

Abstract
How intrinsically disordered proteins and regions evade degradation by cellular machinery evolved to recognize unfolded and misfolded chains remains a vexing question. One potential means by which this can occur is the disorder is transient in nature. That is, the disorder exists just long enough for it to be bound by a partner biomolecule and fold. A review of 30 y of studies of calmodulin’s activation of calcineurin suggests that the regulatory domain of this vital phosphatase is a transiently disordered region. During activation, the regulatory domain progresses from a folded state, to disordered, followed by folding upon being bound by calmodulin. The transient disordered state of this domain is part of a critical intermediate state that facilitates the rapid binding of calmodulin. Building upon “fly-casting” as a means of facilitating partner binding, the mechanism by which calcineurin undergoes activation and subsequent deactivation could be considered “catch and release.”
Keywords: proteases, chaperones, degradation, misfolding, calmodulin, intrinsically disordered region, phosphatase
Introduction
With a few exceptions, it is now generally accepted that intrinsically disordered proteins (IDPs) and disordered regions (IDRs) are common and often functionally important.1-6 As a field, the study of intrinsic disorder is thriving. Progress is being made on determining the sequence determinants of disorder,7,8 the spectrum of conformational ensembles occupied by IDPs and IDRs,6,9-11 as well as functional roles.2,5,12
A somewhat vexing unanswered question, however, is how do disordered chains survive within the cellular environment? Organisms have evolved multiple mechanisms for dealing with unfolded and misfolded protein chains. These range from chaperones,13 to the ubiquitination and proteasomal degradation system.14 How is it that IDPs and IDRs avoid these mechanisms?
One simple answer is perhaps they are not disordered within the cellular environment. Crowded conditions such as those in vivo are believed to promote or stabilize folded structures.15,16 Such conditions would disfavor disorder. This may well be the case for some IDPs and IDRs that have to date only been studied in vitro, or are no more than predictions. On the other hand, there is a growing body of evidence that other IDPs are indeed disordered in the cellular environment. One example is the small disordered protein α-synuclein.17
The very sequence characteristics of IDPs and IDRs that favor their disordered nature—poor in hydrophobes, enriched in polar and charged residues7,8—may also be responsible for their avoidance of chaperones, as well as their ability to remain soluble. Unfolded and misfolded proteins are generally recognized by cells due to exposure of hydrophobic side chains.13 The paucity of such residues may well be what keeps IDPs and IDRs from being bound by chaperones. How many disordered regions avoid ubiquitination and degradation by the proteasome14 is unclear, particularly given evidence that some level of disorder is required for efficient proteasomal degradation.18,19
Another possibility, as noted recently by Janin and Sternberg,20 is that chains that are disordered in vitro may simply be lacking a binding partner. There are multiple examples of IDPs and IDRs that undergo a disorder-to-order transition, i.e., fold, when bound by another biomolecule.4,21,22 Janin and Sternberg argue that many IDPs and IDRs are in fact “proteins waiting for partners”; proteins that are disordered in vitro, but folded and associated with binding partners in vivo.
Another way to view this latter idea is that disorder can be transient. That is, for some IDPs and IDRs, the disorder exists long enough in vivo to fulfill its function, but not long enough to become a target for degradation or chaperones. In the case of transient disorder, the disordered state should be viewed as an essential component of the protein’s function.
The idea of transient disorder (upside-down functionality or dormant disorder) is not new, and was discussed recently by Uversky.23,24 He makes 2 important points about transient disorder. One is that, since the sequences of IDPs and IDRs are typically poor in hydrophobic and other order-promoting residues, the ordered states bracketing the transient disorder generally need to be stabilized by other factors such as binding partners or other folded domains within the protein. Second, the order-to-disorder transition can be triggered by many different factors.23 For example, the bacterial light sensor PYP undergoes an order-to-disorder transition upon absorbing a blue photon.25,26 The disordered state of PYP is the signaling state, and exists just transiently before refolding into the original ordered state.
The idea of transient disorder is closely related to the recently introduced notions of regulated unfolding27 and conditional disorder.28 The regulated unfolding discussed by Mitrea and Kriwacki27 has been observed in signaling processes and can involve activation of an enzyme or protein as a result of the unfolding of a domain or secondary structure element. This can be induced by factors such as phosphorylation. Conditional disorder has been discussed by Reichmann and Jakob28 in relation to redox proteins. In this case, exposure to oxidants can induce chaperone proteins such as Hsp33 or COX17 to undergo either an order-to-disorder or disorder-to-order transition, leading to their activation. Notably, the disordered state is critical for protein function.
Murzin,29 followed by Bryan and Orban,30 have described what they call metamorphic proteins. This involves proteins, or domains, that switch between 2 distinct conformations, altering function. The conformational switch can occur via a disordered intermediate. The set of transiently disordered proteins can be considered to partially overlap the set of metamorphic proteins. Proteins that undergo order-to-disorder-to-order transitions where the beginning and ending ordered states are the same would not be considered metamorphic. Those where the 2 end states differ would.
The essential Ser/Thr phosphatase calcineurin (CaN) provides an example of transient disorder where the disordered state, short-lived as it appears to be, is vital for enzyme activity. The remainder of this review discusses the activation of CaN and the role played by transient disorder.
