

Summary
Taxanes are a mainstay of treatment for breast cancer, but resistance often develops followed by metastatic disease and mortality. Aiming to reveal the mechanisms underlying taxane resistance, we used breast cancer patient-derived orthoxenografts (PDX). Mimicking clinical behavior, triple-negative breast tumors (TNBCs) from PDX models were more sensitive to docetaxel than luminal tumors, but they progressively acquired resistance upon continuous drug administration. Mechanistically, we found that a CD49f+ chemoresistant population with tumor-initiating ability is present in sensitive tumors and expands during the acquisition of drug resistance. In the absence of the drug, the resistant CD49f+ population shrinks and taxane sensitivity is restored. We describe a transcriptional signature of resistance, predictive of recurrent disease after chemotherapy in TNBC. Together, these findings identify a CD49f+ population enriched in tumor-initiating ability and chemoresistance properties and evidence a drug holiday effect on the acquired resistance to docetaxel in triple-negative breast cancer.
Keywords: TNBC, triple-negative breast cancer, PDX, patient-derived orthoxenografts, chemoresistance, docetaxel, CD49f tumor-initiating cells, drug holidays
Graphical Abstract
Highlights
-
•
PDX models mimic the clinical response to docetaxel in breast cancer patients
-
•
Sensitivity to docetaxel can be regained in metastatic resistant TNBC
-
•
A tumor-initiating CD49f chemoresistant population is present in TNBC
-
•
Docetaxel resistance associates with the expansion of a CD49f+ population in TNBC
To study docetaxel resistance in breast cancer, González-Suárez and colleagues utilize multiple PDX models including sensitive-resistant tumor pairs. The characterization of the tumor-initiating CD49f+ chemoresistant population that expands after acquisition of docetaxel resistance reveals potential biomarkers and novel targets for the treatment of resistant metastatic TNBC disease.
Introduction
Triple-negative breast cancer (TNBC) is a heterogeneous disease with divergent profiles of chemosensitivity and prognosis (Perou et al., 2000, Prat et al., 2010, Shah et al., 2012, Yu et al., 2013). Standard chemotherapy with anthracyclines and taxanes is the mainstay treatment. A subset of TNBCs shows increased chemosensitivity compared with other breast cancer subtypes; however, for a significant number of patients, overall prognosis is poorer, with high risk of early relapse. Once metastases appear the patient median survival is drastically reduced (Andre and Zielinski, 2012). Despite enormous efforts, the cause of resistance to chemotherapy agents, including taxanes, is unclear (Bonnefoi et al., 2011). There remains an urgent unmet need to identify the population of patients that will benefit from taxanes, on one hand, and to determine the mechanisms of resistance, on the other.
There is increasing evidence that in a variety of neoplasia, including breast cancer, only a subset of cancer cells are capable of reconstituting the tumor after transplantation. These cells called cancer stem cells (CSCs) or tumor-initiating cells (TICs), have the ability to self-renew and regenerate tumor heterogeneity (Al-Hajj et al., 2003) and show intrinsic resistance to conventional chemotherapies, leading to recurrence or metastasis. In fact, breast tumors from patients who received neoadjuvant chemotherapy are substantially enriched for CSCs compared with tumors of untreated patients (Yu et al., 2007), suggesting that anti-cancer agents kill the bulk of tumor cells, but spare the CSCs (Dean et al., 2005). In breast cancer, a variety of markers (CD44, CD24, EpCAM, CD49f, CD133/2, CD10, and ALDH activity) have been shown to identify CSCs (Al-Hajj et al., 2003, Bachelard-Cascales et al., 2008, Li et al., 2008, Lim et al., 2009, Stingl et al., 2006). However, it is still unclear whether all these markers are appropriate for the different breast cancer subtypes, and further studies are necessary to identify the population of TICs and their functionality in each type of tumors.
The lack of appropriate tools and models has hindered our efforts to gain insight into the mechanisms of drug resistance. The best approach to investigate acquired resistance requires analysis of primary or metastatic samples collected before and after recurrence, but these paired sensitive/resistant samples are often difficult to obtain. To advance our knowledge in clinical breast cancer and the molecular mechanisms of resistance, we have generated breast cancer patient-derived orthoxenografts (PDXs), which allow the amplification and perpetuation of human tumors by serial passages. Our panel of breast cancer PDXs recapitulates the heterogeneity of the clinical disease and constitutes a unique tool for studying the biological mechanisms of clinical response to taxanes and acquisition of resistance. We demonstrate that a CD49f+ cell population with tumor-initiating ability and increased resistance to taxanes is present in the initially sensitive TNBC tumors and expands during continued exposure to the drug in vivo, contributing to taxane resistance and tumor recurrence. Remarkably, the transcriptional differences observed between the CD49f+ population of sensitive and resistant tumors accompany and may contribute to the acquisition of chemoresistance. Finally, we demonstrate that docetaxel sensitivity is recovered in the absence of the drug and associates with changes in the CD49f+ population.
Results
PDX Models Resemble Human Tumors of Origin in Early Passages
PDX were generated as described (DeRose et al., 2011, Zhang et al., 2013; Table S1). Increased tumor rates and shorter latency to tumor formation was observed in samples derived from pleural effusions compared with tumor pieces. The TNBC engrafted better than luminal tumors and all palpable tumors derived from grade 3 human samples (Figures 1A, S1A, and S1B). Of the mammary glands, 52% with no palpable tumor contained human mammary epithelium, mostly normal ducts and grade 1 intraductal carcinoma indicating engraftment of these low-grade lesions (Figures S1C and S1D; Table S1). Tumor lines (Table 1, yellow in Table S1) were maintained by consecutive rounds of transplantation, and include two TNBC models derived from pleural effusions, the second one a BRCA1 mutant (IDB-01, IDB-02); two luminal/HER2-negative models (IDB-03 and IDB-04) derived from tumor pieces and pleural effusion, respectively; and one (IDB-05) derived from a tumor piece of a triple-positive (ER+ PR+ HER2+) breast cancer. In most models, shorter latency and faster tumor growth were observed in late passages (Figures 1B, 1C, S1E, and S1F). Thus, as demonstrated previously (DeRose et al., 2011, Dobrolecki et al., 2016, Zhang et al., 2013), establishment of PDX models was associated with increased tumor aggressiveness and poor prognosis.
Figure 1.

Generation and Characterization of PDX Models of Human Breast Cancer
(A) Percentage of palpable tumors that engrafted relative to total number of independent patient samples, classified by subtype and source. The total number of original patient samples is indicated and mice that did not survive for at least 60 days after surgery were excluded.
(B) Tumor latency in IDB-01 at the indicated passages. Total number of tumors (n), mean, SD, and t test p values are shown. ∗∗∗∗p < 0.0001.
