

Antiarrhythmics and proarrhythmics targeting ERG K+ channels act oppositely to tune parkinsonian brain rhythms and motor behaviors.
Keywords: antiarrhythmics, brain rhythm disorders, burst discharge, ERG channels, Parkinson’s disease, subthalamic nucleus
Abstract
ERG K+ channels have long been known to play a crucial role in shaping cardiac action potentials and, thus, appropriate heart rhythms. The functional role of ERG channels in the central nervous system, however, remains elusive. We demonstrated that ERG channels exist in subthalamic neurons and have similar gating characteristics to those in the heart. ERG channels contribute crucially not only to the setting of membrane potential and, consequently, the firing modes, but also to the configuration of burst discharges and, consequently, the firing frequency and automaticity of the subthalamic neurons. Moreover, modulation of subthalamic discharges via ERG channels effectively modulates locomotor behaviors. ERG channel inhibitors ameliorate parkinsonian symptoms, whereas enhancers render normal animals hypokinetic. Thus, ERG K+ channels could be vital to the regulation of both cardiac and neuronal rhythms and may constitute an important pathophysiological basis and pharmacotherapeutic target for the growing list of neurological disorders related to “brain arrhythmias.”
INTRODUCTION
Parkinson’s disease (PD) is closely related to functional anomalies in the corticobasal ganglia loop due to dopamine deficiency (1–4). Increase of burst firings in the subthalamic nucleus (STN) of the corticobasal ganglia network is a key electrophysiological feature in patients and animal models of PD (5–7). Accordingly, deep brain stimulation (DBS) at the STN has been an effective treatment for PD (8, 9). The causal relation between locomotor symptoms of PD and increased STN burst discharges has been demonstrated recently, as different maneuvers (for example, local applications of T-type Ca2+ channel inhibitors or constant negative electric currents) decreasing STN burst discharges would consistently alleviate parkinsonian motor symptoms (10, 11). Moreover, injection of constant currents of opposite polarity (that is, positive currents) into the STN by itself is sufficient to cause parkinsonian locomotor deficits in normal rats without dopaminergic lesions (11). In addition to motor symptoms, electrophysiological dysfunction of the STN has been correlated with neuropsychiatric changes (for example, cognitive and emotional abnormalities), which could also be ameliorated or aggravated by DBS (12–15). These findings support a view that PD is a motor as well as a nonmotor disease with deranged neural rhythms or “brain arrhythmias,” which could, therefore, be corrected by physical or chemical maneuvers targeting relevant ion channels (see Discussion for more details) (16).
The human ether-à-go-go–related gene (hERG; the human homolog of rodent ERG) encodes the rapid delayed rectifier K+ channel (IKr) that contributes to the repolarization phase in cardiac action potential (17–19). Genetically, mutation- or, clinically, drug-induced dysfunction of hERG K+ currents could lead to abnormal cardiac repolarization and long QT syndrome, a common cause of cardiac arrhythmias (19–22). The ERG K+ channel subfamily has unusual gating properties with relatively fast inactivation on top of slow activation and, thus, minimal conductance during the depolarization (plateau) phase of a cardiac action potential. When repolarization begins, the ERG channel rapidly recovers from inactivation yet slowly deactivates, giving rise to large tail currents and thus ensuring rapid and robust repolarization of action potential in cardiomyocytes (19, 22, 23).
Thus, studies on the roles of ERG channels in cellular excitability have been mainly focused on cardiomyocytes. However, ERG channels are also widely expressed in the central nervous system, including the STN (19, 24, 25). Many central neurons are capable of firing in the burst (oscillatory) mode, which possibly acts as a specific code in neural information flow and contributes to nonutilitarian dynamic stabilization of the circuits (26–28). The burst mode comprises apparently self-sustaining repetitive groups of high-frequency spikes, usually on top of a depolarizing plateau. The burst plateau necessitates relatively long-lasting inward cationic currents to sustain and also slowly activating hyperpolarizing currents to be terminated appropriately. The neuron may then be stabilized at the resting phase by the extraordinarily high K+ conductances for another appropriate interval, a sequential scenario reminiscent of the case in cardiomyocytes. In this regard, the distinctive large tail current of the ERG K+ channel may make it an ideal apparatus for the late hyperpolarizing currents to end a burst and thus determine the length of each burst as well as the timing of subsequent bursts. The increased burst rates as well as prolonged burst plateaus associated with PD (29–32) may therefore implicate a critical role for ERG channels. However, the physiological and pathophysiological roles of ERG channels in the brain, and even their impacts on neuronal burst discharges, have remained poorly understood. Although scattered findings of neuronal ERG K+ conductances were reported, they were usually characterized in rather arbitrarily defined conditions and thus seem to have inconsistent effects on membrane excitability (see Discussion) (33–37). Here, we demonstrate that ERG K+ channels play a determinant role in cellular discharge patterns in STN neurons. In addition, antiarrhythmic agents that block ERG channels would effectively reduce STN bursts and thus ameliorate motor deficits in PD rats. Our data provide direct evidence for the pivotal role of ERG K+ channels in burst firing and parkinsonian locomotor deficits and may illuminate novel approaches for the pharmacotherapy of PD as well as other brain diseases with deranged neural rhythms.
RESULTS
Characterization of ERG K+ currents in subthalamic neurons
Immunohistochemical staining studies have shown significant expression of ERG K+ channel proteins in the rodent STN (24, 25), but biophysical and functional attributes of ERG K+ currents have remained uncharacterized. We demonstrated the existence of ERG K+ currents in STN neurons by taking advantage of E-4031 ([dihydrochlorideN-[4-[[1-[2-(6-Methyl-2-pyridinyl) ethyl]-4-piperidinyl] carbonyl] phenyl] methanesulfonamide dihydrochloride) and dofetilide, which are class III antiarrhythmics specifically targeting ERG channels (38, 39). The voltage-dependent E-4031– and dofetilide-sensitive currents characterized in acutely dissociated STN neurons from juvenile rats closely resemble each other, suggesting a shared target of the two different drugs (Fig. 1, A and B, and refer to fig. S1 for the original currents before subtraction). The drug-inhibited current can be recovered by washing off the drugs (insets of Fig. 1, A and B). The essentially superimposable currents obtained from subtraction of currents recorded in the presence of the drug from the currents before the drug or after washout of the drug for both E-4031 and dofetilide exclude the possibility of major confounding factors such as current rundown. In addition, E-4031– and dofetilide-sensitive currents display very similar hook tail currents (insets of Fig. 1, A and B), which reflect fast recovery from inactivation relative to deactivation (17, 23). Moreover, the amplitude of the tail current is dependent on the prepulse depolarization voltages and approaches its maximal level when the prepulse voltage is above 0 mV. We then examined whether these subthalamic ERG K+ currents have similar biophysical properties to those of the previously reported cases. The V0.5 (the voltage producing half-maximal activation) and k (the slope factor) values of the activation curves are very similar between the E-4031– and dofetilide-sensitive currents (Fig. 1, C and D) and are consistent with the previously reported values (for example, V0.5 between −15 and −30 mV and k values between 6 and 17 in most cases with different external K+ concentrations) characterized from purified ERG clones or other native cells (17, 34, 40–46). In lower external K+ concentration (2 to 5.5 mM), most of the reported V0.5 values are around −30 mV (41–43, 45, 47), which is also consistent with V0.5 of −27.2 ± 3.6 mV (n = 7) with 4 mM external K+ in our hand. One of the major functional roles of ERG K+ currents in cardiomyocytes is contributed by the large tail currents upon membrane repolarization. E-4031– and dofetilide-sensitive currents also display similar large “resurgent” inward currents with a slow monoexponential decay upon repolarization (Fig. 1, A, B, E, and F). Because of much faster deinactivation (recovery from inactivation) than deactivation, the tail current decay time constant approximately reflects the deactivation kinetics. The deactivation kinetics is not dependent on the preceding depolarization voltages (Fig. 1, G and H) but is dependent on the repolarizing potentials (Fig. 1, E and F). Again, E-4031– and dofetilide-sensitive currents show quantitatively similar kinetics and voltage dependence of deactivation, resembling those reported previously for heterologously expressed ERG channels (17, 34, 40–44).
Fig. 1. Characterization of ERG K+ currents in acutely dissociated subthalamic neurons.
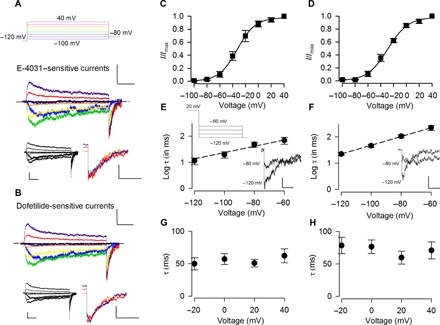
(A and B) Representative ERG K+ channel inhibitor E-4031–subtracted (A) and dofetilide-subtracted (B) currents (see original currents in fig. S1). The voltage protocol is shown on (A) (top). The E-4031–subtracted (600 nM) or dofetilide-subtracted (100 nM) K+ currents are elicited by 1-s depolarizing pulses to different voltages from a holding potential of −120 mV with 150 mM K+ in both extracellular and intracellular solutions. Each depolarizing pulse is immediately followed by a repolarization at −80 mV for 1 s to obtain tail current. Scale bars, 500 pA/200 ms. The bottom-left inset shows the currents after subtraction of the currents in drug from those after washout of the drug. Scale bars, 500 pA/200 ms. The bottom-right inset shows enlarged tail currents evoked at −80 mV after prepulse potentials of 0 to 40 mV. Scale bars, 400 pA/60 ms. The dashed lines indicate the zero-current level. (C and D) Activation curves of E-4031–sensitive (C) and dofetilide-sensitive (D) currents. Peak tail currents are measured and normalized to the maximal tail current produced with a preceding depolarization at 40 mV. The normalized peak tail currents (I/Imax) are then plotted against the voltages (Vt) of the preceding depolarization and fitted with a Boltzmann function I/Imax = 1/(1 + exp([V0.5 − Vt]/k)) with V0.5 (the voltage producing half-maximal activation) of −32 and −29 mV and k (slope factor) of 14 and 15 for E-4031–sensitive (C; n = 6) and dofetilide-sensitive (D; n = 6) currents, respectively. (E and F) Repolarization voltage dependence of deactivation for E-4031– and dofetilide-sensitive K+ currents. Tail currents are elicited at different repolarization potentials immediately after a depolarization at 20 mV. The decay of the tail current is fitted with a monoexponential function. The logarithm of the time constants (τ) is then plotted against the repolarization potential and fitted with a linear function y = 2.57 + 0.01x and y = 3.35 + 0.02x for E-4031–sensitive (E; n = 4) and dofetilide-sensitive (F; n = 6) currents, respectively. The insets show representative tail currents upon repolarization. Scale bars, 200 pA/20 ms (E-4031–sensitive currents) and 400 pA/20 ms (dofetilide-sensitive currents). (G and H) The time constant of the tail current decay at −80 mV is independent of the voltage of the preceding depolarization for E-4031–sensitive (G; n = 5) and dofetilide-sensitive (H; n = 3) K+ currents. Animals used: p9 to p15 rats.
