2, cisterna magna, intrathecal, microRNA, AAV, viral vector, Mecp2−/y
Graphical Abstract
Introduction
Rett Syndrome (RTT) is an X-linked neurodevelopmental disorder associated with severe motor abnormalities and reduced lifespan in a proportion of patients.1, 2, 3 RTT is caused by loss-of-function mutations in methyl CpG-binding protein 2 (MECP2),4 a transcription regulator that is highly expressed in neurons.5, 6 Inactivating mutations in MeCP2 can alter the expression of many genes and ultimately disrupt neuronal morphology and circuitry.2, 7, 8, 9, 10 Adeno-associated virus serotype 9 (AAV9) has been used to transfer wild-type (WT) MECP2 to cells in vivo, extending survival and attenuating neurological deficits in mouse models of RTT.11, 12 These models include hemizygous Mecp2−/y males as well as heterozygous Mecp2+/− females that are mosaic for MeCP2 expression.11, 12 Due to their limited survival (∼9 to 10 weeks) and severe phenotypes, Mecp2−/y mice are often used for early-stage assessment of putative pharmacological and genetic treatments for RTT.11, 12
One study, using a self-complementary (sc) AAV9/MeP229-hMECP2-myc-BGHpA vector (hereafter termed the first-generation vector AAV9/hMECP2(v1)), extended the survival of Mecp2−/y mice injected intravenously (IV) between postnatal day 28 (PND28) and PND35.11 This IV gene therapy, however, resulted in high transgene expression in the liver as well as significantly elevated levels of alanine aminotransferase (ALT), an indicator of liver toxicity.11 After IV treatment with 1 × 1011 viral genomes (vg) AAV9/hMECP2(v1), the level of total MeCP2 expression (measured as copies MECP2 cDNA per β-actin cDNA) in WT liver was ∼6x greater than that observed within the brain.11 This liver tropism and hepatic transgene expression observed after IV treatment can be attributed to the AAV9 capsid13 and the use of a small endogenous Mecp2 promoter fragment (MeP229),11, 14 respectively. We therefore concluded that AAV9/hMECP2(v1) was a successful “proof-of-concept” vector whose design and administration route would need to be optimized before it could be considered for human translation.11
To bias transgene expression away from the liver, we decided to assess the safety and efficacy of AAV9/hMECP2(v1) after injection into the cerebrospinal fluid (CSF) of Mecp2−/y and WT mice. In the current study, we identify deleterious side effects (e.g., abnormal gait and hindlimb clasping) observed after cisterna magna (ICM) delivery of AAV9/hMECP2(v1). Importantly, these side effects were not observed after treatment with a control vector delivering EGFP. To address the limitations of the previously published vector, we designed a second-generation vector (AAV9/hMECP2(v2)) that, in Mecp2−/y mice, increases weight (relative to that of saline-treated Mecp2−/y littermates) and extends survival without aggravating behavioral phenotypes. In addition, the effective dose of AAV9/hMECP2(v2) is well tolerated in WT mice after ICM administration.
Results
IntraCSF Administration of AAV9/hMECP2(v1) Extends Survival of Mecp2−/y Mice
To bias transduction toward the central nervous system (CNS), we decided to shift from an IV to an intraCSF route of administration. Prior to conducting extensive safety and efficacy studies, we assessed neuronal tropism and transduction efficiency 3 to 4 weeks after treating Mecp2−/y mice with 1 × 1012 vg AAV9/hMECP2(v1) via the ICM (Figures 1A and 1B). Consistent with the reported CNS expression profile of the MeP229 promoter,14 we observed predominantly neuronal expression of MeCP2-myc after ICM treatment of juvenile mice, with multiple brain regions showing a modest percentage (∼20%–40%) of transduced anti-myc immunopositive cells (Figures 1A and 1B).
Figure 1.

ICM Delivery of AAV9/hMECP2(v1) Extends the Lifespan of Mecp2−/y Mice, but with Deleterious Side Effects
(A) One month after ICM injection of 1 × 1012 vg/mouse, modest transduction efficiency is observed throughout the brains of Mecp2−/y mice. Above are confocal images of myc+ cells (indicated by white arrows) in the hippocampus, brain stem, and thalamus. Merged images show anti-myc immunoreactivity (green), anti-NeuN immunoreactivity (red), and DAPI+ nuclei (blue). (B) Left: transduction efficiencies calculated for three brain regions. Right: neuronal tropism calculated for three brain regions. n = 2 injected Mecp2−/y mice, with five to six slices imaged per brain region. (C) AAV9/hMECP2(v1) significantly extends the median lifespan of Mecp2−/y mice from 57 days to 84 days. #p = 0.01 (Gehan-Breslow-Wilcoxon test) for treated Mecp2−/y mice (1 × 1012 vg/mouse, ICM) versus controls (0 vg/mouse, ICM). The legend in (C) also applies to (D)–(G). (C–E) Approximate injection time point indicated by black arrows. (D) AAV9/hMECP2(v1) (red) decreased body weight after ICM injection in 4- to 5-week-old mice. (E) 1 × 1012 vg AAV9/hMECP2(v1) (red) worsens aggregate behavior scores in Mecp2−/y mice after ICM administration. (D and E) Shaded area indicates time during which scores for ICM cohorts (saline versus 1 × 1012 vg) were significantly different. *p < 0.05. (F and G) Specifically, 1 × 1012 vg AAV9/hMECP2(v1) (ICM) aggravates limb clasping and abnormal gait in Mecp2−/y mice. p values (paired t test) for treated Mecp2−/y mice versus their pre-treatment scores: **p ≤ 0.0002; *p ≤ 0.002. p values (unpaired t test) comparing virus- and saline-treated mice at a fixed time point: #p ≤ 0.0006. (H) Examples of virus-treated mice with limb clasping (mouse IDs 43, 47). The older saline-treated and AAV9/GFP-treated Mecp2−/y mice do not yet have hindlimb clasping. Blue arrows point to curled toes and limbs held closely against the body. (B and D–G) Data points are mean ± SEM.