(C) Tumor growth in IDB-01, calculated as L × I (mm × mm)/100 versus time (weeks). Each line represents a representative tumor.
(D) Unsupervised clustering using the PAM50 genes across the PDX models, human tumors of origin, and 1,834 human breast cancer clinical samples (Prat et al., 2015b). The type of sample and the subtype call of each sample are shown. Each square represents the relative transcript abundance. All PDX tumors were from passage 5.
See also Table S1; Figures S1 and S2.
Table 1.
Main Characteristics of Human Tumor of Origin and Mouse Grafts in Five Established IDB Models
| Model | IDB-01 (TNBC) | IDB-02 (TNBC) | IDB-03 (Luminal) | IDB-04 (Luminal) | IDB-05 (HER2+) | |
|---|---|---|---|---|---|---|
| Mouse | phenotype early passage | ER-PR-HER2-CK5/6+ CK18+ p53- |
ER-PR-HER2-CK5/6+ CK18+ p53- BRCA1 mut |
ER+ PR+ HER2-CK5/6-CK18+ p53+BRCA2 mut |
ER+ PR+ HER2-CK5/6-CK18+ p53- |
ER+ PR+ HER2+ CK5/6-CK18+ p53- |
| phenotype late passage | no change | no change | loss of ER and PR | no change | no change | |
| passage | 13 | 8 | 16 | 7 | 9 | |
| latency (p5) (days) | 19 | 42 | 18 | 63 | 27 | |
| growth without hormone pellets | yes | yes | yes | yes | no | |
| Local relapse (%) | 17.24 (n = 116) | 6.94 (n = 144) | 20.79 (n = 178) | 23.40 (n = 94) | 8.54 (n = 94) | |
| axillary metastasis (%) | 10 | 10 | 46 | 0 | 8 | |
| lymph ND metastasis (%) | 14 | 33 | 41.2 | 0 | 50 | |
| lung metastasis (%) | 20 | 20 | 10 | 0 | 20 | |
| metastasis to other sites | ND | Yes (brain, axillary, subcutaneous) | Yes (bone, kidney) | Yes (liver) | ND | |
| Human | subtype | TNBC grade 3 | TNBC grade 3 | luminal grade 3 | luminal grade 3 | HER2+ grade 3 |
| IHC | ER-PR-HER2-CK5/6+ CK18+ p53- |
ER-PR-HER2-CK5/6+ CK18+ p53- BRCA1mut |
ER+ PR+ HER2-CK5/6-CK18+ p53+ |
ER+ PR+ HER2-CK5/6-CK18+ p53- |
ER+ PR+ HER2+ CK5/6-CK18+ p53- |
|
| source | pleural | pleural | tumor pieces | pleural | tumor pieces | |
| treatment | FEC, docetaxel, capecitabine | FEC, docetaxel | not treated | paclitaxel, carboplatin, capecitabine | not treated |
ER, estrogen receptor; PR, progesterone receptor; CK, cytokeratin, FEC, triple treatment composed of 5-fluorouracil, epirubicin and cyclophosphamide.
Frequency of tumor relapse per mammary gland (local relapse) and metastasis is indicated in each model. Only mice that survived for at least 60 days after primary tumor excision with no relapse/metastasis were considered as relapse/metastasis free. All metastases were confirmed by pathologists. ND, not determined. See also Table S1.
Expression analyses of markers used in the clinical setting for histopathological tumor classification and selection of treatment (ER, PR, HER2, CK5/6, CK18, and p53), in parental human tumors and PDX tumors at early (0–1) and late passages (4–8) demonstrate that PDX retain most human characteristics in the early passages, but occasional changes are observed in some models (Table 1; Figures S1G and S2A). ER and PR mRNA and protein expression was detected in tumors from all passages of the luminal models IDB-04 and IDB-05 (Figures S2A and S1G), but only IDB-05 required estrogen/progesterone pellets to grow (Figure S1H). IDB-03, ER+ and PR+ in the patient, lost ER and PR expression in the PDX and a population of p53+ cells was enriched (Table 1; Figure S2A and S1G). After surgically resection of tumors, most models developed local relapses and metastases to clinical relevant sites (Table 1; Figure S2B).
Next, we performed intrinsic subtyping of our 5 PDX models and their corresponding human tumors of origin using the PAM50 subtype predictor (Parker et al., 2009), and clustered these samples with 1,834 breast tumor samples representing all subtypes (Prat et al., 2015b). Mimicking the intrinsic subtypes of their corresponding human tumors, the two TNBC models were identified as basal-like, IDB-04 (HR+/HER2–) as luminal B, and the HER2+ IDB-05 as HER2 enriched (HER2-E). Interestingly, the human tumor of origin for IDB-03 was identified as luminal B but the PDX was identified as HER2-E by PAM50 without HER2 overexpression (Figure 1D). As reported in similar PDX collections (Dobrolecki et al., 2016), our mouse grafts retain initial human tumor characteristics, but some models change during serial passages in mice, which may reflect evolution of the clinical disease.
Basal-like PDX Are Initially Sensitive to Docetaxel but Acquire Resistance after Continuous Exposure to the Drug In Vivo
Next, we tested the sensitivity of orthotopic mouse models to docetaxel, one of the most commonly used chemotherapeutics in breast cancer and other solid tumors (Figure S3A). According to docetaxel response, tumors were classified as sensitive when the treatment induced complete tumor regression; partially sensitive when the treatment interfered with tumor growth inducing complete regression in some tumors but not in others; and resistant when tumors continued growing despite docetaxel treatment. In line with these criteria, luminal tumors from IDB-03 and IDB-04 were resistant to docetaxel, the TNBC IDB-01 model was sensitive (IDB-01S), and the TNBC IDB-02 was partially sensitive to the drug (Figures 2A, 2B, and S3B). Despite the initial pathological complete response, all IDB-01 tumors started growing again 30–60 days after treatment interruption. In the second round of treatment, more doses of docetaxel were required to eliminate tumors and a more heterogeneous response between individual tumors was observed (partially sensitive tumors). This behavior was accentuated during consecutive docetaxel treatments and the tumors became resistant in passage 3 (Figures 2B and 2C). Resistance was retained for at least two passages in the absence of docetaxel, as IDB-01-resistant tumors (IDB-01R, passage 5) grew at comparable growth rates irrespective of docetaxel treatment (Figure 2D). Importantly, tumors growing without the selective pressure of docetaxel partially regained sensitivity after five passages (IDB-01R, passage 8), which demonstrates that taxane resistance can be reverted (Figure 2D). No differences in latency to tumor formation or tumor growth were observed between IDB-01S (sensitive tumors of origin) and the resistant ones, IDB-01R, derived from them (Figures S3C and S3D). Gene expression analyses of IDB-01S and IDB-01R tumors identified a signature (22 downregulated genes in IDB-01R) that was predictive of residual disease after anthracycline/taxane-based therapy in 166 patients with basal-like disease (GSE25066) (Hatzis et al., 2011) and poor survival in the TCGA dataset, highlighting the clinical relevance of our sensitive and resistant PDX pairs (Figures 2E, 2F, and S3E). In IDB-02 an already heterogeneous response was observed after the first doses, and docetaxel treatment could not be interrupted in most mice (Figures 2B, 2C, and S3B). Tumors started growing very fast after interruption of the treatment and became resistant in passage 2. A third passage and additional docetaxel treatments did not change tumor growth, demonstrating that tumors had acquired resistance to docetaxel, which was retained for at least two passages (Figures 2B–2D). IDB-02R resistant tumors showed similar latency as IDB-02S sensitive tumors but grew significantly faster (Figures S3C and S3D). These results demonstrate that our triple-negative PDX tumors are more sensitive to docetaxel than the luminal ones. In the clinic, a better response to chemotherapy is observed in TNBC compared with luminal tumors, and in some studies taxanes have been shown to be superior to anthracyclines in this subtype (Kim et al., 2010, Martin et al., 2011). Moreover, initially sensitive PDX tumors gradually became less responsive to docetaxel and acquired resistance after continuous exposure to the drug, mimicking the clinical scenario.