Characterization of subthalamic ERG K+ currents with specific channel activators
Consistent with the findings above, PD-118057 ([2-(4-[2-(3, 4-dichloro-phenyl)-ethyl]-phenylamino)-benzoic acid]), a specific enhancer of ERG channel currents (48, 49), significantly increases voltage-dependent K+ currents in STN neurons (fig. S2). PD-118057–sensitive (–enhanced) currents display a characteristic slow activation upon depolarization and a hook tail upon repolarization (fig. S2, A and B). The activation curve for PD-118057–sensitive currents (fig. S2C) is fairly close to that for ERG inhibitor–sensitive currents documented above (Fig. 1, C and D), except for a small shift in V0.5, which may be ascribable to the gating changes caused by PD-118057 (48). It has been suggested that PD-118057 binds to the pore helix to attenuate fast (P-type) inactivation (48). This may explain the less prominent decay of macroscopic PD-118057–sensitive currents upon depolarization (fig. S2B) than that of E-4031– or dofetilide-sensitive currents (Fig. 1, A and B). The slightly slowed recovery from inactivation by PD-118057 also allows a more reliable measurement of the time to peak of tail current (fig. S2D). The time to peak of the tail current increases with more depolarized prepulse potential, consistent with a voltage-dependent inactivation process that is characteristic of the ERG channel (19). On the other hand, the decay time constant of the tail current, which approximately reflects the deactivation kinetics, is dependent on repolarization but not the preceding depolarization voltages (fig. S2, E and F). The decay time constant and the voltage dependence in fig. S2F are fairly close to those obtained from ERG inhibitor–sensitive currents (Fig. 1, E and F), consistent with the unaltered ERG deactivation kinetics by PD-118057 reported previously (48, 49).
Characterization of the ERG inhibitor–sensitive K+ currents in the presence of ERG activators
Several studies have suggested that PD-118057 is a gating modifier (47–50), although PD-118057 does not seem to affect most characterized gating kinetics of ERG channels (49). On the other hand, E-4031 is generally accepted as an open-pore blocker of the ERG channel (39, 51). We characterized E-4031–subtracted currents in the presence of PD-118057 (fig. S3). The E-4031–subtracted currents in the presence of PD-118057 still carry basic characters of ERG K+ currents, namely, slow activation and deactivation kinetics and large hook tail current (fig. S3B). It is notable that the time to peak of the tail current is more similar to the case of the E-4031–sensitive currents in the absence of PD-118057 (Fig. 1A) than that of PD-118057–sensitive currents (fig. S2B). The half-maximal activation (V0.5) of E-4031–sensitive currents in PD-118057 is −29 mV, and the slope factor is 14 (fig. S3C). These values are again closer to those of E-4031–sensitive currents (in the absence of PD-118057; Fig. 1C) than to those of PD-118057–sensitive currents (fig. S2C). These findings are consistent with a view that binding of PD-118057 in the activated channel may preclude E-4031 from binding. Thus, the biophysical properties of E-4031–subtracted currents in the absence and the presence of PD-118057 would be similar. The binding sites for PD-118057 and for methanesulfonanilide compounds such as E-4031 have been suggested to reside in close proximity in the pore helix and the sixth transmembrane segment (S6) (38, 48, 52–55), and there are reports that PD-118057 may interact with L646 and E-4031 with I647 of S6 in hERG channels (48, 53). In this regard, the preclusion may be more efficient when the channel is activated from the closed state and less efficient with the channel conformation producing tail current. Therefore, the E-4031–subtracted currents in the presence of PD-118057 would have a smaller current during depolarization and a larger tail current during repolarization (fig. S3B; compared to the E-4031–subtracted currents in the absence of PD-118057 in Fig. 1A). Again, the deactivation kinetics is dependent on the repolarizing potentials but remains independent of the depolarizing potential for E-4031–sensitive currents in the presence of PD-118057 (fig. S3, D and E). The biophysical parameters concerning deactivation are also quantitatively similar between E-4031–sensitive currents in the presence and absence of PD-118057.
Inhibition of subthalamic burst discharges by ERG channel blockers
Recent studies have demonstrated a causal relation between parkinsonian locomotor symptoms and burst discharges in the STN (10, 11). The ion channels contributing to the burst discharges are not fully characterized, but sizable hyperpolarizing currents responsible for the termination of each burst (plateau) seem to be necessary. Because electrophysiological and pharmacological evidence consistently indicates the existence of ERG K+ currents in subthalamic neurons (Fig. 1 and figs. S1 to S3), we investigated the possible role of ERG K+ currents in the activities of subthalamic neurons and, thus, the locomotor behaviors of animals. Generally, higher concentrations of ERG inhibitors or activators are needed in brain slices or in vivo conditions than for acutely dissociated neurons because of different extents of drug penetration or retention. In addition, accurate and detailed characterization of neuronal discharges would be preferably done in acute brain slices from young adult mice (see Materials and Methods). We have also repeated the key experiments on young adult rat brain slices to check for consistency. Extracellular recording, which keeps the cell membrane and cytosolic factors intact, shows that subthalamic neurons exhibit spontaneous activities of regular tonic (spiking) as well as burst discharges in young adult mouse brain slices (Fig. 2) (10, 56). Both inhibitors of the ERG K+ channel, E-4031 and dofetilide, reduce spontaneous burst rates (average burst counts per minute) or even turn burst discharges into tonic spikes [Fig. 2, A and B; the activity of ~10% neurons can even be silenced by high concentrations of E-4031 and dofetilide, presumably because of depolarization block at the plateau potential (see below and fig. S4A)]. E-4031 and dofetilide selectively diminish burst discharges but spare spontaneously firings in a regular spiking mode (Fig. 2, C, D, and I to L; see also fig. S5, where distinct effects of the ERG inhibitor on a pair of simultaneously recorded neurons with one firing in the tonic and the other in the burst modes are shown). The “burst inhibition” effects are E-4031 concentration–dependent and reversible (Fig. 2, A and E to H). These results suggest that ERG channels play an important role in the genesis of burst discharges in STN neurons.
Fig. 2. Extracellular recording of spontaneous discharges modulated by ERG inhibitors in acute STN (subthalamic) slices.
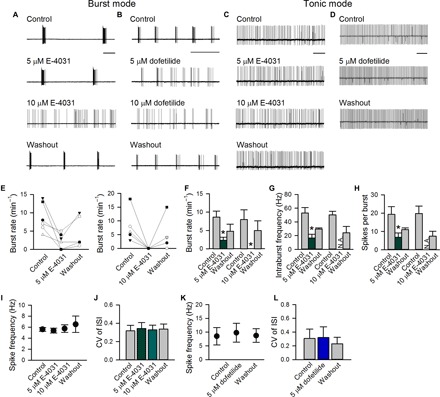
(A to D) Representative cell-attached recordings show that both E-4031 and dofetilide reversibly reduce burst discharges in a dose-dependent fashion (A and B) but do not affect tonic discharges (C and D) (see also fig. S5). (E to H) For the burst mode of discharges, burst rates (average burst counts per minute) (E and F), intraburst spike frequency (G), and the number of spikes per burst (H) before, during, and after application of 5 μM (n = 6) or 10 μM (n = 5) E-4031 are analyzed. *P < 0.05 compared with control, paired two-tailed Student’s t test. N.A., not applicable (no bursts for analysis in that condition). There is no statistically significant difference between “control” and “washout.” *P = 0.13, 0.06, and 0.13 for (F), (G), and (H), respectively, in the study of recovery from 5 μM E-4031 treatment; *P = 0.11, 0.16, and 0.16 for (F), (G), and (H), respectively, in the study of recovery from 10 μM E-4031 treatment. (I to L) For the tonic mode of discharges, single-spike frequency and coefficient of variance (CV) of the interspike intervals (ISIs) before, during, and after E-4031 (I and J; n = 4 in 5 μM and n = 5 in 10 μM) or dofetilide (K and L); n = 3 in 5 μM). Scale bars, 2 s. Animals used: p18 to p26 mice.
Impaired repolarization of the burst plateau by ERG channel blockers
To study how ERG K+ currents modulate burst discharges of the STN in more detail, we performed whole-cell current-clamp recordings that directly measure membrane potential in acute young adult mouse STN slices (Fig. 3). Similar findings to those from extracellular recordings were obtained. E-4031 and dofetilide reversibly reduce spontaneous burst, but not tonic, discharges (Fig. 3, A and B). We also tested a less-specific ERG channel inhibitor, astemizole (1-[(4-fluorophenyl)methyl]-N-[1-[2-(4-methoxyphenyl)ethyl]-4-piperidinyl]-1H-benzimidazol-2-amine) (57), and found very similar effects (Fig. 3, A and B). Moreover, similar findings with E-4031 are obtained at higher temperatures and in slices from young adult rats (figs. S4A, S6A, and S7). Further quantitative analyses show that E-4031 significantly reduces the burst rate (average burst counts per minute), the intraburst spike frequency, and the spike number per burst (Fig. 3, C to E). In contrast, the frequency of spikes and coefficients of variance of the interspike intervals in neurons that fire in the tonic (spike) mode are not altered by E-4031 or dofetilide (Fig. 3, H to K). In cardiomyocytes, the ERG K+ channel chiefly contributes to the large hyperpolarizing force that determines the duration of the plateau phase of an action potential (17, 22, 23). Consistently, superposition of the bursts before and after the application of either E-4031 or dofetilide also suggests a marked change in the repolarization phase of the burst plateau by these ERG inhibitors (insets of Fig. 3A). This is as if E-4031 and dofetilide specifically inhibit repolarization of the burst plateau and keep the membrane potential depolarized for a longer period, and thus, the burst mode could no longer sustain (see also figs. S4A and S9A). The baseline membrane potential is slightly (and reversibly) depolarized by all three ERG inhibitors if the cell fires in a burst mode in control (Fig. 3A). Quantitative analyses also show that E-4031 decreases the afterhyperpolarization (AHP, following the end of a burst) as well as the lowest potential (Fig. 3, F and G). These findings suggest that the ERG channel critically contributes to the hyperpolarizing force that timely terminates a burst (and also to the genesis of the next burst by revival of the channels inactivated during the burst) in STN neurons, reminiscent of the action of the channel in cardiomyocytes.
Fig. 3. Whole-cell patch-clamp recording of spontaneous neuronal activity modulated by different ERG inhibitors in acute STN slices.