To evaluate the efficacy of intraCSF MECP2 gene therapy in juvenile Mecp2−/y mice (PND28-35), we injected 1 × 1012 vg AAV9/hMECP2(v1) via the ICM route (a similar intraCSF dataset for lumbar intrathecal [IT] injections is provided in Figure S1). At this dose, AAV9/hMECP2(v1) extended the median survival of Mecp2−/y mice by 27 days or 47% (p = 0.01, Gehan-Breslow-Wilcoxon test) (Figure 1C).
In addition to reduced survival, Mecp2−/y mice exhibit a well-characterized array of neurological phenotypes (e.g., hindlimb clasping, abnormal gait, and abnormal breathing) and reduced bodyweight compared to their WT littermates.11, 15, 16 Body weight and RTT-like phenotypes for Mecp2−/y mice were recorded prior to injection and weekly thereafter using an established observational aggregate phenotypic severity scoring system.15 At 7 to 8 weeks of age, Mecp2−/y mice treated with 1 × 1012 vg/mouse (ICM) had lower body weights than did saline-treated Mecp2−/y mice (Figure 1D). At this dose, RTT-like phenotype severity scores were significantly increased in ICM-treated mice (p ≤ 0.05, starting at 6 weeks of age) (Figure 1E). In particular, two parameters (limb clasping and gait) were significantly affected at 8 weeks of age (versus pre-ICM clasping and gait scores at 4 weeks of age and versus scores for saline-treated mice at 8 weeks of age) (Figures 1F and 1G). Because most saline-treated Mecp2−/y mice did not survive beyond 9 weeks, the pairwise analyses in Figures 1F and 1G were limited to data observed at 4 and 8 weeks of age. A control vector AAV9/MeP229-EGFP (1 × 1012 vg/mouse, ICM) did not appear to hasten the development of hindlimb clasping in Mecp2−/y mice, indicating that the adverse limb/gait changes were likely caused by MeCP2-myc overexpression (Figure 1H).
Peripheral Transgene Expression Is Observed after IntraCSF Delivery of AAV9/hMECP2(v1)
IV administration of AAV9/hMECP2(v1) (1 × 1011 vg) has previously been shown to cause liver toxicity in WT mice.11 To determine if the transgene was expressed in the liver and other peripheral organs after ICM administration, we conducted biodistribution and gene expression analyses on samples from Mecp2−/y mice treated with 1 × 1012 vg of AAV9/hMECP2(v1). Intracisternally delivered virus escaped to the periphery, with ∼30 viral genome copies detected per liver cell (Figure 2A). This observation is consistent with published data demonstrating peripheral transgene biodistribution after intraCSF administration of AAV.13, 17 In general, one or more viral genome copies were present per cell across multiple organs (Figure 2A). Gene expression was highest in the liver and brain (∼1 MECP2 cDNA copy per β-actin cDNA in Figure 2B; see Figure S2 for liver toxicity data). By delivering vector directly into the CSF, instead of systemically, we were able to diminish the levels of transgene expression in liver tissue relative to those observed in brain tissue (previously published systemic delivery yielded ∼6× greater total expression in the liver than in the brain after IV treatment with 1 × 1011 vg11) (Figure 2B).
Figure 2.

Peripheral Transgene Expression Is Observed after ICM Administration of AAV9/hMECP2(v1) in Mecp2−/y Mice
(A) Vector biodistribution in Mecp2−/y mice. (B) Transgene expression in Mecp2−/y mice. (A and B) n = 3 saline-treated mice and n = 5 AAV9/hMECP2(v1)-treated mice. Data are mean ± SEM. CC, cervical spinal cord; LC, lumbar spinal cord; TC, thoracic spinal cord.
IntraCSF Administration of AAV9/hMECP2(v1) Induces Deleterious Effects in WT Mice
To thoroughly evaluate the safety of intracisternally delivered AAV9/hMECP2(v1), we also treated WT mice in parallel with treated Mecp2−/y mice. The virus-treated (1 × 1012 vg/mouse; ICM) WT cohort had a significantly shorter lifespan compared to that of saline-treated WT mice (p = 0.04) (Figure 3A). At this dose, a significant decrease in body weight was observed between 9 and 20 weeks of age for WT mice treated ICM (Figure 3B). Importantly, administration of AAV9/hMECP2(v1) led to a significant and persistent increase (worsening) in aggregate phenotype scores (Figure 3C). In general, WT mice treated with 1 × 1012 vg AAV9/hMECP2(v1) (ICM) demonstrated a significant increase in severity scores for hindlimb clasping and abnormal gait within 2 weeks after injection (Figures 3D and 3E; Movie S1). Similarly, WT mice treated intrathecally with 1 × 1012 vg AAV9/hMECP2(v1) experienced weight loss and a significant increase in aggregate phenotype severity scores, with significant scores for limb clasping observed at 12 weeks of age (relative to pre-treatment scores) (Figure S1).
Figure 3.

ICM Delivery of AAV9/hMECP2(v1) Induces Behavioral Abnormalities in WT Mice
(A) AAV9/hMECP2(v1) shortens WT lifespan. Because WT and Mecp2−/y mice were tested in parallel, Figures 3 and 1 present the same data for saline-treated controls. #p = 0.04 (Gehan-Breslow-Wilcoxon test) for treated mice (WT, 1 × 1012 vg, ICM) versus controls (WT, 0 vg, ICM). (A–C) Injection time point is indicated by black arrows. (B and C) Shaded areas indicate times during which saline- and virus-treated cohorts had significantly different body weights or behavior scores; *p < 0.05. (B) 1 × 1012 vg (ICM) AAV9/hMECP2(v1) decreases body weight. Green dashed line at 25 weeks indicates the time point at which 50% of mice treated with 1 × 1012 vg AAV9/hMECP2(v1) (ICM) died. An increased mean weight for survivors is observed after this point. (C) In WT mice, 1 × 1012 vg AAV9/hMECP2(v1) induces behavioral abnormalities that are similar to those observed in Mecp2−/y mice (compare to Figure 1). Abnormalities persist until death. (D and E) AAV9/hMECP2(v1) (ICM) aggravates limb clasping and abnormal gait in WT mice. For clarity, only data for saline-treated and 1 × 1012 vg v1-treated mice are shown. p values (paired t test) for virus-treated WT mice versus their pre-treatment scores: **p = 0.001; *p = 0.04. p values (unpaired t test) comparing virus- and saline-treated mice at a fixed time point: ##p ≤ 0.0001; #p = 0.02. (B–E) Data points are mean ± SEM.
