

Abstract
Compound A (CpdA), a plant-derived phenyl aziridine precursor, was recently characterized as a fully dissociated nonsteroidal antiinflammatory agent, acting via activation of the glucocorticoid receptor, thereby down-modulating nuclear factor-κB-mediated transactivation, but not supporting glucocorticoid response element-driven gene expression. The present study demonstrates the effectiveness of CpdA in inhibiting the disease progress in experimental autoimmune encephalomyelitis (EAE), a well-characterized animal model of multiple sclerosis. CpdA treatment of mice, both early and at the peak of the disease, markedly suppressed the clinical symptoms of EAE induced by myelin oligodendrocyte glycoprotein peptide immunization. Attenuation of the clinical symptoms of EAE by CpdA was accompanied by reduced leukocyte infiltration in the spinal cord, reduced expression of inflammatory cytokines and chemokines, and reduced neuronal damage and demyelination. In vivo CpdA therapy suppressed the encephalogenicity of myelin oligodendrocyte glycoprotein peptide-specific T cells. Moreover, CpdA was able to inhibit TNF- and lipopolysaccharide-induced nuclear factor-κB activation in primary microglial cells in vitro, in a differential mechanistic manner as compared with dexamethasone. Finally, in EAE mice the therapeutic effect of CpdA, in contrast to that of dexamethasone, occurred in the absence of hyperinsulinemia and in the absence of a suppressive effect on the hypothalamic-pituitary-adrenal axis. Based on these results, we propose CpdA as a compound with promising antiinflammatory characteristics useful for therapeutic intervention in multiple sclerosis and other neuroinflammatory diseases.
The study demonstrates the therapeutic potential of Compound A (CpdA), an anti-inflammatory Glucocorticoid Receptor modulator, in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis.
Multiple sclerosis (MS) is the most common chronic inflammatory disease of the central nervous system (CNS). The cause of degeneration in MS remains largely enigmatic but is generally considered to be the result of an autoimmune inflammatory reaction leading to demyelination and axonal damage in the CNS (1, 2, 3). The disease usually begins in young adulthood and develops progressively. Axonal demyelination, tissue damage, including loss of neurons and oligodendrocytes, astrogliosis, and remyelination accompany the inflammatory changes. Experimental autoimmune encephalomyelitis (EAE) is a commonly accepted animal model for MS and can be induced in susceptible rodents and other animals by immunization with myelin antigens, such as myelin oligodendrocyte glycoprotein (MOG) (4). Recent research indicates that the early stages of MS and EAE are mediated by autoimmune T cells. T cell reactivity to myelin antigens or other CNS antigen represents one of the multiple disease-activating steps. Focal changes in blood brain barrier (BBB) permeability and an enhanced expression of cell adhesion molecules also contribute to disease activity, allowing the trafficking of lymphocytes from the periphery to the CNS (2). Lymphocytes infiltrating the CNS are the initiator and early effector cells inducing EAE, but infiltrating macrophages, dendritic cells, and resident glial cells, in particular microglial cells, are the ultimate effector cells releasing cytotoxic cytokines and chemokines, thereby amplifying neuroinflammation. Astrocytes are also activated in EAE and MS and may contribute to changes in BBB permeability through their production of cytokines, chemokines, and cell adhesion molecules (2, 5).
The nuclear factor-κB (NF-κB) signaling cascade plays a critical role in the regulation of immune and inflammatory responses and has been implicated in the pathogenesis of autoimmune demyelinating diseases, such as MS, and other neurodegenerative disorders (6, 7). NF-κB activation in peripheral immune cells is essential for the induction of EAE pathology (8, 9). In vivo administration of a peptide that disrupts the integrity of the IκB kinase complex and blocks NF-κB activation protected mice from EAE (10), showing that systemic NF-κB inhibition is protective for EAE pathogenesis. A number of genes regulated by NF-κB are involved in leukocyte extravasation (vascular adhesion molecule-1, intracellular adhesion molecule-1, E-Selectin, chemokines) and their effector functions (proinflammatory cytokines, inducible nitric oxide synthase, Cox-2) (9). Mice with CNS-restricted deletion of NF-κB also develop a considerably milder clinical course of EAE pathology (11), suggesting that the NF-κB activation pathway in cells of the CNS plays a predominant pathogenic role in EAE. CNS-restricted NF-κB inhibition exerts its protective effects in EAE by preventing the expression of proinflammatory genes by resident cells of the CNS (11). Together, these studies demonstrate that NF-κB inhibition, both in peripheral immune cells and in the CNS, is protective in EAE, suggesting that pharmacological targeting of the NF-κB pathway might have a therapeutic effect in MS.
Conventional antiinflammatory drugs can be generally divided in steroidal and nonsteroidal agents. Both groups mediate, at least partially, their antiinflammatory effect through interference with NF-κB signaling. At present, glucocorticoids (GCs) still remain the most effective drugs for the treatment of inflammatory disorders such as MS (12, 13). Dexamethasone (DEX), a steroidal glucocorticoid receptor (GR) ligand, is one of the best characterized and most effective GCs. GR stimulation affects gene expression through either direct DNA binding or cross talk with other transcription factors, such as NF-κB. This cross talk forms the basis of the beneficial antiinflammatory potential of GCs, whereas concomitant side effects are mainly the consequence of its direct transactivating capacities (14, 15). GCs have pleiotropic effects; therefore, in the long run excess exogenous GCs impair many anabolic processes that are critical for human health. Classical side effects include diabetes, fat dysregulations, a swollen moon face, osteoporosis, skin disorders, and muscle wasting. Therefore, recent research has focused on the identification of so-called “dissociated” GR ligands, which still elicit the antiinflammatory effects of GCs but exhibit reduced side effects. In this respect, Compound A (CpdA), or 2-(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride, has recently been categorized as a novel nonsteroidal selective GR modulator, with strong antiinflammatory capacities but with an improved side effect profile in terms of the development of hyperglycemia/hyperinsulinemia (16, 17). CpdA is a stable analog of the hydroxyl phenyl aziridine precursor found in the Namibian shrub Salsola tuberculatiformis Botschantzev (16). CpdA mediates GR-dependent transrepression of NF-κB-dependent gene expression but does not stimulate GR transactivation (16). The detailed underlying mechanism for the differential effects of CpdA in comparison with classical GCs is thought to involve a different GR conformation, a different GR phosphorylation status, and the exclusive formation of GR monomers. GR and GR domain mutant reconstitution experiments further showed that the inhibitory effect of CpdA on NF-κB depends on the presence of an intact ligand-binding domain of GR (16, 17). Recently, from a virtual docking analysis it became clear that CpdA and GC hormones share binding cavities in the GR ligand-binding domain and that they form hydrogen bonds with the same amino acids (18). In vivo, CpdA was already used in a zymosan-induced arthritis model in mice where it is able to prevent the onset of paw swelling (16), and in the collagen-induced arthritis model for rheumatoid arthritis where it inhibits the clinical course and disease progression, without inducing hyperinsulinemia (17).
