

Summary
Mouse embryonic stem cells (ESCs) are maintained in serum with leukemia inhibitory factor (LIF) to maintain self-renewal and pluripotency. Recently, a 2i culture method was reported using a combination of MEK inhibition (MEKi) and GSK3 inhibition (GSK3i) with LIF to maintain ESCs in a naive ground state. How 2i maintains a ground state of ESCs remains elusive. Here we show that MEKi and GSK3i maintain the ESC ground state by downregulating global DNA methylation through two distinct mechanisms. MEK1 phosphorylates JMJD2C for ubiquitin-mediated protein degradation. Therefore, MEKi increased JMJD2C protein levels but decreased DNMT3 expression. JMJD2C promotes TET1 activity to increase 5-hydroxymethylcytosine (5hmC) levels. GSK3i suppressed DNMT3 expression, thereby decreasing DNA methylation without affecting 5hmC levels. Furthermore, 2i increased PRDM14 expression to inhibit DNMT3A/B protein expression by promoting G9a-mediated DNMT3A/B protein degradation. Collectively, 2i allows ESCs to maintain a naive ground state through JMJD2C-dependent TET1 activation and PRDM14/G9a-mediated DNMT3A/B protein degradation.
Keywords: 2i, MEKi, GSK3i, methylation, phosphorylation, JMJD2C, TET1, PRDM14, G9a
Graphical Abstract
Highlights
-
•
MEKi increases JMJD2C protein levels and decreases DNMT3 expression in ESCs
-
•
JMJD2C promotes TET1 hydroxylase activity to increase global 5hmC levels
-
•
GSK3i decreases global DNA methylation without affecting 5hmC levels
-
•
2i-induced PRDM14 expression promotes G9a-mediated DNMT3A/B protein degradation
In this article, Kim and colleagues show that MEKi and GSK3i maintain the ESC ground state by downregulating global DNA methylation through two distinct mechanisms. MEKi increased JMJD2C protein levels, thereby enhancing TET1 activity, whereas GSK3i decreased DNMT3 family expression. Therefore, they uncover that DNA demethylation pathways under 2i conditions are governed by the JMJD2C/TET1 and PRDM14/G9a/DNMT3 complexes.
Introduction
Early mouse zygotes differentiate in the trophectoderm and inner cell mass (ICM). Mouse embryonic stem cells (ESCs) are derived from the ICM in mouse embryos (Evans and Kaufman, 1981, Martin, 1981). ESCs are usually maintained in serum supplemented with leukemia inhibitory factor (LIF), which keeps them in a pluripotent state capable of self-renewal (Smith et al., 1988, Williams et al., 1988) through the activation of JAK-STAT3 signaling (Matsuda et al., 1999, Niwa et al., 1998). Serum/LIF generates a heterogeneous population of ESCs by causing auto-inductive stimulation of the MAPK/ERK pathway by fibroblast growth factor (FGF) 4 (Kunath et al., 2007, Stavridis et al., 2007, Ying et al., 2008). Ying et al. (2008) proposed that LIF and bone morphogenetic protein signals act downstream of the ERK pathway to block ESC commitment. To maintain a ground state in ESCs, they used three selective small-molecule inhibitors, SU5402, PD0325901, and CHIR99021, to inhibit FGF receptor (FGFR) tyrosine kinases, the ERK pathway, and Wnt signaling, respectively (Ying et al., 2008). Later, the use of two inhibitors, PD0325901 and CHIR99021 (called 2i), with LIF to block the MAPK/ERK and glycogen synthase kinase 3β (GSK3β) pathways was postulated to be sufficient to maintain the ESC ground state (Silva et al., 2008). ESCs cultivated in a serum-free 2i medium with LIF (2i ESCs) exhibit greater pluripotent gene expression than ESCs cultivated in serum with LIF (serum ESCs). Further, 2i ESCs homogeneously express NANOG, which potentiates pluripotent gene transcription by creating a permissive chromatin structure (Marks et al., 2012, Marks and Stunnenberg, 2014, Miyanari and Torres-Padilla, 2012, Silva et al., 2009). In addition, 2i leads to genome-wide DNA hypomethylation due to reduced expression of the DNA methyltransferase 3 (DNMT3) family (Bagci and Fisher, 2013, Leitch et al., 2013). The mechanism by which 2i creates a permissive chromatin structure and downregulates DNMT3 expression remains undefined.
DNA methylation by the DNMT family is a heritable epigenetic modification involved in gene silencing, imprinting, and retrotransposon suppression (Baylin, 2005, Jin et al., 2011). In mammals, there are four major members of the DNMT family: DNMT1, DNMT3A, DNMT3B, and DNMT3L. DNMT3A and DNMT3B share similar domain structures: an N-terminal variable region, followed by a PWWP domain, a cysteine-rich zinc-binding domain, and a C-terminal catalytic domain. DNMT3L has no catalytic activity by itself because it lacks the C-terminal catalytic domain (Subramaniam et al., 2014). The DNMT family is dynamically regulated during mouse development (Smith et al., 2012). Global DNA hypomethylation by Dnmt1−/− and Dnmt3a−/−,3b−/− in ESCs blocks differentiation and induces histone hyperacetylation (Lei et al., 1996, Okano et al., 1999).
In ESCs, G9a, a nuclear histone lysine methyltransferase (HMT) that methylates histones H1, H3K9, and H3K27 (Tachibana et al., 2001, Wu et al., 2011), recruits DNMT3A/B independently of its HMT activity (Epsztejn-Litman et al., 2008). G9a contains a SET domain with ankyrin repeats and mediates the transfer of one to three methyl groups from S-adenosylmethionine to the amino group of a target lysine (Esteve et al., 2005), resulting in gene silencing (Dillon et al., 2005, Tachibana et al., 2001). G9a also methylates non-histone proteins, such as p53, WIZ, CDYL1, ACINUS, REPTIN, MYOD, and DNMT1, to regulate chromatin structure and transcriptional machinery (Jung et al., 2015, Ling et al., 2012, Rathert et al., 2008). The mechanism underlying G9a-mediated DNMT3A/B regulation in ESCs is not well understood.
JmjC domain-containing histone demethylase 2C (JMJD2C) is a histone lysine demethylase (HDM) specific for histones H3K9me3, H3K9me2, and H3K36me3 (Cloos et al., 2006, Klose et al., 2006, Whetstine et al., 2006). HDMs are histone-modifying enzymes with biological functions opposite to those of HMTs. JMJD2C removes the repressive histone-methylation at the promoters of pluripotent genes in ESCs. The JMJD2C-mediated reduction of H3K9me3 levels and the resulting alteration of chromatin structure are essential for maintaining ESC pluripotency (Das et al., 2014, Loh et al., 2007, Pedersen et al., 2014). Recent studies demonstrated that JMJD2B and JMJD2C are necessary for ESC self-renewal and induce the generation of pluripotent stem cells (Das et al., 2014, Pedersen et al., 2014). Therefore, JMJD2C acts to maintain ESC self-renewal and pluripotency by lowering H3K9me3 levels (Chen et al., 2013a, Loh et al., 2007, Ng and Surani, 2011).