Effective Doses of a Second-Generation Vector Appear to Be Well-Tolerated in WT and Mecp2−/y Mice
The severe adverse effects observed in both Mecp2−/y and WT mice led us to conclude that the AAV9/hMECP2(v1) vector is not a viable treatment option for RTT. We suspect that many of these symptoms may result from improper hMeCP2-myc transgene regulation for two reasons. First, identical treatment with a control vector encoding EGFP (expressed under the truncated MeCP2 promoter MeP229) does not cause these adverse effects (Figure 1H; Movie S1). Second, transgenic mouse models of loss or overexpression of MeCP2 have been shown to exhibit some overlap in phenotypic abnormalities, including kyphosis, clasping, and decreased locomotor activity.18, 19, 20, 21, 22 Thus, it would be strategic for a second-generation MECP2 gene therapy to incorporate additional endogenous control elements to potentially allow for better regulation of transgene expression in a transduced cell.
The MeP229 core promoter fragment lacks a number of putative regulatory elements (REs) predicted to lie upstream in the MECP2 promoter region.23, 24 These REs may be important in cell-type-specific regulation of MeCP2 expression. The opportunity to engineer additional endogenous repressive elements into the gene expression cassette was particularly appealing in light of the broad peripheral vector distribution observed after ICM injection (Figure 2). Therefore, we designed a second-generation vector containing an extended promoter (mMeP426), a modified 3′ UTR incorporating the highly conserved distal polyadenylation signal of MECP2, and a panel of microRNA (miRNA)-binding sites specific to miRNAs endogenous to, and known to interact with, the MECP2 3′ UTR (Figures 4A and S3).25, 26, 27, 28, 29, 30 Numerous other vector design variations to the hMECP2(v1) cassette have been carefully tested by our colleagues, leading to the development of the hMECP2(v2) cassette. These other vector designs are described in a companion paper by Gadalla et al.31 in this issue of Molecular Therapy – Methods & Clinical Development. For the remaining data described herein, we focus on intraCSF administration of AAV9/hMECP2(v1) and AAV9/hMECP2(v2). Because IT and ICM injections of AAV9/hMECP2(v1) yielded similar results in WT mice (Figures 3 and S1), we decided to streamline our methods and evaluated AAV9/hMECP2(v2) via ICM administration only.
Figure 4.

IntraCSF Delivery of AAV9/hMECP2(v2) Extends the Lifespan of Mecp2−/y Mice
(A) Cartoon of modified gene expression cassette. Figures 1, 3, and 4 present the same saline-treated control data. (B) 1 × 1010 and 1 × 1011 vg AAV9/hMECP2(v2) extend the median lifespan of Mecp2−/y mice from 57 days to 81 days (#p = 0.01) and 80 days (#p = 0.001), respectively. (B–D) Approximate injection time point indicated by black arrows. (C) Left: gray arrows indicate times at which 1 × 1010 vg AAV9/hMECP2(v2) significantly increases the mean body weight of Mecp2−/y mice. Although the mean weight for Mecp2−/y mice (1 × 1010 vg AAV9/hMECP2(v2)) continued to increase after 8 weeks of age, significant differences (versus the weight of saline-treated mice) were not observed presumably due to the early deaths of saline-treated mice. Right: 1 × 1011 to 1 × 1012 vg AAV9/hMECP2(v2) decrease the mean body weight of WT mice. Shaded areas indicate times during which saline- and virus-treated cohorts had significantly different body weights. To improve clarity, weight data for saline-treated WT mice was offset by +1 g. (D) 1 × 1010 or 1 × 1011 vg modified vector does not rescue aggregate behavior scores in Mecp2−/y mice after intraCSF administration, and at 1 × 1012, it worsened behavioral scores in Mecp2−/y and WT mice (p ≤ 0.05 beginning at 6 and 5 weeks of age for treated Mecp2−/y and WT mice [respectively] versus saline controls). (C and D) Data points are mean ± SEM.
After ICM administration of AAV9/hMECP2(v2), we observed a significant increase in Mecp2−/y median survival (p ≤ 0.01, Gehan-Breslow-Wilcoxon test; 42% or 40% increase in median lifespan for mice treated ICM with 1 × 1010 or 1 × 1011 vg/mouse, respectively) (Figure 4B). Importantly, these doses were 10- to 100-fold less than the effective dose of the first-generation vector AAV9/hMECP2(v1) (Figure 1C).
Although 1 × 1010 and 1 × 1011 vg of AAV9/hMECP2(v2) were both effective at extending lifespan, dose-dependent effects on body weight were observed for treated Mecp2−/y and WT mice. AAV9/hMECP2(v2) administered at 1 × 1010 vg/mouse (ICM) increased Mecp2−/y body weight relative to that of saline-treated Mecp2−/y mice at 2 and 4 weeks post-injection (p ≤ 0.05, t test comparing saline- and virus-treated mice at each time point) (Figure 4C). Importantly, 1 × 1010 vg did not affect the mean body weight of treated WT mice. Because WT cohorts treated with saline or 1 × 1010 vg had nearly identical mean body weights, data for saline-treated mice (Figure 4C) were offset to improve clarity (see Figure 4 caption). WT animals treated with 1 × 1011 to 1 × 1012 vg experienced decreased body weight beginning 1 month post-injection (relative to the weight of saline-treated WT mice) (Figure 4C).