To address the potential of CpdA for the treatment of neuroinflammation, we evaluated the antiinflammatory capacities of CpdA on brain inflammation and MOG peptide-induced EAE in mice. Our results demonstrate that administration of CpdA, either at the onset or at the peak of the disease, is able to repress inflammation and the consequent demyelination. Both treatment protocols resulted in a significant suppression of clinical behavioral signs of neuronal damage, concomitant with a reduced NF-κB activity and expression of NF-κB-dependent cytokines and chemokines in the CNS. CpdA treatment also led to reduced inflammatory cell infiltration into the spinal cord, and an overall decrease in neuronal damage and demyelination. Ex vivo, peripheral T cells derived from CpdA-treated EAE mice showed impaired recall immune responses to MOG. Moreover, in isolated primary microglial cells CpdA suppressed inflammatory cytokine mRNA expression and inhibited TNF- and lipopolysaccharide (LPS)-induced NF-κB activation via interfering with the nuclear translocation of NF-κB p65. As expected, DEX was able to also transcriptionally inhibit TNF-induced cytokine expression in primary microglia; however, this result was effected without interfering with the nuclear translocation of p65. Our data thus reveal that in the same tissue type, two structurally different GR modulators can interfere with the NF-κB pathway through differential mechanisms. Finally, as reported before for collagen-induced arthritis mice, CpdA treatment, in contrast to DEX, also did not cause hyperinsulinemia in EAE mice. Chronic CpdA treatment did not result in suppression of the hypothalamic-pituitary-adrenal (HPA) axis, in contrast to DEX treatment. Neither compound displayed malignant side effects with respect to liver toxicity, as verified by measuring the serum levels of aspartate transaminase (AST) and alanine transaminase (ALT) enzymes. Altogether, these results indicate that the ameliorated EAE pathology in CpdA-treated mice is caused, at least partially, by an impairment of peripheral T cell responses and the local inhibition of NF-κB-dependent inflammation and neuronal damage in the CNS, in a manner that mechanistically differs from the action mechanism of DEX.
Results
CpdA administration ameliorates clinical symptoms and disease severity of EAE
EAE-induced neuroinflammation and neurobehavioral deficits are suppressed by GC treatment, and high-dose GC treatment is a common therapeutic strategy for treating relapses in MS (12, 19). DEX, a synthetic GC and strong agonist for GR, is known to alleviate clinical symptoms of EAE, mainly through induction of T-cell apoptosis and suppression of inflammation via inhibiting NF-κB-dependent gene expression. To assess the antiinflammatory properties of the dissociated GR modulator CpdA in EAE, we immunized mice with MOG peptide (MOG35–55) and injected them either with DEX (as a positive control) or with CpdA every second day starting from the onset of acute disease (i.e. d 10 after immunization). Clinical symptoms were monitored daily until d 25 after immunization. Control mice in the PBS-treated group (Fig. 1A) followed a typical disease course and developed signs of severe paralysis, with an incidence of 100% and reaching a mean maximal clinical score of 3.7 (Table 1). In contrast, CpdA-treated mice developed a considerably less severe disease showing only mild paralysis and reached a mean maximal clinical score of 2.5. As expected, the profile of EAE induction in DEX-treated mice only showed minor clinical signs of disease (Fig. 1A). In a second type of experiment we challenged mice with DEX or CpdA starting early (d 2) after MOG peptide immunization. DEX- and CpdA-treated mice were again protected against development of EAE pathology, in contrast to control PBS-treated mice, which all developed signs of paralysis (Fig. 1B and Table 2). To test whether CpdA administration was capable of ameliorating ongoing EAE at a late acute phase of the disease, treatment was initiated at d 17 after immunization when mice had an average clinical score of 4. CpdA treatment enabled a significant recovery in the clinical course of EAE (Fig. 1C). Together, these results clearly suggest that CpdA can suppress disease progression when administered either early (at the onset of the acute disease) or late (at the peak of the disease).
Fig. 1.