PR/SET domain 14 (PRDM14) plays a role in the maintenance of the core pluripotent circuitry in ESCs by reducing protein levels of the DNMT3 family (Grabole et al., 2013, Hackett et al., 2013a, Yamaji et al., 2013). PRDM14 is a key regulator in the specification of primordial germ cells (PGCs) during mouse development (Yamaji et al., 2008). PRDM14 is a PR domain-containing (PRDM) transcription factor that contains a PR domain, a modified SET domain, and six tandemly repeated zinc fingers (Nakaki and Saitou, 2014). Downregulation of the DNMT3 family by PRDM14 is important for establishing genome-wide DNA hypomethylation in 2i culture conditions (Grabole et al., 2013, Hackett et al., 2013a, Yamaji et al., 2013). The mechanism by which PRDM14 regulates DNMT3 expression remains undefined.
The ten-eleven translocation (TET) family of dioxygenases promotes DNA demethylation in ESCs and PGCs (Ito et al., 2010, Koh et al., 2011, Vincent et al., 2013). The TET family converts 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine through a series of active DNA demethylation reactions (Gu et al., 2011, Hackett et al., 2013b, Tahiliani et al., 2009). Therefore, the TET family reduces genome-wide DNA methylation through the production of unmethylated cytosine aided by thymine-DNA glycosylase (Kohli and Zhang, 2013, Shen et al., 2013). In ESCs, TET1 is highly expressed and is required for self-renewal (Ito et al., 2010). The mechanism by which 2i treatment enhances TET1 activity is not yet known.
The purpose of this study was to elucidate two distinct epigenetic mechanisms by which 2i maintains the naive ground state in ESCs. 2i upregulated DNA hydroxymethylation by JMJD2C-mediated TET1 potentiation and decreased genome-wide DNA methylation through PRDM14/G9a-dependent DNMT3A/B protein degradation.
Results
MEK Inhibition Stabilizes JMJD2C Proteins and Decreases H3K9me3 Levels
Murine ESCs have been maintained in serum with LIF to maintain their self-renewal (Smith et al., 1988, Williams et al., 1988). However, Ying et al. (2008) reported that a culture method maintained a ground state in ESCs. This medium was supplemented with three small-molecule inhibitors, PD0325901, CHIR99021, and SU5042, which inhibit MAPK/ERK, GSK3β, and FGFR signaling, respectively. Subsequently, they showed that 2i, PD0325901, and CHIR99021, with LIF (2i/LIF condition) is sufficient to sustain a ground state in ESCs (Silva et al., 2008). Therefore, we have examined how each of the three inhibitors influence the maintenance of a naive state in ESCs.
LIF treatment affects JAK-STAT as well as MAPK/ERK signaling for the maintenance of ESC self-renewal (Cherepkova et al., 2016, Onishi and Zandstra, 2015). In agreement with that, we found that LIF increased alkaline phosphatase (AP) activity, which is indicative of increased stemness (Figure 1A). Therefore, we limited the addition of LIF in our experimental conditions to discriminate between LIF-mediated effects and inhibitor-mediated effects. In the absence of LIF, MEK inhibition (MEKi) by PD0325901 produced intense AP staining with compact ESC morphology. GSK3 inhibition (GSK3i) by CHIR99021 resulted in strong AP staining characterized by a dome-shaped ESC colony morphology. FGFR inhibition (FGFRi) by SU5402 had little effect compared with LIF (Figure 1A). A two-inhibitor combination (PD0325901 and CHIR99021) and a three-inhibitor combination (PD0325901, CHIR99021, and SU5402) produced robust AP staining of ESCs with a compact colony morphology.
Figure 1.

MEK Inhibition Regulates JMJD2C Stabilization and Decreases H3K9me3 Levels
(A) AP staining shows that combinatorial treatments of three small-molecule inhibitors induce pluripotency in ESCs. Scale bar, 200 μm.
(B) Western blots show that PD0325901 increases JMJD2C protein levels and decreases H3K9me3 levels. ESCs were cultured in feeder-free and LIF-free conditions with inhibitors for 24 and 48 hr.
(C) PD0325901 and/or CHIR99021 induce changes of histone H3 methylation in feeder-free and LIF-free ESCs.
(D) MEKi with PD0325901 decreases H3K9me3 levels in ESCs, but not ERKi with FR180204 in western blots.
(E) Combinatorial 2i treatments change gene expression in ESCs. Data come from the GEO DataSet (GEO: GSE43597). The 2i treatments upregulate Prdm14 and downregulate the Dnmt3 family.
(F) qRT-PCR (n = 4 independent experiments) shows that PD0325901 decreases Jmjd2c mRNA levels compared with the DMSO control.
(G) Western blots show that PD0325901 increases JMJD2C, OCT4, and SOX2 protein levels but decreases H3K9me3 levels (n = 3 independent experiments).
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Changes in histone H3 methylation status affect gene expression patterns in a variety of cells (Black et al., 2012). We therefore investigated whether H3K9me3 status is altered by three small-molecule inhibitors. Neither GSK3i nor FGFRi had any effects on H3K9me3 levels, but MEKi reduced H3K9me3 levels. Likewise, 2i (MEKi/GSK3i) decreased H3K9me3 levels (Figure 1B). Interestingly, protein levels of JMJD2C, which is a histone H3K9me3-, H3K9me2-, and H3K36me3-specific histone demethylase (Cloos et al., 2006, Klose et al., 2006, Whetstine et al., 2006), were increased by MEKi after 48 hr (Figure 1B). MEKi also decreased H3K9me2 and H3K36me3 levels (Figures 1C and S1), suggesting that MEKi might keep low H3K9me2/3 levels by the increased JMJD2C levels.
The MAPK/ERK pathway delivers an intracellular signaling by successive kinase reactions, RAS-RAF-MEK-ERK. Therefore, the MEK1/2 inhibitor PD0325901 suppresses MEK1/2 activity, successively reducing downstream ERK1/2 activity. The observation made with PD0325901 could be due to the inhibition of either MEK1/2 or ERK1/2. Therefore, we used the ERK selective inhibitor FR180204 to determine which kinase is responsible for decreasing H3K9me3 levels. Indeed, MEKi, but not ERKi specifically decreased H3K9me3 levels (Figure 1D), indicating that PD0325901-mediated H3K9me3 reduction originates from the selective inhibition of MEK1/2 activity, but not that of ERK1/2 activity.
We used GEO: GSE43597 in the GEO DataSet to explore alterations in gene expression profiles caused by 2i treatments (Zhang et al., 2013). Although the 2i treatment did not alter the expression of most genes in the dataset (Figure 1E and Table S1), it reduced the transcript levels of Dnmt3a, Dnmt3b, Dnmt3l, and Tet1, and increased the transcript levels of Prdm14, Prdm1, Klf2, and Nanog. Because MEKi resulted in high JMJD2C protein levels (Figure 1B), we examined Jmjd2c transcript levels by qRT-PCR. MEKi did not significantly change the transcript levels of Oct4 but increased those of Sox2 and decreased those of Jmjd2c (Figure 1F). We next examined the protein levels in MEKi-treated ESCs (Figure 1G). MEKi increased OCT4 protein levels, probably by increasing OCT4 protein stability, and SOX2 protein levels, probably by transcriptional induction. MEKi also increased JMJD2C, suggesting the presence of a post-translational modification. Together, our results demonstrate that MEKi increases JMJD2C levels that reduce the repressive H3K9me3 marks at the promoter regions of pluripotent genes in ESCs.
