

Abstract
Mutations in TDP-43 cause amyotrophic lateral sclerosis (ALS), a fatal paralytic disease characterized by degeneration and premature death of motor neurons. The contribution of mutant TDP-43-mediated damage within motor neurons was evaluated using mice expressing a conditional allele of an ALS-causing TDP-43 mutant (Q331K) whose broad expression throughout the central nervous system mimics endogenous TDP-43. TDP-43Q331K mice develop age- and mutant-dependent motor deficits from degeneration and death of motor neurons. Cre-recombinase-mediated excision of the TDP-43Q331K gene from motor neurons is shown to delay onset of motor symptoms and appearance of TDP-43-mediated aberrant nuclear morphology, and abrogate subsequent death of motor neurons. However, reduction of mutant TDP-43 selectively in motor neurons did not prevent age-dependent degeneration of axons and neuromuscular junction loss, nor did it attenuate astrogliosis or microgliosis. Thus, disease mechanism is non-cell autonomous with mutant TDP-43 expressed in motor neurons determining disease onset but progression defined by mutant acting within other cell types.
Electronic supplementary material
The online version of this article (doi:10.1007/s00401-017-1698-6) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis (ALS), TDP-43, Frontotemporal dementia (FTD), RanGAP1, Motor neuron, Non-cell autonomous, Neurodegeneration, Mouse model
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are progressive adult-onset neurodegenerative diseases with overlapping clinical and pathological features [34]. In both human patients and animal models of disease, motor neurons appear to be selectively vulnerable to age-dependent degeneration, despite nearly ubiquitous expression of an ALS-associated mutation in multiple cell types. Many hypotheses have been proposed to explain the selective vulnerability of motor neurons [34, 63], including a reduced level of relevant chaperone proteins that allows the accumulation of misfolded protein in motor neurons, relative to other cell types [15, 26, 62].
Expression in mice of ALS-linked mutations in ubiquitously expressed superoxide dismutase (SOD1) produces progressive motor neuron disease. Using either Cre-recombinase mediated genetic approaches or delivery of a short hairpin RNA (shRNA) [16] or a microRNA [66] to lower mutant SOD1 synthesis, the age of disease onset is delayed by reduction of mutant SOD1 expression within motor neurons [9, 69] and precursors of oligodendrocytes [30], while rate of disease progression is delayed by reducing mutant SOD1 synthesized by microglia [5, 9] or astrocytes [16, 67, 69]. A direct non-cell autonomous toxicity from astrocytes towards motor neurons has also been shown in multiple in vitro co-culture systems using astrocytes expressing mutant SOD1 or derived from ALS patients [7, 14, 21, 39, 50].
A majority (~90%) of ALS cases occurs sporadically with no known familial pattern of inheritance or associated gene mutation. Importantly, pathological comparisons between sporadic and inherited cases have revealed a common biochemical signature of ubiquitinated lesions containing TAR DNA binding protein (TDP-43) [1, 44], with the appearance of TDP-43-positive inclusions in almost all ALS cases, except in those caused by mutation in SOD1 or FUS [36, 56]. The discovery of ALS-linked mutations in TDP-43 within a subset of familial cases demonstrated a causative role for this protein in disease pathogenesis [12, 60].
Similar to the ubiquitously expressed SOD1, TDP-43 is expressed in most cell types. Whether TDP-43 mis-accumulation leads to pathology affecting the same cell types as those affected by SOD1 mutants is not known. Previous studies have provided conflicting evidence for a potential non-cell autonomous contribution of mutant TDP-43 to motor neuron degeneration. Neuronal inducible overexpression of TDP-43M337V was sufficient to cause rapidly fatal disease in rats [23], demonstrating an apparent cell-intrinsic component to toxicity mediated by high levels of TDP-43. However, the same team reported that restricted overexpression of TDP-43M337V induced in astrocytes was also sufficient to cause motor neuron degeneration in vivo [64] and led to upregulation of neurotoxic factors in vitro [22]. Despite this, astrocytes derived from glial precursor cells isolated from mice expressing TDP-43A315T at threefold higher than endogenous TDP-43 in the central nervous system were not toxic to wild-type motor neurons in an in vitro co-culture assay [20]. Since TDP-43 protein aggregates appear in the majority of sporadic ALS cases, understanding the cell type specific contributions of TDP-43 to disease pathogenesis has broad implications that could direct therapeutic design for more than the subset of familial patients carrying specific mutations.
Our prior work established that ALS-linked mutant TDP-43Q331K causes progressive age-dependent motor neuron degeneration without a requirement for nuclear-cytosolic relocalization or protein aggregation [2]. Rather, moderate level expression of mutant TDP-43Q331K led to widespread splicing defects throughout the spinal cord and brain. Mutant TDP-43Q331K expressing animals developed age-dependent lower motor neuron disease, progressive decline in motor performance, and reduced hindlimb strength, accompanied by ~30% loss in innervated neuromuscular junctions, motor axons, and motor neurons.
Given the motor defects observed in mice expressing mutant TDP-43Q331K broadly throughout the central nervous system, we have now tested whether mutant TDP-43Q331K synthesized within motor neurons drives disease onset or progression. Selective excision of the mutant encoding TDP-43Q331K transgene from motor neurons is found to delay age of disease onset with no effect on disease progression after onset despite complete preservation of motor neuron numbers even at late ages. Glial cell activation was unaffected, consistent with mutant TDP-43 expression within glial cells and/or other surrounding neurons contributing to non-cell autonomous motor neuron degeneration and death.
Materials and methods
Generation of transgenic mice expressing mutant TDP-43Q331K throughout the central nervous system, with levels selectively lowered in motor neurons
Transgenic mice which express a deletable human TDP-43 transgene carrying a mutation at Q331K broadly within the central nervous system were described in [2] and deposited at Jackson Laboratories as B6N.Cg-Tg(Prnp-TARDBP*Q331K)103Dwc/J. For selective expression of Cre recombinase in motor neurons, VChAT-Cre mice in a C57Bl6 background [40, 69] were bred to the floxed TDP-43Q331K line 103. To assess the specificity of Cre activity within motor neurons, mice were bred to Rosa-26 reporter mice which ubiquitously express a β-galactosidase transgene that can only be translated upon Cre-mediated recombination to remove a premature stop cassette, also in a C57Bl6 background [59, 69]. Breeding schemes paired TDP-43Q331K animals with double transgenic Rosa-26/VChAT-Cre animals to produce litters with triple transgenic offspring. Upon tissue collection at various ages, genotypes for all animals were reconfirmed using fresh tail biopsies.
RNA extraction and RT-qPCR
Total RNA was isolated using Trizol (Invitrogen) extraction, and mRNA levels were determined by qRT-PCR using the iQSYBR Green supermix (Bio-Rad).
Protein extraction and immunoblotting
Tissues were homogenized in a glass dounce homogenizer in lysis buffer (800 μL per spinal cord) (20 mM Hepes pH 7.4, 5 mM MgCl2, 150 mM KCl, 0.5 mM DTT, with freshly added protease inhibitors and RNAseOUT). An aliquot of the lysate was set aside for RNA extraction using trizol, while the rest was processed for protein extraction. Lysates were centrifuged at 2000 g for 10 min to remove myelin, the lysis buffer was supplemented with NP-40 for a final concentration of 1% NP-40, and then centrifuged at high speed, 15,000 rpm, for 15 min. Equal amounts of each sample (30 μg protein) were loaded onto a 12.5% Tris–HCl gel (Bio-Rad, Criterion). Following transfer to nitrocellulose, the immunoblot was sequentially probed with an antibody that recognizes both human and mouse TDP-43 (Proteintech #12892, 1:1000), the Myc-tag (Millipore 4A6, 1:1000), or Hsp90 (Cell Signaling C45G5, 1:1000).