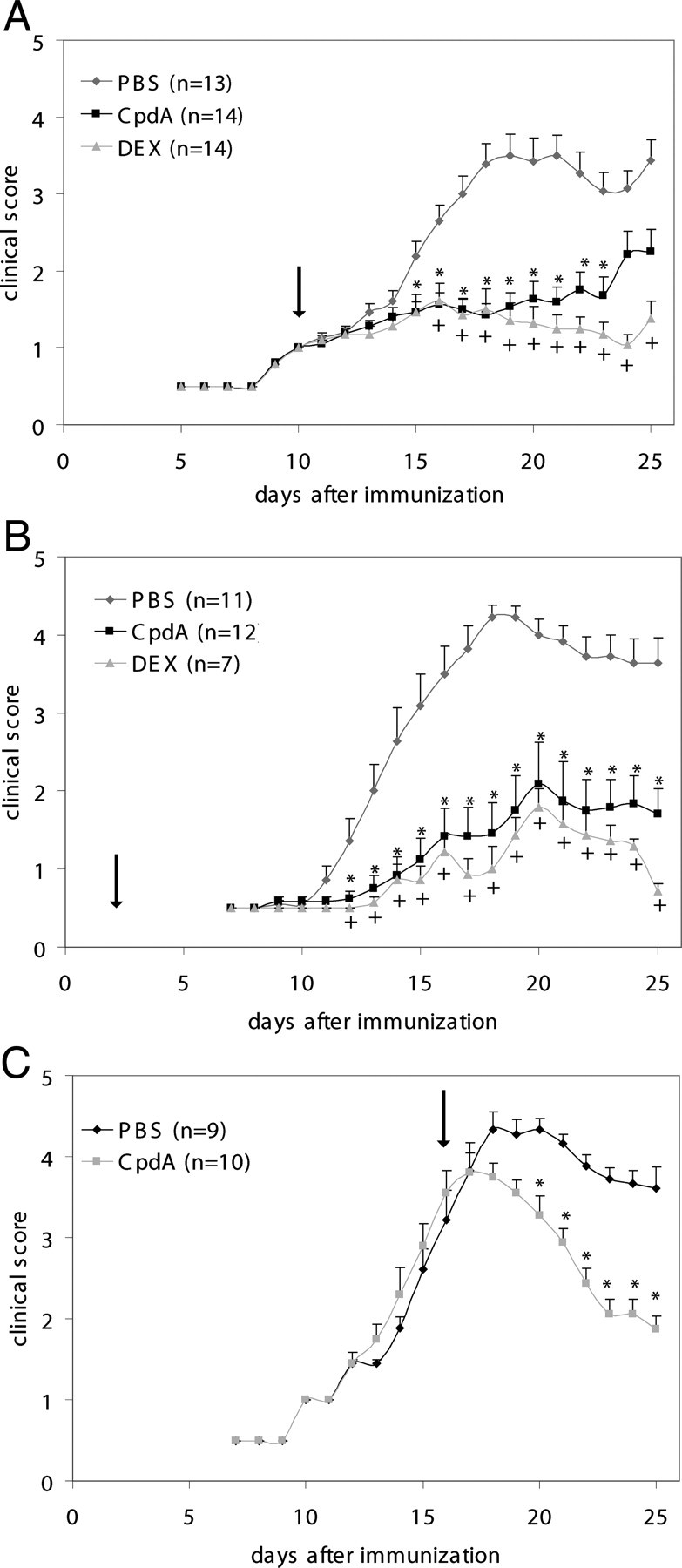
CpdA administration ameliorates clinical symptoms and disease severity of EAE. EAE was induced in male C57BL/6 mice, and clinical symptoms were scored in PBS-, CpdA-, and DEX-treated mice after immunization with MOG peptide. A, PBS, CpdA, and DEX are administered (ip injection) every second day from the onset of acute disease (d 10 on after immunization, as indicated by the arrow). B, PBS, CpdA, and DEX are administered every second day starting from d 2 on after immunization. C, PBS and CpdA are administered every second day starting at the peak of the disease (clinical score 4). Results are displayed as mean values ± sem. *, P < 0.05 (PBS vs. CpdA), and +, P < 0.05 (PBS vs. DEX).
Table 1.
Clinical features of MOG-induced EAE in mice treated with PBS, CpdA, or DEX every second day from d 10 on after immunization
| Treatment | Incidence | Mean maximal clinical score |
|---|---|---|
| PBS | 13/13 (100%) | 3.7 ± 0.3 |
| CpdA | 11/14 (79%) | 2.5 ± 0.3 |
| DEX | 3/14 (21%) | 1.7 ± 0.3 |
Table 2.
Clinical features of MOG-induced EAE in mice treated with PBS, CpdA, or DEX every second day from d 2 on after immunization
| Treatment | Incidence | Mean maximal clinical score |
|---|---|---|
| PBS | 11/11 (100%) | 4.5 ± 0.1 |
| CpdA | 6/12 (50%) | 2.4 ± 0.5 |
| DEX | 2/7 (28%) | 2.1 ± 0.1 |
CpdA administration reduces the infiltration of inflammatory cells into the spinal cord of EAE mice
Clinical examination of mice during the course of EAE revealed that CpdA treatment resulted in a substantially milder disease. To investigate the underlying reason for the improved clinical picture of these mice, we performed immunohistological examination of spinal cord sections from PBS-, CpdA-, and DEX-treated mice 25 d after immunization (Fig. 2, A and B). In control PBS-treated mice, large numbers of MAC3+ macrophages, CD3+ T cells, and B220+ B cells (sections not shown) were detected in the meninges and in the spinal cord parenchyma, accompanied by histological signs of severe demyelination in the white matter as shown by luxol fast blue (LFB) staining, and signs of axonal damage as visualized by immunohistochemical staining for the amyloid precursor protein (APP). In contrast, in spinal cords from CpdA-treated mice, the numbers of inflammatory cells were substantially reduced, whereas myelin remained largely intact and only a few signs of axonal damage could be detected. Analysis of DEX-treated mice led to largely comparable results with diminished infiltration of immune cells in the spinal cord and only mild signs of myelin destruction and axonal damage, confirming the clinical observation that these mice are largely protected from EAE. These results demonstrate that administration of CpdA, similar to the strong GR agonist DEX, protects mice from EAE pathology by reducing the influx of inflammatory cells into the CNS.
Fig. 2.

Reduced inflammation and demyelination in the CNS of CpdA-treated mice. PBS, CpdA, and DEX was administered every second day from the onset of acute disease (d 10 after immunization). A, Histological profiles of spinal cords from PBS-, CpdA-, and DEX-treated mice 25 d after EAE induction. Sections from the lumbar spinal cord were examined for infiltrating macrophages (MAC3, brown) and T cells (CD3, brown) by immunohistochemistry, demyelination by LFB (blue) staining, and axonal damage by APP (brown) immunohistochemistry. Scale bar, 100 μm. B, Quantification of spinal cord infiltrates, demyelination, and axonal damage from histological sections shown in panel A. Numbers of infiltrating T cells (CD3), B cells (B220), and macrophages (MAC3) in PBS-, CpdA-, and DEX-treated mice after EAE induction. Results are displayed as mean values ± sem. *, P < 0.05 (PBS vs. CpdA).