MEK1 Phosphorylates JMJD2C to Promote Ubiquitin-Mediated Protein Degradation
We examined whether MEK1 affects JMJD2C activity via phosphorylation. Purified GST-MEK1 interacted with endogenous JMJD2C directly in ESC lysate (Figure 2A). Because MEK1 is a Ser/Thr and Tyr dual-specificity kinase (Roskoski, 2012), we examined whether MEK1 phosphorylates JMJD2C directly. When we used anti-phosphoserine/threonine antibody to detect phospho-JMJD2C, we did not detect any positive signal (data not shown). An anti-phosphotyrosine 4G10 antibody produced a strong positive signal (Figure 2B), supporting the idea that JMJD2C is a substrate of MEK1 and undergoes phosphorylation at Tyr residues.
Figure 2.

MEK1 Interacts with and Phosphorylates JMJD2C, Which Undergoes Phosphorylation-Dependent Degradation through the Ubiquitin-Pproteasome Pathway
(A) GST pull-down assay shows that MEK1 associates with endogenous JMJD2C.
(B) An in vitro MEK1 kinase assay performed with mouse anti-phosphotyrosine-specific 4G10 antibody shows that MEK1 phosphorylates tyrosine residues of JMJD2C.
(C) MEK1 binds to the JmjN, JmjC, 2×PHD, and 2×TUDOR domains in JMJD2C.
(D) The Y177 residue in JMJD2C is a putative site for phosphorylation by MEK1.
(E and F) In vitro MEK1 kinase assays show that (E) MEK1 phosphorylates the JmjC domain in JMJD2C, and (F) MEK1 phosphorylates wild-type JmjC but not Y177F mutant JmjC. The red asterisk indicates a phosphorylated domain by MEK1.
(G) MEK1 expression increases JMJD2C ubiquitination, but MEKi by PD0325901 suppresses JMJD2C ubiquitination.
(H) MEK1 increases the ubiquitination of wild-type JMJD2C but not Y177F mutant JMJD2C.
(I) MEK1 increases wild-type JmjC ubiquitination.
(J) In the presence of endogenous MEK1, Y177F mutant JmjC is not ubiquitinated compared with wild-type JmjC.
JMJD2C consists of four domains, including jumonji N (JmjN) (amino acids 1–140), JmjC (amino acids 141–310), 2×plant homeodomain (PHD) (amino acids 671–868), and 2×TUDOR (amino acids 869–1,054). GST-MEK1 was associated with four mutants containing JmjN, JmjC, 2×PHD, and 2×TUDOR, respectively (Figure 2C). MEK1 phosphorylates a consensus motif containing the amino acid sequence T-X-Y (Cacace et al., 1999). To determine which Tyr residues of JMJD2C are phosphorylated by MEK1, we searched for T-X-Y motifs within JMJD2C. We identified a highly conserved T-P-Y motif at residues 175–177 in the JmjC domain (Figure 2D), which was specifically phosphorylated by MEK1 (Figure 2E). MEK1 phosphorylated the wild-type JmjC domain but not the Y177F mutant JmjC domain (Figure 2F), indicating that Mek1 phosphorylates Y177.
In line with the observation that MEKi increases JMJD2C protein levels (Figure 1G), the MEK1 expression increased JMJD2C ubiquitination, whereas PD0325901 suppressed that effect (Figure 2G). Moreover, the wild-type JMJD2C was more intensely ubiquitinated than a non-phospho mutant, Y177F JMJD2C, by MEK1 (Figure 2H). MEK1 expression increased the ubiquitination of wild-type GST-JmjC (Figure 2I). Likewise, endogenous MEK1/2 ubiquitinated the wild-type JmjC mutant more than the Y177F JmjC mutant (Figure 2J). Collectively, our results support the conclusion that Y177 is a phosphodegron for JMJD2C degradation.
MEKi Decreases DNA Methylation Levels via DNMT3A/B Reduction and Increases DNA Hydroxymethylation Levels via JMJD2C-Associated TET1 Activation
Accumulating evidence suggests that ESCs cultured in 2i/LIF have genome-wide DNA hypomethylation compared with ESCs cultured in serum/LIF (Leitch et al., 2013, Marks and Stunnenberg, 2014). The activities of two opposing enzyme families, DNMT and TET, govern genome-wide methylation. PD0325901-treated ESCs had low transcript levels of Dnmt3 family and Tet1, but unchanged transcript levels of Tet2 (Figure 3A). PD0325901 seemed to increase TET1 protein levels (Figure 3B), suggesting post-translational modification (Bauer et al., 2015, Chen et al., 2013b, Yu et al., 2013). Consistent with the reductions in transcript levels, DNMT3A/B protein levels were also decreased (Figure 3B), suggesting that MEKi-induced DNMT3A/B reduction is in part due to transcriptional suppression.
Figure 3.

MEK Inhibition Leads to a Decrease of 5mC Levels and an Increase of 5hmC Levels via DNMT3A/B Reduction and JMJD2C-Mediated TET1 Activation, Respectively
(A) qRT-PCR (n = 4 independent experiments) shows that compared with DMSO control, PD0325901 decreases transcript levels of the Dnmt3 family and Tet1 but does not change Tet2 expression. ESCs were cultured in feeder-free and LIF-free conditions with PD0325901 for 48 hr.
(B) PD0325901 decreases DNMT3A/B protein levels but increases TET1 protein levels in ESCs cultured in feeder-free and LIF-free conditions (n = 3 independent experiments).
(C) Dot blots show that PD0325901 decreases 5mC levels but increases 5hmC levels in ESCs cultured in feeder-free and LIF-free conditions with PD0325901 for 48 hr.
(D) JMJD2C interacts with TET1 and TET2 in a pull-down assay.
(E) JMJD2C associates with TET1 in ESCs in a co-immunoprecipitation experiment.
(F) Jmjd2c-KO ESCs do not express endogenous JMJD2C compared with wild-type ESCs.
(G) PD0325901 and/or CHIR99021 decrease DNMT3B levels, but not H3K9me3 levels, in Jmjd2c KO cells compared with control CCE cells. ESCs were cultured in feeder-free and LIF-free conditions.
(H) Dot blots show that JMJD2C deficiency leads to reduction of 5mC levels by PD0325901 in both control and Jmjd2c-KO ESCs. On the contrary, 5hmC levels are unchanged in Jmjd2c-KO ESCs compared to the control cells.
(I) AP staining shows that the specific JMJD2 inhibitor 5-carboxyl-8-HQ disrupts the pluripotent state induced by PD0325901. Scale bar, 200 μm.
∗∗p < 0.01 and ∗∗∗p < 0.001.
DNMT3A/B regulate self-renewal in some stem cells by de novo DNA methylation (Tadokoro et al., 2007, Tsumura et al., 2006). We measured DNA methylation levels by dot-blot assay. Consistent with previous results (Leitch et al., 2013, Marks and Stunnenberg, 2014), MEKi significantly reduced 5mC levels while increasing 5hmC levels (Figure 3C), suggesting that JMJD2C associates with TET1 to enhance methylcytosine dioxygenase activity. Both TET1 and TET2 interacted with JMJD2C (Figure 3D). A co-immunoprecipitation experiment showed that JMJD2C bound to TET1 in ESCs (Figure 3E). Moreover, JMJD2C bound to the catalytic domain of TET1 (Figure S2).