Immunofluorescence
Tissue preparation for immunofluorescence was performed as previously described [2]. Anesthetized animals were transcardially perfused with 4% (vol/vol) paraformaldehyde in phosphate-buffered saline pH 7.4, then tissues were cryopreserved in 30% (wt/vol) sucrose, and embedded in TissueTek OCT (Sakura). Following cryosectioning of spinal cord into 30 μm-thick sections, free floating sections were selected from the same well per animal for each experiment and immunostained with the indicated antibodies. In brief, free floating sections were washed in PBS three times before incubation in blocking solution containing PBS with 1.5% (wt/vol) BSA and 0.5% Tween-20 for 1 h at room temperature. Sections were incubated in primary antibodies (detailed in Supplementary Materials) in PBS, 0.3% (wt/vol) Triton-X 100 overnight at room temperature, and then washed three times in PBS before incubation with secondary antibodies in PBS, 0.3% (wt/vol) Triton-X 100 for 1 hr at room temperature. After incubation with DAPI (1 μg/ml) and a final wash with PBS, sections were mounted onto glass slides and allowed to dry overnight before coverslipping with ProLong Gold anti-fade mounting media (Invitrogen). For detection of primary antibodies, donkey anti-rabbit, anti-mouse, or anti-goat Cy3, Cy5, or Alexa-488 conjugated antibodies (Jackson ImmunoResearch) were used at a 1:500 dilution.
Immunohistochemistry
Tissue sections were cut from blocks of formalin-fixed paraffin embedded ALS tissue, obtained from Dr. John Ravits’ bank collection (control patient #83 and #65). The spinal cord sections of patients harboring TDP-43 mutations (Pt #284 and #203) as well as control #150 were obtained from the MRC London Neurodegenerative Diseases Brain Bank (King’s College London) (see table below). Seven µm-thick tissue sections were stained with an antibody against RanGAP1 (ab92360) from Abcam (Cambridge, MA, USA). Slides were stained on a Ventana Discovery Ultra (Ventana Medical Systems, Tucson, AZ, USA). Antigen retrieval was performed using Tris–EDTA based cell conditioning solution (CC1) for 40 min at 95 °C. The primary antibody was incubated on the sections at 1:300 dilution for 32 min at 37 °C followed by UltraMap (Ventana) and DAB detection. Slides were rinsed, dehydrated through alcohol and xylene, and coverslipped. Imaging was performed on Nanozoomer slide scanner (Hamamatsu) at the UCSD microscopy core.
| Patient # | Age at death (years) | Sex | Diagnosis | Genotype | Post-mortem delay + interval (h) | Reference |
|---|---|---|---|---|---|---|
| 65 | 82 | M | NORMAL | N/A | 4* | |
| 83 | 63 | F | NORMAL | N/A | 4* | |
| A150/01 | 40 | M | NORMAL | N/A | 40 | |
| A284/13 | 57 | M | ALS | TARDBP M337V | 74 | [60] |
| A203/12 | 23 | F | ALS | TARDBP Y374X + FUS P525L | 125 | [32] |
* Post-mortem interval only (post-mortem delay not available)
Animal behavior
Animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committee of the University of California San Diego (IACUC protocol #S00225), and were in compliance with the Association for Assessment of Laboratory Animal Care guidelines for animal use. For behavior experiments, TDP-43Q331K and TDP-43Q331K/VChAT-Cre animals were compared to contemporaneously produced littermates, either containing or lacking the Rosa26-LacZ reporter.
Accelerating rotarod
To test motor performance, cohorts of age-matched, gender-matched transgenic animals and littermate controls were tested for the time to fall on an accelerating rotarod apparatus (UGO Basile; 2–40 rpm). Animals were tested over three trials with a maximum time of 300 s per trial at each age (3, 6, 10–12, 15–16, ≥18 months). Latency to fall was recorded in seconds once the mice fell from the bar, or rotated once around the bar, according to previous studies [2, 10, 37]. Following a 1-day training session, motor performance was assessed in three trials each day over the course of three consecutive days. The data shown are the average of nine values per animal across the three sessions, ±SEM from n ≥ 4 animals per group at each time point. Animals were studied longitudinally over time, and the graph in Fig. 2a (and Supplementary Fig. 2a) depicts the same animals followed from 3- to 15–18 months of age, with a reduced n per group over time as animals were collected for tissue analysis.
Fig. 2.
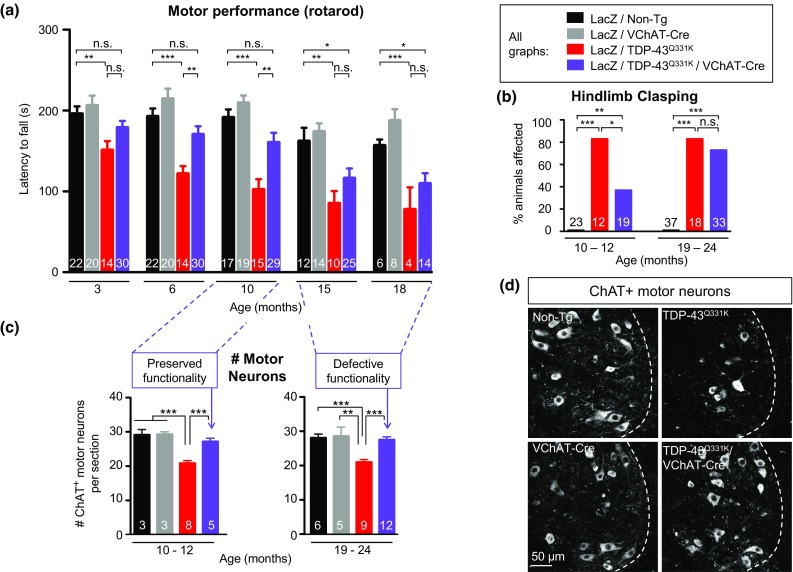
Reduction of mutant TDP-43 within motor neurons delays onset of motor impairment in TDP-43Q331K mice, but not disease progression, despite age-dependent preservation of motor neuron numbers. a Motor performance (using rotarod) of TDP-43Q331K/VChAT-Cre animals was significantly improved compared to TDP-43Q331K animals at 6 and 10 months of age, but declined by 15 months reaching poor motor performance close to TDP-43Q331K. Data are shown as average ± SEM with n = 10–30 mice per group for ages 3–15 months and n = 4–14 animals per genotype at 18 months. *P < 0.05, **P < 0.005, and ***P < 0.0001 using an unpaired t test. b The percentage of animals displaying a clasping phenotype was protected in TDP-43Q331K/VChAT-Cre animals at 10 months, but not at 19 months of age (at 10 months, n > 12 animals, and at 19 months, n > 18, in TDP-43Q331K and TDP-43Q331K/VChAT-Cre animals, respectively). Statistical analysis was performed using Fisher’s exact test (*P = 0.025, **P < 0.005, ***P < 0.0001). c, d Motor neuron loss was rescued in TDP-43Q331K/VChAT-Cre animals compared to TDP-43Q331K animals. c Quantification of ChAT positive motor neurons (average per section ± SEM, n ≥ 3 in controls, and n ≥ 5 animals in TDP-43Q331K and TDP-43Q331K/VChAT-Cre groups; ***P ≤ 0.0006 and **P = 0.004). d Representative confocal micrograph of ChAT-positive α-motor neurons in lumbar spinal cords from TDP-43Q331K transgenic mice at 19 months of age. Scale bar 50 µm. n ≥ 2 in controls, and n ≥ 3 animals in TDP-43Q331K and TDP-43Q331K/VChAT-Cre groups
Analysis of hindlimb clasping
An observer blinded to the genotypes scored animals as being either affected or unaffected by hindlimb clasping. Mice were classified as displaying hindlimb clasping if they displayed retraction of the hindlimbs after being lifted by the tail for 30 s or less. Prior to tissue collection, all animals were video recorded to document hindlimb clasping and other phenotypes. A percentage of animals affected at 10–12 or 19–24 months is shown in Fig. 2b, with n ≥ 12 per group.