Impaired proinflammatory gene expression in CNS from CpdA-treated mice
To examine whether CpdA administration affects the expression of proinflammatory genes during EAE, we used quantitative real-time PCR analysis to determine the expression of several cytokines and chemokines that have been implicated in EAE pathogenesis. All cytokines (Fig. 3A) and chemokines (Fig. 3B) tested were strongly up-regulated in the CNS of control mice at 16 and 22 d after immunization, but their induction was significantly reduced in CpdA-treated mice, the most pronounced effect being apparent at d 16. These results demonstrate that CpdA interferes with the induction of proinflammatory gene expression in cells of the CNS during EAE. Similar observations were made in DEX-treated mice (data not shown). Taken together, these findings suggest that inhibition of NF-κB signaling by CpdA leads to decreased expression of key mediators involved in CNS inflammation.
Fig. 3.

Decreased proinflammatory gene expression and impaired NF-κB DNA-binding in CNS from CpdA-treated mice. PBS and CpdA were administered every second day from the onset of acute disease (d 10 after immunization). A and B, Quantitative measurement of the indicated cytokine and chemokine mRNA expression in spinal cord from PBS- (n = 2) and CpdA-treated (n = 2) mice 10 d (= before CpdA treatment), 16 d, and 22 d after immunization, and from nonimmunized control mice (n = 2). P < 0.05 (PBS vs. CpdA) for all cytokines and chemokines tested. C, ChIP analysis combined with qPCR was used to assay recruitment of p65 to the IL-6 gene promoter. The quantity of p65 (bound fraction) detected on the IL-6 promoter is shown with a correction of the SYBR green qPCR signal for input control, as percent bound over input. The IgG control indicates the threshold levels of aspecific binding. *, P < 0.05.
CpdA treatment inhibits NF-κB p65 promoter recruitment onto IL-6 in the CNS
NF-κB regulates the expression of multiple proinflammatory mediators including cytokines, chemokines, and adhesion molecules. We therefore tested whether administration of CpdA affects activation of NF-κB in the CNS upon induction of EAE by addressing NF-κB p65 recruitment onto the IL-6 promoter via chromatin immunoprecipitation analysis on spinal cord tissue. Strong induction of NF-κB p65 binding was detected in the spinal cord of control mice 25 d after immunization with MOG peptide (Fig. 3C). In contrast, NF-κB DNA-binding activity was strongly reduced in the spinal cord of immunized CpdA-treated mice as compared with the immunized controls, strongly suggesting that CpdA treatment leads to impaired NF-κB activation in the CNS upon induction of EAE.
CpdA inhibits peripheral immune responses
The efficient activation of T lymphocytes is a critical requirement for the induction of CNS inflammation and pathology in EAE. To address whether or not CpdA is able to repress peripheral immune responses, we examined the generation of MOG35–55-specific T cells in mice treated with CpdA. We isolated lymphocytes from control and CpdA-treated mice 10 d after immunization with MOG peptide and tested their in vitro recall responses to a secondary exposure to the MOG peptide. T cells derived from CpdA-treated mice showed impaired recall responses to the MOG peptide compared with controls, as assessed by the measurement of cellular proliferation (Fig. 4A) and of the production of IL-2, IL-17, and interferon (IFN)-γ (Fig. 4B), suggesting that the ameliorated EAE pathology in CpdA-treated mice is caused, at least partially, by an impairment of peripheral T-cell responses.
Fig. 4.
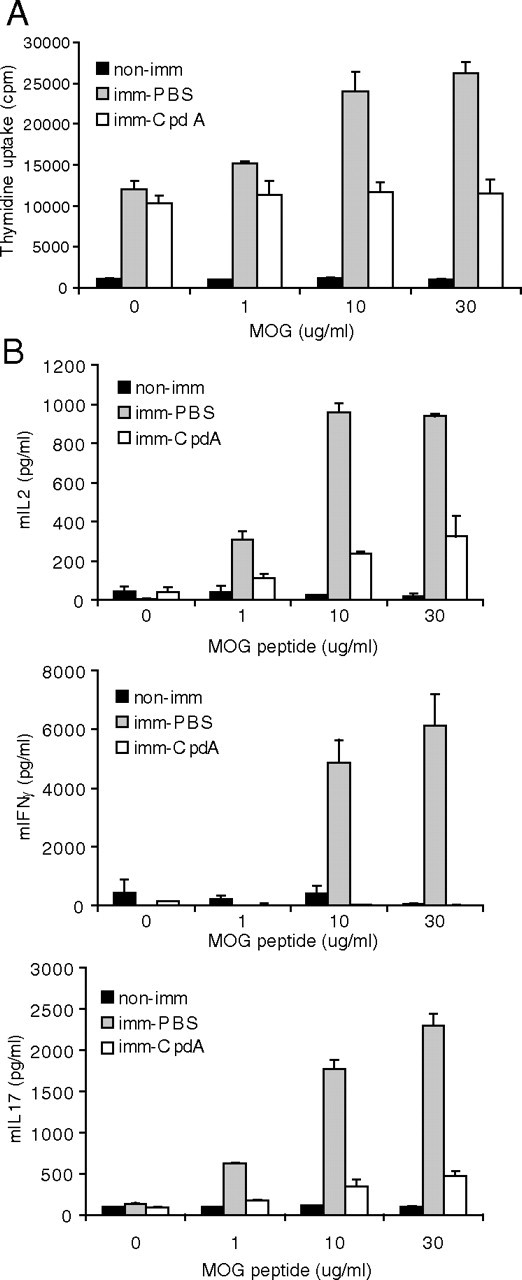
Impaired peripheral immune responses in CpdA-treated mice. Splenocytes from MOG peptide-immunized control (PBS-treated) and CpdA-treated mice were cultured and stimulated with the indicated concentrations of MOG peptide. A, Splenocyte cell proliferation was assessed by measurement of [3H]thymidine incorporation by liquid scintillation counting. Results are expressed as counts per min of triplicate cultures. B, Culture supernatants were collected 48 h after MOG peptide stimulation and assayed for IL-2, IFN-γ, and IL-17 by ELISA. Results are shown as mean ± sem. imm, Immunized.