At 1 × 1012 vg (ICM), AAV9/hMECP2(v2) significantly increased aggregate phenotype severity by increasing severity scores specifically for hindlimb clasping and abnormal gait in both Mecp2−/y and WT mice (Figures 4D and S4). At 1 × 1010 to 1 × 1011 vg (ICM), AAV9/hMECP2(v2) had no effect on the mean aggregate scores of WT and Mecp2−/y mice (Figure 4D). This lack of phenotypic rescue in Mecp2−/y mice was surprising in light of previously published data showing that neonatal intracranial injections of AAV9/CBA-hMeCP2-myc-SV40pA could ameliorate the onset of RTT-like abnormalities, including motor dysfunction.11 These results may be due to a number of factors (modest transduction efficiency, age of injection, etc.). To better sort out potential treatment effects in more detail, we conducted additional experiments in randomized, paired littermates to eliminate cross-litter variability. Historically, we have observed variability in the phenotypic severity of Mecp2−/y mice from different litters as well as variability in the age at which severe symptoms appear. Thus, we were concerned that noise in the mean behavior scores could obscure meaningful phenotypic differences within pairs of saline- and virus-treated littermates. We therefore decided to run pairwise analyses of saline- and virus-treated Mecp2−/y littermates (Figure S5). In short, AAV9/hMECP2(v2) (1 × 1010 to 1 × 1011 vg) significantly increased the peak body weight and growth rate (post-treatment) of Mecp2−/y mice relative to that of their saline-treated Mecp2−/y siblings (Figure S5; p = 0.0003 and 0.02, respectively). In addition, the overall health of virus-treated Mecp2−/y mice appeared to decline more gradually (Figure S5; n = 8 pairs, p = 0.03). Finally, early observations on treated mice suggested some possible benefits that may not be captured using a standardized RTT scoring system.15 To document this qualitatively, we video-recorded pairs of saline- and virus-treated littermates weekly. Videos for these littermate pairs are provided in the Supplemental Information (Movies S2, S3, and S4). A striking feature of these videos is the difference in spontaneous movement between saline- and virus-treated mice. Unlike their saline-treated littermates, AAV9/hMECP2(v2)-treated mice often walked, climbed, explored their surroundings, and/or approached the camera without prompting. Ultimately, however, all treated mice eventually experienced a decline in overall health and succumbed.
To investigate liver function after ICM administration of 1 × 1012 vg AAV9/hMECP2(v2), we quantified levels of albumin, ALT, aspartate aminotransferase (AST), and alkaline phosphatase (ALKP) in the blood serum of treated Mecp2−/y animals (Figure 5). Levels of each marker were indistinguishable between saline- and virus-treated cohorts (p = 0.8, 0.4, 0.4, and 0.9, respectively; Figure 5A). Biodistribution and gene expression data from treated Mecp2−/y mice showed that virus escaped to the periphery after ICM injection (∼33 viral genome copies per liver cell; ∼0.07 MeCP2 cDNA copies per β-actin cDNA; Figures 5B and 5C).
Figure 5.

Peripheral Transgene Expression without Hepatic Toxicity Is Observed after ICM Administration of AAV9/hMECP2(v2) in Mecp2−/y mice
(A) Blood serum levels of liver toxicity indicators in saline and virus-treated Mecp2−/y mice. No significant differences were observed. p = 0.8 for albumin; p = 0.4 for ALT; p = 0.4 for AST; p = 0.9 for ALKP. n = 5 mice per group. (B) Vector biodistribution in Mecp2−/y mice. High levels of vector DNA are observed in liver cells (relative to background signal). (C) Transgene expression in Mecp2−/y mice. (B and C) n = 3 saline-treated mice and 4 AAV9/hMECP2(v2)-treated mice (1 × 1012 vg/mouse). (A–C) Data are mean ± SEM. CC, cervical spinal cord; LC, lumbar spinal cord; TC, thoracic spinal cord.
Hypothetically, the divergent safety and efficacy profiles of intracisternally administered AAV9/hMECP2(v1) and AAV9/hMECP2(v2) could potentially be due to differences in how these two viral genomes regulate transgene expression in the brain, liver, and/or other organs. After all, viral genomes were found throughout the entire body 3 to 4 weeks after ICM injection (Figures 2, 5, and S2). In Mecp2−/y mice, AAV9/hMECP2(v2) generated fewer cDNA copies per viral genome in the heart, liver, and spinal cord (Figure 6). Interestingly, a 10-fold dose increase (from 1 × 1011 vg to 1 × 1012 vg) for AAV9/hMECP2(v2) resulted in decreased normalized transgene expression in the heart and liver, suggesting the presence of a dose-dependent inhibitory feedback mechanism in these tissues (1E11 vg AAV9/hMECP2(v2)/mouse: **p ≤ 0.004 for heart, liver, and lumbar spinal cord; *p = 0.04 for cervical spinal cord; 1E12 vg AAV9/hMECP2(v2)/mouse: ##p = 0.001 for liver; #p ≤ 0.05 for heart, cervical spinal cord, and thoracic spinal cord) (Figure 6). Immunofluorescence analyses also showed that transgene expression is more tightly regulated by AAV9/hMECP2(v2) (versus that of AAV9/hMECP2(v1)) in WT liver tissue after ICM administration (1 × 1011 vg/mouse; Figure 7A). Interestingly, ICM administration of AAV9/hMECP2(v2) (1 × 1011 vg/mouse) resulted in more anti-myc immunopositive hepatic cells in Mecp2−/y mice than in WT mice (1.2% versus 0.2%, respectively; p = 0.04; Figures 7A–7C). Mecp2−/y mice treated with 1 × 1011 AAV9/hMECP2(v1) reached their natural endpoints before these immunofluorescent analyses were conducted. Figure 6, however, shows that the ratio of transcript to viral genome is more tightly regulated by AAV9/hMECP2(v2) (versus that of AAV9/hMECP2(v1)) in Mecp2−/y liver tissue. Finally, our examination of brain tissue provided fewer clues explaining the improved safety of AAV9/hMECP2(v2). On a WT background, 1 × 1011 vg of either AAV9/hMECP2(v1) or AAV9/hMECP2(v2) resulted in low apparent transduction efficiencies, with predominantly neuronal tropism (Figures 7D–7G).