Quantification of lower motor neurons
ChAT-positive ventral horn motor neurons were quantified from at least 20 sections per animal, spaced 360 μm apart throughout the lumbar spinal cord. Data were acquired from n > 3 animals per genotype at 10–12 or 19–24 months of age and are shown as the average of the total number of motor neurons counted divided by the number of sections for each animal, ±SEM.
Morphometric analysis and quantification of L5 root motor axons
Mice were perfused transcardially with 4% paraformaldehyde in 0.1 M Sorenson’s phosphate buffer, pH 7.4. Roots from lumbar level 5 of the spinal cord (L5) were dissected from n > 3 animals per genotype at 10–12 months and at 19–24 months of age, and preserved in fixative at 4 °C until embedding. Roots were embedded in Epon-Araldite as previously described in [47]. Thick sections (0.75 μm) were prepared and stained with toluidine blue for analysis by light microscopy. For animals in each age group, cross sections of axons were analyzed using BioQuant software and axonal diameters were graphed as a size distribution curve.
Quantification of neuromuscular junctions and morphology
Floating 40 μm-thick longitudinal sections of gastrocnemius muscle were incubated in blocking solution [PBS, 0.5% vol/vol Tween 20, 1.5% wt/vol BSA] for 4 h at room temperature. Sections were then incubated with polyclonal synaptophysin antibody (ThermoFisher, SP11, 1:50 dilution) in PBS, 0.3% Triton-X 100 at room temperature overnight. The following day, sections were washed in PBS and incubated with donkey anti-rabbit Cy3 (Jackson ImmunoResearch) and α-bungarotoxin Alexa488 (ThermoFisher, B13422) at 1:500 for 1 h, and then with fluoromyelin red (ThermoFisher, F34652) at 1:300 for 30 min. The sections were further washed with PBS, mounted, and dried overnight before coverslipping. For each animal, about ten sections were analyzed for quantification of neuromuscular junctions (shown as average ± SEM). Morphology was scored from >300 NMJs per animal, and endplates that appeared fragmented or small were scored as abnormal. An observer blinded to animal genotype performed quantification and morphology scoring. Representative images were acquired across a 25-μm Z-stack acquired at 20× magnification using a Nikon Eclipse laser scanning confocal microscope, and maximum projection images were obtained using Nikon Elements software (shown in Fig. 3c).
Fig. 3.
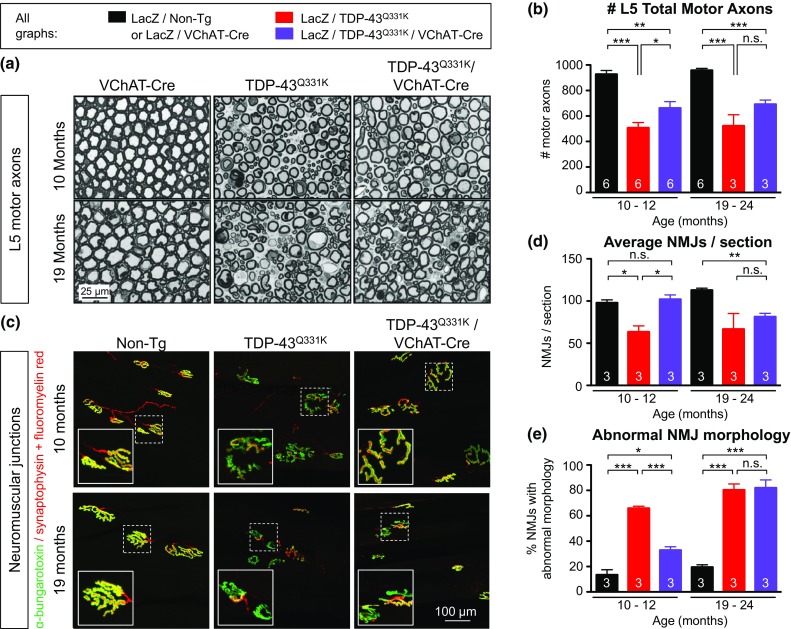
Reduction of mutant TDP-43 within motor neurons only partially delays degeneration of motor axons and neuromuscular junctions. a Representative micrograph of motor axons in the lumbar L5 motor root at 10 or 19 months of age. Scale bar 25 µm. b Quantification of total motor axons in the lumbar L5 motor root at 10 or 19 months of age (average ± SEM, n = 6 in controls, and n ≥ 3 animals in TDP-43Q331K and TDP-43Q331K/VChAT-Cre groups; *P < 0.04, **P < 0.009, and ***P = 0.0001 using an unpaired t test). c Representative neuromuscular junctions from non-transgenic, TDP-43Q331K and TDP-43Q331K/VChAT-Cre animals. Muscle acetylcholine receptor endplates were labeled with α-bungarotoxin, and motor axons (synaptic motor axon terminals and myelinated axons) were marked by synaptophysin antibody and fluoromyelin red, respectively. Scale bar 100 µm. d Quantification of average neuromuscular junctions per section ± SEM, n = 3; *P < 0.04, **P < 0.003. e Assessment of neuromuscular junction morphology during aging. Morphology was scored in >300 NMJs per animal, and abnormal morphology included fragmented or small NMJs; average ± SEM, n = 3; *P < 0.02, ***P < 0.0006
Confocal microscopy and image analysis
All confocal images were acquired on a Nikon Eclipse laser scanning confocal microscope using Nikon Elements software. For each immunostaining experiment, confocal acquisition parameters were determined optimally below saturation level and kept consistent across all samples. Images used for quantification in Figs. 1c, e, 4b, c, and 5f, g were acquired at 20× magnification. Representative images in Figs. 4a, 5a–e (right panels) were acquired at 60× magnification.
Fig. 1.

Reduction of mutant TDP-43 in motor neurons without altering total transgene levels. a Scheme of the three transgenic mouse lines used in this study: a floxed construct expressing mutant TDP-43Q331K throughout the nervous system driven by the prion promoter, VChAT-Cre targeting Cre to motor neurons, and the Rosa26 reporter to detect Cre activity. b Enzymatic Cre activity occurs specifically within lumbar spinal cord motor neurons, as shown by representative micrographs illustrating immunostaining with antibodies against β-galactosidase (β-gal) and ChAT in 2-month-old animals. Scale bar 200 µm. c Quantification of ChAT positive motor neurons that express β-gal in (b). Cre-excision occurred in about 50% of motor neurons in both VChAT-Cre and TDP-43Q331K/VChAT-Cre groups (n ≥ 3 animals per group). d Representative confocal micrographs of the ventral horn of lumbar spinal cord from 2-month-old animals showing TDP-43Q331K in motor neurons using antibodies against the transgene encoded Myc tag and ChAT. Scale bar 200 µm for all images, with enlarged inset panels on the right. e The percentage of ChAT positive cells containing TDP-43Q331K (white arrows) was quantified from n ≥ 3 animals per genotype. About 45% of motor neurons show reduced TDP-43Q331K expression in TDP-43Q331K/VChAT-Cre animals. f, g Total levels of TDP-43Q331K are similar between TDP-43Q331K and TDP-43Q331K/VChAT-Cre groups at 2 months of age, supporting the selective expression of Cre within motor neurons. f mRNA levels of human and mouse TDP-43 were assessed by qRT-PCR using species-specific primers, shown as fold change over controls (Non-Tg and VChAT-Cre) (n ≥ 5 per group). g Protein levels of human and mouse TDP-43 in total spinal cord lysates were determined by immunoblotting with an antibody recognizing both human and mouse TDP-43
Fig. 4.