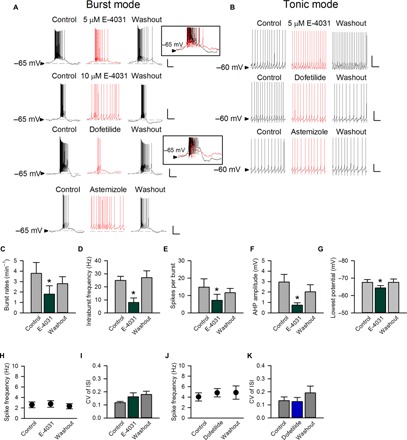
(A) Top: E-4031 (5 and 10 μM) decreases burst discharges and the intraburst spike frequency and even turns bursts into single spikes in a higher concentration (see also fig. S6A). Inset: Superimposed bursts in the absence (black) and the presence (red) of 5 μM E-4031 demonstrate a decreased AHP in the latter condition (see also fig. S9A). Continuous recording of spontaneous activity in a representative neuron is provided in fig. S8. Bottom: 5 μM dofetilide as well as 10 nM astemizole reduce burst discharges. Inset: Superimposed bursts in the absence (black) and presence (red) of 5 μM dofetilide once again demonstrate a decreased AHP by dofetilide. (B) The spontaneous tonic activity is not affected by E-4031, dofetilide, and astemizole. (C to G) For the burst mode, the effects of E-4031 (5 μM) on the burst rate (C), intraburst spike frequency (D), the number of spikes per burst (E), postburst AHP amplitude (F), and the lowest potential (G) are analyzed (each n = 5). (H to K) The effects of E-4031 (5 μM; H and I) or dofetilide (5 μM; J and K) on single-spike frequency and coefficient of variance of the interspike intervals (n = 4 for dofetilide and n = 8 for the others). *P < 0.05 compared to control, paired two-tailed Student’s t test. Scale bars, 20 mV/1 s. Animals used: p18 to p26 mice.
Switch from tonic to burst discharges with a hyperpolarizing force provided by ERG channel activators
Figure 3 shows that the ERG K+ channel blockers suppress burst discharges by inhibition of repolarization from the burst plateau in STN neurons. We then explored whether ERG channel activators would have opposite effects on subthalamic discharges. Given that the firing mode of an STN neuron is tightly correlated with the baseline membrane potential (56), we first studied the STN neurons with a baseline membrane potential of about −60 mV, where the neurons usually maintain spontaneous tonic discharges in the acute young adult mouse brain slices (Fig. 4). Application of the ERG activator PD-118057 readily switches the firing pattern from the tonic to the burst modes (Fig. 4, A and D to F; see also fig. S4B) and thus largely alters the coefficients of variance of the interspike intervals compared to the cases by E-4031 or dofetilide (Fig. 4J). Notably, the switch of discharge modes by PD-118057 is accompanied with membrane hyperpolarization, which is quantitatively well correlated with the tendency of burst and with PD-118057 concentration (Fig. 4, A, C, D, and G to I). Similar findings with PD-118057 are obtained at higher temperatures and in slices from young adult rats (Fig. 6A and figs. S4B and S7). A relatively low concentration (~0.5 μM) of PD-118057 was used to discern the subtle changes in membrane potential and to prevent excessive hyperpolarization that may consequently cease neuronal discharges. Both firing mode–switching and membrane-hyperpolarizing effects of PD-118057 are readily abolished by the addition of the ERG channel blocker E-4031 (Fig. 4B; see also fig. S4B), strongly supporting that both effects are ascribable to the actions on the ERG channels. We have recorded a E-4031–inhibited current in the presence of PD-118057 (fig. S3). It is characterized by relatively small outward currents upon depolarization and extraordinarily large tails. In this regard, the abolition of the PD-118057 effect by E-4031 in Fig. 4B would strongly implicate that the ERG tail currents may play a major role in the alteration of the discharge pattern. Together, these findings indicate that ERG channel activators appear to provide additional hyperpolarizing currents, which would switch the tonic mode of the STN neuronal discharges into the burst mode.
Fig. 4. The effect of ERG channel activator on tonic discharges of subthalamic neurons.
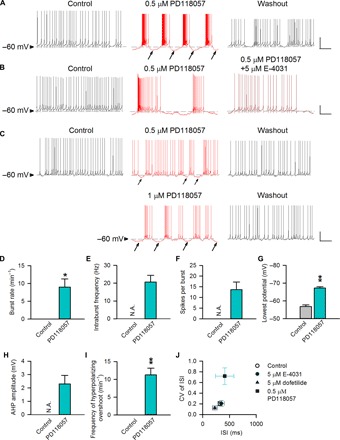
(A to C) Three representative subthalamic neurons that fire in spontaneous tonic activity at a “resting” membrane potential of about −60 mV are shown. The horizontal dashed lines indicate the level of −60 mV. Continuous recording of spontaneous activity in a representative neuron from a rat is provided in fig. S8. (A) PD-118057 (0.5 μM) reversibly hyperpolarizes the membrane (marked by arrows) and turns the tonic spikes into burst discharges (see also Fig. 6A and fig. S4B). (B) The firing mode switching as well as the hyperpolarizing effects of 0.5 μM PD-118057 are abolished by the coapplication of the ERG channel inhibitor E-4031 (5 μM). (C) The tendency toward bursts is correlated with the concentration of PD-118057 as well as its effect on membrane hyperpolarization (marked by arrows). (D to I) There are prominent effects of PD-118057 (0.5 μM) on the burst rate (D), intraburst spike frequency (E), spikes per burst (F), the lowest membrane potential (G), AHP amplitude (H), and the frequency of hyperpolarizing overshoot (I; the hyperpolarization period more negative than the resting membrane potential) (each n = 4). (J) The effects of E-4031 [5 μM; data from Fig. 3 (n = 8)], dofetilide [5 μM; data from Fig. 3 (n = 4)], and PD-118057 (0.5 μM; n = 4) on the interspike intervals and the coefficient of variance of the interspike intervals are compared. *P < 0.05; **P < 0.01 compared to control, paired two-tailed Student’s t test. N.A., not applicable (no bursts for analysis in that condition). Scale bars, 20 mV/2 s. Animals used: p18 to p26 mice.
Fig. 6. Remedy of locomotor deficits of parkinsonian rats by ERG channel blockers.
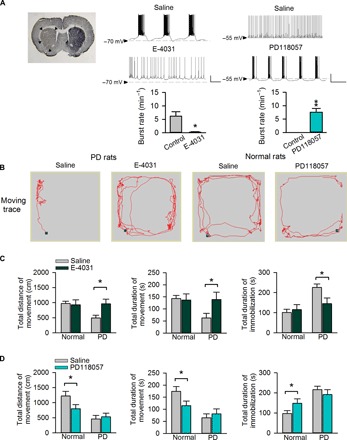
(A) Left: Tyrosine hydroxylase immunohistochemistry shows unilateral dopaminergic deficiency (indicated by arrow heads) in the striatum of parkinsonian (PD) rat coronal brain sections. Middle and right: Whole-cell recording in STN slices from either PD rats [where subthalamic neurons tend to fire in the burst mode (middle); refer to more cases in fig. S4] or normal rats [where subthalamic neurons more frequently fire in the tonic mode (right)] in the absence and presence of 5 μM E-4031 (n = 5; middle) and 0.5 μM PD-118057 (n = 7; right), respectively. Scale bars, 20 mV/2 s. *P < 0.05; **P < 0.01, paired two-tailed Student’s t test. Also refer to in vivo electrophysiological data in fig. S11. (B) Representative locomotor traces of a PD (left) or a normal (right) rat in an arena with administration of normal saline and 200 μM E-4031 (left) or PD-118057 (right), respectively. (C and D) Changes in locomotor activities after direct microinjection of E-4031 or PD-118057 into the STN. The changes in the total distance of movement (left), total duration of movement (middle), and total duration of immobilization (right) in normal or parkinsonian (PD) rats with administration of either 200 μM E-4031 (C; n = 10 for both normal and PD groups) or 200 μM PD-118057 (D; n = 9 for both normal and PD groups) are compared with those injected with normal saline. *P < 0.05, two-way mixed-design analysis of variance (ANOVA). Application of E-4031 markedly improves locomotor activities of PD (but not normal) rats in (C). In contrast, PD-118057 significantly decreases locomotor activities of normal (but not PD) rats in (D). See also fig. S10. Animals used: p37 to p40 rats for brain slice recording; 2- to 3-month-old rats (weighing 260 to 380 g) for other experiments.
Burst discharges shortened or abolished by ERG channel activators
We next recorded STN neurons at about −65 mV, a more hyperpolarized membrane potential where the neurons tend to fire in spontaneous burst activities in acute young adult mouse brain slices (Fig. 5) (56). We found that the ERG activator PD-118057 slightly hyperpolarizes the plateau phase in those neurons with a prolonged plateau for each burst (Fig. 5A). As a result, the duration of each burst is shortened (and thus, the spike number per burst is decreased) as if PD-118057 disrupts the plateau by hyperpolarization (Fig. 5, G and H). Similar findings can be obtained in slices from young adult rats (fig. S6B). The shortening of burst or plateau duration is particularly evident in neurons with longer plateau, such as those from parkinsonian rats, or when the spikes superimposed on the plateau are blocked with tetrodotoxin (figs. S4B and S9B). The burst rate, the intraburst spike frequency, the lowest potential, and the amplitude of AHP are not significantly altered (Fig. 5, E, F, I, and J). However, in neurons that fire in short bursts, PD-118057 may either show no significant effect or abolish the bursts by membrane hyperpolarization (Fig. 5, B to D). The preserved burst discharges in the presence of PD-118057 are readily abolished (with a more depolarized baseline membrane potential) by the addition of the ERG channel blocker E-4031 (Fig. 5B), suggesting that the ERG conductance still contributes to these short bursts.
Fig. 5. The effect of the ERG activator on burst discharges of subthalamic neurons recorded at a membrane potential of about −65 mV.
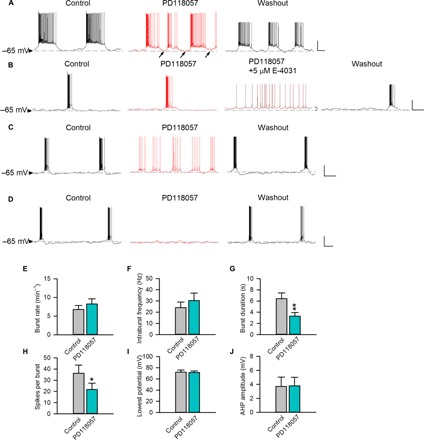
The horizontal dashed lines indicate the level of −65 mV. (A) A representative neuron that spontaneously fires in bursts with relatively long plateau is shown. PD-118057 (0.5 μM) evidently hyperpolarizes the burst plateau and also shortens burst duration [see analyses in (E) to (J), n = 4; see also figs. S4B and S6B]. (B to D) Three representative neurons that spontaneously fire in relatively short bursts are shown [n = 8, 8, and 4 for (B), (C), and (D), respectively]. Note that the baseline membrane potential is not markedly altered by PD-118057 (0.5 μM) in any case but is slightly depolarized by E-4031 (5 μM). (B) The burst pattern remains in 0.5 μM PD-118057 but is abolished by the addition of 5 μM E-4031. (C and D) PD-118057 (0.5 μM) hyperpolarizes the plateau and abolishes bursts. (E to J) In subthalamic neurons having burst duration >3 s, there are no significant effects of PD-118057 (0.5 μM) on burst rates (E), intraburst spike frequency (F), the lowest potential (I), and AHP amplitude (J). However, PD-118057 significantly shortens the burst duration (G; see also fig. S9B) and decreases the number of spikes per burst (H) (n = 4; *P < 0.05; **P < 0.01 compared to control, paired two-tailed Student’s t test). Scale bars, 20 mV/2 s. Animals used: p18 to p26 mice.