Figure 6.

AAV9/hMECP2(v2) Provides Tighter Regulation of Transgene Expression
In Mecp2−/y mice, the second-generation vector genome generates fewer cDNA copies per viral genome in specific organs. n = 4 to 5 mice per treatment. **p ≤ 0.004 for heart, liver, and lumbar spinal cord; *p = 0.04 for cervical spinal cord; ##p = 0.001 for liver; #p ≤ 0.05 for heart, cervical spinal cord, and thoracic spinal cord. For clarity, p values are only shown for comparisons against mice treated with 1 × 1012 vg AAV9/hMECP2(v1). Data points are mean ± SEM. CC, cervical spinal cord; LC, lumbar spinal cord; TC, thoracic spinal cord.
Figure 7.

The hMECP2(v2) Viral Genome Tightly Regulates Transgene Expression in WT Liver Tissue after ICM Administration
(A) In WT mice, the hMECP2(v2) viral genome tightly regulates transgene expression in the liver. Arrows point to myc+ cells. (B and C) hMECP2(v2) drives transgene expression in MeCP2 null liver tissue. (C) Percentage of myc+ liver cells (versus all DAPI+ liver cells). n = 3–5 mice and 9–15 imaged sections per group. (D–G) AAV9/hMECP2(v1) and AAV9/hMECP2(v2) yield similar transduction efficiencies and neuronal tropism in treated WT mice. (A–G) A single dose (1 × 1011 vg/mouse) was evaluated for AAV9/hMECP2(v1) and AAV9/hMECP2(v2). (C–G) Data points are mean ± SEM.
Discussion
IV administration of AAV9/hMECP2(v1) has been previously shown to extend the lifespan of Mecp2−/y mice, but overexpression of MeCP2 in the liver induced liver toxicity.11 We have now shown that ICM administration of AAV9/hMECP2(v1) extends the lifespan of Mecp2−/y mice, with a more favorable biodistribution toward CNS transduction compared to IV. Importantly, however, intracisternal administration of 1 × 1012 vg AAV9/hMECP2(v1) still resulted in potentially problematic peripheral transgene expression (Figure 2), a trend toward elevated blood serum toxicity indicators (Figure S2), and deleterious neurological side effects (Figures 1 and 3), which were not seen in mice treated with an AAV9/EGFP control vector. Increased severity of hindlimb clasping and abnormal gait were unexpected, given the absence of these side effects in previously published MeCP2 gene transfer studies.11, 12, 32 In light of these data, our conclusion is that AAV9/hMECP2(v1) cannot move forward in translational studies. Thus, we developed a second-generation MeCP2 viral genome that included additional putative regulatory elements from the endogenous MECP2 promoter and 3′ UTR. By modifying the viral genome, we were able to extend Mecp2−/y survival with a 10- to 100-fold lower dose compared to that used for the AAV9/hMECP2(v1) vector (Figures 1 and 4). This dose range is especially beneficial for a gene therapy because it increases the likelihood of administering a safe, effective dose in vivo. This low dose (1 × 1010 to 1 × 1011 vg injected ICM) is well tolerated by the liver and does not increase severity scores for limb clasping and abnormal gait in treated Mecp2−/y or WT mice (Figures 5 and S4). Our second-generation vector significantly improved peak body weight and acute growth rate (post-treatment) in Mecp2−/y mice (Figure S5). The mechanism underlying the increased body weight is unclear. In addition, our second-generation vector significantly attenuated the deterioration in overall health of MeCP2-null mice (Figure S5). Our overall conclusion is that the AAV9/hMECP2(v2) design provides a substantial improvement over the original design we previously published,11 conferring some benefit to Mecp2−/y mice over a 10-fold dynamic range (1 × 1010 to 1 × 1011 vg injected ICM), which is also well-tolerated.
The mechanism behind the improved therapeutic index of AAV9/hMECP2(v2) versus AAV9/hMECP2(v1) is unclear, although our data points toward it providing generally more regulated expression. We currently know that the modified viral genome reduces transgene expression (as detected by immunofluorescence) in WT liver tissue at the dose (1 × 1011 vg) and route in our study (Figure 7). In addition, in Mecp2−/y mice, AAV9/hMECP2(v2) appears to regulate transgene expression more tightly (than that of AAV9/hMECP2(v1)), with fewer cDNA copies generated per viral genome in the heart, liver, and spinal cord (AAV9/hMECP2(v1) compared against AAV9/hMECP2(v2)) (Figure 6). Indeed, when we evaluated the liver safety profile of 1 × 1012 vg AAV9/hMECP2(v2), we observed no difference in the mean levels of liver toxicity markers (versus those of saline-treated mice; Figure 5). Tighter regulation of transgene expression is especially important in light of the broad peripheral biodistribution observed for both vectors after ICM administration (Figures 2 and 5). This tighter regulation is consistent with the inclusion of additional silencing elements in the second-generation vector. However, the exact mechanism (such as a specific miRNA target) underlying the tighter transgene regulation for AAV9/hMECP2(v2) remains to be determined.
Importantly, neither AAV9/hMECP2(v1) nor AAV9/hMECP2(v2) were able to completely and permanently reverse the symptoms of RTT in mice. Furthermore, at higher doses (1 × 1012 vg injected ICM), the AAV9/hMECP2(v2) vector induced deleterious behavioral effects in WT mice, indicating an upper tolerated dose between 1 × 1011 and 1 × 1012 vg in mice by this route. Thus, third-generation vectors should be developed with a goal of expanding this upper tolerated dosing threshold and/or increasing the therapeutic benefit at lower doses. Given the dosing limits determined in mice and the known side effects associated with overdosing, dose-ranging studies in large animals that are carefully monitored for side effects would be warranted before human translation should be considered. In closing, this study suggests that the inclusion of regulatory elements that help control levels of transgene expression in the CNS and periphery may prove to be critical for intraCSF delivery of MeCP2 gene therapy.