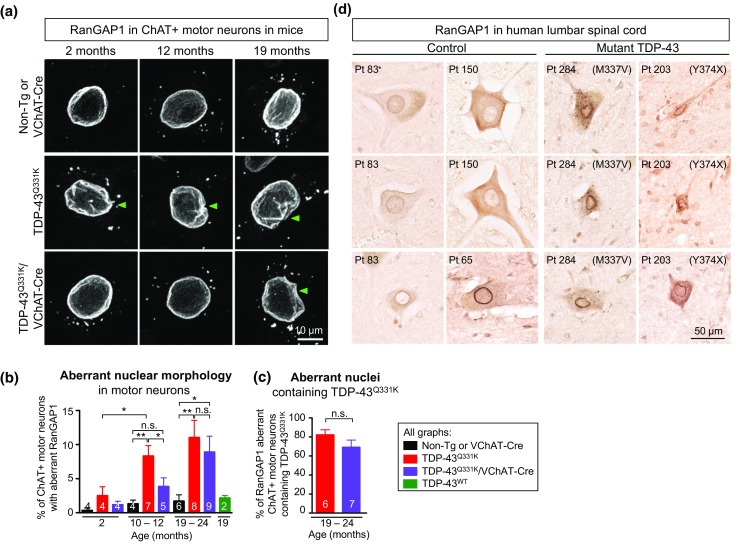
The TDP-43 mutant-dependent nuclear morphology defects in motor neurons of mice and ALS patients harboring TDP-43 mutations are delayed upon reduction of mutant TDP-43 within mouse motor neurons. a, b Age-dependent increased nuclear morphology aberrations in motor neurons from mutant TDP-43Q331K mice, which was significantly delayed but not prevented with disease progression upon motor neuron specific excision of the transgene in TDP-43Q331K/VChAT-Cre animals. a Representative confocal micrographs of nuclear membrane morphology in lumbar spinal cord motor neurons, revealed by RanGAP1 immunostaining in ChAT-positive cells. Green arrowheads indicate examples of motor neurons with nuclear aberrations. Scale bar 10 µm. b Quantification of ChAT positive motor neurons with aberrant nuclear morphology revealed by RanGAP1 immunostaining (average ± SEM, n ≥ 4 animals per genotype at each timepoint). c Quantification of aberrant RanGAP1 containing nuclei that are TDP-43Q331K positive in lumbar spinal cord motor neurons at 19–24 months (n ≥ 6 animals from each group in non-transgenic, TDP-43Q331K, and TDP-43Q331K/VChAT-Cre). d Aberrant nuclear ring shape in motor neurons in the lumbar spinal cord anterior horn of ALS patients harboring TDP-43 mutations. RanGAP1 immunostaining shows an irregular nuclear membrane in motor neurons from mutant TDP-43 patients (n = 2), compared to large round nuclei in motor neurons from control patients (n = 3). Scale bar 50 µm
Fig. 5.
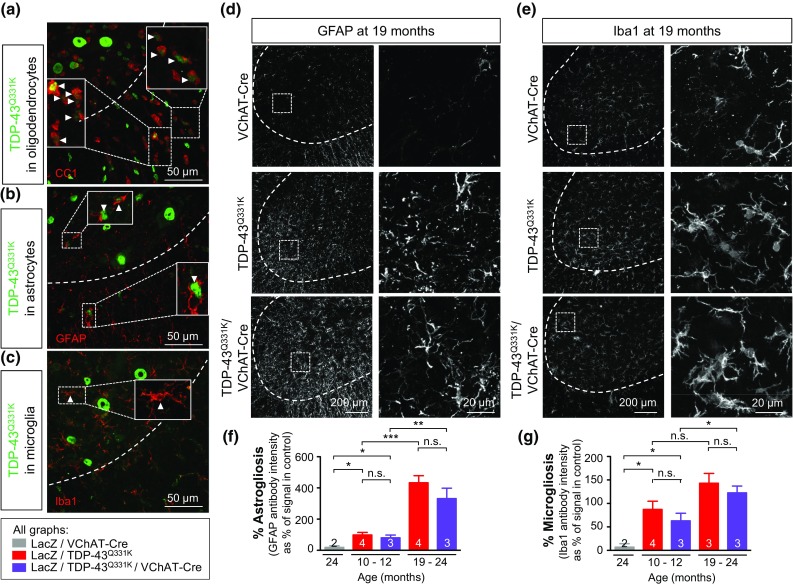
A non-cell autonomous contribution of mutant TDP-43Q331K to disease progression: reduction of mutant TDP-43Q331K within motor neurons does not alter neuroinflammation during disease progression. a Broad expression of the human TDP-43Q331K transgene in glial cells including oligodendrocytes and astrocytes, but not microglia. Oligodendrocytes (CC1 antibody in (a), astrocytes (GFAP antibody in (b), and microglia (Iba1 antibody in (c) were identified by immunofluorescence in spinal cords from 12-month-old TDP-43Q331K animals using antibodies for cell type-specific markers. Arrows highlight glial cells (in red) containing TDP-43Q331K positive nuclei (in green). Dashed outlines correspond to the boundary between gray and white matter. Scale bar 50 μm. d, e Representative micrographs of lumbar spinal cords from VChAT-Cre, TDP-43Q331K and TDP-43Q331K/VChAT-Cre animals at late stages of disease processed for immunofluorescence using an antibody detecting activated astrocytes (GFAP) (d) or activated microglia (Iba1) (e). Scale bar 200 μm, with enlarged inset panels on the right. f, g Astrogliosis and microgliosis were quantified by measuring GFAP (f) or Iba1 (g) fluorescence intensity from confocal images in the lumbar spinal cord of 12 or 19-month-old animals (n ≥ 3 in TDP-43Q331K and TDP-43Q331K/VChAT-Cre; *P < 0.05, **P < 0.005, and ***P < 0.0001 using an unpaired t test)
VChAT-Cre excision efficiency determined by β-gal/ChAT or Myc/ChAT staining
Co-localization of β-gal or Myc with ChAT staining was determined from multiple 10 μm-thick Z-stack images of the lumbar spinal cord acquired at 20× magnification on a Nikon confocal microscope. The percent of ChAT-positive cells containing β-gal staining were counted from >10 maximum projection images taken across the lumbar region, scoring at least 200 cells per animal in n ≥ 3 mice per group. The percent of ChAT positive cells containing bright nuclear Myc (human TDP-43Q331K) staining was analyzed using Volocity software to quantify fluorescence intensity of Myc within each ChAT positive cell. An average of 230 ChAT positive cells were counted per animal and scored as either Myc positive or negative relative to a standard Myc fluorescence intensity threshold across samples. To determine percentages of both β-gal/ChAT and Myc/ChAT overlap, n ≥ 3 animals were analyzed per group at each time point.
Extent of nuclear morphology aberrations based on RanGAP1/Myc/ChAT staining
To assess nuclear morphology in motor neurons, lumbar spinal cord sections were immunostained with RanGAP1, Myc, and ChAT antibodies. Identical observations were made for RanGAP1 aberrations using two different antibodies for RanGAP1 (rabbit polyclonal H-180 and goat polyclonal N-19 from Santa Cruz Biotechnology) and Myc (mouse monoclonal 4A6 from Millipore or rabbit polyclonal from Sigma). For each animal, between 5 and 9 sections of the lumbar spinal cord were imaged using a 20× objective for acquisition across a 10 μm-thick Z-stack. Maximum projection images were scored by an observer blinded to genotypes. All ChAT positive motor neurons were first counted in each image, then scored as containing normal or aberrant nuclear morphology, based on RanGAP1 staining, and scored as positive or negative for Myc levels (transgene) from four to six sections per animal (n > 50 cells each, n > 4 animals per group). Nuclear morphological aberrations were defined by lack of circularity of the nucleus and/or presence of invaginations or wrinkles around the nuclear perimeter using the RanGAP1 antibody. Representative images for Fig. 4a and Supplementary Fig. 3b were acquired at 60× magnification across a 15 μm-thick Z-stack, and a maximum projection image is shown.
Astrogliosis and microgliosis determined by GFAP or Iba1 fluorescence intensity
For quantification of GFAP and Iba1 staining, at least four 20× magnification Z-stack images were analyzed throughout the ventral horn of each animal. Nikon Elements Software was used to determine the average fluorescence intensity (A.U.) across a region of interest around the gray matter in maximum projection images. Values were normalized to the average value in control animals at 24 months and shown as the average ± SEM.