The Case of Calcineurin
Calcineurin function and structure
CaN is a calcium-sensitive phosphatase that is highly conserved through the eukaryotes.31 It is the only phosphatase known to be activated by the calcium-sensing protein calmodulin (CaM).31-33 CaN is known to play pivotal roles in a number of processes in mammals, including development and function of the central nervous system, cardiac development, and activation of T cells within the adaptive immune system. Within the higher eukaryotes, CaN is known to dephosphorylate numerous proteins. Some of the better known of these targets are the family of nuclear factors activating T cells (NFATs), and the microtubule-binding protein τ.31,32,34
CaN is a heterodimer that consists of a 58–64 kDa A chain and a 19 kDa B chain (Fig. 1).31-33 The A chain (CnA) is made up of the catalytic domain, a B chain binding helix (BBH), a regulatory domain (RD), autoinhibitory domain (AID), and a C-terminal tail (CT). CaM is known to bind to the RD of CaN, removing the AID from the active site cleft. There are 3 isoforms of mammalian CnA; the neuronal α isoform, ubiquitous β isoform, and the γ isoform which is predominantly expressed in testes. The latter 2 isoforms differ from the α isoform primarily through N-terminal polyproline and C-terminal basic extensions, respectively. The B chain (CnB) is structurally homologous to CaM, comprised of 4 calcium-binding EF-hands, 2 in each lobe.32,35 There are 2 mammalian isoforms of the B chain, CnB1 and CnB2. CnB1 is found associated with the CnA α and β isoforms, and CnB2 with the γ isoform. Lower eukaryotes tend to have fewer isoforms of CaN.31
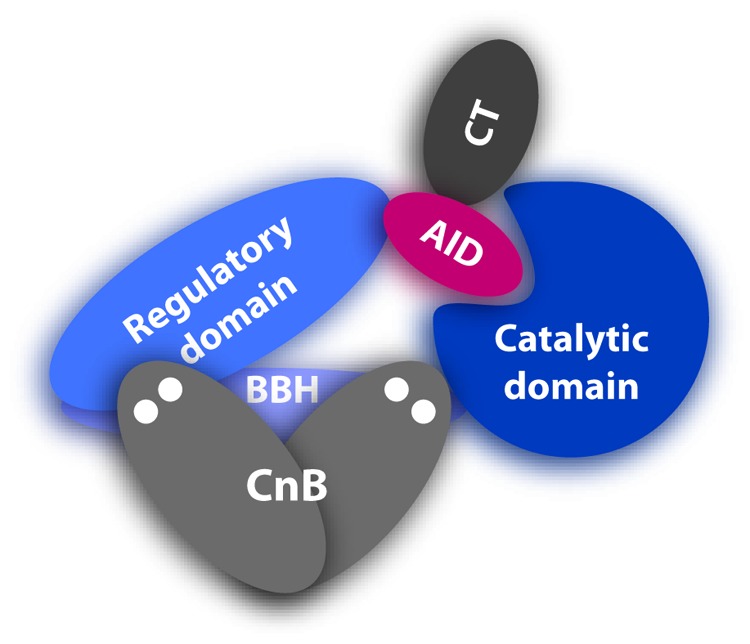
Figure 1. Domain structure of CaN. Catalytic domain is shown in dark blue, the BBH in purple-blue, RD in mid-blue, AID in red, CT in dark gray, and CnB in mid-gray. Calcium bound to CnB is represented by the 4 white dots.
The 3-dimensional structure of CaN (human αCnA:CnB1) was first solved by Kissinger et al. in 1995.36 This 2.1 Å resolution structure (PDB structure 1AUI) is shown in Figure 2. The catalytic domain, BBH, AID, and CnB are all clearly seen. Missing from this structure are the first 13 residues, the entire 95 residue RD (residues A:374-A:468) and the 35 residue CT (residues A:487-A:521). There was insufficient electron density to determine the structures of these regions. Multiple other structures of CaN have subsequently been determined, all of which are also missing these regions (for examples see refs. 37–41). Notably, Kissinger et al. solved the structure of calcium-loaded CaN in the absence of CaM.36 As will be discussed, there is ample evidence that the RD is disordered in this state. To date, no structures of activated CaN, with CaM bound, have been published.
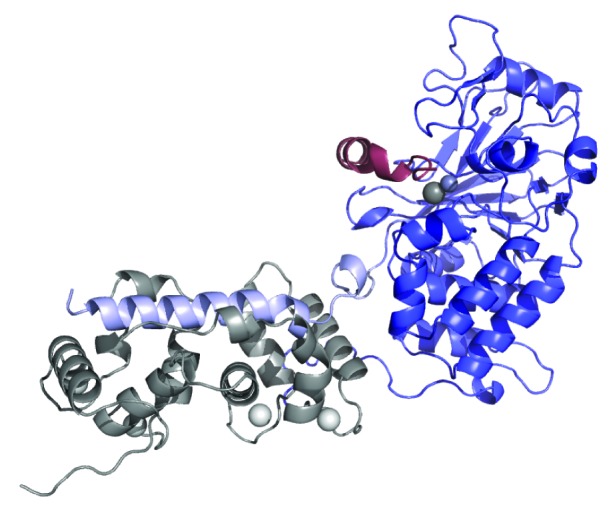
Figure 2. Ribbon diagram of CaN crystal structure (PDB ID 1AUI).36 CnA catalytic domain shown in blue, BBH in lilac, and AID in red. Catalytic Zn-Fe pair shown as mid-gray balls. First 16 residues of CnA, RD, and CT were missing from electron density and are not shown. CnB chain shown in gray with bound calcium ions depicted as white balls.
The disordered regulatory domain
The lack of electron density observed for the RD in CaN structure determination by X-ray crystallography indicates that this 95 residue domain occupies multiple conformations in the crystals.36 One possible explanation for this is that the RD is an intrinsically disordered region (IDR). Of course lack of electron density alone is not strong evidence for disorder. Data suggesting that the RD is indeed an IDR was published a number of years prior to the solution of the CaN structure. Manalan and Klee demonstrated that bovine-brain derived αCaN (the CnA α/B1 isoform), in the presence of calcium but absence of CaM, could be activated by limited tryptic digestion.42 In this experiment, the ~60kDa CnA chain was rapidly digested, leaving a ~45kDa active fragment. The CnB chain was not degraded. Notably, the ~45kDa CnA fragment was found to be fully active in the absence of CaM, suggesting that the RD had been at least partially digested. Indeed, the RD through to the C-terminus of αCnA is replete potential trypsin cut sites (Fig. 3). Similar data were obtained by Hubbard and Klee in limited proteolytic digestion experiments employing clostripain.43 The relatively rapid digestion of the RD would suggest that it is readily accessible to proteases, implying that it is very dynamic or disordered.