JMJD2C Deficiency Disrupts ESC Pluripotency through Aberrant Methylation of DNA and Histones
We generated Jmjd2c knockout (KO) ESCs to study how JMJD2C is involved in the TET1-mediated DNA demethylation pathway (Figure S3). Wild-type ESCs (J1 and CCE) expressed endogenous JMJD2C, whereas Jmjd2c KO cells did not express JMJD2C proteins (Figure 3F). We examined whether JMJD2C deficiency influences the 2i-mediated maintenance of a ground state. When parental CCE and Jmjd2c KO cells were treated with PD0325901 alone or with 2i, the H3K9me3 levels were decreased in the CCE cells but not in the Jmjd2c KO cells (Figure 3G). Nonetheless, both PD0325901 alone and 2i decreased DNMT3B levels in both CCE and Jmjd2c KO cells, indicating that DNMT3 expression is independent of JMJD2C. Unlike Tet1 transcript levels, TET1 protein levels were increased in both CCE cells and Jmjd2c KO cells in response to PD0325901 (Figure 3G). Consistent with those results (Figure 3C), MEKi decreased 5mC levels in both CCE cells and Jmjd2c KO cells (Figure 3H, see 5mC). In contrast, MEKi increased 5hmC levels in CCE cells but not in Jmjd2c KO cells (Figure 3H, see 5hmC), indicating that active TET1-mediated DNA demethylation requires JMJD2C. In addition, the specific JMJD2 inhibitor 5-carboxyl-8-HQ abolished the MEKi-mediated AP staining and ESC-like colony morphology (Figure 3I).
GSK3 Inhibition Decreases 5mC Levels through DNMT3A/B Reduction
We next elucidated the molecular mechanism underlying the maintenance of a ground state in ESCs by GSK3i, which sustains the naive state by activating Wnt signaling (Miki et al., 2011, Wray and Hartmann, 2012). GSK3i decreased the transcript and protein levels of the DNMT3 family (Figures 4A and 4B). GSK3α/β and MEK1/2 are involved in disparate signaling pathways; however, our data demonstrate that they have partly shared effects, because GSK3i also suppressed Dnmt3 levels. In contrast, GSK3i downregulated Tet1 transcription in addition to decreasing TET1 protein levels. In contrast to the effect of MEKi, GSK3i modestly decreased JMJD2C protein levels (Figures 1B and 4B). GSK3i decreased global DNA methylation levels without altering DNA hydroxymethylation levels (Figure 4C), suggesting that GSK3i-mediated DNA hypomethylation results partly from the downregulation of DNMT3 expression.
Figure 4.

GSK3 Inhibition Decreases 5mC Levels, but Not 5hmC Levels, through Passive DNA Demethylation
(A) qRT-PCR (n = 4 independent experiments) shows that GSK3 inhibition by CHIR99021 decreases transcript levels of the Dnmt3 family and Tet1, but does not alter Tet2 transcript levels compared with the DMSO control.
(B) Western blots show that CHIR99021 reduces DNMT3A/B and TET1 protein levels without changing JMJD2C levels (n = 3 independent experiments).
(C) Dot blots show that CHIR99021 decreases 5mC levels, but not 5hmC levels, in ESCs cultured in feeder-free and LIF-free conditions with CHIR99021 for 48 hr.
(D) qRT-PCR (n = 4 independent experiments) shows that the 2i treatments upregulate Prdm14 transcript levels.
(E) PRDM14 expression in 293T cells decreases DNMT3B levels in a dose-dependent fashion (top panel). PRDM14 expression in ESCs decreases DNMT3A/B levels in ESCs (middle panel). Knockdown of Prdm14 by shRNA increases DNMT3A/B levels in ESCs (bottom panel).
(F) GST pull-down assays show that PRDM14 expression increases the ubiquitination of DNMT3A/B through protein-protein interactions; PRDM14 does not increase DNMT1 ubiquitination.
∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
PRDM14 Downregulates DNMT3A/B Protein Expression
To find a factor that accelerates DNMT3A/B protein degradation, we examined GEO: GSE43597 in the GEO DataSet (Figure 1E and Table S1). Among the genes altered by the 2i conditions, PRDM14 is unique, because it was identified as a key regulator for maintaining ESC pluripotency (Yamaji et al., 2013). Other studies showed that PRDM14 downregulates Dnmt3 expression (Grabole et al., 2013, Hackett et al., 2013a, Yamaji et al., 2013). Consistent with those observations, all combinatorial 2i treatments increased Prdm14 expression but decreased Dnmt3 family expression (Figure 4D).
We investigated the role of PRDM14 to see if it facilitates DNMT3A/B protein degradation. PRDM14 expression in 293T cells decreased endogenous DNMT3B protein levels in a dose-dependent fashion (Figure 4E, upper panel), implying that PRDM14 downregulates DNMT3B by translational control. Similarly, PRDM14 overexpression in ESCs significantly decreased DNMT3A/B protein levels (Figure 4E, middle panel). Conversely, Prdm14 knockdown by small hairpin RNA (shRNA) upregulated DNMT3A/B protein levels (Figures 4E and S4A), indicating that PRDM14 downregulates DNMT3A/B expression.
Because PRDM14 decreases the expression of Dnmt3a/b, but not that of Dnmt1 (Grabole et al., 2013, Yamaji et al., 2013), we examined the possibility that PRDM14 reduces DNMT3A/B protein levels by promoting ubiquitination-dependent degradation. PRDM14 increased the ubiquitination of DNMT3A/B, but not that of DNMT1 (Figure 4F, see IB: HA). Consistent with that observation, exogenous PRDM14 bound to DNMT3A/B, but not to DNMT1 (Figure 4F, see IB: GFP), indicating that DNMT3A/B protein degradation requires a direct interaction between PRDM14 and DNMT3A/B. Endogenous PRDM14 associated with DNMT3A (Figure S4B) and DNMT3B (Figure S4C). Taken together, our findings strongly indicate that PRDM14 is a key factor in the repression of DNMT3A/B expression and the control of the ESC ground state.
PRDM14 Has G9a Methyltransferase Activity toward DNMT3A/B
We predicted that PRDM14 contains methyltransferase activity toward the DNMT family because of the presence of a PR domain that is a modified version of the HMT SET domain. Contrary to our expectation, PRDM14 did not have obvious methyltransferase activity toward the DNMT family (Figure 5A). We detected only non-specific signals, which we speculated were due to endogenous G9a bound to the DNMT proteins. As expected, PRDM14 interacted with endogenous G9a in an immunoprecipitation assay (Figure 5B) and a pull-down assay (Figure S5).
Figure 5.

PDRM14-Mediated G9a/DNMT3A Complex Formation Promotes DNMT3A Protein Degradation
(A) DNMT1, DNMT3A2, and DNMT3B in vitro PRDM14 methyltransferase assay shows that PRDM14 has no methyltransferase activity toward the DNMT family.
(B) Immunoprecipitation assay shows that endogenous PRDM14 associates with G9a.