Amelioration of parkinsonian locomotor deficits by inhibition of ERG K+ channels
Several lines of evidence have demonstrated a causal relation between subthalamic burst discharges and parkinsonian locomotor symptoms (10, 11, 16, 58). Because ERG channels could profoundly modulate subthalamic bursts, we investigated the effect of ERG channel modulators on locomotor behaviors in adult rats (Fig. 6). We directly applied E-4031 into STN via a chronically implanted cannula and examined the effect on locomotor behaviors of 6-hydroxydopamine (6-OHDA)–lesioned (parkinsonian) rats. With normal saline injection, the locomotor behavior of the parkinsonian (PD) rat group (Fig. 6, B to D) did not significantly differ from that of the same group of rats having microinjection cannula connected but receiving no treatment [with total movement distance of 517 ± 91 cm, total immobilization duration of 187 ± 32, and total movement duration of 76 ± 13 s; n = 8 (data not shown in the figure)]. In contrast, microinjection of the ERG channel inhibitor E-4031 (Fig. 6, B and C), but not of the ERG channel activator PD-118057 (Fig. 6, B and D), directly into STN markedly improves all locomotor behavior parameters tested (see also fig. S10). E-4031 even recovers the total movement distance as well as the total movement and immobilization duration of PD rats to the level of normal rats (Fig. 6C). Moreover, E-4031 also improves the forelimb use and motor asymmetry of PD rats, as assessed by the cylinder test (fig. S10) (59–61). Consistent with the behavioral findings, whole-cell electrophysiological recordings in young adult rat brain slices demonstrate that subthalamic discharges in the PD rats are usually bursts with definite longer plateau and readily switched from the burst to the tonic (single-spike) mode by E-4031 (Fig. 6A and fig. S4A). In vivo extracellular recordings from adult rats also confirm that the increased subthalamic burst rates associated with the 6-OHDA lesion could be readily corrected by E-4031 (fig. S11). On the other hand, PD-118057, but not E-4031, significantly impairs motor function in the normal rat group (Fig. 6, B to D). In addition, whole-cell recordings show that subthalamic discharges in normal rats can be switched from the tonic to the burst mode by PD-118057 (Fig. 6A and fig. S4B). PD-118057 can even convert the locomotor behavior parameters of normal rats to the level of PD rats (Fig. 6D; see also fig. S10, A to D). These findings together demonstrate the crucial modulation of subthalamic activity and, consequently, animal locomotor behavior, by ERG K+ channels.
DISCUSSION
ERG K+ channels in mammalian central neurons
We have seen that ERG channel inhibitors attenuate burst discharges in the STN and ameliorate locomotor deficits in parkinsonian rats, whereas ERG channel activators tend to promote or maintain burst discharges in the STN and impair motor function in normal rats (Figs. 2 to 6). This is the first demonstration that ERG channels exist in principal neurons of the STN (Fig. 1 and fig. S2) and critically control the neuronal discharge patterns that regulate locomotor behaviors via the corticobasal ganglia network. ERG channels are widely expressed in the mammalian brain, where their functional roles have remained poorly characterized (22, 62, 63). ERG channels have been reported to influence excitability of several central neurons in areas other than the cerebrum, including cerebellar Purkinje neurons (37), developing ventral horn interneurons (33), basal vomeronasal neurons (34), medial vestibular nucleus neurons (36), and midbrain dopamine neurons (35). The contribution of ERG channels to firing activity, however, seems to be quite variable among these neurons. For instance, the application of ERG channel inhibitors may increase (35–37) or decrease (34) the firing frequency and may promote (34, 35) or reduce (33, 36, 37, 64) spike frequency adaptation or depolarization block. The variation may be, at least partly, ascribable to different properties of the neurons and especially the characteristic gating features of ERG channels, as well as the experimental elicitation of neuronal activities by injection of an arbitrarily defined amount of current without discretion of the level of depolarization or the pattern/mode of neuronal discharges (see below). Not only the contribution of native ERG conductance to the excitability of brain neurons, but also the functional as well as pathophysiological role of ERG channels in the brain, remain elusive and has been a long-term question for both scientists and clinicians. The first clue may be related to schizophrenia. The expression of a primate-specific isoform of the ERG K+ channel was recently found to be associated with schizophrenia (34, 65, 66). Earlier works on membrane excitability simulated by injection of rather arbitrarily defined amount of currents or computational modeling have suggested a possible role of the partial block of ERG channels in dopaminergic neurons by antipsychotic drugs, which cause a wide spectrum of cellular effect including increase of firing frequency and bursting activity, promotion of spike frequency adaptation, or even depolarization block [(63, 67, 68); see also (35)]. However, many antipsychotic drugs that induce depolarization block in ventral tegmental area neurons do not have the same effect in substantia nigra pars compacta (SNc) neurons (69), suggesting that multiple mechanisms should be considered in different dopaminergic neurons. Because the contribution of ERG channels to neuronal activity may be critically dependent on the setting of membrane potential, the firing modes, and the delicate configuration of the discharges in specific neurons, the characterization most favorably should be done in spontaneous or more naturally occurring conditions than in arbitrarily defined ones. The mutually supportive cellular-to-behavioral evidence provided in this study demonstrates, for the first time, the causal relation among changes in ERG conductance, naturally occurring spontaneous neuronal activity patterns of established pathophysiological meanings, and brain function/behavior. It would be desirable to explore the presumably much broader physiological and pathophysiological roles of ERG channels in the brain with similar approaches in the future.
An ideal role of ERG K+ channels in the modulation of neuronal burst discharges and relevant membrane potential changes
ERG K+ channels are composed of three isoforms (ERG1 to ERG3) (62, 70) and widely expressed in many cells. Most studies on the physiological functions of ERG channels have been focused on the human homolog of rodent ERG1 (hERG) channels in the heart (21, 22), where the experimental results are much more congruent than in the brain. This difference is probably ascribable to the relatively uniform cellular properties and discharge patterns of the cardiac cells. Suppression of hERG currents results in a prolonged plateau in cardiac action potential and, thus, long-QT syndromes or even lethal cardiac arrhythmia (17, 19, 20). On the other hand, gain-of-function mutation in hERG channels or pharmacological activation of the channels leads to shortened cardiac action potential or short-QT syndromes (21, 71). All of these effects are consistent with the characteristics of ERG channel gating, which is unique for the fast inactivation on top of the slow activation, strong voltage dependence of development of as well as recovery from inactivation, and large resurgent tail currents upon membrane repolarization (that is, recovery from inactivation via the open state) (19, 23). That is, ERG channels would tend to be activated with long- and strong-enough depolarization, but allow little current flow because of the rapid transition into the inactivated state. Upon repolarization, rapid recovery of the inactivated channel via the open state gives rise to large slowly decaying resurgent ERG currents, prohibiting premature depolarization of the cell. Therefore, ERG channels could be critical for setting the duration of both the prolonged depolarization (plateau) phase and the following repolarization phase, and thus the timing and frequency of repetitive discharges (17, 22, 23, 64, 72). Many central neurons are capable of firing in repeated bursts or the oscillatory mode. Burst discharges comprise groups of high-frequency spikes most likely superimposed on a prolonged depolarization plateau, which is reminiscent of the plateau of a cardiac action potential. It is thus conceivable that neuronal ERG channels could be activated during burst discharges and then critically regulate the duration and frequency of the bursts (see below).
Fine-tuning of the corticosubcortical reentrant loop function by ERG K+ channels
We have seen that the hyperpolarization phase after a burst slows down with a lower dosage of ERG channel inhibitors and that the burst discharges are turned into spikes on top of a more depolarized baseline membrane potential with higher dosage or longer perfusion of the inhibitors (Fig. 3). It seems that the inhibitor decreases the repolarizing force provided by ERG channels at the end of a burst, resulting in a lengthened plateau depolarization reminiscent of the findings in cardiac cells (22). On the other hand, the ERG channel inhibitors do not affect tonic (spike) discharges (Figs. 2 and 3), consistent with the idea that the drug effect is evident only when there is significant activation (and inactivation) of ERG channels by long-enough plateau depolarization. The role of ERG channels is also supported by the effect of the ERG channel activator PD-118057, which has been shown to be both anti- and proarrhythmic by shortening cardiac action potentials (49, 71). The effect of PD-118057 is ascribable to slowed inactivation and, thus, more hyperpolarizing K+ effluxes in the depolarization phase (48). Accordingly, PD-118057 causes membrane hyperpolarization and switches tonic to burst discharges in a concentration-dependent manner (Fig. 4), chiefly because more “active” conductances (for example, Na+ and T-type Ca2+ channels) are made available by hyperpolarization for the next burst generation (10, 11, 73). In neurons that fire in spontaneous long bursts, PD-118057–induced hyperpolarization could prematurely end the plateau (Fig. 5A and fig. S4B) (22, 49, 71). However, in neurons that fire in relatively short bursts, PD-118057 may increase ERG K+ conductance to the extent that abolishes the burst discharges (Fig. 5, C and D). Consistently, we showed that the whole-cell ERG currents are characterized by a large and slowly decaying tail (Fig. 1 and fig. S2). The large amplitude of the tail current signals its effectiveness in ending a burst (see also E-4031–inhibited extraordinarily large tail current in the presence of PD-118057 in fig. S3). On the other hand, the slow decay signals a major role in the determination of the depth and length of the interburst interval. In this regard, it is noted that subthalamic adenosine 5′-triphosphate (ATP)–sensitive K+ (K-ATP) channels may also be involved in the shaping of burst discharges (74). K-ATP channel function could be facilitated by glutamatergic transmission [which, for example, may activate NMDA (N-methyl-d-aspartate) receptor channels to provide the long-lasting depolarizing currents for a burst] or up-regulated with 6-OHDA lesions (75–77). Together, our findings indicate that neuronal ERG channels, with their unique gating behaviors, may assert a powerful yet delicate control in the configuration of burst discharges as well as the interburst intervals. Because the length and frequency of burst discharges, or the relative proportion and timing of burst versus tonic discharges, in the STN are tightly correlated with the function of the corticosubcortical reentrant loops and, consequently, locomotor behaviors (1, 4, 7), modulation of ERG channels (and, probably, also other slowly activating K+ channels) may be an effective way to change motor activities and other functional presentations relevant to the reentrant loops (Fig. 6).