Figure 3. Sequence of the RD (dark blue), AID (red), and CT (dark gray) from human CnA. The CaM binding region is denoted by the blue-shaded box. Potential trypsin cleavage sites are noted by arrows.
Dunker and coworkers explored the potentially disordered nature of the CnA RD early in their development of disorder predictors.44 The RD sequence is rich in polar residues and relatively poor in hydrophobes, hallmarks of intrinsically disordered sequences. A more PONDR prediction44,45 suggests the RD, AID and CT are all disordered (Fig. 4). Although the AID was clearly ordered in the crystal structure (Fig. 2), as noted, electron density for the RD and CT was missing, supporting the prediction.
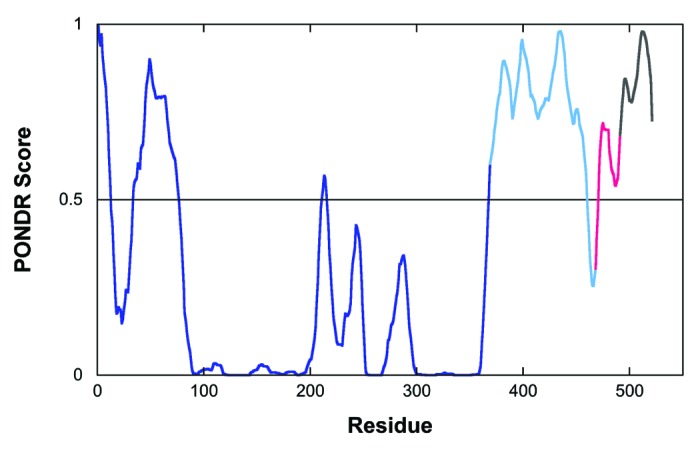
Figure 4. PONDR prediction for CnA.44,45 The catalytic domain and BBH are shown in dark blue, RD in mid-blue, AID in red, and CT in dark gray.
More recently, Rumi-Masante et al. expressed and purified a construct consisting of the RD, AID, and CT (RD-AID-CT) from αCnA.46 These authors used fluorescence techniques to show there was no detectable interaction between this construct and an αCnA truncated immediately prior to the RD, in the presence of sufficient calcium that the CnB calcium binding sites would be fully occupied. They then went on to employ circular dichroism (CD) spectroscopy and hydrogen-deuterium exchange/mass spectrometry (HXMS) to confirm that the RD-AID-CT construct was devoid of stable secondary structure in solution, i.e., it is disordered.
Notably, all of the experimental data supporting the hypothesis that the RD is disordered in full-length CaN was collected in the presence of sufficient calcium that the calcium-binding sites of CnB would be fully occupied.
Inactive calcineurin
The concentration of calcium ions in “resting” cells has been estimated to be ~100nM.47 The atomic-level structure of CaN as it exists at this concentration, or in the absence, of calcium has not yet been solved. However, extensive limited proteolysis work by Klee and coworkers, coupled with the work of Perrino and coworkers, has provided a model for this state.42,43,48-50 In the presence of calcium, but absence of CaM, the RD of CaN is readily digested.42,43 However, at resting intracellular calcium concentrations, the RD is largely protected.48 Under these conditions, the calcium-binding sites of CnB would not be fully loaded, although it remains tightly bound to CnA.51 This is likely due to both interactions between CnB and the BBH, and the extensive interface between the C-terminal lobe of CnB and the catalytic domain of CnA (Fig. 2). After prolonged exposure to protease (trypsin or chymotrypsin), some cleavage between the CaM binding region and the AID is observed. In addition, cleavages in the N-terminal lobe and the linker region between the 2 lobes of CnB are detected. Based on these data, Yang and Klee have suggested that, in the inactive CaN state, the RD is folded onto the BBH and C-terminal lobe of CnB.48 Furthermore, the N-terminal lobe is no longer bound to the BBH.
Further evidence for the RD being folded onto the BBH and CnB C-terminal lobe was provided by Perrino et al.50 They found that a CaN truncated immediately C-terminal to the BBH was fully active in the absence of calcium, but a CaN truncated immediately after the CaM binding region was only partially active. This latter construct required addition of calcium and CaM to become fully active, suggesting the CaM binding region interacted with the BBH and CnB in the absence of calcium. Perrino followed up on this observation and demonstrated that the RD region between the CaM binding region and the AID contained elements that inhibited activity, indicating interaction between these elements and the remainder of CaN in the absence of calcium.49
That elements binding to the BBH and CnB C-terminal lobe inhibit CaN activity is not surprising. This region contains the LxVP substrate binding site.49,52 Many, although not all, of CaN’s substrates possess sequence motifs that bind to this site. Included among these is the RII peptide, derived from the regulatory domain of cAMP-dependent protein kinase.53 Phosphorylated RII peptide is a commonly used substrate in CaN activity assays, including those performed by Perrino and coworkers.49,50 Furthermore, CaN is the target for the immunosupressants FK506 and cyclosporine. These bind to the BBH and CnB C-terminal lobe region of CaN, preventing substrate access to the LxVP binding site.36,37,54
Activation of calcineurin by calcium and calmodulin
As noted above, CaN is activated by the calcium-sensing protein CaM.31-33 But CaM alone is not sufficient. CnB is essential for CaN activity—removal of this subunit results in inactive CnA even in the presence of CaM.55-57 Stemmer and Klee determined that the binding of calcium-loaded CaM to CaN increased the Vmax of the phosphatase, but had no effect on Km.58 The binding of calcium by CnB was required to observe a decrease in Km (increased substrate affinity) compared with the low-calcium, inactive state of CaN. Thus, activation of CaN requires the concerted effort of both calcium-binding proteins, CaM and CnB.