(C) DNMT3A2 ubiquitination is proportional to the expression of G9a > PRDM14 > Control > PRDM1.
(D) PRDM14 increases DNMT3A2 ubiquitination, but UNC0638 inhibits PRDM14-mediated DNMT3A2 degradation.
(E) Knockdown of endogenous G9a by the shRNA decreases PRDM14-mediated DNMT3A2 ubiquitination.
(F and G) Knockout of G9a and GLP disrupts 2i-mediated reduction of DNMT3A/B and induction of PRDM14 determined by (F) western blots and (G) qRT-PCR (n = 4 independent experiments) in G9a and GLP double-knockout 248-5 ESCs compared with control TT2 cells. ∗∗∗p < 0.001.
Recently, we reported that G9a activates MYOD protein degradation in a methylation-dependent manner (Jung et al., 2015). We examined whether G9a activates DNMT3A/B protein degradation. The extent of DNMT3A2 ubiquitination was dependent on G9a > PRDM14 > control > PRDM1 expression (Figure 5C, see IB: HA). We detected an association of DNMT3A2 with PRDM14 and G9a, but not with PRDM1 (Figure 5C, see IB: GFP/GST). We observed the similar associations for DNMT3B (Figure S6A). Overall, the degree of PRDM14-mediated DNMT3A/B ubiquitination was lower than that of G9a-mediated DNMT3A/B ubiquitination, suggesting that PRDM14-mediated DNMT3A/B degradation is required for G9a recruitment to the PRDM14/DMNT3A/B complex in ESCs.
G9a inhibition attenuates PRDM14-mediated DNMT3/B degradation. The G9a-specific inhibitor UNC0638 suppressed PRDM14-mediated DNMT3A/B degradation (Figures 5D and S6B). Likewise, G9a silencing by shRNA decreased DNMT3A/B ubiquitination (Figures 5E and S6C). The 2i culture conditions induced a high level of Prdm14 expression (Figures 1E and 4D). Therefore, we hypothesized that PRDM14 functions as a scaffold to aid the assembly between DNMT3A/B and G9a. To address that, we used the mouse 248-5 ESC line, which does not express G9a and G9a-like protein (GLP). Compared with wild-type TT2 cells, the 248-5 cells exhibited alterations in the 2i-mediated reduction of DNMT3A/B and induction of PRDM14 determined by the express levels of proteins (Figure 5F) and mRNAs (Figure 5G), suggesting that PRDM14-mediated DNMT3 protein degradation requires G9a and GLP recruitment. Taken together, our results show that PRDM14 does not have methyltransferase activity; however, it serves as a scaffold to promote DNMT3A/B protein degradation by G9a.
G9a Methylates DNMT3A/B
The DNMT3A family consists of the DNMT3A and DNMT3A2 isoforms. Dnmt3a2 transcription is initiated from a downstream intronic promoter. As a result, DNMT3A2 lacks the 219 N-terminal amino acid residues of the full-length protein (Chen et al., 2002). The K44 residue in DNMT3A is a G9a methylation site (Chang et al., 2011). Although DNMT3A2 lacks the K44 residue, G9a methylated DNMT3A2, DNMT3B, and DNMT1 (Figure 6A), indicating the presence of another methylation site besides K44 in DNMT3A.
Figure 6.

G9a Directly Methylates DNMT3A2, DNMT3B, and DNMT1
(A) In vitro G9a methyltransferase assay shows that G9a methylates DNMT3A2, DNMT3B, and DNMT1. G9a also methylates DNMT3A2, an alternative splicing form, which lacks the K44 residue.
(B–D) In vitro G9a methyltransferase assay shows that G9a methylates (B) the region of amino acids 126–250 in DNMT3A2, (C) the region of amino acids 241–340 in DNMT3B, and (D) lysine residues at the consensus RK methylation site in DNMT3A2 and DNMT3B. The red asterisks are the methylated proteins.
(E) A schematic diagram of potential sites for methylation by G9a in DNMT3A, DNMT3A2, and DNMT3B.
To determine potential methylation sites in DNMT3A2, we conducted an in vitro G9a methyltransferase assay. G9a methylated full-length DNMT3A2 and an N-terminal mutant DNMT3A2126 -250 (Figure 6B). Similarly, G9a methylated full-length DNMT3B as well as an N-terminal mutant, DNMT3B241-340 (Figure 6C). The PWWP domain in DNMT3A, DNMT3A2, and DNMT3B contains a conserved G9a methylation motif with RK residues (Figure 6E). As anticipated, G9a only methylated a GST-fusion protein containing the PAKKPRKSTTEKP residues in DNMT3A2 (Figure 6D). Collectively, our data show that G9a methylates DNMT3A/B, and the PWWP domain in DNMT3A/B is a G9a methylation site. Those findings contradict a previous report that K44 in full-length DNMT3A is the sole residue methylated by G9a (Chang et al., 2011).
G9a/GLP Promotes DNMT3A/B Degradation through Methylation-Dependent Ubiquitination
GLP belongs to the same family of histone lysine N-methyltransferases as G9a (Shinkai and Tachibana, 2011). Therefore, we expected GLP to increase DNMT3A2 ubiquitination. Indeed, GLP increased DNMT3A2 ubiquitination similarly to G9a (Figure 7A), indicating that G9a and GLP facilitate DNMT3A2 and DNMT3B ubiquitination in a methylation-dependent manner. The G9a/GLP inhibitor UNC0638 decreased DNMT3A2 ubiquitination (Figure 7B) and increased DNMT3A protein levels in a dose-dependent fashion (Figure 7C). Likewise, UNC0638 increased 5mC levels (Figure 7D). The silencing of endogenous G9a by shRNAs resulted in an increase of DNMT3A protein levels concomitant with a reduction of G9a levels (Figure S7A), indicating that DNMT3A protein levels are regulated by methylation status. In 248-5 cells, DNMT3A and DNMT3A2 levels were elevated compared with those in control TT2 cells (Figures 5F and S7B). Overall, our results strongly suggest that G9a/GLP regulates global DNA methylation status through DNMT3A/B protein degradation.
Figure 7.

G9a and GLP Control DNMT3 Degradation in a Methylation-Dependent Manner
(A and B) GST pull-down assays show that (A) GLP and G9a play a key role in DNMT3A2 ubiquitination, and (B) G9a inhibition by the G9a/GLP inhibitor UNC0638 decreases DNMT3A2 ubiquitination.
(C) Western blots show that UNC0638 increases endogenous DNMT3A protein levels in a dose-dependent manner (n = 3 independent experiments).
(D) Dot blots show that G9a inhibition by UNC0638 increases DNA methylation levels.