Materials and Methods
Vectors
The complete names for hMECP2(v1) and hMECP2(v2) are self-complementary MeP229-human MeCP2-myc-BGHpA11 and self-complementary MeP426-human MeCP2-myc-RDH1pA, respectively. Both gene expression cassettes encode the e1 isoform of MeCP2, which is composed of exons 1, 3, and 4.24, 33 The MeP426 promoter includes additional putative regulatory elements that are absent from the shorter truncated promoter MeP229.23 RDH1pA is a synthetic 3′ UTR containing 110 bp of the highly conserved MeCP2 distal polyadenylation signal34 and an upstream miRNA-binding panel containing sites for three additional miRNAs endogenous to the MECP2 3′ UTR: miR-19, miR-22, and miR-132.25, 28, 29, 30 Sequences for MeP426 and RDH1pA are provided in Figure S3. The self-complementaryAAV9/MeP229-GFP-SV40pA vector has been previously described.11
Animals
Animal studies at University of North Carolina (UNC) Chapel Hill were conducted according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC). Mice were weaned at PND28 and were provided food and water ad libitum. Both WT and Mecp2−/y mice were weaned at PND28 so that Mecp2−/y mice could achieve a viable weight (∼10 g) before weaning. Euthanasia criteria have been previously described.11
Virus Production
Vectors were produced by triple transfection, iodixanol gradient centrifugation, and ion exchange chromatography, as described for the UNC Vector Core.35 Purified vectors were dialyzed in PBS (350 mM final NaCl concentration) containing 5% D-sorbitol and stored at −80°C until use. Thawed aliquots were subsequently stored at 4°C. A filter-sterilized solution of PBS (350 mM final NaCl concentration) containing 5% D-sorbitol was used as vehicle and virus dilution buffer.
Injections
Mice were randomized into treatment groups and injected (ICM or IT, vector or vehicle treated) between 28 and 35 days of age. For littermate comparisons, vehicle and virus treatments were randomly assigned without any prior knowledge regarding each sibling’s body weight or general physical condition. Percutaneous IT injections were performed on non-anesthetized mice as described.36 Mice assigned to ICM cohorts were injected intraperitoneally with a sterile-filtered solution of 1.25% Avertin (also known as tribromoethanol; 0.02 mL/g body weight) dissolved in 1x PBS prior to inhalational isoflurane anesthesia. Mice remained on inhalational anesthesia for the duration of the surgery, which was completed using a sterile technique. An incision was made in the scalp, exposing the skull and neck muscles. A 50-μL Hamilton syringe was used to deliver a 10-μL bolus injection into the cisterna magna. Vetbond tissue adhesive (3M) was then used to close the incision. Injected mice were returned to a recovery cage, where they remained until they were ambulatory. Injected mice had access to acetaminophen (∼200 mg/kg body weight) in their drinking water (∼100 mL) for the next 48 hr.
Behavior Scoring
Behavior scoring was completed by an observer blinded to treatment according to previously published guidelines.15 In short, mobility, gait, hindlimb clasping, breathing, tremor, and general appearance were each assessed on a scale of 0–2, with 12 being the maximum aggregate score attainable. Videos of mice provided as supplemental data were taken by an observer that was not blinded to treatment.
Immunofluorescent Analyses
Mice were perfused 3 to 4 weeks post-injection with 1x PBS, followed by filtered 4% paraformaldehyde (PFA) (in 1× PBS, 2 mM NaOH, pH 7.4). Brains were then fixed for an additional 48 hr at 4°C. A vibrating microtome (Leica VT 1000S) was used to prepare 40-μM sections from liver and brain tissue. Sections were washed in 50% ethanol, followed by three washes in 0.3 M PBS with 0.3% Triton X-100. Sections were then transferred to 10 mM sodium citrate (pH 6, 85°C, 30 min) for antigen retrieval, and then washed (three times) prior to blocking for 1 hr at room temperature in 5% goat serum (in 0.3 M PBS, 0.3% Triton X-100). Tissue sections were then incubated with primary antibodies at 4°C for 48 hr. Primary antibody mixtures were chicken anti-myc antibody (Novus; 1:500) mixed with either rabbit anti-mouse MeCP2 (Cell Signaling; 1:500) or rabbit anti-mouse NeuN (Millipore; 1:500) in 5% goat serum. Sections were then washed three times and incubated with secondary antibodies for at least 4 hr at room temperature (goat anti-chicken Alexa Fluor 488 [1:1,000]; goat anti-rabbit Alexa Fluor 594 [1:1,000] in 0% goat serum). After immunolabeling, sections were incubated with DAPI (1:2,000, v/v) in 0.3 M PBS and 0.3% Triton X-100 for 1 hr at room temperature. Sections were washed 3x prior to mounting with ProLong Gold Antifade Mountant with DAPI. Neuronal tropism (or % NeuN+ cells) = . Transduction efficiencies were calculated as the percentage of total nuclei that are also myc+. Apparent transduction efficiencies (or % myc+ cells) = . Cells within an 80x field of view were counted, with multiple brain sections imaged per mouse.
Microscopy
All immunolabeled sections were imaged with a Zeiss 710 confocal microscope at the UNC Confocal and Multiphoton Imaging Facility.
Biodistribution and Gene Expression
Biodistribution of viral genomes and gene expression analyses (using mRNA as a source target) for saline- and virus-treated Mecp2−/y mice were performed according to previously published methods11 using primers specific for human MeCP2. Primer sequences were published by Gadalla et al.11 Tissue was harvested 3 to 4 weeks post-ICM. To assess how tightly transgene expression was regulated, we calculated the following ratio per organ per mouse: . We then calculated the mean ratio across four to five mice per cohort.
Blood Serum Collection
Blood serum was collected 3 to 4 weeks post-ICM according to previously published methods.11 Serum testing of blinded samples was completed by the Animal Clinical Chemistry Core at UNC.
Statistical Analyses
Graphpad Prism was used to generate Kaplan-Meier survival plots and calculate p values for survival cohorts. Unpaired t tests or paired t tests were performed as appropriate for all other statistical calculations.