CpdA inhibits TNF- and LPS-induced NF-κB nuclear translocation in primary microglial cells, but not in astrocytes
In addition to interfering with peripheral immune responses, the activity of CpdA in inhibiting EAE pathology might also be due to the impairment of an NF-κB-dependent proinflammatory activity in resident cells of the CNS, especially in conditions when CpdA is administered at the peak of the disease (Fig. 1C). Microglial cells are considered to be key players for the induction of the (auto)immune pathology in the CNS during MS and EAE (20). Astrocytes are also activated in EAE and MS, thereby contributing to cytotoxic gene expression and pathology (11, 21, 22). Therefore, a defective activation of microglial cells or astrocytes could also account for the protection from EAE in CpdA-treated mice. We first addressed the effect of CpdA on TNF- or LPS-induced NF-κB activation in cultured primary microglial cells. NF-κB was strongly activated in response to TNF or LPS, as shown by a clear nuclear translocation of p65 (Fig. 5A and supplemental Fig. 1, A and C, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). In contrast, pretreatment of microglia with CpdA completely inhibited the nuclear accumulation of NF-κB, strongly suggesting an efficient antiinflammatory potential of CpdA in microglial cells. In sharp contrast herewith, in primary astrocytes a normal in vitro NF-κB translocation pattern is apparent upon TNF and IL-1β stimulation in the presence of CpdA (Fig. 5B and supplemental Fig. 1, B and D). In line with these observations, CpdA completely blocked LPS-induced IκBα phosphorylation in microglial cells, but not IL1β-induced IκBα phosphorylation in astrocytes (Fig. 5C). Intriguingly, DEX does not inhibit NF-κB activation in microglial cells and astrocytes (Fig. 5, A and B and supplemental Fig. 1), which is in contrast to what is observed for CpdA in microglia. Similar observations were made on immortalized macrophage cell lines in which CpdA inhibits NF-κB translocation in contrast to DEX (data not shown). Both DEX and CpdA, however, efficiently block activated cytokine gene expression in primary microglia and in primary astrocytes, as demonstrated for TNF-induced IL-1β and IL-1β-induced TNF expression, respectively (Fig. 5, E and F). Similar results were obtained for monocyte chemotactic protein-1 (MCP-1) and TNF cytokine gene expression in primary microglia and IL-1β and MCP-1 cytokine gene expression in primary astrocytes (data not shown). Therefore, these results indicate that DEX and CpdA use a differential mechanism for blocking NF-κB-driven gene expression in microglia. Together, these results suggest that the protection from EAE pathology in CpdA-treated mice occurs by inhibiting the induction of NF-κB-dependent proinflammatory genes via a cytoplasmic mechanism in microglial cells, but via a nuclear mechanism in astrocytes.
Fig. 5.
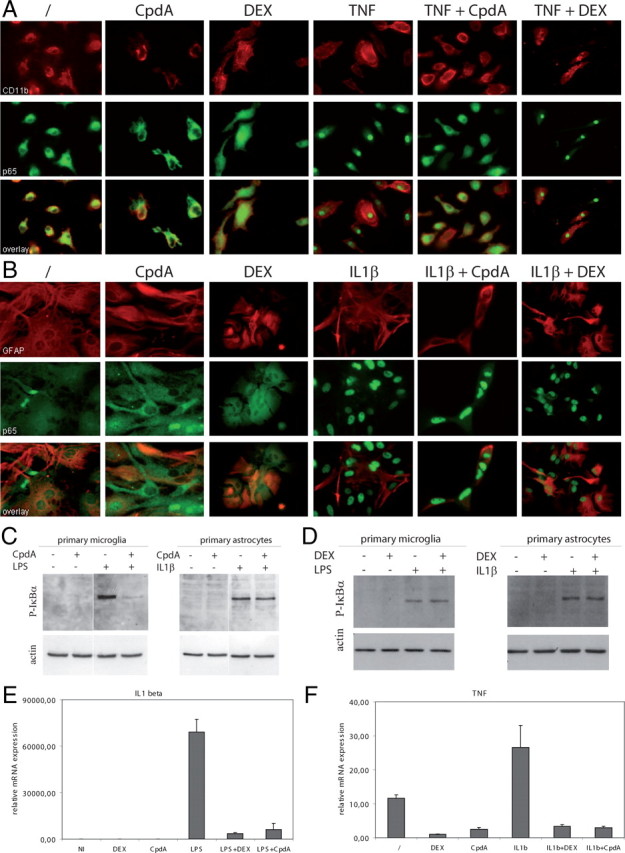
Primary microglia, but not astrocytes, show impaired IκBα phosphorylation and NF-κB nuclear translocation after CpdA treatment in vitro. A, Primary microglial cultures from C57BL/6 mice were pretreated with PBS, CpdA, or DEX for 45 min and stimulated for 20 min with TNF and stained for CD11b (red) and p65 (green). Original magnification, ×400. Images are representative of two independent experiments. B, Primary astrocyte cultures from C57BL/6 mice were pretreated with PBS, CpdA, or DEX for 45 min and stimulated for 20 min with IL-1β and stained for GFAP (red) and p65 (green). Original magnification, ×400. C, Immunoblot analysis for phosporylated IκBα expression in cytoplasmic extracts from primary microglia and astrocytes either pretreated or not pretreated with CpdA. D, Immunoblot analysis for phosporylated IκBα expression in cytoplasmic extracts from primary microglia and astrocytes either pretreated or not pretreated with DEX. E, Quantitative measurement of IL-1β mRNA expression in primary microglial cells pretreated with CpdA or DEX followed by TNF stimulation for 6 h. F, Quantitative measurement of TNF mRNA expression in primary astrocytes pretreated with CpdA or DEX followed by IL-1β stimulation for 6 h.
CpdA, in contrast to DEX, does not induce GRE-driven gene expression
GC-activated GR is able to bind onto GREs on specific gene promoters and, as such, activates target genes involved in the metabolism of sugar, protein, fat, muscle, and bone (23). CpdA acts as a selective GR modulator, with strong antiinflammatory capacities but lacking the potential to transactivate. This was confirmed by analyzing GRE-driven gene transcription on primary microglia and astrocytes in the presence of CpdA or DEX. mRNA levels of GC-induced leucine zipper (GILZ), FK506 binding protein 51 (FKBP51), and MAPK phosphatase 1 (MKP1) were strongly induced by DEX treatment both in microglia and astrocytes (Fig. 6). CpdA, however, did not induce GILZ, FKBP51, or MKP1 mRNA in both cell types, confirming its dissociated nature.