The crystal structure of CaN shown in Figure 2, which depicts CnB in the calcium-loaded state, includes the AID bound to the active site cleft, and thus represents a potentially inactive state. However, Klee, Stemmer, and coworkers have shown that CaN is truly inactive only at low calcium concentrations, similar to resting intracellular concentrations.48,51,58,59 Addition of calcium leads to a partial activation CaN in the absence of CaM. Although the AID appears to remain in the active site cleft in this state (Fig. 2), the binding seems weak enough to allow for some activity. A peptide corresponding to the AID has been shown to inhibit calcium-load CaN activity with an IC50 in the low micromolar range, consistent with this hypothesis.49,50,60,61
Gallagher et al. determined the affinity of each binding site in CnB for calcium (Table 1).62 The 2 sites in the N-terminal lobe have markedly weaker affinities than the 2 in the C-terminal lobe, and would be calcium free at resting cellular levels (~100nM).47 The latter, high-affinity lobe is the one that remains bound to the BBH at resting calcium concentrations, with site 4 calcium-loaded even under these conditions.48 The lower affinity N-terminal lobe acts as a switch. At resting calcium concentrations it is not bound to the BBH. This allows the RD to remain in a folded state docked onto the BBH and C-terminal lobe of CnB. As calcium levels rise, the N-terminal lobe binds both calcium and to the BBH, releasing the RD which subsequently becomes disordered.
Table 1. Macroscopic calcium binding affinities (μM) for each of the 4 sites in CnB.*.
| K1 | K2 | K3 | K4 | |
| CnB | 11 | 81 | 0.15 | 0.03 |
Taken from Gallagher et al.62
Full activation of calcium-loaded CaN is achieved only when calcium-loaded CaM is bound, or the RD and AID are removed via proteolysis.32,42 The binding of CaM appears to cause the RD to undergo a disorder-to-order transition.46 CaM is known to bind to an approximately 24 residue region in the RD (Fig. 1). Much like many CaM binding sites, this region is enriched in basic residues. Although there are crystallographic data suggesting CaM binds to its binding region in CaN with a unique 2:2 stoichiometry,63,64 hydrodynamic data strongly support a 1:1 stoichiometry.65,66 The most common mode of CaM binding to basic substrate sequences is for CaM to wrap around the binding site, inducing α-helical structure.67 This is depicted in Figure 5 for CaM bound to its binding region from myosin light chain kinase.68 The basic nature of the CaM binding region in CaN, plus published hydrodynamic data, suggest that CaM binds to CaN in this common mode.
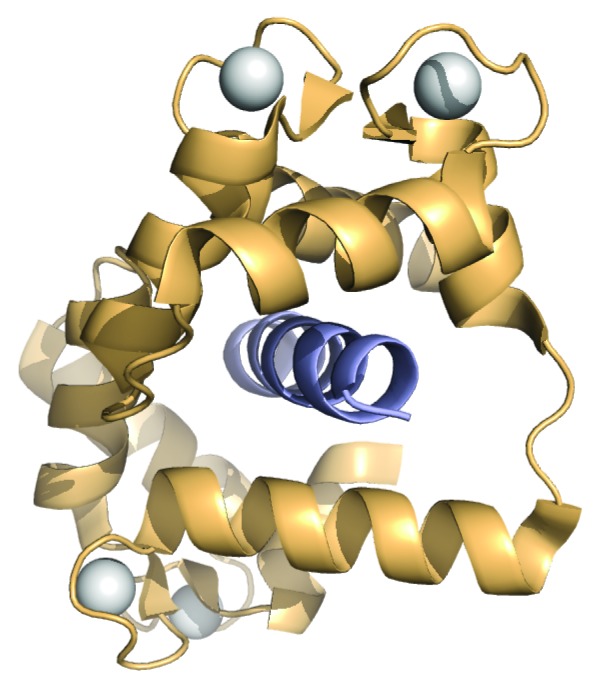
Figure 5. Archetypal CaM binding mode represented by ribbon diagram of the crystal structure of CaM bound to its binding region from myosin light chain kinase (PDB ID 1CDL).68 CaM is shown in pale orange, bound peptide in mid-blue, and calcium ions as white spheres.
The folding of the RD upon CaM binding involves much more than just the CaM binding region. Limited proteolysis experiments by Klee and coworkers demonstrated that the entire RD was protected upon CaM binding.42,43 Following up on that observation, Rumi-Masante et al. used CD to show that the RD gained approximately twice as much α-helical structure upon binding a single CaM as would be expected if just the CaM binding region were folding.46 They went on to use HXMS to identify a region between the CaM binding region and the AID that gained structure. In all ~50 residues of the RD fold into α-helices, making this the largest CaM-induced folding observed to date.
CaM-induced folding of the RD provides the impetus to remove the AID from CaN’s active site. Thus full activation of the phosphatase requires both CnB binding calcium, and for calcium-loaded CaM to bind to the RD.
Transient nature of the disordered state
That the disordered nature of CaN’s RD is transient is strongly suggested by consideration of the relative affinities of CnB (Table 1) and of CaM (Table 2) for calcium. At resting intracellular concentrations of calcium (~100nM), only calcium binding site 4 in CnB would be calcium-loaded. CaM, on the other hand, has an overall much higher affinity for calcium. CaM would be fully loaded with calcium before CnB. Therefore, CaM would be poised to bind the CaN RD as soon as it was released from its folded, inactive state and becomes disordered. The binding of CaM to the RD of CaN is very tight with a Kd estimated to be in the low picomolar range.66 This makes CaN one of CaM’s highest affinity substrates. Quintana et al. have estimated the on-rate to be 4.6 x 107 M.−1s−1, indicating that calcium-loaded CaM binds to CaN rapidly.69 Given that the RD appears to fold upon CaM binding,42,43,46 and assuming CaM and CaN are in the same locality within the cell, this all indicates that the disordered state of the RD exists on average for a tiny fraction of a second. It is very much a transient state.