Figure 2.

TNBC PDX Tumors were Sensitive to Docetaxel and Acquired Resistance after Continuous Treatment, whereas the Luminal Tumors were Resistant
(A) Representative kinetics of tumor growth during docetaxel treatment. Docetaxel treatment (20 mg/kg i.p., once per week) started when tumors reached 6 × 6 mm. Each line illustrates a representative tumor (passages 4–14).
(B) Percentage of sensitive, partially sensitive or resistant tumors of each model to docetaxel. Total number of tumors (n) and passage are indicated.
(C) Representative kinetics of tumor growth during acquisition of resistance to docetaxel in the basal-like IDB-01 and IDB-02. Each line represents one tumor and each color represents an independent sensitive tumor of origin. Ps, passage treated with docetaxel. Red circles indicate the tumors that were transplanted.
(D) Representative kinetics of tumor growth during docetaxel treatment after acquisition of resistance to taxanes. Each line represents one tumor. IDB-01R tumors were analyzed after growing for two and five passages, respectively, in the absence of docetaxel.
(E) Supervised expression analysis of the genes found differentially expressed between IDB-01R and IDB-01S tumors. Each square represents the relative transcript abundance.
(F) Association of IDB-01 resistant signature with chemotherapy response in 166 patients with basal-like breast cancer (Hatzis et al., 2011). Response was measured as pathological complete response (pCR) or residual disease (RD). Mean values, box and whiskers (min to max) and t test p values are shown.
(A, C, and D) Arrows represent docetaxel doses. See also Figure S3.
Docetaxel Acquired Resistance Is Accompanied by an Increase in the CD49f+ Population
It has been shown that chemotherapy efficiently eliminates the bulk tumor cells but spares the CSC population (Li et al., 2008). Thus, we analyzed the expression of markers previously shown to identify CSCs in our PDX tumor collection including paired sensitive and resistant tumors from IDB-01 and IDB-02 (Figure S4A-B). Variability in marker expression was detected between models with the same histological and molecular subtype. Docetaxel-resistant luminal tumors (IDB-03 and IDB-04) showed the highest percentages of EpCAM, CD49f, and CD24 cells, but the CD133 population was scarce. IDB-03 contained an abundant CD44+ population and ALDH activity, and is the only one expressing CD10. A CD133+ population was found in basal-like and HER2+ PDX. The CD44+ CD24– population, shown to identify human breast CSCs (Al-Hajj et al., 2003), was only detected in the TNBC IDB-02 (Figures S4B and S4C).
No significant changes in the expression of CD44, CD24, CD133, or CD10 were found between sensitive and resistant TNBC paired samples, neither in IDB-01 nor in IDB-02. The CD44+ CD24– population remained barely detectable in the chemoresistant models, and the ALDH+ population, based on ALDH enzymatic activity, was also comparable between paired sensitive and resistant tumors (Figure 3A). In contrast, the frequency of CD49f+ cells significantly increased in TNBC-resistant tumors compared with paired sensitive ones in both models. A significant increase in the frequency of EpCAM+ cells was also observed in IDB-01R compared with IDB-01S tumors (Figure 3A). Resistant tumors from IDB-01 and IDB-02 showed significantly higher mRNA expression levels of CD49f (ITGA6) but not EpCAM, than the corresponding sensitive tumors (Figure 3B).
Figure 3.

The CD49f+ Population Is Enriched after Acquisition of Resistance to Docetaxel
(A) Frequency of indicated markers within the H2Kd– population in IDB-01 and IDB-02, sensitive and resistant tumors analyzed by flow cytometry at passage 3, at least 5 days after the last docetaxel treatment. Total number of tumors analyzed (n) mean values, SDs and t test p values are shown. ∗0.01 < p < 0.05; ∗∗0.001 < p < 0.01.
(B) Box and whiskers (min to max) graph showing expression levels of CD49f and EpCAM mRNA relative to PPiA in additional sensitive and resistant tumors measured by qRT-PCR. Determinations were done in triplicate and means are used. t test p values for significant differences are shown. ∗∗0.001 < p < 0.01.
(C) Box and whiskers (min to max) graph showing association of CD49f and EpCAM with chemotherapy response in 74 patients with basal-like breast cancer EORTC (Bonnefoi et al., 2011). Response was measured as pathological complete response (pCR) or residual disease (RD). t test p values are shown.
(D) Kaplan-Meier analysis of overall survival of ER-tumors all treated with chemotherapy using CD49f and EpCAM mRNA expression in the clinical dataset (GSE16446) from the TOP TRIAL (Desmedt et al., 2011).
See also Figures S3 and S4.
We next sought to investigate the clinical relevance of our findings by analyzing different clinical datasets. In basal-like tumors from the EORTC 10994/BIG-1-00 clinical trial (Bonnefoi et al., 2011), higher expression of CD49f and EpCAM was associated with a non-pathological complete response (non-pCR) after chemotherapy (Figure 3C). Using GOBO, the Gene expression-based Outcome for Breast cancer Online tool (Ringner et al., 2011), high expression levels of EpCAM and CD49f combined predicted a reduction in distal metastasis-free survival in basal-like tumors (Figure S3F). Associations with poor overall survival were obtained for CD49f, but not EpCAM, in other ER-negative or basal-like tumor samples after chemotherapy treatment (Clarke et al., 2013, Desmedt et al., 2011) (Figures 3D, S3G, and S3H). These results demonstrate that, whereas CD44+ CD24– and ALDH activity are not altered, the percentage of the CD49f+ population significantly increases during the acquisition of resistance to docetaxel in basal-like breast cancer.