Statistical analyses
Differences between two groups were determined by an unpaired t test, using GraphPad Prism Software. The threshold for significance was set as P ≤ 0.05. Alternatively, Fisher’s exact test was used to determine significance for the percentage of animals affected by hindlimb clasping (Fig. 2b).
Results
Motor neuron-specific reduction of mutant TDP-43 in mice expressing mutant TDP-43 broadly throughout the central nervous system
Transgenic mice expressing a Myc-tagged ALS-linked Q331K mutant human TDP-43 at 1.2 times the normal level of TDP-43 throughout the central nervous system, including in motor neurons, oligodendrocytes and astrocytes, were previously shown to develop progressive adult-onset motor neuron disease [2]. The TDP-43Q331K transgene (flanked by loxP sites) is inactivatable by action of the Cre recombinase. To test the degree to which motor phenotypes were caused by cell intrinsic damage from mutant TDP-43 expressed within motor neurons, the TDP-43Q331K animals were bred to VChAT-Cre mice that have previously been shown to postnatally express Cre recombinase selectively within motor neurons [40, 69] (Fig. 1a). Excision efficiency and specificity of the TDP-43Q331K transgene was initially determined using the Rosa26 reporter strain which ubiquitously expresses a β-galactosidase transgene preceded by a floxed stop cassette and only expresses β-galactosidase in cells in which Cre recombinase has previously been activated [59]. By 2 months of age, Cre-mediated recombination in TDP-43Q331K/VChAT-Cre/Rosa26 mice was seen in 50% of choline acetyltransferase (ChAT)-positive motor neurons throughout the lumbar spinal cord (Fig. 1b, c, Supplementary Fig. 1a, b), consistent with previous reports [40]. Also as expected, Cre-activated excision was selectively found only within motor neurons, with β-galactosidase activity restricted to ventral horn motor neurons (Supplementary Fig. 1a).
Finally, by evaluating the expression pattern of mutant TDP-43 in the ventral horn of lumbar spinal cords, excision efficiency of the actual TDP-43Q331K transgene was measured to be 45% (Fig. 1d, e, Supplementary Fig. 1c, d) in TDP-43Q331K/VChAT-Cre animals. Excision was selective to motor neurons, as no excision of TDP-43Q331K was found in sensory neurons in the dorsal horn or glial cells (Supplementary Fig. 1e). VChAT-Cre inactivation of the TDP-43Q331K transgene did not significantly alter overall levels of mutant TDP-43 expression within the central nervous system (CNS), as determined by measuring TDP-43 mRNA (Fig. 1f) and protein (Fig. 1g) levels in total spinal cord extracts. Consistent with previous reports establishing a mechanism for TDP-43 autoregulation [3, 4, 48], the levels of endogenous mouse TDP-43 were reduced compared to non-transgenic animals.
Motor neuron-specific reduction of mutant TDP-43 levels delays onset of motor deficits
To determine the contribution of mutant TDP-43-dependent damage within motor neurons to disease pathogenesis, motor performance was evaluated longitudinally. Age-dependent motor deficits in TDP-43Q331K mice (relative to non-transgenic littermates) initiated prior to 3 months of age and the deficits increased during aging (Fig. 2a), as previously reported [2]. In contrast, the motor performance of TDP-43Q331K/VChAT-Cre animals was nearly unaffected up to 10 months of age (Fig. 2a, Supplementary Fig. 2a, with a latency to fall of 160 s, compared to 191 s in non-transgenic animals, n = 29 and 17, respectively). Reduction of TDP-43Q331K synthesis within motor neurons delayed the appearance of a hindlimb clasping motor phenotype that developed in >80% of TDP-43Q331K animals by 10 months (Fig. 2b). Thus, motor neuron-specific reduction of mutant TDP-43 delays onset of significant motor deficits.
Reduction of mutant TDP-43 within motor neurons prevents age-dependent motor neuron death, but only modestly delays degeneration of motor axons and neuromuscular junctions
Although disease initiation was delayed, disease in the TDP-43Q331K/VChAT-Cre animals reached a similar disease state to the TDP-43Q331K mice by 18 months of age (Fig. 2a, Supplementary Fig. 2a). Nevertheless, eliminating TDP-43Q331K synthesis within individual motor neurons almost completely prevented the 30% loss of lumbar spinal cord α-motor neurons (27 ± 1 ChAT-positive motor neurons per section in TDP-43Q331K/VChAT-Cre, n = 12, compared with 21 ± 1 ChAT-positive motor neurons in TDP-43Q331K, n = 9, P < 0.001 and 28 ± 1 ChAT-positive motor neurons in non-transgenic littermates, n = 6) (Fig. 2c, d).
Despite complete rescue of motor neuron cell bodies, however, degeneration of motor axons (measured in L5 roots) was only modestly inhibited in TDP-43Q331K/VChAT-Cre animals compared to TDP-43Q331K mice, both at onset and at later stages of disease (averages of 662 and 508 motor axons, respectively, compared to 921 in non-transgenic controls, n = 6, P < 0.001 for TDP-43Q331K compared to controls, or P < 0.04 for TDP-43Q331K/VChAT-Cre compared to TDP-43Q331K at 10 months) (Fig. 3a, b, Supplementary Fig. 2b, c). Examination of the remaining motor axons revealed degenerating axons, vacuolization, and myelin defects in both TDP-43Q331K and TDP-43Q331K/VChAT-Cre animals (Fig. 3a), with a modest protection in TDP-43Q331K/VChAT-Cre animals at onset, but this benefit was not sustained with aging (Fig. 3a). Mutant TDP-43-dependent loss of the large-caliber α-motor axons (with axonal size >5.5 µm) was only partially alleviated at 10 months in TDP-43Q331K/VChAT-Cre and did not reach numbers close to those in non-transgenic animals (Supplementary Fig. 2b). Furthermore, the age-dependent atrophy of the remaining α-motor axons was similar to that in TDP-43Q331K (Supplementary Fig. 2c).
Quantification of postsynaptic neuromuscular junctions in the gastrocnemius muscle using α-bungarotoxin staining revealed a significant degree of protection in TDP-43Q331K/VChAT-Cre animals at 10–12 months, in contrast to the 35% reduction in the number of neuromuscular endplates seen in TDP-43Q331K animals (Fig. 3d, n = 3, P < 0.02 for TDP-43Q331K compared to controls, and P < 0.02 for TDP-43Q331K/VChAT-Cre compared to TDP-43Q331K). However, this protection was not sustained at 19–24 months (Fig. 3c, d, n = 3, P < 0.003 for TDP-43Q331K/VChAT-Cre compared to controls). Morphological examination of neuromuscular junctions further indicated abnormalities including fragmented or small endplates in TDP-43Q331K animals (Fig. 3e), which were significantly attenuated at 10–12 months but not prevented at later stages of disease in TDP-43Q331K/VChAT-Cre animals (Fig. 3e, 33% of NMJs with aberrant morphology in TDP-43Q331K/VChAT-Cre compared to 65% in TDP-43Q331K, n = 3, P < 0.0005 at 10–12 months of age). Therefore, motor neuron-specific reduction of mutant TDP-43Q331K was not sufficient to abrogate defects in motor axons, neuromuscular junctions, and behavioral performance at late stages of disease, despite nearly complete preservation of motor neuron cell bodies.
TDP-43Q331K expression within motor neurons contributes to nuclear aberrations
Defects in nuclear membrane structure or nucleocytoplasmic transport have been reported in SOD1-mediated toxicity in the CNS of ALS patients [33] and mouse models [42, 70], and more recently in disease caused by the hexanucleotide repeat expansion in C9orf72 [17, 27, 71, 72]. Given the diminished motor performance in aged TDP-43Q331K/VChAT-Cre animals despite absence of motor neuron death, we examined the integrity of the nuclear membrane in the remaining motor neurons. Age-dependent aberrations in nuclear morphology, including nuclear membrane invaginations (Fig. 4a, green arrows), were observed as early as 2 months of age in lumbar spinal cord motor neurons of TDP-43Q331K animals compared to non-transgenic mice (n = 4) (Fig. 4a, b, Supplementary Fig. 3a and movie 1).