Table 2. Macroscopic calcium binding affinities (μM) for each of the 4 sites in CaM.*.
| K1 | K2 | K3 | K4 | |
| CaM | 0.20 | 0.80 | 0.07 | 0.04 |
Taken from Maune et al.74
Despite its transient nature, the disordered state of CaN is essential. Calcium-loaded CaM must engulf its binding region in the RD, wrapping around it to induce helical structure (Fig. 5). To do so requires that the binding region be readily accessible. Being a part of a disordered domain would enhance the accessibility of this region. Dunker and coworkers have predicted that CaM binding regions in general will be within IDRs for this very reason.70 The CaM binding region itself need not necessarily be disordered,71 although in the case of CaN it is.46
The disordered state of the RD, albeit transient, not only makes the CaM binding region more accessible, but could also enhance the rate of binding. In a recent review, Kiefhaber et al. noted that the “fly-casting” mechanism, in which an IDR undergoes a disorder-to-order transition upon being bound by a partner, is characterized by an apparent on-rate > 1x107 M.−1 s.-1 72 The binding of CaM to CaN appears to be in this regime.69 According to the fly-casting model, the rate of a protein binding to an IDP or IDR is enhanced as a result of the increased capture radius of the disordered chain.73 Whether the binding of CaM to the RD of CaN induces folding after binding, or is the result of conformational selection, is not clear and can only be determined by a detailed study of the kinetics.72
That the RD of CaN is folded, unfolds as calcium levels increase, and is then induced into a different folded state by the binding of CaM means that it spans more than 1 of the spatiotemporal structure categories defined by Uversky.24 Going from the inactive to active state, as calcium levels rise, the as-yet-unidentified folded regions of the RD first act as an unfoldons, becoming disordered. In the subsequent CaM-induced folding, the regions of the RD that fold (e.g., the CaM binding region) can be considered inducible foldons. To what extent the unfoldon and inducible foldon regions within the RD overlap is currently unknown.
The above discussion leads to the model for CaN activation by CaM shown in Figure 6. At resting calcium levels in the cell CaN is in an inactive state characterized by the N-terminal lobe of CnB being in the apo state, not bound to the BBH of CnA. Limited proteolysis data suggests the RD is folded down onto the BBH in CnA and the C-terminal lobe of CnB.42,43,48 As intracellular levels rise, first CaM will bind calcium and undergo the conformational changes required for it to be in the correct state for binding to substrates such as CaN. CnB then binds calcium, causing its N-terminal lobe to bind to the BBH, releasing the RD into a disordered state. Calcium-loaded CaM can then rapidly bind to the RD, causing it to fold and remove the AID from the active site of CaN, leading to full activation of the phosphatase.46 In keeping with the disordered state of the RD conforming to the fly-casting model of Shoemaker et al.,73 the process by which CaN is activated by binding CaM, and then deactivated by its release, could be considered a “catch and release” mechanism.

Figure 6. Model for the activation of CaN by CaM via a transiently disordered intermediate state. (A) CaN in the low-calcium inactive state with the N-terminal lobe of CnB detached from the BBH, and the RD folded onto CnB and the BBH. As intracellular calcium levels rise, first (B) CaM binds calcium and adopts a binding competent conformation, then (C) CnB binds calcium, its N-terminal lobe binds to the BBH, releasing the RD into a disordered state. Finally, (D) CaM binds to the RD causing it to fold and remove the AID from the catalytic site, thereby fully activating the phosphatase.
Conclusion
The data discussed above, culled from 30 y of literature on the activation of CaN by CaM, strongly supports the hypothesis that the RD is disordered only transiently in vivo. More experiments are of course necessary to determine if this is truly the case. The transient disorder proposed for CaN provides an explanation for how some IDPs and IDRs might survive the cellular machinery evolved to detect and degrade unfolded and misfolded proteins. CaN is not the only example of transient disorder. As noted above, the bacterial light sensor PYP also possesses a transient disordered state essential for function.25,26 One should also note that transient disorder is likely but one of several, if not many, ways by which IDPs and IDRs evade degradation in the cell.
Glossary
Abbreviations:
- IDP
intrinsically disordered protein
- IDR
intrinsically disordered region
- CaN
calcineurin
- CnA
calcineurin A chain
- CnB
calcineurin B chain
- CaM
calmodulin
- BBH
B chain binding helix
- RD
regulatory domain
- AID
autoinhibitory domain
- CT
C-terminal domain
- CD
circular dichroism spectrapolarimetry
- HXMS
hydrogen-deuterium exchange with mass spectrometry
- NFAT
nuclear factor activating T cells
Citation: Creamer T. Transient disorder: Calcineurin as an example. Intrinsically Disordered Proteins 2013; 1:e26412
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a grant to Creamer TP from the NSF (MCB-0843551).