Fig. 6.

CpdA, in contrast to DEX, does not induce GRE-driven gene expression. A, Primary microglial cultures from C57BL/6 mice were treated with solvent (/), CpdA, or DEX for 24 h and analyzed by quantitative pPCR for GILZ, FKBP51, and MKP1. B, Primary astrocyte cultures from C57BL/6 mice were treated with solvent (/), CpdA, or DEX for 6 h and analyzed by quantitative pPCR for GILZ, FKBP51, and MKP1.
CpdA, in contrast to DEX, does not induce diabetogenic or HPA axis-suppressive side effects in vivo
In addition to their beneficial antiinflammatory potential, GCs have concomitant side effects, of which some have been shown to result from the direct transactivating capacities of the GR (14, 15). CpdA, able to actively support GR monomer formation, was previously shown to display an improved side effect profile in terms of the development of hyperglycemia/hyperinsulinemia (16, 17). To exclude any additional catabolic side effect of a long-term CpdA treatment during EAE, body weight loss was closely monitored from mice treated as described in Fig. 1B. Neither CpdA nor DEX treatment induced significant additional body weight losses during EAE (Fig. 7A). On the contrary, both groups show a milder body weight loss compared with the diseased PBS-treated mice because both are significantly protected from disease development after MOG-peptide immunization. At d 25 after immunization, all mice were bled and analyzed for blood glucose and serum insulin levels. Neither CpdA nor DEX induced hyperglycemia (data not shown); insulin levels, however, were strongly induced by DEX but not by CpdA in EAE mice (Fig. 7B). The absence of an effect of DEX on the regulation of blood glucose levels can be explained by the compensatory action of insulin. Similar observations were made in a previous study (16, 17). To assess a concomitant potential systemic side effect with respect to suppression of the (HPA) axis, corticosterone levels were measured after chronic treatment of the EAE mice with CpdA in comparison with DEX (Fig. 7C). As expected, DEX treatment gave rise to significantly lower levels of corticosterone as compared with PBS-treated mice. This is due to the well-known GC-induced feedback mechanisms at the central level, preventing the release of endogenous corticosterone. Chronic CpdA treatment, however, did not affect corticosterone levels as compared with PBS-treated mice, suggesting a normal physiological HPA axis regulation. Finally, liver enzyme AST and ALT levels were measured in serum to assess possible toxic side effects of CpdA and DEX during EAE. As shown in Fig. 7D, no significant AST or ALT levels were detected in serum from both CpdA- and DEX-treated animals. Taken together, these data demonstrate the absence of hyperglycemia/hyperinsulinemia and of HPA axis suppression in CpdA treatment conditions and exclude liver toxicity questions concerning this compound.
Fig. 7.

CpdA, in contrast to DEX, does not induce diabetogenic or HPA axis-suppressive side effects in vivo. EAE was induced in male C57BL/6 mice. PBS, CpdA, and DEX are administered every second day starting from d 2 onward after immunization. A, Percentage body weight loss measured during EAE. B, Serum was collected 25 d after MOG peptide stimulation and assayed for insulin by ELISA. Results are shown as mean ± sem. *, P < 0.05. C, Serum was collected 25 d after MOG peptide stimulation and assayed for corticosterone via a corticosterone immunoassay. Results are shown as mean ± sem. D, Serum was collected 25 d after MOG peptide immunization, and levels of AST and ALT enzymes were measured. Results are shown as mean ± sem.
Discussion
EAE is widely recognized as a representative animal model for MS, to study signaling pathways regulating neuroinflammation and to identify potential therapeutic targets. As in many other inflammatory diseases, the transcription factor NF-κB is detrimental in regulating inflammation in MS and EAE, both in the initiation and propagation of the inflammatory processes, making it an interesting target for therapeutic intervention. At present, GCs are still among the most commonly prescribed drugs worldwide for the treatment of autoimmune disorders, such as MS (12, 13). In addition to their beneficial antiinflammatory properties, however, GCs have unwanted side effects, of which some are mainly the consequence of their transactivating capacities. Indeed, GC-activated GR is able to bind as a homodimer onto specific promoter recognition sequences, termed “glucocorticoid response elements” (GRE), and as such activates target genes involved in the metabolism of sugar, protein, fat, muscle, and bone (23). Target genes that are negatively regulated by GR, via the transrepression or so-called tethering mechanism, include a plethora of cytokine genes. This most often involves the negative interference of, presumably monomeric, GR with the activity of other DNA-bound transcription factors, including NF-κB (15). The latter mechanism explains most of their antiinflammatory properties, although a number of recently characterized genes that are being up-regulated by GR have been proposed to also display distinct antiinflammatory roles. In this respect, the best known is the inhibitor protein of NF-κB, IκBα, but also GILZ, MKP-1, lipocortin-1, secretory leukoprotease inhibitor, type II IL-1 (decoy), annexin A1, IL-10, docking protein 1, Dexras, p11/calpactin-binding protein, and tristetraprolin, all able to inhibit various stages of cytokine signaling, synthesis, secretion, and activity, have been added to this list (24, 25). It is recognized that prominent undesired effects, such as diabetes mellitus, dyslipidemia, thymus atrophy, muscle atrophy, and glaucoma induction depend on GR-mediated activation of gene expression (23, 26), whereas other side effects have been suggested to have transrepression mechanisms, or both transactivation- and transrepression-dependent mechanistics lying at their basis, e.g. HPA axis suppression, growth retardation, and osteoporosis (27).