A Chemoresistant CD49f+ Population Is Present in Most TNBC Tumors
We hypothesized that a chemoresistant CD49f+ population is present in the original sensitive tumors. To test this hypothesis we analyzed CD49f mRNA expression in IDB-01S and IDB-02S tumors after two to three doses of docetaxel treatment when tumors were shrinking, and found a significant increase in CD49f mRNA expression in the residual disease of both PDX tumors (Figure 4A). Next, we evaluated by flow cytometry the percentage of cells expressing CD49f in residual disease and found that the frequency of CD49f+ cells in residual disease of IDB-01S after docetaxel treatment increases by 20%; these levels are comparable with those of resistant IDB-01R tumors, indicating that the surviving population is enriched in CD49f+ cells (Figure 4B). Importantly, in IDB-01R tumors that regained sensitivity to taxanes after growing in the absence of docetaxel (passage 8), the frequency of the CD49f+ population decreases again to basal levels, similar to those found in sensitive tumors of origin (Figure 4B).
Figure 4.

CD49f Expression Increases in Residual Disease of Most TNBC PDX Tumors after Treatment with Docetaxel, but Not on Resistant Tumors
(A) Scheme of short-term docetaxel treatment and CD49f mRNA expression levels in sensitive tumors from IDB-01S and IDB-02S after short-term treatment with docetaxel (DTX) and in untreated controls (CT). Each dot represents one tumor. ∗0.01 < p < 0.05; ∗∗0.001 < p < 0.01.
(B) Frequency of CD49f+ cells within H2Kd– in IDB-01S tumors after two to three doses of docetaxel and in IDB-01R tumors that have been treated with docetaxel (at least 5 days after last treatment) or growing in the absence of docetaxel for two and five passages. ∗0.01 < p < 0.05; ∗∗0.001 < p < 0.01.
(C and D) Docetaxel-sensitive tumors (C) and docetaxel-resistant tumors (D). Top panels: tumor size of the indicated PDX tumors treated with docetaxel (20 mg/kg, arrows) and corresponding controls relative to the size at the first day of treatment. n = total number of tumors. ∗∗∗∗p < 0.0001. Bottom panels: CD49f mRNA expression levels in PDX tumors after short-term treatment with docetaxel and in untreated controls. Each dot represents one tumor. ∗0.01 < p < 0.05; ∗∗0.001 < p < 0.01; ∗∗∗0.001 < p < 0.0001.
(A–D) Mean values, SEM, and t test p values are shown in all cases.
See also Figure S5.
To evaluate whether a chemoresistant CD49f+ population could be found in other TNBC tumors, we analyzed CD49f expression after short-term in vivo treatment with docetaxel in 12 additional TNBC PDX tumors derived from patient samples (Bruna et al., 2016, DeRose et al., 2011). Four of these PDX tumors were resistant to docetaxel (no differences in tumor growth after docetaxel treatment), and eight showed different grades of sensitivity to the drug (tumors either shrank or showed tumor growth stabilization after two to four doses of docetaxel). After docetaxel treatment, an increase in CD49f mRNA expression levels was observed in residual disease of five out of the eight TNBC-sensitive tumors treated, whereas in resistant tumors CD49f expression remained unaltered (Figures 4C, 4D, and S5A). No changes in the expression of the most common partners of CD49f, CD29 (ITGB1) and CD104 (ITGB4), were observed between sensitive, resistant and residual disease in TNBC tumors (Figure S5B-D). The increase in CD49f expression in residual tumors suggests that CD49f+ chemoresistant cells are present in docetaxel-sensitive tumors and get enriched in residual disease.
In addition, we analyzed CD49f mRNA expression in five independent TNBC cell lines after 72 h of treatment with increasing concentrations of docetaxel. Different cell lines showed different grades of sensitivity to taxanes but, in four out of the five cell lines tested, a significant increase in CD49f mRNA expression was found in cells that survive docetaxel treatment compared with the untreated ones (Figure 5A). No changes in CD49f expression were observed at shorter time points with negligible cell death, suggesting that docetaxel does not induce CD49f expression and that the observed increase in residual disease, most probably represents the survival of a pre-existing CD49f+ population (Figure 5B). Higher levels of CD49f mRNA after paclitaxel treatment were also observed in some cell lines (Figure S5E). No changes in docetaxel sensitivity were observed in MDA-MB-436 cells upon stable reduction of CD49f expression with two independent short hairpin RNA constructs, ruling out a functional role for CD49f itself in chemoresistance of these cells (Figures 5C–5E and S5F).
Figure 5.

CD49f Expression Increases in Surviving TNBC Cells after Treatment with Docetaxel
(A and B) Top panels: percentage of surviving cells treated with docetaxel for 72 h (A) or 8 h (B). Bottom panels: CD49f mRNA expression levels in the indicated TNBC cell lines treated with docetaxel relative to untreated controls. ∗0.01 < p < 0.05; ∗∗0.001 < p < 0.01; ∗∗∗0.001 < p < 0.0001.
(C and D) CD49f mRNA expression levels (C) and CD49f protein expression measured by flow cytometry (D) in cells stably infected with two independent shCD49f knockdown constructs and control vector (pGIPZ).
(E) Percentage of surviving shCD49f-infected and control pGIPZ-infected cells treated with indicated doses of docetaxel for 72 hr.
RT-PCR Determinations were done in triplicate and means are used in the calculations. Mean values of three independent experiments, SEM, and t test p values for the higher concentrations are shown. See also Figure S5.
Together these results demonstrate that higher expression of CD49f was observed in residual disease after docetaxel treatment for most TNBC-sensitive models (seven out of ten PDX models and four out of five cell lines), suggesting that despite the heterogeneity of the TNBC subtype a chemoresistant CD49f+ population is present in most TNBC.
CD49f+/hi Cells Show Enhanced Tumor-Initiating Ability and Resistance to Docetaxel
Next, we asked whether chemoresistant CD49f+ cells showed a higher tumor-initiating potential than CD49f− cells and could be responsible for tumor recurrence. Using fluorescence-activated cell sorting (FACS), we sorted the higher and lower quartile of tumor cells based on CD49f expression from IDB-01S and IDB-02S tumors and functionally tested their tumor-initiating potential (Figure 6A). Indeed, only CD49f+/hi cells, but not the CD49f− in the IDB-01S model, were able to give rise to tumors when re-implanted in mice (Figure 6B). Tumors derived from the IDB-01S-CD49f+/hi cells contained a more abundant CD49f+ population, but also CD49f– cells demonstrating that the tumor-initiating CD49f+/hi cells were able to give rise to non-TICs CD49f− cells (Figure 6C). Docetaxel attenuated growth in tumors derived from IDB-01S-CD49f+/hi cells, but tumors were still palpable after ten doses of docetaxel, in contrast to sensitive tumors of origin IDB-01S that were not detectable after four doses (Figure 6D). Thus, IDB-01S-CD49f+/hi derived tumors are more resistant to docetaxel than the original IDB-01S tumors.