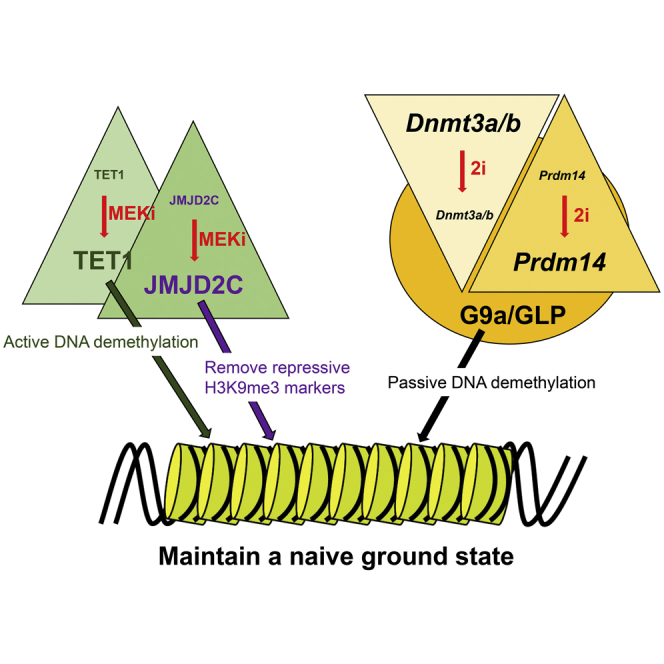
(E) A model showing that the 2i condition maintains ESCs in a naive ground state through two axes of distinct protein complexes: JMJD2C-enhanced TET1 potentiation and PRDM14/G9A-dependent DNMT3A/B protein degradation.
∗p < 0.05 and ∗∗p < 0.01.
Discussion
ESCs cultured in 2i/LIF exhibit unique features including the elevated expression of pluripotent genes, the suppression of genes mediating cell differentiation, and global DNA hypomethylation. Thus, 2i ESCs sustain a ground state of pluripotency similar to that of the ICM. To elucidate the molecular mechanism by which 2i (MEKi/GSK3i) maintains a naive ground state in ESCs, we devised a treatment method of MEKi and GSK3i, both separately and in combination, without LIF. This approach allowed us to uncover that MEKi increased JMJD2C protein levels, thereby enhancing TET1 activity, whereas GSK3i decreased DNMT3 family expression. Our results clearly demonstrate that active and passive DNA demethylation pathways under 2i conditions are governed by the JMJD2C/TET and PRDM14/G9a/DNMT3 complexes, respectively.
A recent series of findings indicated that DNA methylation in 2i ESCs correlates with H3K27me3 (Marks et al., 2012) and H3K9me3 levels (Habibi et al., 2013). JMJD2C is an HDM specific for H3K9me3, H3K9me2, and H3K36me3 (Cloos et al., 2006, Klose et al., 2006, Whetstine et al., 2006), and plays important roles in the maintenance of self-renewal and pluripotency in ESCs (Das et al., 2014, Loh et al., 2007, Pedersen et al., 2014, Wang et al., 2010). MEKi increased JMJD2C protein levels, which was accompanied with H3K9me3 reduction and downregulated Jmjd2c transcript levels, indicating the existence of a post-translational modification.
Because MEK1/2 is a dual-specificity kinase (Roskoski, 2012), there must be a T-X-Y motif present in JMJD2C. We identified a T-P-Y motif in the JmjC domain. Our findings attribute the phosphorylation of full-length JMJD2C by MEK1 to the presence of the T-P-Y motif in the JmjC domain. The phosphorylation of some proteins is coupled to protein degradation through ubiquitination (Huang et al., 2012, Hunter, 2007, Swaney et al., 2013). Hence, the phosphorylation of the Y177 residue in the T-P-Y motif of JMJD2C may play a role in recruiting an unknown E3 ligase to degrade JMJD2C. MEK1 expression increased JMJD2C ubiquitination; however, MEKi suppressed JMJD2C ubiquitination. The Y177F mutant JMJD2C was less ubiquitinated than wild-type JMJD2C in the presence of MEK1. Therefore, we propose that Y177 is a phosphodegron for JMJD2C protein degradation in ESCs.
We speculated that MEKi might decrease DNA methylation levels through the downregulation of the DNMT3 family. MEKi significantly diminished global DNA methylation levels and, surprisingly, was sufficient to induce high 5hmC levels. Because the TET family of dioxygenases that catalyze the conversion of 5mC into 5hmC plays important roles in the induction of DNA hypomethylation (Hill et al., 2014, Kohli and Zhang, 2013), we hypothesized that JMJD2C might interact with TET family members. Thus, the absence of Jmjd2c may disrupt MEKi-induced DNA hydroxymethylation. Indeed, when Jmjd2c was knocked out, 5hmC levels were not altered by MEKi, suggesting that TET1 activity toward 5mC requires JMJD2C. Consequently, MEKi decreases 5mC levels via DNMT3A/B reduction and increases 5hmC levels via the enhancement of TET1 activity. Our findings support the idea that MEKi promotes a ground state in ESCs by lowering global DNA methylation through two different mechanisms: passive DNA demethylation via reduction of the DNMT3 family and active DNA demethylation via JMJD2C-mediated TET1 activation.
GSK3i reduced Dnmt3 family transcript levels, thus decreasing DNMT3A/B protein levels. Remarkably, GSK3i decreased global 5mC levels without altering 5hmC levels, indicating that GSK3i regulates DNA demethylation by a mechanism different than that of MEKi. Thus, our results show that GSK3i-induced DNA demethylation does not involve active DNA demethylation by the TET family.
PRDM14 regulates the maintenance of naive pluripotency in ESCs through a dual mechanism: the inhibition of FGFR signaling and the repression of Dnmt3 expression (Grabole et al., 2013, Hackett et al., 2013a, Yamaji et al., 2013). Although PRDM14 contains a PR/SET domain, we could not detect PRDM14 methyltransferase activity toward the DNMT family. Instead, we discovered that G9a interacts with PRDM14. Consistent with recent reports (Grabole et al., 2013, Hackett et al., 2013a, Yamaji et al., 2013), PRDM14 suppressed the expression levels of DNMT3A/B and, moreover, interacted with DNMT3A/B but not with DNMT1. G9a interacted with and methylated the DNMT3 family. Based on those findings, we postulated that PRDM14 might serve as a scaffold to accommodate both G9a and DNMT3A/B and facilitate G9a-mediated DNMT3A/B protein degradation when PRDM14 is strongly induced in ESCs under 2i/LIF conditions. In accordance with that idea, DNMT3A/B levels in G9a/GLP double-KO cells were high under normal conditions and were less affected than those in wild-type cells under 2i conditions. We propose that DNMT3A/B methylation by G9a leads to the degradation of those proteins through a mechanism similar to the one we reported previously (Jung et al., 2015). Therefore, DNMT3A/B may contain a methyl degron targeted by G9a. This idea is supported by our findings that 248-5 cells express high levels of DNMT3A/B mRNA and protein compared with wild-type TT2 cells.
MEK1/2 and GSK3α/β could have many phosphorylation targets in ESCs. Thus, we focused on the roles of targets changed by PD0325901 and CHIR99021. However, researchers have largely ignored protein modifications induced by 2i thus far. 2i may affect the protein and mRNA levels of those targets differently, as we observed with JMJD2C, OCT4, and TET1. In agreement with that conclusion, UHRF1 was decreased on the protein level during the serum-to-2i transition (von Meyenn et al., 2016). We could discriminate between the effects of the inhibitors, either alone or in combination, on transcriptional and translational control; thus, the DNMT3 family was reduced by 2i through transcriptional suppression and protein degradation. Our findings suggest that PD0325901 alone might be sufficient to maintain a ground state in ESCs. Likewise, PD0325901 alone induced the formation of PGC-like cells from ESCs (Kimura et al., 2014). But, CHIR99021 alone sustained an ESC-like morphology (Figure 1A), probably due to suppression of Wnt signaling (Atlasi et al., 2013). Thus, the combination of MEKi and GSK3i must have a synergistic effect on the maintenance of ESCs in a naive ground state. Taken together, our data support a model wherein 2i maintains the ground state in ESCs through JMJD2C-enhanced TET1 activation and PRDM14/G9a-dependent DNMT3A/B protein degradation (Figure 7E).