CpdA, a plant derivative acting as a dissociated GR ligand, was recently identified and characterized for its antiinflammatory properties in vitro and in vivo (16, 17). CpdA blocks NF-κB-driven gene expression but does not support GRE-driven gene expression. CpdA was further shown to markedly inhibit joint inflammation in a collagen-induced mouse model for rheumatoid arthritis, without inducing a concomitant hyperinsulinemia, which is unlike DEX. CpdA, in contrast to DEX, actively stimulates the formation of GR monomers, which helps to explain its dissociated behavior (17). Importantly, we recently showed in synovial fibroblasts that CpdA is less likely to evoke therapy resistance, because, in contrast to classical GCs, it does not lead to homologous GR down-regulation (28). Because the expression level of GR has recently been directly correlated to the GC-dependent therapeutic efficacy in EAE (29), the lack of receptor down-regulation with CpdA may provide an additional therapeutic advantage over classical GCs. In addition to CpdA, also other compounds have recently been characterized and reported to display a dissociated profile, not only in vitro but also in vivo. These include AL-438 (30), ZK 216348 (31, 32), and ZK 245186 (26). These results demonstrate that continuous efforts are being made to reach an improved clinical therapeutic benefit, following the strategy of a dissociated receptor activity.
In the current study, we set out to investigate whether the plant-derived dissociated GR ligand CpdA was able to suppress inflammation in EAE. We showed that the clinical symptoms of EAE were significantly relieved in mice treated with CpdA. This was accompanied by a reduced infiltration of inflammatory cells into the spinal cord, reduced expression of inflammatory cytokines and chemokines, and reduced neuronal damage and demyelination. Especially the ability of CpdA to repress clinical symptoms of EAE when administered at the peak of the disease not only demonstrates its potent antineuroinflammatory potential, but also strongly supports its usefulness for therapeutic intervention of MS and possibly other autoimmune and inflammatory diseases afflicting the CNS. Our data further imply that the antiinflammatory effects of liganded GR in EAE are most likely not mediated via GR transactivation mechanisms but are mainly supported by GR transrepression mechanisms. Two lines of evidence support this hypothesis. First, classical GRE-dependent target genes are found not to be up-regulated with CpdA upon treatment of microglia or astrocytes with DEX or CpdA (Fig. 6). Second, analysis of the same set of genes both in the DEX- and CpdA-treated groups in the EAE experiment, also did not show any up-regulatory effect with DEX (data not shown), although the cytokine repression was functional (Fig. 3). This is in line with recent findings describing that MKP-1 (DUSP1) knockout mice, which show an enhanced susceptibility to anaphylaxis, remain sensitive to GCs, implying that the antiinflammatory action of GCs occurs largely independent of the up-regulation of this GRE-driven gene (33). As expected from previous findings using a collagen-induced arthritis model, we found that CpdA also did not evoke hyperinsulinemia in the EAE model. Because we wondered also about other typical side effects including HPA axis suppression we now also determined the serum corticosterone levels. Quite surprisingly, we found that CpdA did not suppress the HPA axis, a phenomenon that depends on GR-mediated transrepression of hypothalamic CRH or pituitary ACTH and POMC genes (34). This result may imply a narrower window of transcription factor selectivity with CpdA, as compared with DEX. Low levels of liver enzymes AST and ALT indicated that a chronic CpdA treatment does not result in liver toxicity.
Activation of the adaptive immune system and the induction of myelin-specific autoreactive T cells is an obligatory requirement for the induction of EAE. The effectiveness of CpdA in suppressing characteristic signs of EAE indicates that it affects this early stage in the immunological cascade leading to EAE. Indeed, we showed that CpdA clearly represses peripheral immune responses and the generation of MOG peptide-responsive T cells, as demonstrated by strongly reduced lymphocyte proliferation and a reduced production of Th1 and Th17 cytokines upon restimulation with MOG peptide antigen. It has been proposed that also the innate immune system plays an important role in EAE, by providing a permissive cytokine microenvironment that potentiates the immune response locally within the CNS. This role has mainly been attributed to microglia, which express multiple cytokines and chemokines within the CNS (35). As a novel aspect of the inflammatory mechanism, we showed that CpdA suppresses NF-κB nuclear translocation in cultured primary microglial cells, but not in primary astrocytes. This does not yet definitely exclude an involvement of astrocytes, as it has already been recognized before that the mechanism of cytokine gene repression through activated GR may occur through different mechanisms in different cells (14). Mechanistically, this differential result was explained by a concomitant CpdA-mediated block of LPS-induced IκBα phosphorylation in microglial cells, but not of IL1β-induced IκBα phosphorylation in astrocytes. Because phosphorylation of IκBα leads to its subsequent degradation, preceding the release and nuclear translocation of NF-κB, a block in the phosphorylation of IκBα would explain the gene-repressive effect on NF-κB-dependent genes. Intriguingly, the classical GC DEX does not support a block of nuclear translocation of NF-κB in astrocytes or in microglial cells. DEX and CpdA thus seem to use a differential mechanism for blocking NF-κB-driven gene expression in microglial cells. Concerning the action mechanism of classical GCs, we have demonstrated previously that DEX uses a nuclear mechanism to block NF-κB-driven gene expression in fibroblasts (36). Depending on the cell type, various mechanisms may account for the inhibitory activity of GCs on NF-κB-driven gene expression, ranging from a loss of NF-κB from the DNA but maintaining a nuclear phenotype or a loss of NF-κB from the DNA coupled to the translocation of NF-κB out of the nucleus, to an interference mechanism between the activated GR and NF-κB whereby NF-κB remains bound onto DNA but has a hampered transactivation function (14, 15). Therefore, both in primary microglia and astrocytes, DEX most probably blocks NF-κB transactivation via a nuclear mechanism. Concerning the primary microglia, because of the CpdA-induced block in the translocation of NF-κB, it is clear that these cells are a target for CpdA-mediated gene repression, rather via a cytoplasmic mechanism. In agreement herewith, in vivo NF-κB DNA-binding activity onto the IL-6 promoter, as determined by chromatin immunoprecipitation (ChIP) analysis, was considerably impaired in spinal cord tissue after treatment with CpdA as compared with PBS-treated mice that were at the peak of the disease. These findings add on to a recent study by Reichardt and co-workers (29), who reported on peripheral T cells as crucial therapeutic targets for GCs in EAE, and suggest an additional role for microglial cells as valuable targets for GR-driven antiinflammatory effects in EAE, and possibly also MS. Although it is possible that oligodendrocytes and neurons also contribute to the endogenous proinflammatory capacity of the CNS in EAE, and also astrocytes through a completely nuclear mechanism, our results indicate that microglial inhibition of NF-κB by CpdA contributes to the protection of mice against MOG-induced inflammation and demyelination in EAE, via a cytosolic blocking step.