In contrast, motor neuron-specific reduction of mutant TDP-43Q331K significantly delayed the appearance of RanGAP1 aberrations in ChAT positive motor neurons of TDP-43Q331K/VChAT-Cre animals (Fig. 4a, b; 8.5% in TDP-43Q331K vs 3.8%, in TDP-43Q331K/VChAT-Cre, n = 7 vs 5 animals, respectively, P = 0.043). Nevertheless, by 19 months, the extent of motor neurons with aberrant nuclear morphology (scored as shown in Fig. 4a, green arrows), in the TDP-43Q331K/VChAT-Cre mice was almost equivalent to that of TDP-43Q331K animals (Fig. 4b; 11 and 9%, respectively; n ≥ 8), thus correlating with severity of the motor performance (Fig. 2a). Most of the motor neurons with RanGAP1 aberrations (including invaginations in the nuclear membrane–Fig. 4a, Supplementary Fig. 3a; movie 1) expressed TDP-43Q331K (Fig. 4c, Supplementary Fig. 3b, green arrow). Nevertheless, one-fourth of the motor neurons with aberrations in RanGAP1 lacked mutant TDP-43 (Supplementary Fig. 3b, white arrows). In animals expressing wild-type human TDP-43 at total levels close to endogenous in a normal mouse [2], nearly all motor neurons maintained normal nuclei at 19 months of age (Fig. 4b, green bar), as in non-transgenic animals (Supplementary Fig. 3b). Cytosolic foci containing RanGAP1 were present in young animals, and appeared to increase in size during aging (Fig. 4a), but the number of motor neurons containing foci did not differ between genotypes or during disease progression (Fig. 4a, Supplementary Fig. 3c).
Defects in nuclear membrane morphology were also found in lumbar spinal cords of ALS patients harboring a mutation in TDP-43 (Fig. 4d). Immunohistochemistry for RanGAP1 in the anterior horn revealed a smaller irregular ring shape around nuclei in mutant TDP-43 patients (n = 2), compared to large round nuclear membrane in motor neurons from controls (n = 3). Consistent with observations in animals (Fig. 4a), cytosolic RanGAP1-positive foci were observed in both patients and controls, and an increase in their size appears to correlate with increased age (Fig. 4d). Together, these data suggest that nuclear membrane morphology, as visualized by RanGAP1 staining, can distinguish healthy motor neurons from degenerating motor neurons, and these defects correlate with poor functionality of surviving motor neurons in TDP-43Q331K/VChAT-Cre animals at late stages of disease.
Reduction of mutant TDP-43Q331K within motor neurons does not alter neuroinflammation
TDP-43Q331K was widely expressed in the CNS and accumulated in most oligodendrocytes (Fig. 5a, arrows) and astrocytes (Fig. 5b, arrows), but not microglia (Fig. 5c) (scored by immunofluorescence using the cell-type specific markers CC1, GFAP and Iba1, respectively). Remarkably, age-dependent activation of astrocytes (Fig. 5d) and microglia (Fig. 5e) in ventral horn of spinal cords of TDP-43Q331K animals (scored by GFAP and Iba1 immunoreactivity, respectively; Fig. 5f, g) was indistinguishable irrespective of whether mutant TDP-43 was expressed by motor neurons or not, both at young and old ages.
Discussion
One of the key discoveries of the past decade is that ALS-like disease pathogenesis from ubiquitously expressed mutant SOD1 is non-cell autonomous [9]. Mutant SOD1 synthesis within motor neurons [9, 69] and precursors of oligodendrocytes [30] drives disease onset, but mutant-mediated damage within astrocytes [67, 69] and microglia [5, 9] drives rapid disease progression. To this, we have now established that ALS-mutant linked TDP-43 mutations expressed at moderate levels in a pattern mimicking endogenous TDP-43 also cause toxicity in a non-cell autonomous manner. Eliminating mutant TDP-43Q331K synthesis in a proportion of motor neurons delayed disease onset, reduced aberrant nuclear morphology in those neurons at early disease stages, and almost eliminated age-dependent accelerated death of those motor neurons. Nevertheless, despite nearly complete rescue of motor neuron loss, deficits in motor performance at later disease stages—accompanied by motor axon degeneration, neuromuscular junction loss, and increased nuclear morphology aberrations within motor neurons—were unaffected, as was the timing of sustained activation of both astrocytes and microglia.
Our findings add to the growing body of evidence uncoupling motor neuron cell death from overall ALS disease course. In mutant SOD1 mouse models, genetic interventions which successfully prevented motor neuron cell death either by modulating levels of apoptotic factors [19, 54], elevating chaperones [52], or by rescuing mitochondrial calcium buffering capacity [47], were ineffective in altering disease course or prolonging lifespan. Similarly, rescuing motor neuron death in mutant FUS-mediated disease does prevent motor deficits and axonal damage during aging [55].
A convergence of recent evidence has implicated errors in nucleo/cytoplasmic transport as a component of normal aging [11] as well as several neurodegenerative disorders [13], including ALS/FTD [8, 49], Huntington’s disease (Cleveland and Lagier-Tourenne, personal communication) [35, 43, 68], and other repeat expansion models [38, 51]. Studies in yeast [27] and flies [61] have identified components of nuclear transport as modifiers of toxicity in models expressing mutant TDP-43, or the C9orf72 hexanucleotide repeat expansion [17, 71]. Altered distribution of importins has been described in motor neurons of mice expressing ALS-linked mutation in SOD1 [70], as have aberrations in RanGAP1 localization in C9orf72 patients [71] and irregularities in the nuclear membrane of cells with TDP-43 pathology in sporadic ALS patients [33]. Mutations in the mRNA nuclear export factor Gle1 have also been identified in rare cases of ALS [28], and recently expression of TDP-43 fragments prone to aggregation has been reported to cause mRNA retention in the nuclei of cultured cells [68].
To these earlier efforts, our findings suggest that TDP-43 aggregation is not required to drive nuclear morphology aberrations as accumulation of mutant TDP-43 to only about the normal level of endogenously expressed TDP-43 in both neurons and glial cells drives progressive motor neuron disease with specific loss of a third of lower motor neurons, despite the absence of cytosolic accumulation of protein aggregates (nuclear or cytoplasmic) and loss of nuclear TDP-43 at any age [2]. Thus, our data provide in vivo evidence that 1) ALS-linked mutant TDP-43 causes age-dependent defects in nuclear membrane morphology (as indicated by RanGAP1 staining) not only in mouse motor neurons (correlating with motor functionality), but also in ALS patients harboring TDP-43 mutations, and 2) motor neuron-specific reduction of mutant TDP-43 significantly delays the appearance of such defects and of motor performance deficits. It is noteworthy that nuclear aberrations have also been reported in other ALS model systems [17, 27, 33, 42, 70–72], as well as other neurodegenerative diseases including Huntington’s disease (Cleveland and Lagier-Tourenne, personal communication), suggesting that nuclear morphology aberrations could be a general hallmark of neurodegeneration, possibly indicative of dysfunctional neurons before their degeneration. How ALS-linked mutations in TDP-43 provoke alterations in RanGAP1 distribution is not established, and additional efforts are now needed to determine the underlying mechanism(s) and to test whether those are widely applicable to sporadic disease.