Figure 6.

CD49f+ Population Is Enriched in Tumor-Initiating Cells with Increased Resistance to Docetaxel
(A) Scheme of functional experiments.
(B and E) Table showing limiting dilution assay of CD49f+/hi and CD49f− tumor cells from IDB-01 (B) and IDB-02 (E) cells. Tumor-initiating cell frequency (with confidence intervals) for each group was calculated by ELDA; chi-square values and associated probabilities are shown.
(C and G) Frequency of CD49f+ cells in tumors derived from indicated cells. Mean values, SEM, and significant t test p values are shown. ∗∗0.001 < p < 0.01; ∗∗∗0.001 < p < 0.0001.
(D and H) Kinetics of tumor growth during docetaxel treatment in tumors derived from indicated cells. Mean values, SEM, and t test p values are shown. ∗∗0.001 < p < 0.01; ∗∗∗∗p <0.0001.
(F) Latency of tumors derived from the injection of the indicated number of IDB-02S-CD49f+/hi and 02S-CD49f− tumor cells. Mean values, SEM and significant t test p values are shown. ∗∗0.001 < p < 0.01.
(I) Unsupervised analysis of all CD49f sorted samples from IDB-01S and -01R tumors using 105 breast cancer-related genes. The type of sample and tumor are shown below the array tree. Each square represents the relative transcript abundance.
See also Figure S6.
In IDB-02, where tumors were partially sensitive to docetaxel and contained a higher proportion of CD49f+ cells, both CD49f+/hi and CD49f− cells gave rise to tumors. However, limiting dilution assays and extreme limiting dilution analyses (ELDA) revealed that the CD49f+/hi population showed a 5-fold increase in tumor-initiating ability compared with the CD49f− cells (Figure 6E). In addition, the CD49f+/hi cells gave rise to tumors with shorter latency than CD49f− cells (Figure 6F). CD49f+ cells were more abundant in CD49f+/hi than in tumors derived from CD49f−, but tumors from both groups contained CD49f+ and CD49f− cells (Figure 6G), demonstrating that CD49f− cells can also give rise to CD49f+ cells. Again, IDB-02S-CD49f−/lo-derived tumors were more sensitive to docetaxel than the ones derived from IDB-02S-CD49f+ cells (Figure 6H).
Unsupervised gene expression profiling of FACS-sorted CD49f+/hi and CD49f− cells from IDB-01S and -01R, using 105 breast cancer-selected genes, revealed two main clusters which broadly represents the CD49f+ and CD49f− populations (Figures 6I and S6A). Compared with CD49f− cells, CD49f+ cells showed downregulation of keratins, claudins and CDH3, and upregulation of SFRP1, MIA and proliferation-related genes (UBE2C, CDC6 and CDC20) (Figure 6I). Further gene expression analyses revealed significant transcriptome differences between CD49f+/hi cells from resistant and sensitive tumors, including enhanced decrease in tight junction proteins, claudins, and CDH3, which may suggest a more claudin-low phenotype (Prat et al., 2010). Downregulation of tumor suppressors (e.g., PTEN and RAB25) is also observed in CD49f+ cells from resistant tumors (Figure S6A). Interestingly, CD49f+/hi cells showed increased proliferation by gene expression analysis than CD49f− cells, especially within sensitive tumors (Figure S6B). Among the two CD49f+ signatures, the IDB-01R/CD49f+ signature was found to predict residual disease following anthracycline/taxane-based therapy in breast tumors (GSE25066), concordant with our preclinical observations (Figure S6C). On the other hand, the IDB-01S/CD49f+ signature was found to predict pathological complete response (pCR) following anthracycline/taxane-based therapy, likely due to the large difference in proliferation between CD49f+ and CD49f− cells in IDB-01S tumors (Figures S6B and S6C) (Hatzis et al., 2011). The IDB-01S/CD49f+ signature was associated with lower recurrence-free survival in an additional dataset of breast cancer patients (Prat et al., 2010) (Figure S6D). Together, these results demonstrate that sensitive tumors of origin contain a tumorigenic and docetaxel-resistant CD49f+ population that changes and expands during the acquisition of taxane resistance; whereas in the absence of the drug, the CD49f+ chemoresistant population shrinks and taxane sensitivity is restored.
Discussion
Patient-derived xenograft (PDX) models have emerged as an important intermediate tool between basic research and clinical trials to expedite the translation of basic research findings into effective therapies for patients. We have generated a panel of PDX models that recapitulates the heterogeneity of human breast tumors. Initial collections of breast PDX were reported to remain phenotypically identical to human tumors during serial passages (DeRose et al., 2011, Zhang et al., 2013). However, in agreement with our findings, there is increasing evidence that tumors in PDX are not “static” and can evolve, as observed in patients (Eirew et al., 2015).
Our PDX models constitute a unique tool to investigate resistance in cancer as they mimic clinical responses: TNBC tumors are more sensitive to chemotherapy than the luminal tumors, confirming previous clinical results (Berry et al., 2006, Colleoni et al., 2004, Guarneri et al., 2006, Martin et al., 2011), and even initially sensitive tumors develop resistance upon continuous exposure to taxanes. Both basal-like tumors (IDB-01 and IDB-02) derived from metastatic samples that were heavily exposed to multiple treatments including taxanes showed minimal clinical response. Strikingly, sensitivity to docetaxel was restored upon xenografting and was retained for months. Moreover, we observed that in PDX tumors with acquired resistance, sensitivity is partially restored when maintained in the absence of the drug. This regain of sensitivity, the so-called “drug holiday,” has been described for targeted therapies in melanoma (Das Thakur et al., 2013, Sun et al., 2014). We now demonstrate that the same is true for cytotoxics such as docetaxel, with important implications for clinical decisions and drug scheduling, as resistant metastatic disease may benefit from intermittent docetaxel treatment.