Author Contributions
S.E.S. injected mice, analyzed data, conducted immunofluorescence analyses, and prepared the manuscript text and figures. K.K.E.G. optimized the immunofluorescence protocol, consulted on the project, and provided feedback on the manuscript. R.D.H. designed AAV9/hMECP2(v2). C.H. scored behavior blind to genotype and treatment. D.C. processed tissue samples for biodistribution and gene expression. V.Z. validated primer design and PCR conditions for biodistribution and gene expression analyses and carried out those analyses. S.R.C., M.E.S.B., and S.J.G. led the collaboration, provided guidance on experimental designs, analyzed data, and helped with manuscript preparation.
Conflicts of Interest
S.J.G. declares a conflict of interest with Asklepios Biopharma, from which he has received patent royalties for IP that are not used in this study.
Acknowledgments
This work was funded by NIH grant 4T32HD040127-15 (to S.E.S.) and a grant from the Rett Syndrome Research Trust (to S.J.G., S.R.C., and M.E.S.B.). Indirect administrative support for S.J.G. was provided by Research to Prevent Blindness to the UNC-CH Department of Ophthalmology. The authors thank the UNC Animal Histopathology and Laboratory Medicine Core Facility (which is supported in part by an NCI Center Core Support grant 2P30CA016086-40 to the UNC Lineberger Comprehensive Cancer Center) and the UNC Confocal and Multiphoton Imaging Facility (supported by NINDS Center grant P30 NS045892) for assistance with blood serum testing and imaging, respectively. We thank Alejandra Rozenberg and Mary Kate Crawford for technical assistance with IT injections and colony maintenance, respectively. We thank Bethany Wagner and Emma Hoffman for proofreading this manuscript. We are grateful for constructive discussions of the project direction and results with Dr. Brian Kaspar (Nationwide Children’s Hospital) and Dr. Gail Mandel (Oregon Health and Science University) as part of a consortium focused on developing gene therapy for Rett syndrome.
Footnotes
Supplemental Information includes five figures and four movies and can be found with this article online at http://dx.doi.org/10.1016/j.omtm.2017.04.006.
Supplemental Information
WT mice were injected ICM between 4 and 5 weeks of age. At 6 months of age, the saline-treated mouse in this video appears alert and has good mobility and gait. At 6 months of age, the AAV9/hMECP2(v1)-treated mouse in this video appears alert but has difficulty using his hind legs correctly. At 6 months of age, the AAV9/EGFP-treated mouse has normal mobility and gait. The absence of hindlimb clasping in the AAV9/EGFP-treated mouse was photographed at this age and is featured at the end of the video. See also Figure 3.
{kind=link}
Both littermates were injected on PND30 and were video-recorded on PND52. The saline-treated mouse has poor mobility and shows little spontaneous movement during the duration of the video. The virus-treated sibling exhibits more spontaneous movement than his saline-treated Mecp2−/y littermate. Note that the virus-treated littermate approaches the camera. This behavior could be an indicator of improved sociability. See also Figure 4.
{kind=link}
These siblings were injected on PND31 and video-recorded on PND51. The saline-treated mouse walks slowly and explores in response to prompting. Note that he does not explore the camera. His virus-treated Mecp2−/y littermate walks, climbs, explores, and approaches the camera without prompting. See also Figure 4.
{kind=link}
These siblings were injected on PND28 and video-recorded on PND40. The saline-treated mouse immediately walks to his hut. The reason for this behavior (i.e., aversion to light or aversion to open spaces) is unclear. His virus-treated Mecp2−/y littermate is more active. See also Figure 4.
{kind=link}
References
- 1.Chapleau C.A., Lane J., Larimore J., Li W., Pozzo-Miller L., Percy A.K. Recent progress in Rett syndrome and MeCP2 dysfunction: assessment of potential treatment options. Future Neurol. 2013;8 doi: 10.2217/fnl.12.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feldman D., Banerjee A., Sur M. Developmental dynamics of Rett syndrome. Neural Plast. 2016;2016:6154080. doi: 10.1155/2016/6154080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirby R.S., Lane J.B., Childers J., Skinner S.A., Annese F., Barrish J.O., Glaze D.G., Macleod P., Percy A.K. Longevity in Rett syndrome: analysis of the North American Database. J. Pediatr. 2010;156:135–138.e1. doi: 10.1016/j.jpeds.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 5.Shahbazian M.D., Antalffy B., Armstrong D.L., Zoghbi H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- 6.Lyst M.J., Bird A. Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet. 2015;16:261–275. doi: 10.1038/nrg3897. [DOI] [PubMed] [Google Scholar]
- 7.Katz D.M. Brain-derived neurotrophic factor and Rett syndrome. Handb. Exp. Pharmacol. 2014;220:481–495. doi: 10.1007/978-3-642-45106-5_18. [DOI] [PubMed] [Google Scholar]
- 8.Bedogni F., Rossi R.L., Galli F., Cobolli Gigli C., Gandaglia A., Kilstrup-Nielsen C., Landsberger N. Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 2014;46:187–201. doi: 10.1016/j.neubiorev.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Jordan C., Li H.H., Kwan H.C., Francke U. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med. Genet. 2007;8:36. doi: 10.1186/1471-2350-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y.T., Goffin D., Johnson B.S., Zhou Z. Loss of MeCP2 function is associated with distinct gene expression changes in the striatum. Neurobiol. Dis. 2013;59:257–266. doi: 10.1016/j.nbd.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gadalla K.K., Bailey M.E., Spike R.C., Ross P.D., Woodard K.T., Kalburgi S.N., Bachaboina L., Deng J.V., West A.E., Samulski R.J. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol. Ther. 2013;21:18–30. doi: 10.1038/mt.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garg S.K., Lioy D.T., Cheval H., McGann J.C., Bissonnette J.M., Murtha M.J., Foust K.D., Kaspar B.K., Bird A., Mandel G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci. 2013;33:13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray S.J., Nagabhushan Kalburgi S., McCown T.J., Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray S.J., Foti S.B., Schwartz J.W., Bachaboina L., Taylor-Blake B., Coleman J., Ehlers M.D., Zylka M.J., McCown T.J., Samulski R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guy J., Gan J., Selfridge J., Cobb S., Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen R.Z., Akbarian S., Tudor M., Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 17.Schuster D.J., Dykstra J.A., Riedl M.S., Kitto K.F., Honda C.N., McIvor R.S., Fairbanks C.A., Vulchanova L. Visualization of spinal afferent innervation in the mouse colon by AAV8-mediated GFP expression. Neurogastroenterol. Motil. 