Physiological and clinical implications
Quite a few central neurons are capable of firing in two different modes: the burst (oscillatory) and the tonic (transfer). The tonic mode comprises irregular or semiregular single spikes that are more responsive to synaptic inputs and, consequently, a more faithful transfer of messages in corresponding networks. On the other hand, the burst mode comprises recurrent grouped discharges that are much less responsive to external stimuli but may be autonomously repetitive (self-regenerative) and strong enough to drive the correlative networks into synchronized actions (78–80), greatly contributing to nonutilitarian dynamic recruitment and/or stabilization of the circuits (26–28). The corticosubcortical reentrant loops involve extended thalamocortical networks that also involve the basal ganglia. Same as in the thalamocortical networks where rhythmic oscillations are assumed to disconnect the external sensory stimuli from cortical processing, the oscillating GPe (external globus pallidus)/STN network may impair information coding by the basal ganglia and thus result in the cardinal symptoms of PD (16, 81, 82). That is, when burst discharges become more prevalent because of more hyperpolarized resting membrane potentials (56), the cortical input may be less faithfully followed by the STN neuronal responses. In addition to motor control, the function of the STN involves emotion, motivation, and cognition (12, 13). In this regard, it is notable that PD is not only a motor disease. Many patients with PD also suffer from nonmotor symptoms (12–15, 83, 84). The anatomical plan of the circuits in the corticosubthalamic pathway allows STN to modulate motor, associative, and limbic information (12, 85–87). That is, burst discharges in the STN may act as a brake of not only motor but also nonmotor loops, interfering with corticosubthalamic information flow. DBS at the STN may release this brake and result in disinhibition of motor, cognitive, and emotional behaviors (12, 13), and thus lead to an improvement of motor deficits and obsessions/compulsions or acceleration of necessary slow processes for decision-making (a potential cause of impulse control disorders) (8, 9, 12–15, 84, 88). These attributes are very much analogous to the cases of thalamic relay neurons and the corticothalamic network (81, 89). The initiation of burst discharges in the thalamus relies on the activation of adequate T-type Ca2+ channels (73, 89). Consistently, the indispensable role of T channels in the STN burst discharges and, thus, locomotor symptoms in PD have been demonstrated (10, 11). Because ERG channels, in principle, could critically configure both the burst itself and the interburst phase whereas T-type channels would chiefly affect only the genesis of burst discharges, ERG channels may potentially provide more extensive and delicate regulation of STN firing patterns and behavioral consequences. As an important electrophysiological signature for both motor and nonmotor behavioral control, different configurations and frequencies of bursts would implicate different likelihoods of a postsynaptic neuron to follow presynaptic firing patterns and different scales of the oscillating networks. We have noted that ERG activators can be pro-parkinsonian on normal rats, whereas ERG inhibitors can be anti-parkinsonian on the PD rat model (Fig. 6). Moreover, the same concentrations of ERG activators and inhibitors produce very similar changes for similar discharge patterns in normal and PD brain slices. These results suggest that there are significant ERG conductances in both normal and PD animals and that the marked increase of burst discharges with prolonged plateaus in PD animals could facilitate functional activation of ERG channels (“recruit” more ERG conductance). In addition, PD is a disease of dopamine deficiency (90–94), which may then set the membrane potential more hyperpolarized and consequently switch the firing mode from tonic spikes to bursts (95–99). In PD animals, we have found that local application of ERG channel inhibitors effectively ameliorates locomotor symptoms via modulation of STN bursts. Because ERG channels are also expressed in the other neurons, such as dopaminergic neurons (35, 68, 69), and because dopamine may have a wide range of action in neuromodulation as well as motor and behavioral control, it would be desirable to fine-tune the burst mode of discharges in the STN and the other nuclei with different dosages of ERG inhibitors or activators, and monitor the behavioral consequences concomitantly in the future. One may therefore gain more insight into the potential therapeutic effect and possible adverse events of ERG channel modulators in different clinical disorders.
MATERIALS AND METHODS
Study design
Information about sample sizes, data inclusion/exclusion criteria, replicates, research subjects, experimental design, blinding, and statistics for different experiments were included in the following sections or the figure legends whenever applicable.
Explanation for animals used
We have done electrophysiological characterizations in acutely dissociated neurons, acute brain slices, and anesthetized animals, as well as behavioral studies in free-running animals, all using Wistar rats to obtain mutually supportive data from cellular to behavioral levels. Animals of different ages were used for different experimental configurations because of technical reasons. Accurate and detailed assessment of the biophysical properties of ion channels and membrane potential changes to the level that our study presents needs to be done in acutely dissociated neurons and brain slices from animals of younger ages. In particular, reliable drug-subtracted ERG currents can only be recorded in acutely dissociated neurons from juvenile rats. On the other hand, all in vivo data were done in adult Wistar rats (weighing 260 to 380 g and aged 2 to 3 months). In addition, neuronal discharges were studied in detail in acute brain slices from young adult mice, which have relatively smaller brain size and thus the advantage of easier manipulation and faster/efficient exchange of the drug in the bath reservoir. The consistency among the data from cellular to behavioral levels and from rats to mice, and the consistency among the data using different inhibitors as well as activators in different configurations, would well support the validity of the approach. Other details for the animals used for each experiment are given below. Moreover, the species and ages of the animals are specified in the figure legends.
Brain slice preparation
Parasagittal brain slices (250 to 270 μm thick to facilitate visualization of neurons) that contain the STN were prepared from C57/Bl6 mice of both sexes (aged p18 to p27) or from 6-OHDA–lesioned male Wistar rats (aged p32 to p46). Before preparation of both brain slices and dissociated neurons (see below), there is no experiment or manipulation done on the animals. The whole brain was rapidly removed and placed into ice-cold oxygenated (95% O2/5% CO2) choline-based solution (87 mM NaCl, 25 mM NaHCO3, 37.5 mM choline cloride, 25 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 7 mM MgCl2, and 0.5 mM CaCl2). Brain slices were then cut on a vibratome (Leica VT1200S, Leica) and sequentially recovered at 30°C in the oxygenated choline-based solution for 20 min and then in the oxygenated saline mimicking the physiological extracellular solution (125 mM NaCl, 26 mM NaHCO3, 25 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, and 2 mM CaCl2) before electrophysiological recordings.
Electrophysiological recordings in brain slices
Before recording, a slice was put on a recording chamber, where stable bath flow of oxygenated (95% O2/5% CO2) saline (125 mM NaCl, 26 mM NaHCO3, 25 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, and 2 mM CaCl2) in the absence or presence of different pharmacological agents at a constant flow rate of ~5 ml/min was maintained by a peristaltic pump (Gilson Medical Electric). Individual STN neurons were identified by 60× water immersion objective on the upright microscope (BX51WI, Olympus). Extracellular or whole-cell current-clamp recordings from STN neurons were performed at room temperature (~25°C). Recording electrodes were prepared from thin-walled borosilicate glass capillaries (Harvard Apparatus; 1.65-mm outer diameter, 1.28-mm inner diameter) and pulled by Brown-Flaming micropipette puller (DMZ-Zeitz-Puller). For extracellular recordings, electrodes were filled with 0.9% NaCl and had a resistance of 3 to 4 megohm. Whole-cell recordings used glass pipettes (~2 megohm) containing the following internal solution: 116 mM KMeSO4, 6 mM KCl, 2 mM NaCl, 20 mM Hepes, 0.5 mM EGTA, 4 mM MgATP, 0.3 mM NaGTP, 10 mM NaPO4 creatine (pH 7.25 with KOH). Data were acquired by a sampling rate of 20 kHz with an analog/digital interface (Digidata 1440, MDS Analytical Technologies) and filtered at 2 kHz with a MultiClamp 700B amplifier (MDS Analytical Technologies). E-4031 was dissolved in distilled water, and astemizole, PD-118057, and dofetilide were dissolved in dimethyl sulfoxide (DMSO) as stock. All drugs used in this study were purchased from Tocris, stored in −20°C, and diluted 1:1000 into the bath reservoir immediately before application. Bath application of 1:1000 DMSO did not result in an alteration of any responses under investigation. In most cases, the good-quality current-clamp recordings would last for 60 min or longer. For most cells, at least 10 min was needed to achieve a significant washout of effects by ERG channel activators or inhibitors except E-4031. The reversal of the E-4031 effect is even slower, and a significant washout usually takes more than 20 min. The washout effect was assessed when the discharge activity is recovered and stable for at least 3 min after perfusion of saline. Refer to the representative continuous recordings before, during, and after application of drugs in fig. S8.
Electrophysiological recordings in acutely dissociated STN neurons
For the preparation of acutely dissociated STN neurons, 370-μm-thick brain slices containing STN were first obtained from Wistar rats aged p9 to p15 and incubated at 32.5°C in the oxygenated (95% O2/5% CO2) choline-based solution for 12 min after cutting. The slices were then treated with protease XXIII (1 mg/ml; Sigma) and bovine serum albumin (1 mg/ml) in the oxygenated choline-based solution for 3 and 0.5 min, respectively, and thoroughly washed with the oxygenated choline-based solution at 32.5°C. To avoid air bubbles, we dissolved both protease XXIII and bovine serum albumin immediately before use and temporarily stopped the oxygenation during enzyme and albumin treatments (pH 7.3 to 7.4 throughout the incubation). After treatment, the slices were transferred to a fresh oxygenated choline-based solution in room temperature. Immediately before electrophysiological recording, an STN chunk was removed and triturated into single neurons in the dissociation solution containing 82 mM Na2SO4, 30 mM K2SO4, 10 mM Hepes, and 3 mM MgCl2 (pH 7.4). The fire-polished borosilicate micropipettes for whole-cell voltage-clamp recording had resistances of ~1 megohm after filling with the intracellular solution [75 mM KF, 75 mM KCl, 5 mM EGTA, 10 mM Hepes, 0.3 mM GTP (guanosine 5′-triphosphate), 10 mM phosphocreatine, and 4 mM MgATP (at pH 7.25)]. When a whole-cell configuration was established in Tyrode’s solution [150 mM NaCl, 4 mM KCl, 10 mM Hepes, 2 mM MgCl2, and 2 mM CaCl2 (at pH 7.4)], the cell was lifted from the bottom and positioned in front of an array of flow pipes (Microcapillary, Drummond Scientific Company; 1 μl, 64 mm), which emitted a high-potassium external solution [150 mM KCl, 4 mM NaCl, 10 mM Hepes, 2 mM MgCl2, 2 mM CaCl2, and 0.0003 mM tetrodotoxin (at pH 7.4)] in the absence or presence of different pharmacological agents targeting on ERG channels (for example, E-4031, dofetilide, or PD-118057). The external solution was not changed until the currents were stable for at least three sweeps. Currents were recorded at room temperature (~25°C) with a MultiClamp 700B amplifier, low-pass–filtered at 2 kHz, digitized at 100-μs intervals, and stored using the pCLAMP software via a Digidata 1550 interface (MDS Analytical Technologies). The drug-sensitive currents were obtained by subtracting the currents in the presence of the drug from those in the absence of the drug.