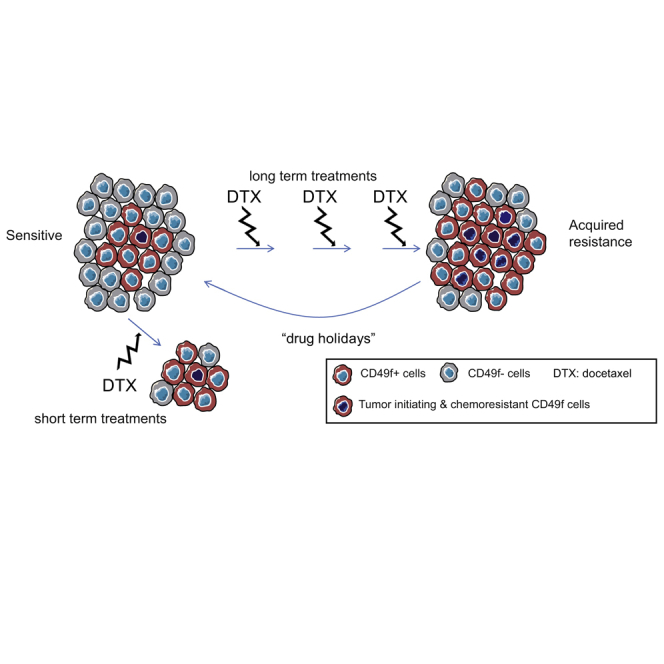
Our data demonstrate that a pre-existing and chemoresistant CD49f+ subpopulation is present in most sensitive TNBC, expands during long-term therapy, and has the ability to generate novel tumors contributing to recurrence and acquisition of chemoresistance (as shown in the graphical abstract), and importantly that this population shrinks again in the absence of taxanes, restoring drug sensitivity. Previous reports have also shown the increased tumor-initiating ability of CD49f+ cells in breast and other solid tumors (Haraguchi et al., 2013, Lo et al., 2012, Meyer et al., 2010, Vassilopoulos et al., 2014). These findings do not imply that the CD49f+ cells are the CSC in TNBC, but demonstrate that the CD49f+ population is associated with taxane resistance.
Aiming to further characterize the chemoresistant CD49f+ population, an unbiased approach was undertaken. Gene expression analysis revealed important differences, not only between CD49f+ and CD49f− cells, but also between CD49f+ cells from sensitive and resistant tumors. These changes may suggest that the chemoresistant CD49f+ population has expanded during the exposure to docetaxel, and can provide novel therapeutic targets for the metastatic chemoresistant basal-like tumors. Given the heterogeneity of the TNBC subtype, the significant increase in CD49f expression observed in residual or stabilized disease of most TNBC cell lines and PDX models is remarkable and indicates that modulation of CD49f positivity as a biomarker of taxane resistance is not a peculiarity of a single PDX model but a general event in TNBC, which can be exploited for clinical benefit.
These findings can be clinically validated in the neoadjuvant setting, evaluating whether an enrichment of the CD49f population is observed in residual disease following taxane-based chemotherapy. However, as the rates of pCR in TNBC are high (30%–40%), a dynamic study of early changes in the CD49f population after the first cycles of taxane treatment and occurrence of pCR could be a better approach. The clinical utility of the biomarker could be tested in a prospective clinical trial in the neoadjuvant setting where patients are randomized based on the biomarker modulation to change treatment or continue with taxane-based therapy. Improvement of clinical outcomes (pCR rates or survival) should be the final objective. The effect of novel drugs can be evaluated in the subgroup of chemoresistant CD49f-enriched TNBC. In clinical series the presence of CD49f+ in breast cancer is associated with a poor clinical outcome (Friedrichs et al., 1995, Ye et al., 2015). Moreover, within several CSC markers (CD44, CD24, ALDH1A3, and CD49f) analyzed by IHC in breast cancer samples, only CD49f retained prognostic value in a multivariate analyses in ER– disease (Ali et al., 2011). Our results provide a functional rationale for the poor outcome associated with CD49f expression in hormone receptor-negative breast cancer. Further studies will reveal whether this population can be manipulated in order to unveil the ever-elusive status of tumor drug resistance and recurrence.
Experimental Procedures
Patient Characteristics and Generation of PDX
IDB PDX were generated by orthotopic transplantation of primary tumor pieces obtained directly after surgery or cancer cells isolated from pleural effusions and transplanted into the fat pad of immunodeficient mice, as described previously (DeRose et al., 2011). The clinical characteristics from original patient samples, the number and strain of recipient mice, and the outcome of the implant are indicated in Table S1 (IDB-01-05 models). All experimental procedures were performed according to Spanish regulations. Informed consent was obtained from all subjects and the study received approval from the institutional Ethics Committee. Additional models were generated following similar procedures (Bruna et al., 2016). All research involving animals was performed at the IDIBELL animal facility in compliance with protocols approved by the IDIBELL Committee on Animal Care and following national and European Union regulations.
Breast Cancer Cell Isolation, Flow Cytometry, and Sorting
Single cells were isolated from tumors as described previously (Smalley, 2010). Single cells were resuspended and blocked with PBS 2% fetal bovine serum (FBS), 2 mM EDTA, and immunoglobulin G blocking reagent for 10 min on ice. Then they were labeled with antibodies against CD24-PE (555428), CD44-APC (559942), EpCAM-FITC (347197), CD10-PECy5 (555376), and CD49f-A647 (562473) (all from BD Pharmingen), CD133/1-PE (130-098-826 from Miltenyi Biotec), and CD49f-APC (FAB13501A from R&D Systems). Mouse cells were excluded in flow cytometry using H2Kd-PECy7 (116622 from BioLegend). Gating was based on “Fluorescence Minus One” controls. Single cells were assessed for their ALDH activity using the ALDEFLUOR assay system (01700 from STEMCELL Technologies) following the manufacturer's procedures. A population of 10,000 living cells was captured in all FACS experiments. FACS analysis and sorting was performed using Gallios and MoFlo (Beckman Coulter) flow cytometers, respectively. Data was analyzed using the FlowJo software (see Figure S4).
Therapeutic and Limiting Dilution Assays
Docetaxel (Hospira/Actavis, 20 mg/kg) was administered intraperitoneally once per week (unless reported otherwise), followed 24 hr later by Fortecortin (Dexametasona, 0.132 mg/kg, Merck). The treatment scheme of resistant variants generation is shown in Figure S3. For orthotopic ELDA, isolated tumor cells were mixed 1:1 with Matrigel Basement Membrane (BD Biosciences) and orthotopically implanted in the inguinal mammary gland of non-obese diabetic/severe combined immunodeficiency females. Tumor development was monitored once per week for a maximum of 25 weeks. In all assays the tumor-initiating potential was defined as the ability to form palpable, growing tumors of ≥2 mm diameter.
Culture and Treatment of Human Breast Cancer Cells
All cell lines were purchased from the American Type Culture Collection (Rockville, MD), except for UACC3199 which was obtained from the Arizona Cancer Center (Tucson, AZ). All cells but HCC1143, which was cultured in RPMI 1640, were maintained in DMEM high glucose, containing 10% FBS (Gibco), L-glutamate (Gibco), and penicillin/streptomycin (PAA Laboratories) at 37°C in 5% CO2. At 60%–70% confluence the indicated concentrations of docetaxel or paclitaxel were added. Cells were collected at the indicated time points and counted with trypan blue to exclude dead ones. All cell lines were routinely tested for mycoplasma, and were shown to be free of contamination.