2013;25:e89–e100. doi: 10.1111/nmo.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodda C., Tantra M., Mollajew R., Arunachalam J.P., Laccone F.A., Can K., Rosenberger A., Mironov S.L., Ehrenreich H., Mannan A.U. Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Am. J. Pathol. 2013;183:195–210. doi: 10.1016/j.ajpath.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 19.Collins A.L., Levenson J.M., Vilaythong A.P., Richman R., Armstrong D.L., Noebels J.L., David Sweatt J., Zoghbi H.Y. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 20.Goffin D., Allen M., Zhang L., Amorim M., Wang I.T., Reyes A.R., Mercado-Berton A., Ong C., Cohen S., Hu L. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 2011;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Na E.S., Nelson E.D., Kavalali E.T., Monteggia L.M. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 2013;38:212–219. doi: 10.1038/npp.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson L., Guy J., McKay L., Brockett E., Spike R.C., Selfridge J., De Sousa D., Merusi C., Riedel G., Bird A. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135:2699–2710. doi: 10.1093/brain/aws096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J., Francke U. Identification of cis-regulatory elements for MECP2 expression. Hum. Mol. Genet. 2006;15:1769–1782. doi: 10.1093/hmg/ddl099. [DOI] [PubMed] [Google Scholar]
- 24.Liyanage V.R., Zachariah R.M., Rastegar M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol. Autism. 2013;4:46. doi: 10.1186/2040-2392-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein M.E., Lioy D.T., Ma L., Impey S., Mandel G., Goodman R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007;10:1513–1514. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- 26.Jovičić A., Roshan R., Moisoi N., Pradervand S., Moser R., Pillai B., Luthi-Carter R. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J. Neurosci. 2013;33:5127–5137. doi: 10.1523/JNEUROSCI.0600-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Y., Huang W., Wani M., Yu X., Ashraf M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS ONE. 2014;9:e88685. doi: 10.1371/journal.pone.0088685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manners M.T., Tian Y., Zhou Z., Ajit S.K. MicroRNAs downregulated in neuropathic pain regulate MeCP2 and BDNF related to pain sensitivity. FEBS Open Bio. 2015;5:733–740. doi: 10.1016/j.fob.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao H., Wen G., Huang Y., Yu X., Chen Q., Afzal T.A., Luong A., Zhu J., Ye S., Zhang L. MicroRNA-22 regulates smooth muscle cell differentiation from stem cells by targeting methyl CpG-binding protein 2. Arterioscler. Thromb. Vasc. Biol. 2015;35:918–929. doi: 10.1161/ATVBAHA.114.305212. [DOI] [PubMed] [Google Scholar]
- 30.Lyu J.W., Yuan B., Cheng T.L., Qiu Z.L., Zhou W.H. Reciprocal regulation of autism-related genes MeCP2 and PTEN via microRNAs. Sci. Rep. 2016;6:20392. doi: 10.1038/srep20392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadalla K.K.E., Vudhironarit T., Hector R.D., Sinnett S., Bahey N.G., Bailey M.E.S., Gray S.J., Cobb S.R. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol. Ther. Methods Clin. Dev. 2017;5:180–190. doi: 10.1016/j.omtm.2017.04.007. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matagne V., Ehinger Y., Saidi L., Borges-Correia A., Barkats M., Bartoli M., Villard L., Roux J.C. A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol. Dis. 2016;99:1–11. doi: 10.1016/j.nbd.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 33.Kriaucionis S., Bird A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004;32:1818–1823. doi: 10.1093/nar/gkh349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newnham C.M., Hall-Pogar T., Liang S., Wu J., Tian B., Hu J., Lutz C.S. Alternative polyadenylation of MeCP2: influence of cis-acting elements and trans-acting factors. RNA Biol. 2010;7:361–372. doi: 10.4161/rna.7.3.11564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clément N., Grieger J.C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016;3:16002. doi: 10.1038/mtm.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gray S.J., Choi V.W., Asokan A., Haberman R.A., McCown T.J., Samulski R.J. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr. Protoc. Neurosci. 2011;57:4.17.1–4.17.30. doi: 10.1002/0471142301.ns0417s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
WT mice were injected ICM between 4 and 5 weeks of age. At 6 months of age, the saline-treated mouse in this video appears alert and has good mobility and gait. At 6 months of age, the AAV9/hMECP2(v1)-treated mouse in this video appears alert but has difficulty using his hind legs correctly. At 6 months of age, the AAV9/EGFP-treated mouse has normal mobility and gait. The absence of hindlimb clasping in the AAV9/EGFP-treated mouse was photographed at this age and is featured at the end of the video. See also Figure 3.
Both littermates were injected on PND30 and were video-recorded on PND52. The saline-treated mouse has poor mobility and shows little spontaneous movement during the duration of the video. The virus-treated sibling exhibits more spontaneous movement than his saline-treated Mecp2−/y littermate. Note that the virus-treated littermate approaches the camera. This behavior could be an indicator of improved sociability. See also Figure 4.
These siblings were injected on PND31 and video-recorded on PND51. The saline-treated mouse walks slowly and explores in response to prompting. Note that he does not explore the camera. His virus-treated Mecp2−/y littermate walks, climbs, explores, and approaches the camera without prompting. See also Figure 4.
These siblings were injected on PND28 and video-recorded on PND40. The saline-treated mouse immediately walks to his hut. The reason for this behavior (i.e., aversion to light or aversion to open spaces) is unclear. His virus-treated Mecp2−/y littermate is more active. See also Figure 4.