References
- 1.Uversky VN, Dunker AK. . The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol Rep 2013; 5:1; http://dx.doi.org/ 10.3410/B5-01; PMID: 23361308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. . Intrinsic disorder and functional proteomics. Biophys J 2007; 92:1439 - 56; http://dx.doi.org/ 10.1529/biophysj.106.094045; PMID: 17158572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tompa P. . Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci 2012; 37:509 - 16; http://dx.doi.org/ 10.1016/j.tibs.2012.08.004; PMID: 22989858 [DOI] [PubMed] [Google Scholar]
- 4.Wright PE, Dyson HJ. . Linking folding and binding. Curr Opin Struct Biol 2009; 19:31 - 8; http://dx.doi.org/ 10.1016/j.sbi.2008.12.003; PMID: 19157855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dyson HJ, Wright PE. . Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 2005; 6:197 - 208; http://dx.doi.org/ 10.1038/nrm1589; PMID: 15738986 [DOI] [PubMed] [Google Scholar]
- 6.Babu MM, Kriwacki RW, Pappu RV. . Structural biology. Versatility from protein disorder. Science 2012; 337:1460 - 1; http://dx.doi.org/ 10.1126/science.1228775; PMID: 22997313 [DOI] [PubMed] [Google Scholar]
- 7.Williams RM, Obradovi Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK. . The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput 2001; •••:89 - 100; PMID: 11262981 [DOI] [PubMed] [Google Scholar]
- 8.Uversky VN, Gillespie JR, Fink AL. . Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins 2000; 41:415 - 27; http://dx.doi.org/; PMID: 11025552 [DOI] [PubMed] [Google Scholar]
- 9.Mao AH, Lyle N, Pappu RV. . Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem J 2013; 449:307 - 18; http://dx.doi.org/ 10.1042/BJ20121346; PMID: 23240611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. . Flavors of protein disorder. Proteins 2003; 52:573 - 84; http://dx.doi.org/ 10.1002/prot.10437; PMID: 12910457 [DOI] [PubMed] [Google Scholar]
- 11.Marsh JA, Teichmann SA, Forman-Kay JD. . Probing the diverse landscape of protein flexibility and binding. Curr Opin Struct Biol 2012; 22:643 - 50; http://dx.doi.org/ 10.1016/j.sbi.2012.08.008; PMID: 22999889 [DOI] [PubMed] [Google Scholar]
- 12.Chen J. . Towards the physical basis of how intrinsic disorder mediates protein function. Arch Biochem Biophys 2012; 524:123 - 31; http://dx.doi.org/ 10.1016/j.abb.2012.04.024; PMID: 22579883 [DOI] [PubMed] [Google Scholar]
- 13.Walter S, Buchner J. . Molecular chaperones--cellular machines for protein folding. Angew Chem Int Ed Engl 2002; 41:1098 - 113; http://dx.doi.org/; PMID: 12491239 [DOI] [PubMed] [Google Scholar]
- 14.Varshavsky A. . Regulated protein degradation. Trends Biochem Sci 2005; 30:283 - 6; http://dx.doi.org/ 10.1016/j.tibs.2005.04.005; PMID: 15950869 [DOI] [PubMed] [Google Scholar]
- 15.Miklos AC, Sarkar M, Wang Y, Pielak GJ. . Protein crowding tunes protein stability. J Am Chem Soc 2011; 133:7116 - 20; http://dx.doi.org/ 10.1021/ja200067p; PMID: 21506571 [DOI] [PubMed] [Google Scholar]
- 16.Braselmann E, Chaney JL, Clark PL. . Folding the proteome. Trends Biochem Sci 2013; 38:337 - 44; http://dx.doi.org/ 10.1016/j.tibs.2013.05.001; PMID: 23764454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binolfi A, Theillet FX, Selenko P. . Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature?. Biochem Soc Trans 2012; 40:950 - 4; http://dx.doi.org/ 10.1042/BST20120096; PMID: 22988846 [DOI] [PubMed] [Google Scholar]
- 18.Kraut DA, Matouschek A. . Proteasomal degradation from internal sites favors partial proteolysis via remote domain stabilization. ACS Chem Biol 2011; 6:1087 - 95; http://dx.doi.org/ 10.1021/cb2002285; PMID: 21815694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prakash S, Inobe T, Hatch AJ, Matouschek A. . Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol 2009; 5:29 - 36; http://dx.doi.org/ 10.1038/nchembio.130; PMID: 19029916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janin J, Sternberg MJE. . Protein flexibility, not disorder, is intrinsic to molecular recognition. F1000 Biol Rep 2013; 5:2; http://dx.doi.org/ 10.3410/B5-02; PMID: 23361309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dyson HJ, Wright PE. . Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 2002; 12:54 - 60; http://dx.doi.org/ 10.1016/S0959-440X(02)00289-0; PMID: 11839490 [DOI] [PubMed] [Google Scholar]
- 22.Kovacs D, Szabo B, Pancsa R, Tompa P. . Intrinsically disordered proteins undergo and assist folding transitions in the proteome. Arch Biochem Biophys 2013; 531:80 - 9; http://dx.doi.org/ 10.1016/j.abb.2012.09.010; PMID: 23142500 [DOI] [PubMed] [Google Scholar]
- 23.Uversky VN. . A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci 2013; 22:693 - 724; http://dx.doi.org/ 10.1002/pro.2261; PMID: 23553817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uversky VN. . Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 2013; 1834:932 - 51; http://dx.doi.org/ 10.1016/j.bbapap.2012.12.008; PMID: 23269364 [DOI] [PubMed] [Google Scholar]
- 25.Rubinstenn G, Vuister GW, Mulder FA, Düx PE, Boelens R, Hellingwerf KJ, Kaptein R. . Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat Struct Biol 1998; 5:568 - 70; http://dx.doi.org/ 10.1038/823; PMID: 9665170 [DOI] [PubMed] [Google Scholar]
- 26.Ramachandran PL, Lovett JE, Carl PJ, Cammarata M, Lee JH, Jung YO, Ihee H, Timmel CR, van Thor JJ. . The short-lived signaling state of the photoactive yellow protein photoreceptor revealed by combined structural probes. J Am Chem Soc 2011; 133:9395 - 404; http://dx.doi.org/ 10.1021/ja200617t; PMID: 21627157 [DOI] [PubMed] [Google Scholar]