Gene Expression-Based Analyses
A minimum of ∼100 ng of total RNA was used to measure the expression of 105 breast cancer-related genes and five house-keeping genes using the nCounter platform (Nanostring Technologies). Data was log base 2 transformed and normalized using five house-keeping genes (ACTB, MRPL19, PSMC4, RPLP0 and SF3A1). The list of 105 genes includes genes from the following three signatures: PAM50 intrinsic subtype predictor (n = 50) (Parker et al., 2009), claudin-low subtype predictor (n = 43) (Prat et al., 2010), 13-VEGF/hypoxia signature (n = 13) (Hu et al., 2009), and eight individual genes that have been found to play an important role in breast cancer (e.g., CD24). Raw gene expression data and signatures can be found in Table S2. All tumors were assigned to an intrinsic molecular subtype of breast cancer (luminal A, luminal B, HER2-enriched, basal-like, and claudin-low) and the normal-like group using the previously reported PAM50 subtype and the claudin-low subtype predictors (Parker et al., 2009, Prat et al., 2010, Prat et al., 2015b).
Gene Expression-Based Signatures
Genes differentially expressed between the two groups were identified using a two-class unpaired Significance Analysis of Microarrays (SAM) (Tusher et al., 2001) and a false discovery rate of <5%. The final signature of up- and/or downregulated genes was then summarized as a single “enrichment/activity score” by multiplying the SAM score of each gene by its expression value in the tested sample and then summing all the values of each sample. Each signature was evaluated in GSE25066, a microarray-based dataset of patients treated with neoadjuvant anthracycline/taxane-based chemotherapy (Hatzis et al., 2011) and the Perou-extended dataset GSE18229 (Prat et al., 2010). This microarray dataset was normalized as described previously (Prat et al., 2015a). Raw data can be found in Table S2.
Statistical Analyses
All data are expressed as mean ± SEM. Statistical comparison was performed by Student's t test using GraphPad Prism version 5.04. p ≤ 0.05 was considered statistically significant. The statistical significance of difference between groups is expressed by asterisks: ∗0.01 < p < 0.05; ∗∗0.001 < p < 0.01; ∗∗∗0.001 < p < 0.0001; ∗∗∗∗p < 0.0001.
Author Contributions
J.G.M., M.P., and L.P., collection and assembly of data, data analysis and interpretation, writing, and final approval of manuscript; J.G.M., identified the enrichment in the CD49f+ population and performed functional assays and analyses of residual disease; M.P., generated the PDX models and docetaxel-resistant variants; I.F., S.V., P.P., H.P.M., A.I., G.Y., P.G., A.U., T.S.M., J.C., I.M., M.E., A.N., V.S., A.P., and S.P., collection and assembly of data and final approval of manuscript; P.M., data analysis and interpretation and final approval of manuscript; A.P. and E.G.S., conception and design, financial support, collection and assembly of data, data analysis and interpretation, writing, and final approval of manuscript.
Acknowledgments
We thank A. Villanueva, C. Saura, C. Cruz, A. Fernández, E. Nadal, M. Martin, and R. Iggo for reagents and helpful advice, A. Welm and Y. DeRose for sharing PDX models, V. Peg, X. Serres, J. Balmaña, J. Pérez, and C. Hierro for providing samples, S. Hernández-Ortega, R. Gil, L. Barberá, and G. Boigues, the Pathology Department of the University Hospital of Bellvitge, the IDIBELL animal facility, histology service and UB-SCT, for technical support, and members of the laboratory for useful discussions and reading of the manuscript. This work was supported by grants to E. González Suárez by the Spanish Ministry of Economy and Competitivity MINECO and from the Health Institute Carlos III (ISCIII) SAF2008-01975, SAF2011-22893, SAF2014-55997, PIE13/00022, co-funded by FEDER funds/European Regional Development Fund (ERDF – a way to build Europe), by a Career Catalyst Grant from the Susan G Komen Foundation CCR13262449 and by funds to A. Prat from ISCIII-PI13/01718, by a Career Catalyst Grant from the Susan G. Komen Foundation, by Banco Bilbao Vizcaya Argentaria (BBVA) Foundation, and by a Sociedad Española de Oncología Médica (SEOM) grant. The PDX from VHIO were supported by a “GHD-pink” research support via the FERO Foundation to V. Serra. V.S. is recipient of ISCIII grants (PI13/01714 and CP14/0028). M.P., J.G.M., and P.P. were recipients of FPU/FPI grants from the MINECO.
Published: April 27, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.03.026.
Supplemental Information
References
- Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H.R., Dawson S.J., Blows F.M., Provenzano E., Pharoah P.D., Caldas C. Cancer stem cell markers in breast cancer: pathological, clinical and prognostic significance. Breast Cancer Res. 2011;13:R118. doi: 10.1186/bcr3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre F., Zielinski C.C. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann. Oncol. 2012;23:vi46–51. doi: 10.1093/annonc/mds195. [DOI] [PubMed] [Google Scholar]
- Bachelard-Cascales E., Chapellier M., Delay E., Pochon G., Voeltzel T., Puisieux A., Caron de Fromentel C., Maguer-Satta V. The CD10 enzyme is a key player to identify and regulate human mammary stem cells. Stem Cells. 2008;28:1081–1088. doi: 10.1002/stem.435. [DOI] [PubMed] [Google Scholar]
- Berry D.A., Cirrincione C., Henderson I.C., Citron M.L., Budman D.R., Goldstein L.J., Martino S., Perez E.A., Muss H.B., Norton L. Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA. 2006;295:1658–1667. doi: 10.1001/jama.295.14.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefoi H., Piccart M., Bogaerts J., Mauriac L., Fumoleau P., Brain E., Petit T., Rouanet P., Jassem J., Blot E. TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1-00): a randomised phase 3 trial. Lancet Oncol. 2011;12:527–539. doi: 10.1016/S1470-2045(11)70094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A., Rueda O.M., Greenwood W., Batra A.S., Callari M., Batra R.N., Pogrebniak K., Sandoval J., Cassidy J.W., Tufegdzic-Vidakovic A. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167:260–274.e22. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke C., Madden S.F., Doolan P., Aherne S.T., Joyce H., O'Driscoll L., Gallagher W.M., Hennessy B.T., Moriarty M., Crown J. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013;34:2300–2308. doi: 10.1093/carcin/bgt208. [DOI] [PubMed] [Google Scholar]
- Colleoni M., Viale G., Zahrieh D., Pruneri G., Gentilini O., Veronesi P., Gelber R.D., Curigliano G., Torrisi R., Luini A. Chemotherapy is more effective in patients with breast cancer not expressing steroid hormone receptors: a study of preoperative treatment. Clin. Cancer Res. 2004;10:6622–6628. doi: 10.1158/1078-0432.CCR-04-0380. [DOI] [PubMed] [Google Scholar]