Prior studies have established non-cell autonomous toxicity of mutant SOD1 from microglial cells [6, 9], oligodendrocytes [29, 30], and astrocytes [67, 69] (reviewed in Ref. [25]). Whether non-cell autonomous neuronal-glial signaling is critical in TDP-43 causing disease is not yet established. While mutant astrocytes (derived from glial precursors isolated from transgenic TDP-43A315T mice [20] or from patient-induced pluripotent stem cells (iPSCs) harboring the TDP-43M337V mutation [57]) were not toxic to co-cultured wild-type mouse motor neurons, conditioned medium from TDP-43A315T mouse astrocytes [53] or microglia treated with recombinant TDP-43 mutants [73] triggered motor neuron death. Additionally, mutant TDP-43-mediated toxicity in mutant TDP-43 motor neurons was not enhanced by co-cultured mutant astrocytes derived from iPSCs [57]. However, while transplanted glial precursor cells expressing mutant TDP-43A315T had no effect on motor neuron death or behavioral phenotypes in wild-type rats [20], astrocyte-specific expression of mutant TDP-43M337V caused rapid fatal motor deficits within a month after induction of the transgene in transgenic rats [64]. Similarly, widespread overexpression of wild-type TDP-43 through an AAV9-mediated viral intravenous delivery in neonatal rats provoked rapid mortality in less than 1 month with only a mild (14%) loss of motor neurons, consistent with TDP-43-dependent alterations in motor neuron functionality independent of cell death, and/or contribution(s) of the surrounding cells including glia [65].
Our evidence strongly supports that mutant TDP-43-mediated disease is non-cell autonomous. While most of the motor neurons with nuclear defects were mutant TDP-43 positive, one-fourth of them did not express the mutant protein, suggesting that damage to those nuclei is mediated by mutant TDP-43 expression in other cell types surrounding the motor neurons. Given the wide expression of mutant TDP-43, and that astrogliosis and microgliosis were not attenuated by mutant TDP-43 gene excision from motor neurons, toxicity must also be mediated from a mutant acting in non-motor neurons as a contributor to motor deficits associated with age. Similarly, FUS-mediated disease in mice where one copy of endogenous FUS was replaced by a mutant form of FUS restricted to the cytoplasm is also non-cell autonomous [55]. Altogether, non-cell autonomous toxicity is likely to be widely applicable in ALS pathogenesis. Perhaps most importantly, therapies specifically targeting motor neurons have promise to delay early stages of disease, but it seems unlikely that they will suffice to extend overall survival, unless non-neuronal cells are also targeted.
The mechanisms underlying non-cell autonomous toxicity mediated by mutant TDP-43 are not known. TDP-43 has been reported to be secreted via exosomes from neurons [24], which could corroborate the hypothesis, so far untested in vivo, that TDP-43 proteinopathy propagates cell to cell [45]. TDP-43 proteins can be detected in CSF of ALS patients [18, 31, 46], suggesting that TDP-43 may be present in the CNS extracellular space. Addition of mutant TDP-43 protein fragments to cultured microglia did induce a receptor-mediated proinflammatory signaling response which was neurotoxic [73] possibly through passive diffusion of TDP-43 [upon cleavage and/or post-translation modification] or release extracellularly during cell death [58]. However, the latter pathway is unlikely as motor neuron death was prevented in our TDP-43Q331K animals even at late ages. One underlying mechanism would be that mutant TDP-43 expressing glial cells and/or other surrounding neurons may release yet unidentified factors that are selectively toxic for motor neurons, as has been proposed in mutant SOD1-mediated disease [39, 41]. Another alternative mechanism for the non-cell autonomous component to disease progression could be altered neuronal connectivity, given the expression of mutant TDP-43 within other neuronal sub-types and widespread splicing defects [2].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (AVI 481035 kb)
Acknowledgements
We thank Ying Jones (University of California San Diego) for technical assistance with plastic thin sectioning and staining, shown in Fig. 3a, and Jennifer Santini (University of California San Diego, Light Microscopy Core, NS047101) for training and resources including the Nanozoomer Slide Scanner and Volocity software. We thank Drs. Christopher Shaw and Claire Troakes (King’s College London) for their help in obtaining the human samples, which were supplied by the London Neurodegenerative Disease Brain Bank, which receives funding from the MRC and is part of the Brains for Dementia Research Programme (jointly funded by Alzheimer’s Research UK and Alzheimer Society). D.D. was a recipient of a post-doctoral fellowship from the NINDS/National Institutes of Health (1-F32-NS073269-01A1). S.S. is a recipient of NIH K99/R00 Award (NS091538-01) and Target ALS Springboard Fellowship. This work was supported by grants to D.W.C. from the National Institutes of Health (R01 NS27036) and Target ALS. D.W.C. and S.D.C. receive salary support from the Ludwig Institute for Cancer Research.
Author contributions
D.D., M.M., K.D. and D.P. designed and performed experiments; D.D., M.M., M.M-D., and A.S. maintained the animal colony and confirmed genotypes; D.D. and S.D.C. designed experiments and analyzed data; S.S., E.A. S.C.L. and J.R. provided key reagents; S.D.C. and D.W.C. provided scientific suggestions and supervision; D.D and S.D.C. wrote the manuscript; All authors reviewed and edited the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s00401-017-1698-6) contains supplementary material, which is available to authorized users.
Contributor Information
Don W. Cleveland, Phone: 858-534-7811, Email: dcleveland@ucsd.edu
Sandrine Da Cruz, Phone: 858-534-4299, Email: sdacruz@ucsd.edu.
References
- 1.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 2.Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, Da Cruz S, Clutario KM, Swing D, Tessarollo L, Marsala M, Shaw CE, Yeo GW, Cleveland DW. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110:E736–E745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avendano-Vazquez SE, Dhir A, Bembich S, Buratti E, Proudfoot N, Baralle FE. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012;26:1679–1684. doi: 10.1101/gad.194829.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4 + T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem. 2008;107:1271–1283. doi: 10.1111/j.1471-4159.2008.05699.x. [DOI] [PubMed] [Google Scholar]
- 8.Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132:159–173. doi: 10.1007/s00401-016-1586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 10.Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther J Am Soc Gene Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21:904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Da Cruz S, Cleveland DW. Disrupted nuclear import-export in neurodegeneration. Science. 2016;351:125–126. doi: 10.1126/science.aad9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de L’Etang AF, Maharjan N, Brana MC, Ruegsegger C, Rehmann R, Goswami A, Roos A, Troost D, Schneider BL, Weis J, Saxena S. Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat Neurosci. 2015;18:227–238. doi: 10.1038/nn.3903. [DOI] [PubMed] [Google Scholar]
- 16.Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H, Meyer K, Schmelzer L, Braun L, Cleveland DW, Kaspar BK. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther J Am Soc Gene Ther. 2013;21:2148–2159. doi: 10.1038/mt.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256:1205–1214. doi: 10.1007/s00415-009-5069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci Off J Soc Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haidet-Phillips AM, Gross SK, Williams T, Tuteja A, Sherman A, Ko M, Jeong YH, Wong PC, Maragakis NJ. Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp Neurol. 2013;250:250–259. doi: 10.1016/j.expneurol.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Huang B, Bi F, Yan LH, Tong J, Huang J, Xia XG, Zhou H. Profiling the genes affected by pathogenic TDP-43 in astrocytes. J Neurochem. 2014;129:932–939. doi: 10.1111/jnc.12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Tong J, Bi F, Zhou H, Xia XG. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Investig. 2012;122:107–118. doi: 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iguchi Y, Eid L, Parent M, Soucy G, Bareil C, Riku Y, Kawai K, Takagi S, Yoshida M, Katsuno M, Sobue G, Julien JP. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain. 2016 doi: 10.1093/brain/aww237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Israelson A, Ditsworth D, Sun S, Song S, Liang J, Hruska-Plochan M, McAlonis-Downes M, Abu-Hamad S, Zoltsman G, Shani T, Maldonado M, Bui A, Navarro M, Zhou H, Marsala M, Kaspar BK, Da Cruz S, Cleveland DW. Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron. 2015;86:218–232. doi: 10.1016/j.neuron.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, Daoud H, Noreau A, Rochefort D, Hince P, Szuto A, Levert A, Vidal S, Andre-Guimont C, Camu W, Bouchard JP, Dupre N, Rouleau GA, Wente SR, Dion PA. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:1363–1373. doi: 10.1093/hmg/ddu545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2 + CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, Mann DM, Allsop D, Nakagawa M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009;117:55–62. doi: 10.1007/s00401-008-0456-1. [DOI] [PubMed] [Google Scholar]