- 27.Mitrea DM, Kriwacki RW. . Regulated unfolding of proteins in signaling. FEBS Lett 2013; 587:1081 - 8; http://dx.doi.org/ 10.1016/j.febslet.2013.02.024; PMID: 23454209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reichmann D, Jakob U. . The roles of conditional disorder in redox proteins. Curr Opin Struct Biol 2013; 23:436 - 42; http://dx.doi.org/ 10.1016/j.sbi.2013.02.006; PMID: 23477949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murzin AG. . Biochemistry. Metamorphic proteins. Science 2008; 320:1725 - 6; http://dx.doi.org/ 10.1126/science.1158868; PMID: 18583598 [DOI] [PubMed] [Google Scholar]
- 30.Bryan PN, Orban J. . Proteins that switch folds. Curr Opin Struct Biol 2010; 20:482 - 8; http://dx.doi.org/ 10.1016/j.sbi.2010.06.002; PMID: 20591649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rusnak F, Mertz P. . Calcineurin: form and function. Physiol Rev 2000; 80:1483 - 521; PMID: 11015619 [DOI] [PubMed] [Google Scholar]
- 32.Klee CB, Ren H, Wang X. . Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem 1998; 273:13367 - 70; http://dx.doi.org/ 10.1074/jbc.273.22.13367; PMID: 9593662 [DOI] [PubMed] [Google Scholar]
- 33.Hogan PG, Li H. . Calcineurin. Curr Biol 2005; 15:R442 - 3; http://dx.doi.org/ 10.1016/j.cub.2005.06.006; PMID: 15964258 [DOI] [PubMed] [Google Scholar]
- 34.Li H, Rao A, Hogan PG. . Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol 2011; 21:91 - 103; http://dx.doi.org/ 10.1016/j.tcb.2010.09.011; PMID: 21115349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anglister J, Grzesiek S, Wang AC, Ren H, Klee CB, Bax A. . 1H, 13C, 15N nuclear magnetic resonance backbone assignments and secondary structure of human calcineurin B. Biochemistry 1994; 33:3540 - 7; http://dx.doi.org/ 10.1021/bi00178a010; PMID: 8142351 [DOI] [PubMed] [Google Scholar]
- 36.Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, et al. . Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature 1995; 378:641 - 4; http://dx.doi.org/ 10.1038/378641a0; PMID: 8524402 [DOI] [PubMed] [Google Scholar]
- 37.Jin L, Harrison SC. . Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci U S A 2002; 99:13522 - 6; http://dx.doi.org/ 10.1073/pnas.212504399; PMID: 12357034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Zhang L, Rao A, Harrison SC, Hogan PG. . Structure of calcineurin in complex with PVIVIT peptide: portrait of a low-affinity signalling interaction. J Mol Biol 2007; 369:1296 - 306; http://dx.doi.org/ 10.1016/j.jmb.2007.04.032; PMID: 17498738 [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi K, Roehrl MHA, Sun Z-YJ, Wagner G. . Structure of the calcineurin-NFAT complex: defining a T cell activation switch using solution NMR and crystal coordinates. Structure 2007; 15:587 - 97; http://dx.doi.org/ 10.1016/j.str.2007.03.015; PMID: 17502104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ke H, Huai Q. . Structures of calcineurin and its complexes with immunophilins-immunosuppressants. Biochem Biophys Res Commun 2003; 311:1095 - 102; http://dx.doi.org/ 10.1016/S0006-291X(03)01537-7; PMID: 14623295 [DOI] [PubMed] [Google Scholar]
- 41.Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. . X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 1995; 82:507 - 22; http://dx.doi.org/ 10.1016/0092-8674(95)90439-5; PMID: 7543369 [DOI] [PubMed] [Google Scholar]
- 42.Manalan AS, Klee CB. . Activation of calcineurin by limited proteolysis. Proc Natl Acad Sci U S A 1983; 80:4291 - 5; http://dx.doi.org/ 10.1073/pnas.80.14.4291; PMID: 6576338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hubbard MJ, Klee CB. . Functional domain structure of calcineurin A: mapping by limited proteolysis. Biochemistry 1989; 28:1868 - 74; http://dx.doi.org/ 10.1021/bi00430a066; PMID: 2541767 [DOI] [PubMed] [Google Scholar]
- 44.Romero O. Dunker K. Sequence Data Analysis for Long Disordered Regions Prediction in the Calcineurin Family. Genome informatics Workshop on Genome Informatics 1997; 8:110–24. [PubMed]
- 45.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. . Sequence complexity of disordered protein. Proteins 2001; 42:38 - 48; http://dx.doi.org/; PMID: 11093259 [DOI] [PubMed] [Google Scholar]
- 46.Rumi-Masante J, Rusinga FI, Lester TE, Dunlap TB, Williams TD, Dunker AK, Weis DD, Creamer TP. . Structural basis for activation of calcineurin by calmodulin. J Mol Biol 2012; 415:307 - 17; http://dx.doi.org/ 10.1016/j.jmb.2011.11.008; PMID: 22100452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clapham DE. . Calcium signaling. Cell 2007; 131:1047 - 58; http://dx.doi.org/ 10.1016/j.cell.2007.11.028; PMID: 18083096 [DOI] [PubMed] [Google Scholar]
- 48.Yang SA, Klee CB. . Low affinity Ca2+-binding sites of calcineurin B mediate conformational changes in calcineurin A. Biochemistry 2000; 39:16147 - 54; http://dx.doi.org/ 10.1021/bi001321q; PMID: 11123943 [DOI] [PubMed] [Google Scholar]
- 49.Perrino BA. . Regulation of calcineurin phosphatase activity by its autoinhibitory domain. Arch Biochem Biophys 1999; 372:159 - 65; http://dx.doi.org/ 10.1006/abbi.1999.1485; PMID: 10562429 [DOI] [PubMed] [Google Scholar]
- 50.Perrino BA, Ng LY, Soderling TR. . Calcium regulation of calcineurin phosphatase activity by its B subunit and calmodulin. Role of the autoinhibitory domain. J Biol Chem 1995; 270:340 - 6; http://dx.doi.org/ 10.1074/jbc.270.1.340; PMID: 7814394 [DOI] [PubMed] [Google Scholar]
- 51.Klee CB, Draetta GF, Hubbard MJ. . Calcineurin. Adv Enzymol Relat Areas Mol Biol 1988; 61:149 - 200; PMID: 2833077 [DOI] [PubMed] [Google Scholar]