Animal model of parkinsonism
Experiments were performed on adult male Wistar rats weighing 260 to 380 g (for behavioral studies) or aged p37 to p39 (for brain slice recordings). Animals (three rats per cage in the vivarium) were housed at constant temperature and humidity under a 12-hour light/dark cycle with free access to food and water. We performed unilateral 6-OHDA lesion in adult rats to establish rodent parkinsonian model. Rats were anesthetized with zoletil/xylazine [zoletil (20 to 40 mg/kg) and xylazine (5 to 10 mg/kg)] (Virbac), mounted in a stereotactic frame (Kopf Instruments), and pretreated with desipramine hydrochloride (25 mg/kg, intraperitoneally) (Sigma) to protect noradrenergic neurons. After a 30-min interval, a hole was drilled above the medial forebrain bundle (MFB) region. A stainless steel cannula was connected by a polyethylene catheter to a Hamilton microsyringe driven by an infusion pump (Harvard Apparatus) and then inserted to the depth indicated in Paxinos and Watson’s stereotactic brain atlas (1996): A, −5.0 mm; L, 2.2 mm; and D, 7.5 mm from bregma. 6-OHDA (8 μg; Sigma) was dissolved in 0.01% ascorbic acid (Sigma), diluted with 2 ml of normal saline, and protected from light. Five minutes after insertion of the steel cannula into the accurate stereotaxic target region, 4 μl of 6-OHDA was infused into the MFB target area over a period of 8 min. After the stop of microinjection, the cannula was then left in place for 10 min before being withdrawn slowly. Rats were tested for rotational behavior 7 to 10 days after surgery by subcutaneous injection of apomorphine (0.05 mg/kg, subcutaneously). Only rats that show consistent turning (more than 25 turns in 5 min) toward the side contralateral to 6-OHDA lesion were considered to have a severe-enough SNc lesion (>90% loss of dopaminergic neurons) and retained for subsequent behavioral studies. The extent of dopaminergic loss in all of these rats was verified by tyrosine hydroxylase immunohistochemistry at the end of the experiments.
Implantation of microinjection cannula for chronic use
Under anesthesia of zoletil/xylazine [zoletil (20 to 40 mg/kg) and xylazine (5 to 10 mg/kg)] (Virbac), the rats were mounted in a stereotactic frame (Kopf Instruments). The skull was exposed after adequate sterilization procedure, and a hole was drilled above the STN coordinates: A, −3.8 mm; L, 2.4 mm; and D, 6.5 mm from bregma. A plastic cylindrical rod holding a stainless steel cannula within (Plastics One) was centered on the hole targeting STN and slowly lowered to the level of the upper part of the STN. The plastic rod was then secured with dental cement, which was attached to three stainless steel screws fixed unto the skull. After the dental cement was solidified and local bleeding was clear, the scalp wound was closed. The motor behavior studies were performed 1 to 2 weeks after the implantation surgery. After the experiments, the location of the implanted cannula tip into the STN was always verified by cresyl violet staining. Data were only collected from the animals with a confirmed location of the cannula.
Animal locomotor behavior study and microinjection of pharmacological agents into STN
On the scheduled date of locomotor behavior study, rats were transferred to a quiet, sound-attenuated room for motor behavior testing at least 2 hours before the test session started. The experiments were performed at fixed hours of the day (in the afternoon, light cycle). The open-field test arena (45 cm × 45 cm × 40 cm) was made of Plexiglas and equipped with a video recording and a video tracking system (EthoVision 3.0). Immediately before the test session, the animal was first connected to the microinjection system by insertion of an inner injection cannula, which was connected to a microsyringe and a microinjection pump (Harvard Apparatus) with polyethylene tubing. Three minutes after the start of microinjection of pharmacological agents (at an infusion speed of 0.5 μl/min), the animal was placed in the center of the testing arena for the motor behavior test, which would last for 5 min. The total injection volume would thus be 4 μl. The same group of rats was subjected to two 5-min test sessions daily, usually with microinjection of normal saline in the first session and the pharmacological agents under investigation in the second session. The pharmacological agents (E-4031 or PD-118057; Fig. 6 and fig. S11) were injected with a concentration of 200 μM (and thus a total injected amount of 0.8 nmol). Because PD-118057 has to be dissolved in DMSO with a solubility limit of 100 mM, 0.2% (1:500) DMSO was used as a vehicle control, which did not result in any significant alteration of behavioral responses under investigation. The results from multiple behavioral tests in the same group of animals remained reasonably constant in the first 10 testing sessions. Four major locomotor behavior parameters [namely, total movement distance, movement duration, duration of immobilization, and rearing times (rearing score)] were documented for the evaluation of motor functions [refer to the study by Tai et al. (10)].
Data analysis
Electrophysiological data were analyzed with pCLAMP 10 (MDS Analytical Technologies), SigmaPlot 12 (Systat Software Inc.), and Excel 2013 (Microsoft) softwares. For whole-cell patch recording on acutely dissociated neurons, we excluded the data from the analysis if the leak currents at a holding potential of −120 mV were larger than −500 pA or if the drug effect cannot be reversed by washing. For brain slice recording, each analysis was based on the data collected and averaged from a 1-min continuous recording of stable spontaneous activity. We excluded the data from the analysis if the leak currents at a holding potential of −70 mV were larger than −50 pA (in the voltage-clamp mode) or if the action potential overshoot was below 0 mV (in the current-clamp mode). The action-potential spikes were detected by the “Threshold Search” tool of pCLAMP. Burst discharges were first identified visually and defined by the interspike interval algorithm, which delimits the interspike interval to be less than 100 ms in at least three consecutive spikes (Figs. 2 to 6 and fig. S7). Using the pCLAMP software, burst analysis was then performed to obtain the burst rates, intraburst frequency, and burst duration. AHP amplitudes were measured as the difference between the averaged baseline (resting) membrane potential and the peak of the postburst hyperpolarization. The lowest membrane potential as well as the frequency of hyperpolarizing overshoot (the hyperpolarization period more negative than the resting membrane potential) during a 1-min continuous stable recording were also measured.
Statistics
Electrophysiological data were analyzed with the pCLAMP 10 (MDS Analytical Technologies), SigmaPlot 12 (Systat Software Inc.), and Excel 2013 (Microsoft) softwares. In vivo animal behavior numerical data and statistical analysis were managed with Excel software (version 2013, Microsoft), Prism software version 6.0 (GraphPad Software), and Statistical Package for the Social Sciences version 19.0 (IBM). The investigators executing animal behavioral studies were blinded to the drugs used (for example, whether they are inhibitors or activators) during the experiments and when assessing the outcome. All data are presented as means ± SEM. The sample size represents the number of animals (that is, each n represents data collected from a different animal). For the comparison between the two groups, either unpaired or paired Student’s t tests were used, assuming the differences between the two populations of relatively small sample sizes are normally distributed. Similar variance was generally obtained between groups compared. For animal behavioral analyses in Fig. 6, two-way mixed-design ANOVA was used, with application of pharmacological agents and 6-OHDA lesion of rats being the within-subject factor and between-subject factor, respectively. Exact statistical tests used are reported in relevant figure legends. All statistical tests were two-sided. Values of P < 0.05 were accepted as indicating significant differences for all comparisons.
Study approval
All animal experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee at Chang Gung University and National Taiwan University Hospital (Taiwan).
Supplementary Material
Acknowledgments
We thank T. Wang, Y.-C. Lai, A.-Y. Chuang, and P.-C. Tseng for their technical support in the animal experiments. We are also grateful to the Neuroscience Research Center of Chang Gung Memorial Hospital, Linkou, Taiwan. Funding: This work was supported by grants MOST103-2311-B-182-001-MY3 (to Y.-C.Y.) and MOST104-2314-B-002-047-MY3 (to C.-H.T.) from the Ministry of Science and Technology, and grant CMRPD3E0261-2 (to Y.-C.Y.) from the Chang Gung Medical Foundation, Taiwan. Author contributions: C.-S.H., G.-H.W., C.-H.T., and C.-C.H. conducted the experiments. C.-S.H., G.-H.W., C.-H.T., C.-C.H., and Y.-C.Y. analyzed the data. C.-S.H., G.-H.W., and Y.-C.Y. wrote the manuscript with the help of C.-H.T. and C.-C.H. Y.-C.Y. conceived the study, designed the experiments, and supervised the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602272/DC1
fig. S1. The original currents of Fig. 1.
fig. S2. Characterization of ERG K+ currents by channel activator PD-118057 in acutely dissociated subthalamic neurons following similar experimental protocols and analyses of Fig. 1.
fig. S3. The effect of PD-118057 on E-4031–sensitive currents.
fig. S4. The effect of the ERG inhibitor and activator on spontaneous firing of subthalamic neurons in acute STN slices from parkinsonian rats.
fig. S5. The distinct effects of the ERG inhibitor dofetilide on a pair of simultaneously recorded neurons, with one firing in the spontaneous tonic mode and the other in the burst mode.
fig. S6. The effect of ERG channel inhibitor and activator on burst discharges in rat STN slices.
fig. S7. The effect of ERG channel inhibitor and activator on the spontaneous firing activity of subthalamic neurons at low and high temperatures.
fig. S8. Continuous recording of spontaneous firing activity in acute STN slices before, during, and after application of 5 μM E-4031 (A) or 0.5 μM PD-118057 (B).
fig. S9. The effect of ERG channel inhibitor and activator on the plateau depolarization in the presence of tetrodotoxin.
fig. S10. The effect of ERG channel modulation on rearing scores and the asymmetric limb use.
fig. S11. Inhibition of ERG channels ameliorates abnormal burst discharges in parkinsonian rats.