- 32.King A, Troakes C, Smith B, Nolan M, Curran O, Vance C, Shaw CE, Al-Sarraj S. ALS-FUS pathology revisited: singleton FUS mutations and an unusual case with both a FUS and TARDBP mutation. Acta Neuropathol Commun. 2015;3:62. doi: 10.1186/s40478-015-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S, Kusaka H. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2009;68:1184–1192. doi: 10.1097/NEN.0b013e3181bc3bec. [DOI] [PubMed] [Google Scholar]
- 34.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, James Shen CK, Marsh JL. Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington’s disease. Hum Mol Genet. 2015;24:1602–1616. doi: 10.1093/hmg/ddu574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 37.Mandillo S, Tucci V, Holter SM, Meziane H, Banchaabouchi MA, Kallnik M, Lad HV, Nolan PM, Ouagazzal AM, Coghill EL, Gale K, Golini E, Jacquot S, Krezel W, Parker A, Riet F, Schneider I, Marazziti D, Auwerx J, Brown SD, Chambon P, Rosenthal N, Tocchini-Valentini G, Wurst W. Reliability, robustness, and reproducibility in mouse behavioral phenotyping: a cross-laboratory study. Physiol Genom. 2008;34:243–255. doi: 10.1152/physiolgenomics.90207.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mapelli L, Canale C, Pesci D, Averaimo S, Guizzardi F, Fortunati V, Falasca L, Piacentini M, Gliozzi A, Relini A, Mazzanti M, Jodice C. Toxic effects of expanded ataxin-1 involve mechanical instability of the nuclear membrane. Biochim Biophys Acta. 2012;1822:906–917. doi: 10.1016/j.bbadis.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Misawa H, Nakata K, Toda K, Matsuura J, Oda Y, Inoue H, Tateno M, Takahashi R. VAChT-Cre. Fast and VAChT-Cre.Slow: postnatal expression of Cre recombinase in somatomotor neurons with different onset. Genesis. 2003;37:44–50. doi: 10.1002/gene.10224. [DOI] [PubMed] [Google Scholar]
- 41.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagara Y, Tateishi T, Yamasaki R, Hayashi S, Kawamura M, Kikuchi H, Iinuma KM, Tanaka M, Iwaki T, Matsushita T, Ohyagi Y, Kira J. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1alpha in amyotrophic lateral sclerosis. Brain Pathol. 2013;23:534–546. doi: 10.1111/bpa.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nekrasov ED, Vigont VA, Klyushnikov SA, Lebedeva OS, Vassina EM, Bogomazova AN, Chestkov IV, Semashko TA, Kiseleva E, Suldina LA, Bobrovsky PA, Zimina OA, Ryazantseva MA, Skopin AY, Illarioshkin SN, Kaznacheyeva EV, Lagarkova MA, Kiselev SL. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol Neurodegener. 2016;11:27. doi: 10.1186/s13024-016-0092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 45.Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H, Hasegawa M. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–134. doi: 10.1016/j.celrep.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 46.Noto Y, Shibuya K, Sato Y, Kanai K, Misawa S, Sawai S, Mori M, Uchiyama T, Isose S, Nasu S, Sekiguchi Y, Fujimaki Y, Kasai T, Tokuda T, Nakagawa M, Kuwabara S. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: specificity, sensitivity, and a possible prognostic value. Amyotroph Later Scler. 2011;12:140–143. doi: 10.3109/17482968.2010.541263. [DOI] [PubMed] [Google Scholar]
- 47.Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci Off J Soc Neurosci. 2013;33:4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prpar Mihevc S, Darovic S, Kovanda A, Bajc Cesnik A, Zupunski V, Rogelj B. Nuclear trafficking in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain. 2017;140:13–26. doi: 10.1093/brain/aww197. [DOI] [PubMed] [Google Scholar]
- 50.Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodriguez R, Hernandez-Hernandez O, Magana JJ, Gonzalez-Ramirez R, Garcia-Lopez ES, Cisneros B. Altered nuclear structure in myotonic dystrophy type 1-derived fibroblasts. Mol Biol Rep. 2015;42:479–488. doi: 10.1007/s11033-014-3791-4. [DOI] [PubMed] [Google Scholar]
- 52.Rohde G, Kermer P, Reed JC, Bahr M, Weishaupt JH. Neuron-specific overexpression of the co-chaperone Bcl-2-associated athanogene-1 in superoxide dismutase 1(G93A)-transgenic mice. Neuroscience. 2008;157:844–849. doi: 10.1016/j.neuroscience.2008.09.055. [DOI] [PubMed] [Google Scholar]
- 53.Rojas F, Cortes N, Abarzua S, Dyrda A, van Zundert B. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front Cell Neurosci. 2014;8:24. doi: 10.3389/fncel.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci Off J Soc Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scekic-Zahirovic J, Oussini HE, Mersmann S, Drenner K, Wagner M, Sun Y, Allmeroth K, Dieterlé S, Sinniger J, Dirrig-Grosch S, René F, Dormann D, Haass C, Ludolph AC, Lagier-Tourenne C, Storkebaum E, Dupuis L (2017) Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta neuropathol 1–20. doi:10.1007/s00401-017-1687-9 [DOI] [PMC free article] [PubMed]
- 56.Scotter EL, Chen HJ, Shaw CE. TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurother J Am Soc Exp Neurother. 2015;12:352–363. doi: 10.1007/s13311-015-0338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, Carrasco MA, Phatnani HP, Shum C, Wilmut I, Maniatis T, Shaw CE, Finkbeiner S, Chandran S. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci USA. 2013;110:4697–4702. doi: 10.1073/pnas.1300398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smethurst P, Sidle KC, Hardy J. Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS) Neuropathol Appl Neurobiol. 2015;41:578–597. doi: 10.1111/nan.12206. [DOI] [PubMed] [Google Scholar]
- 59.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 60.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sreedharan J, Neukomm LJ, Brown Jr RH, Freeman MR. Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr Biol CB. 2015;25:2130–2136. doi: 10.1016/j.cub.2015.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S, Kopelevich A, Kaspar BK, Lagier-Tourenne C, Cleveland DW. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A. 2015;112:E6993–E7002. doi: 10.1073/pnas.1520639112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, Xia XG. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013;32:1917–1926. doi: 10.1038/emboj.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang DB, Dayton RD, Henning PP, Cain CD, Zhao LR, Schrott LM, Orchard EA, Knight DS, Klein RL. Expansive gene transfer in the rat CNS rapidly produces amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol Ther J Am Soc Gene Ther. 2010;18:2064–2074. doi: 10.1038/mt.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H, Yang B, Qiu L, Yang C, Kramer J, Su Q, Guo Y, Brown RH, Gao G, Xu Z. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum Mol Genet. 2013;23:668–681. doi: 10.1093/hmg/ddt454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet. 2011;20:286–293. doi: 10.1093/hmg/ddq463. [DOI] [PubMed] [Google Scholar]
- 68.Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351:173–176. doi: 10.1126/science.aad2033. [DOI] [PubMed] [Google Scholar]
- 69.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Ito H, Wate R, Ohnishi S, Nakano S, Kusaka H. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006;112:673–680. doi: 10.1007/s00401-006-0130-4. [DOI] [PubMed] [Google Scholar]
- 71.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, Lin WL, Garrett A, Stankowski JN, Daughrity L, Tong J, Perkerson EA, Yue M, Chew J, Castanedes-Casey M, Kurti A, Wang ZS, Liesinger AM, Baker JD, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Rademakers R, Boylan KB, Bu G, Link CD, Dickey CA, Rothstein JD, Dickson DW, Fryer JD, Petrucelli L. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–677. doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, Appel SH. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp Neurol. 2015;273:24–35. doi: 10.1016/j.expneurol.2015.07.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1 (AVI 481035 kb)