- 52.Grigoriu S, Bond R, Cossio P, Chen JA, Ly N, Hummer G, Page R, Cyert MS, Peti W. . The molecular mechanism of substrate engagement and immunosuppressant inhibition of calcineurin. PLoS Biol 2013; 11:e1001492; http://dx.doi.org/ 10.1371/journal.pbio.1001492; PMID: 23468591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blumenthal DK, Takio K, Hansen RS, Krebs EG. . Dephosphorylation of cAMP-dependent protein kinase regulatory subunit (type II) by calmodulin-dependent protein phosphatase. Determinants of substrate specificity. J Biol Chem 1986; 261:8140 - 5; PMID: 3013843 [PubMed] [Google Scholar]
- 54.Huai Q, Kim H-Y, Liu Y, Zhao Y, Mondragon A, Liu JO, Ke H. . Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc Natl Acad Sci U S A 2002; 99:12037 - 42; http://dx.doi.org/ 10.1073/pnas.192206699; PMID: 12218175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milan D, Griffith J, Su M, Price ER, McKeon F. . The latch region of calcineurin B is involved in both immunosuppressant-immunophilin complex docking and phosphatase activation. Cell 1994; 79:437 - 47; http://dx.doi.org/ 10.1016/0092-8674(94)90253-4; PMID: 7525078 [DOI] [PubMed] [Google Scholar]
- 56.Sikkink R, Haddy A, MacKelvie S, Mertz P, Litwiller R, Rusnak F. . Calcineurin subunit interactions: mapping the calcineurin B binding domain on calcineurin A. Biochemistry 1995; 34:8348 - 56; http://dx.doi.org/ 10.1021/bi00026a016; PMID: 7599126 [DOI] [PubMed] [Google Scholar]
- 57.Watanabe Y, Perrino BA, Chang BH, Soderling TR. . Identification in the calcineurin A subunit of the domain that binds the regulatory B subunit. J Biol Chem 1995; 270:456 - 60; http://dx.doi.org/ 10.1074/jbc.270.1.456; PMID: 7814411 [DOI] [PubMed] [Google Scholar]
- 58.Stemmer PM, Klee CB. . Dual calcium ion regulation of calcineurin by calmodulin and calcineurin B. Biochemistry 1994; 33:6859 - 66; http://dx.doi.org/ 10.1021/bi00188a015; PMID: 8204620 [DOI] [PubMed] [Google Scholar]
- 59.Feng B, Stemmer PM. . Ca2+ binding site 2 in calcineurin-B modulates calmodulin-dependent calcineurin phosphatase activity. Biochemistry 2001; 40:8808 - 14; http://dx.doi.org/ 10.1021/bi0025161; PMID: 11467941 [DOI] [PubMed] [Google Scholar]
- 60.Hashimoto Y, Perrino BA, Soderling TR. . Identification of an autoinhibitory domain in calcineurin. J Biol Chem 1990; 265:1924 - 7; PMID: 2153670 [PubMed] [Google Scholar]
- 61.Sagoo JK, Fruman DA, Wesselborg S, Walsh CT, Bierer BE. . Competitive inhibition of calcineurin phosphatase activity by its autoinhibitory domain. Biochem J 1996; 320:879 - 84; PMID: 9003375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallagher SC, Gao ZH, Li S, Dyer RB, Trewhella J, Klee CB. . There is communication between all four Ca(2+)-bindings sites of calcineurin B. Biochemistry 2001; 40:12094 - 102; http://dx.doi.org/ 10.1021/bi0025060; PMID: 11580284 [DOI] [PubMed] [Google Scholar]
- 63.Ye Q, Wang H, Zheng J, Wei Q, Jia Z. . The complex structure of calmodulin bound to a calcineurin peptide. Proteins 2008; 73:19 - 27; http://dx.doi.org/ 10.1002/prot.22032; PMID: 18384083 [DOI] [PubMed] [Google Scholar]
- 64.Ye Q, Li X, Wong A, Wei Q, Jia Z. . Structure of calmodulin bound to a calcineurin peptide: a new way of making an old binding mode. Biochemistry 2006; 45:738 - 45; http://dx.doi.org/ 10.1021/bi0521801; PMID: 16411749 [DOI] [PubMed] [Google Scholar]
- 65.Majava V, Kursula P. . Domain swapping and different oligomeric States for the complex between calmodulin and the calmodulin-binding domain of calcineurin a. PLoS One 2009; 4:e5402; http://dx.doi.org/ 10.1371/journal.pone.0005402; PMID: 19404396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Donnell SE, Yu L, Fowler CA, Shea MA. . Recognition of β-calcineurin by the domains of calmodulin: thermodynamic and structural evidence for distinct roles. Proteins 2011; 79:765 - 86; http://dx.doi.org/ 10.1002/prot.22917; PMID: 21287611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoeflich KP, Ikura M. . Calmodulin in action: diversity in target recognition and activation mechanisms. Cell 2002; 108:739 - 42; http://dx.doi.org/ 10.1016/S0092-8674(02)00682-7; PMID: 11955428 [DOI] [PubMed] [Google Scholar]
- 68.Meador WE, Means AR, Quiocho FA. . Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science 1992; 257:1251 - 5; http://dx.doi.org/ 10.1126/science.1519061; PMID: 1519061 [DOI] [PubMed] [Google Scholar]
- 69.Quintana AR, Wang D, Forbes JE, Waxham MN. . Kinetics of calmodulin binding to calcineurin. Biochem Biophys Res Commun 2005; 334:674 - 80; http://dx.doi.org/ 10.1016/j.bbrc.2005.06.152; PMID: 16009337 [DOI] [PubMed] [Google Scholar]
- 70.Radivojac P, Vucetic S, O’Connor TR, Uversky VN, Obradovic Z, Dunker AK. . Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition. Proteins 2006; 63:398 - 410; http://dx.doi.org/ 10.1002/prot.20873; PMID: 16493654 [DOI] [PubMed] [Google Scholar]
- 71.Dunlap TB, Kirk JM, Pena EA, Yoder MS, Creamer TP. . Thermodynamics of binding by calmodulin correlates with target peptide α-helical propensity. Proteins 2013; 81:607 - 12; http://dx.doi.org/ 10.1002/prot.24215; PMID: 23180611 [DOI] [PubMed] [Google Scholar]
- 72.Kiefhaber T, Bachmann A, Jensen KS. . Dynamics and mechanisms of coupled protein folding and binding reactions. Curr Opin Struct Biol 2012; 22:21 - 9; http://dx.doi.org/ 10.1016/j.sbi.2011.09.010; PMID: 22129832 [DOI] [PubMed] [Google Scholar]
- 73.Shoemaker BA, Portman JJ, Wolynes PG. . Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci U S A 2000; 97:8868 - 73; http://dx.doi.org/ 10.1073/pnas.160259697; PMID: 10908673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maune JF, Klee CB, Beckingham K. . Ca2+ binding and conformational change in two series of point mutations to the individual Ca(2+)-binding sites of calmodulin. J Biol Chem 1992; 267:5286 - 95; PMID: 1544911 [PubMed] [Google Scholar]