Experimental Procedures
Cell Culture
The mouse ESC lines J1 (ATCC SCRC-1010), CCE, and Jmjd2c KO were used.
AP Staining
For AP staining, ESCs were washed twice with PBS and fixed them with 4% paraformaldehyde for 5 min at room temperature. We performed staining using the Leukocyte Alkaline Phosphatase Kit (Sigma-Aldrich, 86R) according to the manufacturer's protocol.
Statistics
We presented data as the mean ± SEM. We used GraphPad Prism 6 software to perform Student's t tests for parametric data to compare groups. In the figures, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 denote the statistical significance.
Author Contributions
C.K. conceived the study; K.K. planned the project; Y.S., Y.Y., and S.K. performed the ESC-related experiments; M.K. and A.N. performed the molecular experiments; K.P., C.K., and K.K. analyzed and interpreted the data; C.K., K.P., and Y.S. wrote the manuscript; all authors discussed the results and approved the final manuscript for publication.
Acknowledgments
We are grateful to Dr. Makoto Tachibana for providing mouse TT2 and 248-5 ESCs. This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (no. 2012M3A9B4028738) and the Ministry of Education (no. 2015R1D1A1A01060986).
Published: April 27, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.04.001.
Contributor Information
Chang-Hoon Kim, Email: chakimster@korea.ac.kr.
Kye-Seong Kim, Email: ks66kim@hanyang.ac.kr.
Supplemental Information
References
- Atlasi Y., Noori R., Gaspar C., Franken P., Sacchetti A., Rafati H., Mahmoudi T., Decraene C., Calin G.A., Merrill B.J. Wnt signaling regulates the lineage differentiation potential of mouse embryonic stem cells through Tcf3 down-regulation. PLoS Genet. 2013;9:e1003424. doi: 10.1371/journal.pgen.1003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagci H., Fisher A.G. DNA demethylation in pluripotency and reprogramming: the role of tet proteins and cell division. Cell Stem Cell. 2013;13:265–269. doi: 10.1016/j.stem.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Bauer C., Gobel K., Nagaraj N., Colantuoni C., Wang M., Muller U., Kremmer E., Rottach A., Leonhardt H. Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-linked N-acetylglucosamine transferase (OGT) J. Biol. Chem. 2015;290:4801–4812. doi: 10.1074/jbc.M114.605881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005;2(Suppl 1):S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Black J.C., Van Rechem C., Whetstine J.R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace A.M., Michaud N.R., Therrien M., Mathes K., Copeland T., Rubin G.M., Morrison D.K. Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol. Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Sun L., Kokura K., Horton J.R., Fukuda M., Espejo A., Izumi V., Koomen J.M., Bedford M.T., Zhang X. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011;2:533. doi: 10.1038/ncomms1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Ueda Y., Xie S., Li E. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 2002;277:38746–38754. doi: 10.1074/jbc.M205312200. [DOI] [PubMed] [Google Scholar]
- Chen J., Liu H., Liu J., Qi J., Wei B., Yang J., Liang H., Chen Y., Chen J., Wu Y. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- Chen Q., Chen Y., Bian C., Fujiki R., Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepkova M.Y., Sineva G.S., Pospelov V.A. Leukemia inhibitory factor (LIF) withdrawal activates mTOR signaling pathway in mouse embryonic stem cells through the MEK/ERK/TSC2 pathway. Cell Death Dis. 2016;7:e2050. doi: 10.1038/cddis.2015.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloos P.A., Christensen J., Agger K., Maiolica A., Rappsilber J., Antal T., Hansen K.H., Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- Das P.P., Shao Z., Beyaz S., Apostolou E., Pinello L., De Los Angeles A., O'Brien K., Atsma J.M., Fujiwara Y., Nguyen M. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell. 2014;53:32–48. doi: 10.1016/j.molcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon S.C., Zhang X., Trievel R.C., Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztejn-Litman S., Feldman N., Abu-Remaileh M., Shufaro Y., Gerson A., Ueda J., Deplus R., Fuks F., Shinkai Y., Cedar H. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008;15:1176–1183. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve P.O., Patnaik D., Chin H.G., Benner J., Teitell M.A., Pradhan S. Functional analysis of the N- and C-terminus of mammalian G9a histone H3 methyltransferase. Nucleic Acids Res. 2005;33:3211–3223. doi: 10.1093/nar/gki635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M.J., Kaufman M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Grabole N., Tischler J., Hackett J.A., Kim S., Tang F., Leitch H.G., Magnusdottir E., Surani M.A. Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep. 2013;14:629–637. doi: 10.1038/embor.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T.P., Guo F., Yang H., Wu H.P., Xu G.F., Liu W., Xie Z.G., Shi L., He X., Jin S.G. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- Habibi E., Brinkman A.B., Arand J., Kroeze L.I., Kerstens H.H., Matarese F., Lepikhov K., Gut M., Brun-Heath I., Hubner N.C. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Hackett J.A., Dietmann S., Murakami K., Down T.A., Leitch H.G., Surani M.A. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Rep. 2013;1:518–531. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J.A., Sengupta R., Zylicz J.J., Murakami K., Lee C., Down T.A., Surani M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill P.W., Amouroux R., Hajkova P. DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: an emerging complex story. Genomics. 2014;104:324–333. doi: 10.1016/j.ygeno.2014.08.012. [DOI] [PubMed] [Google Scholar]
- Huang W., Lv X., Liu C., Zha Z., Zhang H., Jiang Y., Xiong Y., Lei Q.Y., Guan K.L. The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J. Biol. Chem. 2012;287:26245–26253. doi: 10.1074/jbc.M112.382036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Ito S., D'Alessio A.C., Taranova O.V., Hong K., Sowers L.C., Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B., Li Y., Robertson K.D. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2:607–617. doi: 10.1177/1947601910393957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung E.S., Sim Y.J., Jeong H.S., Kim S.J., Yun Y.J., Song J.H., Jeon S.H., Choe C., Park K.T., Kim C.H. Jmjd2C increases MyoD transcriptional activity through inhibiting G9a-dependent MyoD degradation. Biochim. Biophys. Acta. 2015;1849:1081–1094. doi: 10.1016/j.bbagrm.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Kimura T., Kaga Y., Ohta H., Odamoto M., Sekita Y., Li K., Yamano N., Fujikawa K., Isotani A., Sasaki N. Induction of primordial germ cell-like cells from mouse embryonic stem cells by ERK signal inhibition. Stem Cells. 2014;32:2668–2678. doi: 10.1002/stem.1781. [DOI] [PubMed] [Google Scholar]