Methods for figs. S10 (E and F) and S11
REFERENCES AND NOTES
- 1.DeLong M. R., Wichmann T., Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 64, 20–24 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Galvan A., Devergnas A., Wichmann T., Alterations in neuronal activity in basal ganglia-thalamocortical circuits in the parkinsonian state. Front. Neuroanat. 9, 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magill P. J., Bolam J. P., Bevan M. D., Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus–globus pallidus network. Neuroscience 106, 313–330 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Wichmann T., Dostrovsky J. O., Pathological basal ganglia activity in movement disorders. Neuroscience 198, 232–244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollerman J. R., Grace A. A., Subthalamic nucleus cell firing in the 6-OHDA-treated rat: Basal activity and response to haloperidol. Brain Res. 590, 291–299 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Hutchison W. D., Allan R. J., Opitz H., Levy R., Dostrovsky J. O., Lang A. E., Lozano A. M., Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson’s disease. Ann. Neurol. 44, 622–628 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Vila M., Perier C., Feger J., Yelnik J., Faucheux B., Ruberg M., Raisman-Vozari R., Agid Y., Hirsch E. C., Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur. J. Neurosci. 12, 337–344 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Benabid A. L., Chabardes S., Mitrofanis J., Pollak P., Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 8, 67–81 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Limousin P., Krack P., Pollak P., Benazzouz A., Ardouin C., Hoffmann D., Benabid A.-L., Electrical stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N. Engl. J. Med. 339, 1105–1111 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Tai C.-H., Yang Y.-C., Pan M.-K., Huang C.-S., Kuo C.-C., Modulation of subthalamic T-type Ca2+ channels remedies locomotor deficits in a rat model of Parkinson disease. J. Clin. Invest. 121, 3289–3305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tai C.-H., Pan M.-K., Lin J. J., Huang C.-S., Yang Y.-C., Kuo C.-C., Subthalamic discharges as a causal determinant of parkinsonian motor deficits. Ann. Neurol. 72, 464–476 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Castrioto A., Lhommée E., Moro E.. Krack P., Mood and behavioural effects of subthalamic stimulation in Parkinson’s disease. Lancet Neurol. 13, 287–305 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Castrioto A., Thobois S., Carnicella S., Maillet A., Krack P., Emotional manifestations of PD: Neurobiological basis. Mov. Disord. 31, 1103–1113 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Damier P., Why do Parkinson’s disease patients sometimes make wrong decisions? J. Parkinsons Dis. 5, 637–642 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Piallat B., Polosan M., Fraix V., Goetz L., David O., Fenoy A., Torres N., Quesada J.-L., Seigneuret E., Pollak P., Krack P., Bougerol T., Benabid A. L., Chabardès S., Subthalamic neuronal firing in obsessive-compulsive disorder and Parkinson disease. Ann. Neurol. 69, 793–802 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Yang Y.-C., Tai C.-H., Pan M.-K., Kuo C.-C., The T-type calcium channel as a new therapeutic target for Parkinson’s disease. Pflugers Arch. 466, 747–755 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Sanguinetti M. C., Jiang C., Curran M. E., Keating M. T., A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Trudeau M. C., Warmke J. W., Ganetzky B., Robertson G. A., HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269, 92–95 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., Hill A. P., hERG K+ channels: Structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Curran M. E., Splawski I., Timothy K. W., Vincen G. M., Green E. D., Keating M. T., A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Sanguinetti M. C., HERG1 channel agonists and cardiac arrhythmia. Curr. Opin. Pharmacol. 15, 22–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanguinetti M. C., Tristani-Firouzi M., hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Smith P. L., Baukrowitz T., Yellen G., The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379, 833–836 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Guasti L., Cilia E., Crociani O., Hofmann G., Polvani S., Becchetti A., Wanke E., Tempia F., Arcangeli A., Expression pattern of the ether-a-go-go-related (ERG) family proteins in the adult mouse central nervous system: Evidence for coassembly of different subunits. J. Comp. Neurol. 491, 157–174 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Papa M., Boscia F., Canitano A., Castaldo P., Sellitti S., Annunziato L., Taglialatela M., Expression pattern of the ether-a-gogo-related (ERG) k+ channel-encoding genes ERG1, ERG2, and ERG3 in the adult rat central nervous system. J. Comp. Neurol. 466, 119–135 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Eggermont J. J., Is there a neural code? Neurosci. Biobehav. Rev. 22, 355–370 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Kavanau J. L., Memory, sleep and the evolution of mechanisms of synaptic efficacy maintenance. Neuroscience 79, 7–44 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Lisman J. E., Bursts as a unit of neural information: Making unreliable synapses reliable. Trends Neurosci. 20, 38–43 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Gale J. T., Shields D. C., Jain F. A., Amirnovin R., Eskandar E. N., Subthalamic nucleus discharge patterns during movement in the normal monkey and Parkinsonian patient. Brain Res. 1260, 15–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magnin M., Morel A., Jeanmonod D., Single-unit analysis of the pallidum, thalamus and subthalamic nucleus in parkinsonian patients. Neuroscience 96, 549–564 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Steigerwald F., Pötter M., Herzog J., Pinsker M., Kopper F., Mehdorn H., Deuschl G., Volkmann J., Neuronal activity of the human subthalamic nucleus in the parkinsonian and nonparkinsonian state. J. Neurophysiol. 100, 2515–2524 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Wichmann T., Soares J., Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J. Neurophysiol. 95, 2120–2133 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Furlan F., Taccola G., Grandolfo M., Guasti L., Arcangeli A., Nistri A., Ballerini L., ERG conductance expression modulates the excitability of ventral horn GABAergic interneurons that control rhythmic oscillations in the developing mouse spinal cord. J. Neurosci. 27, 919–928 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagendorf S., Fluegge D., Engelhardt C., Spehr M., Homeostatic control of sensory output in basal vomeronasal neurons: Activity-dependent expression of ether-à-go-go-related gene potassium channels. J. Neurosci. 29, 206–221 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji H., Tucker K. R., Putzier I., Huertas M. A., Horn J. P., Canavier C. C., Levitan E. S., Shepard P. D., Functional characterization of ether-à-go-go-related gene potassium channels in midbrain dopamine neurons - implications for a role in depolarization block. Eur. J. Neurosci. 36, 2906–2916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pessia M., Servettini I., Panichi R., Guasti L., Grassi S., Arcangeli A., Wanke E., Pettorossi V. E., ERG voltage-gated K+ channels regulate excitability and discharge dynamics of the medial vestibular nucleus neurones. J. Physiol. 586, 4877–4890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sacco T., Bruno A., Wanke E., Tempia F., Functional roles of an ERG current isolated in cerebellar Purkinje neurons. J. Neurophysiol. 90, 1817–1828 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Ficker E., Jarolimek W., Kiehn J., Baumann A., Brown A. M., Molecular determinants of dofetilide block of HERG K+ channels. Circ. Res. 82, 386–395 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Kamiya K., Niwa R., Mitcheson J. S., Sanguinetti M. C., Molecular determinants of hERG channel block. Mol. Pharmacol. 69, 1709–1716 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Huffaker S. J., Chen J., Nicodemus K. K., Sambataro F., Yang F., Mattay V., Lipska B. K., Hyde T. M., Song J., Rujescu D., Giegling I., Mayilyan K., Proust M. J., Soghoyan A., Caforio G., Callicott J. H., Bertolino A., Meyer-Lindenberg A., Chang J., Ji Y., Egan M. F., Goldberg T. E., Kleinman J. E., Lu B., Weinberger D. R., A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat. Med. 15, 509–518 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbott G. W., Sesti F., Splawski I., Buck M. E., Lehmann M. H., Timothy K. W., Keating M. T., Goldstein S. A. N., MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Einarsen K., Calloe K., Grunnet M., Olesen S.-P., Schmitt N., Functional properties of human neuronal Kv11 channels. Pflugers Arch. 458, 689–700 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Restano-Cassulini R., Korolkova Y. V., Diochot S., Gurrola G., Guasti L., Possani L. D., Lazdunski M., Grishin E. V., Arcangeli A., Wanke E., Species diversity and peptide toxins blocking selectivity of ether-à-go-go-related gene subfamily K+ channels in the central nervous system. Mol. Pharmacol. 69, 1673–1683 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Sanguinetti M. C., Jurkiewicz N. K., Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96, 195–215 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sale H., Wang J., O’Hara T. J., Tester D. J., Phartiyal P., He J.-Q., Rudy Y., Ackerman M. J., Robertson G. A., Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ. Res. 103, e81–e95 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weerapura M., Hébert T. E., Nattel S., Dofetilide block involves interactions with open and inactivated states of HERG channels. Pflugers Arch. 443, 520–531 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Wu W., Sachse F. B., Gardner A., Sanguinetti M. C., Stoichiometry of altered hERG1 channel gating by small molecule activators. J. Gen. Physiol. 143, 499–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perry M., Sachse F. B., Abbruzzese J., Sanguinetti M. C., PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc. Natl. Acad. Sci. U.S.A. 106, 20075–20080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou J., Augelli-Szafran C. E., Bradley J. A., Chen X., Koci B. J., Volberg W. A., Sun Z., Cordes J. S., Novel potent human ether-à-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol. Pharmacol. 68, 876–884 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Mao H., Lu X., Karush J. M., Huang X., Yang X., Ba Y., Wang Y., Liu N., Zhou J., Lian J., Pharmacologic approach to defective protein trafficking in the E637K-hERG mutant with PD-118057 and thapsigargin. PLOS ONE 8, e65481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stork D., Timin E. N., Berjukow S., Huber C., Hohaus A., Auer M., Hering S., State dependent dissociation of HERG channel inhibitors. Br. J. Pharmacol. 151, 1368–1376 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herzberg I. M., Trudeau M. C., Robertson G. A., Transfer of rapid inactivation and sensitivity to the class III antiarrhythmic drug E-4031 from HERG to M-eag channels. J. Physiol. 511, 3–14 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishii K., Kondo K., Takahashi M., Kimura M., Endoh M., An amino acid residue whose change by mutation affects drug binding to the HERG channel. FEBS Lett. 506, 191–195 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Lees-Miller J. P., Duan Y., Teng G. Q., Duff H. J., Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: Involvement of S6 sites. Mol. Pharmacol. 57, 367–374 (2000). [PubMed] [Google Scholar]