- Das Thakur M., Salangsang F., Landman A.S., Sellers W.R., Pryer N.K., Levesque M.P., Dummer R., McMahon M., Stuart D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M., Fojo T., Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- DeRose Y.S., Wang G., Lin Y.C., Bernard P.S., Buys S.S., Ebbert M.T., Factor R., Matsen C., Milash B.A., Nelson E. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011;17:1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt C., Di Leo A., de Azambuja E., Larsimont D., Haibe-Kains B., Selleslags J., Delaloge S., Duhem C., Kains J.P., Carly B. Multifactorial approach to predicting resistance to anthracyclines. J. Clin. Oncol. 2011;29:1578–1586. doi: 10.1200/JCO.2010.31.2231. [DOI] [PubMed] [Google Scholar]
- Dobrolecki L.E., Airhart S.D., Alferez D.G., Aparicio S., Behbod F., Bentires-Alj M., Brisken C., Bult C.J., Cai S., Clarke R.B. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016;35:547–573. doi: 10.1007/s10555-016-9653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirew P., Steif A., Khattra J., Ha G., Yap D., Farahani H., Gelmon K., Chia S., Mar C., Wan A. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518:422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrichs K., Ruiz P., Franke F., Gille I., Terpe H.J., Imhof B.A. High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res. 1995;55:901–906. [PubMed] [Google Scholar]
- Guarneri V., Broglio K., Kau S.W., Cristofanilli M., Buzdar A.U., Valero V., Buchholz T., Meric F., Middleton L., Hortobagyi G.N., Gonzalez-Angulo A.M. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J. Clin. Oncol. 2006;24:1037–1044. doi: 10.1200/JCO.2005.02.6914. [DOI] [PubMed] [Google Scholar]
- Haraguchi N., Ishii H., Mimori K., Ohta K., Uemura M., Nishimura J., Hata T., Takemasa I., Mizushima T., Yamamoto H. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int. J. Oncol. 2013;43:425–430. doi: 10.3892/ijo.2013.1955. [DOI] [PubMed] [Google Scholar]
- Hatzis C., Pusztai L., Valero V., Booser D.J., Esserman L., Lluch A., Vidaurre T., Holmes F., Souchon E., Wang H. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA. 2011;305:1873–1881. doi: 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Fan C., Livasy C., He X., Oh D.S., Ewend M.G., Carey L.A., Subramanian S., West R., Ikpatt F. A compact VEGF signature associated with distant metastases and poor outcomes. BMC Med. 2009;7:9. doi: 10.1186/1741-7015-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.I., Sohn J., Koo J.S., Park S.H., Park H.S., Park B.W. Molecular subtypes and tumor response to neoadjuvant chemotherapy in patients with locally advanced breast cancer. Oncology. 2010;79:324–330. doi: 10.1159/000322192. [DOI] [PubMed] [Google Scholar]
- Li X., Lewis M.T., Huang J., Gutierrez C., Osborne C.K., Wu M.F., Hilsenbeck S.G., Pavlick A., Zhang X., Chamness G.C. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- Lim E., Vaillant F., Wu D., Forrest N.C., Pal B., Hart A.H., Asselin-Labat M.L., Gyorki D.E., Ward T., Partanen A. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- Lo P.K., Kanojia D., Liu X., Singh U.P., Berger F.G., Wang Q., Chen H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFbeta signaling. Oncogene. 2012;31:2614–2626. doi: 10.1038/onc.2011.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M., Romero A., Cheang M.C., López García-Asenjo J.A., García-Saenz J.A., Oliva B., Román J.M., He X., Casado A., de la Torre J. Genomic predictors of response to doxorubicin versus docetaxel in primary breast cancer. Breast Cancer Res. Treat. 2011;128:127–136. doi: 10.1007/s10549-011-1461-y. [DOI] [PubMed] [Google Scholar]
- Meyer M.J., Fleming J., Lin A.F., Hussnain S.A., Ginsburg E., Vonderhaar B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010;70:4624–4633. doi: 10.1158/0008-5472.CAN-09-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C.M., Sorlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Prat A., Parker J.S., Karginova O., Fan C., Livasy C., Herschkowitz J.I., He X., Perou C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A., Fan C., Fernandez A., Hoadley K.A., Martinello R., Vidal M., Viladot M., Pineda E., Arance A., Munoz M. Response and survival of breast cancer intrinsic subtypes following multi-agent neoadjuvant chemotherapy. BMC Med. 2015;13:303. doi: 10.1186/s12916-015-0540-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A., Pineda E., Adamo B., Galvan P., Fernandez A., Gaba L., Diez M., Viladot M., Arance A., Munoz M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast. 2015;24(Suppl 2):S26–S35. doi: 10.1016/j.breast.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Ringner M., Fredlund E., Hakkinen J., Borg A., Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS One. 2011;6:e17911. doi: 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S.P., Roth A., Goya R., Oloumi A., Ha G., Zhao Y., Turashvili G., Ding J., Tse K., Haffari G. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley M.J. Isolation, culture and analysis of mouse mammary epithelial cells. Methods Mol. Biol. 2010;633:139–170. doi: 10.1007/978-1-59745-019-5_11. [DOI] [PubMed] [Google Scholar]
- Stingl J., Eirew P., Ricketson I., Shackleton M., Vaillant F., Choi D., Li H.I., Eaves C.J. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Sun C., Wang L., Huang S., Heynen G.J., Prahallad A., Robert C., Haanen J., Blank C., Wesseling J., Willems S.M. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–122. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos A., Chisholm C., Lahusen T., Zheng H., Deng C.X. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. 2014;33:5477–5482. doi: 10.1038/onc.2013.516. [DOI] [PubMed] [Google Scholar]
- Ye F., Qiu Y., Li L., Yang L., Cheng F., Zhang H., Wei B., Zhang Z., Sun L., Bu H. The presence of EpCAM(-)/CD49f(+) cells in breast cancer is associated with a poor clinical outcome. J. Breast Cancer. 2015;18:242–248. doi: 10.4048/jbc.2015.18.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Yao H., Zhu P., Zhang X., Pan Q., Gong C., Huang Y., Hu X., Su F., Lieberman J., Song E. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Yu K.D., Zhu R., Zhan M., Rodriguez A.A., Yang W., Wong S., Makris A., Lehmann B.D., Chen X., Mayer I. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin. Cancer Res. 2013;19:2723–2733. doi: 10.1158/1078-0432.CCR-12-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Claerhout S., Prat A., Dobrolecki L.E., Petrovic I., Lai Q., Landis M.D., Wiechmann L., Schiff R., Giuliano M. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.