- Klose R.J., Yamane K., Bae Y., Zhang D., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- Koh K.P., Yabuuchi A., Rao S., Huang Y., Cunniff K., Nardone J., Laiho A., Tahiliani M., Sommer C.A., Mostoslavsky G. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R.M., Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T., Saba-El-Leil M.K., Almousailleakh M., Wray J., Meloche S., Smith A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- Lei H., Oh S.P., Okano M., Juttermann R., Goss K.A., Jaenisch R., Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- Leitch H.G., McEwen K.R., Turp A., Encheva V., Carroll T., Grabole N., Mansfield W., Nashun B., Knezovich J.G., Smith A. Naive pluripotency is associated with global DNA hypomethylation. Nat. Struct. Mol. Biol. 2013;20:311–316. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling B.M., Bharathy N., Chung T.K., Kok W.K., Li S., Tan Y.H., Rao V.K., Gopinadhan S., Sartorelli V., Walsh M.J. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA. 2012;109:841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y.H., Zhang W., Chen X., George J., Ng H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H., Stunnenberg H.G. Transcription regulation and chromatin structure in the pluripotent ground state. Biochim. Biophys. Acta. 2014;1839:129–137. doi: 10.1016/j.bbagrm.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Stewart A.F., Smith A. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T., Nakamura T., Nakao K., Arai T., Katsuki M., Heike T., Yokota T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999;18:4261–4269. doi: 10.1093/emboj/18.15.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T., Yasuda S.Y., Kahn M. Wnt/beta-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011;7:836–846. doi: 10.1007/s12015-011-9275-1. [DOI] [PubMed] [Google Scholar]
- Miyanari Y., Torres-Padilla M.E. Control of ground-state pluripotency by allelic regulation of Nanog. Nature. 2012;483:470–473. doi: 10.1038/nature10807. [DOI] [PubMed] [Google Scholar]
- Nakaki F., Saitou M. PRDM14: a unique regulator for pluripotency and epigenetic reprogramming. Trends Biochem. Sci. 2014;39:289–298. doi: 10.1016/j.tibs.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Ng H.H., Surani M.A. The transcriptional and signalling networks of pluripotency. Nat. Cell Biol. 2011;13:490–496. doi: 10.1038/ncb0511-490. [DOI] [PubMed] [Google Scholar]
- Niwa H., Burdon T., Chambers I., Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Onishi K., Zandstra P.W. LIF signaling in stem cells and development. Development. 2015;142:2230–2236. doi: 10.1242/dev.117598. [DOI] [PubMed] [Google Scholar]
- Pedersen M.T., Agger K., Laugesen A., Johansen J.V., Cloos P.A., Christensen J., Helin K. The demethylase JMJD2C localizes to H3K4me3-positive transcription start sites and is dispensable for embryonic development. Mol. Cell Biol. 2014;34:1031–1045. doi: 10.1128/MCB.00864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P., Dhayalan A., Murakami M., Zhang X., Tamas R., Jurkowska R., Komatsu Y., Shinkai Y., Cheng X., Jeltsch A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr. MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 2012;417:5–10. doi: 10.1016/j.bbrc.2011.11.145. [DOI] [PubMed] [Google Scholar]
- Shen L., Wu H., Diep D., Yamaguchi S., D'Alessio A.C., Fung H.L., Zhang K., Zhang Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y., Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J., Barrandon O., Nichols J., Kawaguchi J., Theunissen T.W., Smith A. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol. 2008;6:e253. doi: 10.1371/journal.pbio.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J., Nichols J., Theunissen T.W., Guo G., van Oosten A.L., Barrandon O., Wray J., Yamanaka S., Chambers I., Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.G., Heath J.K., Donaldson D.D., Wong G.G., Moreau J., Stahl M., Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988;336:688–690. doi: 10.1038/336688a0. [DOI] [PubMed] [Google Scholar]
- Smith Z.D., Chan M.M., Mikkelsen T.S., Gu H., Gnirke A., Regev A., Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–344. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavridis M.P., Lunn J.S., Collins B.J., Storey K.G. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- Subramaniam D., Thombre R., Dhar A., Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:80. doi: 10.3389/fonc.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney D.L., Beltrao P., Starita L., Guo A., Rush J., Fields S., Krogan N.J., Villen J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods. 2013;10:676–682. doi: 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M., Sugimoto K., Fukushima T., Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001;276:25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- Tadokoro Y., Ema H., Okano M., Li E., Nakauchi H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 2007;204:715–722. doi: 10.1084/jem.20060750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumura A., Hayakawa T., Kumaki Y., Takebayashi S., Sakaue M., Matsuoka C., Shimotohno K., Ishikawa F., Li E., Ueda H.R. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- Vincent J.J., Huang Y., Chen P.Y., Feng S., Calvopina J.H., Nee K., Lee S.A., Le T., Yoon A.J., Faull K. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenn F., Iurlaro M., Habibi E., Liu N.Q., Salehzadeh-Yazdi A., Santos F., Petrini E., Milagre I., Yu M., Xie Z. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol. Cell. 2016;62:848–861. doi: 10.1016/j.molcel.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang M., Zhang Y., Kou Z., Han Z., Chen D.Y., Sun Q.Y., Gao S. The histone demethylase JMJD2C is stage-specifically expressed in preimplantation mouse embryos and is required for embryonic development. Biol. Reprod. 2010;82:105–111. doi: 10.1095/biolreprod.109.078055. [DOI] [PubMed] [Google Scholar]
- Whetstine J.R., Nottke A., Lan F., Huarte M., Smolikov S., Chen Z., Spooner E., Li E., Zhang G., Colaiacovo M. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Williams R.L., Hilton D.J., Pease S., Willson T.A., Stewart C.L., Gearing D.P., Wagner E.F., Metcalf D., Nicola N.A., Gough N.M. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336:684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Wray J., Hartmann C. WNTing embryonic stem cells. Trends Cell Biol. 2012;22:159–168. doi: 10.1016/j.tcb.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Wu H., Chen X., Xiong J., Li Y., Li H., Ding X., Liu S., Chen S., Gao S., Zhu B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011;21:365–367. doi: 10.1038/cr.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji M., Seki Y., Kurimoto K., Yabuta Y., Yuasa M., Shigeta M., Yamanaka K., Ohinata Y., Saitou M. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat. Genet. 2008;40:1016–1022. doi: 10.1038/ng.186. [DOI] [PubMed] [Google Scholar]
- Yamaji M., Ueda J., Hayashi K., Ohta H., Yabuta Y., Kurimoto K., Nakato R., Yamada Y., Shirahige K., Saitou M. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell. 2013;12:368–382. doi: 10.1016/j.stem.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Ying Q.L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Zhang Y.L., Pan W.W., Li X.M., Wang Z.W., Ge Z.J., Zhou J.J., Cang Y., Tong C., Sun Q.Y. CRL4 complex regulates mammalian oocyte survival and reprogramming by activation of TET proteins. Science. 2013;342:1518–1521. doi: 10.1126/science.1244587. [DOI] [PubMed] [Google Scholar]
- Zhang X., Peterson K.A., Liu X.S., McMahon A.P., Ohba S. Gene regulatory networks mediating canonical Wnt signal-directed control of pluripotency and differentiation in embryo stem cells. Stem Cells. 2013;31:2667–2679. doi: 10.1002/stem.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.