- 55.Mitcheson J. S., Chen J., Lin M., Culberson C., Sanguinetti M. C., A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 97, 12329–12333 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beurrier C., Congar P., Bioulac B., Hammond C., Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J. Neurosci. 19, 599–609 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suessbrich H., Waldegger S., Lang F., Busch A. E., Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 385, 77–80 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Lobb C. J., Abnormal bursting as a pathophysiological mechanism in Parkinson’s disease. Basal Ganglia 3, 187–195 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kirik D., Rosenblad C., Björklund A., Mandel R. J., Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson’s model: Intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J. Neurosci. 20, 4686–4700 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schallert T., Fleming S. M., Leasure J. L., Tillerson J. L., Bland S. T., CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39, 777–787 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Woodlee M. T., Kane J. R., Chang J., Cormack L. K., Schallert T., Enhanced function in the good forelimb of hemi-parkinson rats: Compensatory adaptation for contralateral postural instability? Exp. Neurol. 211, 511–517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi W., Wymore R. S., Wang H.-S., Pan Z., Cohen I. S., McKinnon D., Dixon J. E., Identification of two nervous system-specific members of the erg potassium channel gene family. J. Neurosci. 17, 9423–9432 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shepard P. D., Canavier C. C., Levitan E. S., Ether-a-go-go-related gene potassium channels: What’s all the buzz about? Schizophr. Bull. 33, 1263–1269 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiesa N., Rosati B., Arcangeli A., Olivotto M., Wanke E., A novel role for HERG K+ channels: Spike-frequency adaptation. J. Physiol. (Lond.) 501, 313–318 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Apud J. A., Zhang F., Decot H., Bigos K. L., Weinberger D. R., Genetic variation in KCNH2 associated with expression in the brain of a unique hERG isoform modulates treatment response in patients with schizophrenia. Am. J. Psychiatry 169, 725–734 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Calcaterra N. E., Hoeppner D. J., Wei H., Jaffe A. E., Maher B. J., Barrow J. C., Schizophrenia-associated hERG channel Kv11.1-3.1 exhibits a unique trafficking deficit that is rescued through proteasome inhibition for high throughput screening. Sci. Rep. 6, 19976 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Canavier C. C., Oprisan S. A., Callaway J. C., Ji H., Shepard P. D., Computational model predicts a role for ERG current in repolarizing plateau potentials in dopamine neurons:Implications for modulation of neuronal activity. J. Neurophysiol. 98, 3006–3022 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Nedergaard S., A Ca2+-independent slow afterhyperpolarization in substantia nigra compacta neurons. Neuroscience 125, 841–852 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Grace A. A., Bunney B. S., Moore H., Todd C. L., Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 20, 31–37 (1997). [DOI] [PubMed] [Google Scholar]
- 70.Warmke J. W., Ganetzky B., A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 91, 3438–3442 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grunnet M., Hansen R. S., Olesen S.-P., hERG1 channel activators: A new anti-arrhythmic principle. Prog. Biophys. Mol. Biol. 98, 347–362 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Clark R. B., Mangoni M. E., Lueger A., Couette B., Nargeot J., Giles W. R., A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 286, H1757–H1766 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Perez-Reyes E., Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 83, 117–161 (2003). [DOI] [PubMed] [Google Scholar]
- 74.Shen K. Z., Johnson S. W., Ca2+ Influx through NMDA-gated channels activates ATP-sensitive K+ currents through a nitric oxide–cGMP pathway in subthalamic neurons. J. Neurosci. 30, 1882–1893 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen K.-Z., Johnson S. W., Chronic dopamine depletion augments the functional expression of K-ATP channels in the rat subthalamic nucleus. Neurosci. Lett. 531, 104–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen K.-Z., Johnson S. W., Group I mGluRs evoke K-ATP current by intracellular Ca2+ mobilization in rat subthalamus neurons. J. Pharmacol. Exp. Ther. 345, 139–150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shen K.-Z., Yakhnitsa V., Munhall A. C., Johnson S. W., AMP kinase regulates K-ATP currents evoked by NMDA receptor stimulation in rat subthalamic nucleus neurons. Neuroscience 274, 138–152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huguenard J. R., McCormick D. A., Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 30, 350–356 (2007). [DOI] [PubMed] [Google Scholar]
- 79.McCormick D. A., Bal T., Sleep and arousal: Thalamocortical mechanisms. Annu. Rev. Neurosci. 20, 185–215 (1997). [DOI] [PubMed] [Google Scholar]
- 80.McCormick D. A., Contreras D., On the cellular and network bases of epileptic seizures. Annu. Rev. Physiol. 63, 815–846 (2001). [DOI] [PubMed] [Google Scholar]
- 81.Bevan M. D., Magill P. J., Terman D., Bolam J. P., Wilson C. J., Move to the rhythm: Oscillations in the subthalamic nucleus–external globus pallidus network. Trends Neurosci. 25, 525–531 (2002). [DOI] [PubMed] [Google Scholar]
- 82.Nambu A., Tachibana Y., Mechanism of parkinsonian neuronal oscillations in the primate basal ganglia: Some considerations based on our recent work. Front. Syst. Neurosci. 8, 74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ardouin C., Chéreau I., Llorca P.-M., Lhommée E., Durif F., Pollak P., Krack P. Groupe Évaluation Comportementale de la Maladie de Parkinson , Assessment of hyper- and hypodopaminergic behaviors in Parkinson’s disease. Rev. Neurol. 165, 845–856 (2009). [DOI] [PubMed] [Google Scholar]
- 84.Lhommée E., Klinger H., Thobois S., Schmitt E., Ardouin C., Bichon A., Kistner A., Fraix V., Xie J., Aya Kombo M., Chabardès S., Seigneuret E., Benabid A.-L., Mertens P., Polo G., Carnicella S., Quesada J.-L., Bosson J.-L., Broussolle E., Pollak P., Krack P., Subthalamic stimulation in Parkinson’s disease: Restoring the balance of motivated behaviours. Brain 135, 1463–1477 (2012). [DOI] [PubMed] [Google Scholar]
- 85.Haynes W. I. A., Haber S. N., The organization of prefrontal-subthalamic inputs in primates provides an anatomical substrate for both functional specificity and integration: Implications for Basal Ganglia models and deep brain stimulation. J. Neurosci. 33, 4804–4814 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mathai A., Smith Y., The corticostriatal and corticosubthalamic pathways: Two entries, one target. So what? Front Syst. Neurosci. 5, 64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cavanagh J. F., Wiecki T. V., Cohen M. X., Figueroa C. M., Samanta J., Sherman S. J., Frank M. J., Subthalamic nucleus stimulation reverses mediofrontal influence over decision threshold. Nat. Neurosci. 14, 1462–1467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mallet L., Polosan M., Jaafari N., Baup N., Welter M.-L., Fontaine D., du Montcel S. T., Yelnik J., Chéreau I., Arbus C., Raoul S., Aouizerate B., Damier P., Chabardès S., Czernecki V., Ardouin C., Krebs M.-O., Bardinet E., Chaynes P., Burbaud P., Cornu P., Derost P., Bougerol T., Bataille B., Mattei V., Dormont D., Devaux B., Vérin M., Houeto J.-L., Pollak P., Benabid A.-L., Agid Y., Krack P., Millet B., Pelissolo A. STOC Study Group , Subthalamic nucleus stimulation in severe obsessive–compulsive disorder. N. Engl. J. Med. 359, 2121–2134 (2008). [DOI] [PubMed] [Google Scholar]
- 89.McCormick D. A., McGinley M. J., Salkoff D. B., Brain state dependent activity in the cortex and thalamus. Curr. Opin. Neurobiol. 31, 133–140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Agid Y., Parkinson’s disease: Pathophysiology. Lancet 337, 1321–1324 (1991). [DOI] [PubMed] [Google Scholar]
- 91.Ehringer H., Hornykiewicz O., Verteilung von noradrenalin und dopamin (3-hydroxytyramin) im gehirn des menschen und ihr verhalten bei erkrankungen des extrapyramidalen systems. Klin. Wochenschr. 38, 1236–1239 (1960). [DOI] [PubMed] [Google Scholar]
- 92.Hornykiewicz O., Basic research on dopamine in Parkinson’s disease and the discovery of the nigrostriatal dopamine pathway: The view of an eyewitness. Neurodegener. Dis. 5, 114–117 (2008). [DOI] [PubMed] [Google Scholar]
- 93.McNeill A., Wu R.-M., Tzen K.-Y., Aguiar P. C., Arbelo J. M., Barone P., Bhatia K., Barsottini O., Bonifati V., Bostantjopoulou S., Bressan R., Cossu G., Cortelli P., Felicio A., Ferraz H. B., Herrera J., Houlden H., Hoexter M., Isla C., Lees A., Lorenzo-Betancor O., Mencacci N. E., Pastor P., Pappata S., Pellecchia M. T., Silveria-Moriyama L., Varrone A., Foltynie T., Schapira A. H. V., Dopaminergic neuronal imaging in genetic Parkinson’s disease: Insights into pathogenesis. PLOS ONE 8, e69190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodriguez-Oroz M. C., Jahanshahi M., Krack P., Litvan I., Macias R., Bezard E., Obeso J. A., Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. Lancet Neurol. 8, 1128–1139 (2009). [DOI] [PubMed] [Google Scholar]
- 95.Baufreton J., Garret M., Rivera A., de la Calle A., Gonon F., Dufy B., Bioulac B., Taupignon A., D5 (not D1) dopamine receptors potentiate burst-firing in neurons of the subthalamic nucleus by modulating an L-type calcium conductance. J. Neurosci. 23, 816–825 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baufreton J., Bevan M. D., D2-like dopamine receptor-mediated modulation of activity-dependent plasticity at GABAergic synapses in the subthalamic nucleus. J. Physiol. 586, 2121–2142 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cragg S. J., Baufreton J., Xue Y., Bolam J. P., Bevan M. D., Synaptic release of dopamine in the subthalamic nucleus. Eur. J. Neurosci. 20, 1788–1802 (2004). [DOI] [PubMed] [Google Scholar]
- 98.Ramanathan S., Tkatch T., Atherton J. F., Wilson C. J., Bevan M. D., D2-like dopamine receptors modulate SKCa channel function in subthalamic nucleus neurons through inhibition of Cav2.2 channels. J. Neurophysiol. 99, 442–459 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Zhu Z.-T., Shen K.-Z., Johnson S. W., Pharmacological identification of inward current evoked by dopamine in rat subthalamic neurons in vitro. Neuropharmacology 42, 772–781 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Taylor H., Schmiedt J. T., Çarçak N., Onat F., Di Giovanni G., Lambert R., Leresche N., Crunelli V., David F., Investigating local and long-range neuronal network dynamics by simultaneous optogenetics, reverse microdialysis and silicon probe recordings in vivo. J. Neurosci. Methods 235, 83–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Castro-Alamancos M. A., Gulati T., Neuromodulators produce distinct activated states in neocortex. J. Neurosci. 34, 12353–12367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602272/DC1
fig. S1. The original currents of Fig. 1.
fig. S2. Characterization of ERG K+ currents by channel activator PD-118057 in acutely dissociated subthalamic neurons following similar experimental protocols and analyses of Fig. 1.
fig. S3. The effect of PD-118057 on E-4031–sensitive currents.
fig. S4. The effect of the ERG inhibitor and activator on spontaneous firing of subthalamic neurons in acute STN slices from parkinsonian rats.
fig. S5. The distinct effects of the ERG inhibitor dofetilide on a pair of simultaneously recorded neurons, with one firing in the spontaneous tonic mode and the other in the burst mode.
fig. S6. The effect of ERG channel inhibitor and activator on burst discharges in rat STN slices.
fig. S7. The effect of ERG channel inhibitor and activator on the spontaneous firing activity of subthalamic neurons at low and high temperatures.
fig. S8. Continuous recording of spontaneous firing activity in acute STN slices before, during, and after application of 5 μM E-4031 (A) or 0.5 μM PD-118057 (B).
fig. S9. The effect of ERG channel inhibitor and activator on the plateau depolarization in the presence of tetrodotoxin.
fig. S10. The effect of ERG channel modulation on rearing scores and the asymmetric limb use.
fig. S11. Inhibition of ERG channels ameliorates abnormal burst discharges in parkinsonian rats.
Methods for figs. S10 (E and F) and S11