Materials and Methods
Mice and reagents
C57BL/6J mice were purchased from Janvier (Laval, France). DEX was purchased from Sigma-Aldrich (St. Louis, MO). CpdA (Alexis Chemical, Carlsbad, CA) was synthesized according to Louw et al. (37), lyophilized, and stored at −70 C. Mice were injected ip with PBS alone, 150 μg CpdA in PBS, or 50 μg water-soluble DEX in PBS, every second day starting either 10 d after MOG immunization or at the peak of the disease, as indicated in the figure legend.
Induction and assessment of EAE
All experiments on mice were performed according to institutional, national, and European animal regulations. Mouse MOG peptide, amino acids 35–55 (MEVGWYRSPFSRVVHLYRNGK), was synthesized by Sigma Genosys (Cambridge, UK). Male mice, 10–15 wk of age, received sc injection of 200 μg MOG peptide in 200 μl sterile PBS emulsified with an equal volume of complete Freund’s adjuvant (Sigma) containing 5 mg/ml Mycobacterium tuberculosis H37Ra (BD Biosciences, San Jose, CA). Mice also received ip 50 ng pertussis toxin (Sigma) in 200 μl sterile PBS, at the time of immunization and 48 h later. Clinical signs of disease were scored, as described before (11), on a scale of 0–6, with 0.5 points for immediate clinical findings as follows: 0 = normal; 1 = weakness of the tail; 2 = complete loss of tail tonicity; 3 = partial hind limb paralysis; 4 = complete hind limb paralysis; 5 = forelimb paralysis or moribund; 6 = death. To eliminate any diagnostic bias, mice were scored blindly.
Histological analysis
Mice were perfused with 4% paraformaldehyde via cardiac puncture of the left ventricle and were postfixed in 10% formalin. Spinal cords were dissected and embedded into paraffin blocks before sectioning and staining with hematoxylin-eosin, LFB for an assessment of the degree of demyelination or with specific antibodies Mac-3 (BD Biosciences), CD3 (Serotec, Raleigh, NC), B220 (BD Biosciences) or APP (Chemicon, Billerica, MA). Histological quantification was performed as described previously (38).
Quantitative real-time PCR
Total RNA from untreated (n =2) and CpdA-treated (n =2) mice was purified from spinal cord tissue 10 d, 16 d, or 22 d after immunization using Trizol Reagent (Invitrogen, Paisley, Scotland, UK). Total RNA (1 μg) was used to synthesize cDNA using SuperScript First-Strand Synthesis System (Invitrogen) and was resuspended in 100 μl H2O. cDNA samples (5 μl) were used for real-time PCR, in a total volume of 20 μl using SYBR Green Reagent (Roche, Indianapolis, IN) and specific primers on a LightCycler 480 (Roche). Real-time PCRs were performed in triplicate. The following mouse-specific primers were used: TNF forward, ACCCTGGTATGAGCCCATATAC; TNF reverse, ACACCCATTCCCTTCACAGAG; IL-1β forward, CACCTCACAAGCAGAGCACAAG; IL-1β reverse, GCATTAGAAACAGTCCAGCCCATAC; IFNγ forward, GCCAAGCGGCTGACTGA; IFNγ reverse, TCAGTGAAGTAAAGGTACAAGCTACAATCT: IP10 forward, GTCACATCAGCTGCTACTC; IP10 reverse, GTGGTTAAGTTCGTGCTTAC; IL6 forward, GAGGATACCACTCCCAACAGACC; IL6 reverse, AAGTGCATCATCGTTGTTCATACA; IL17 forward, GAGAGCTGCCCCTTCACTTTCA; IL17 reverse, TTGGGACCCCTTTACACCTTCTTT; IL23 forward, CCCATTAGGACTTGTGCTGTTCTT; IL-23 reverse, TTTTTCTGATGCTCTGGGTTTTCT; MCP-1 forward, GCATCTGCCCTAAGGTCTTCA; MCP-1 reverse, TGCTTGAGGTGGTTGTGGAA; monocyte inflammatory protein-1 (MIP1)-α forward, CTTGGAGGCAGCGAGGAA; MIP1α reverse, GGCAGCAAACAGCTTATAGGAGAT, FKBP51 forward, TGAGGGCACCAGTAACAATGG, FKBP51 reverse, CAACATCCCTTTGTAGTGGACAT, MKP-1 forward, GAGCTGTGCAGCAAACAGTC, MKP-1 reverse, CTTCCGAGAAGCGTGATAGG, GILZ forward, CCAGTGTGCTCCAGAAAGTGTAAG, GILZ reverse, AGAAGGCTCATTTGGCTCAATCTC. An average of the following household genes was used for normalization: Cyclophilin forward, GCA TAC GGG TCC TGG CAT CTT GTC C; cyclophilin reverse, ATG GTG ATC TTC TTG CTG GTC CTT GC; 36B4 forward, CAT GCT CAA CAT CTC CCC CTT CTC C; 36B3 reverse, GGG AAG GTG TAA TCC GTC TCC ACA G; HPRT forward, CCT AAG ATG AGC GCA AGT TGA A; HPRT reverse, CCA CAG GAC TAG AAC ACC TGC TAA.
Tissue ChIP analysis
Equal amounts of freshly isolated spinal cord tissue (three mice per setup) were homogenized and subjected to cross-linking with 1% formaldehyde for 10 min. Cross-linking was stopped by adding glycine to a final concentration of 125 mm. The tissue was washed in PBS supplemented with 0.5 mm EDTA/EGTA and subsequently snap frozen. After thawing, the fixed homogenates were resuspended in RIPA buffer (50 mm Tris-HCl, pH 8; 140 mm NaCl; 1% Triton X-100; 0.1% Na-deoxycholate; 0.2 mm EDTA; 0.2 mm NaVO3, 10 mm Na-butyrate; 20 mm β-glycerophosphate) supplemented with protease inhibitors and subjected to repeated sonication steps using a Bioruptor (Diagenode, Villers-Le-Bouillet, Belgium) for three times at 7.5 min with in-between cooling steps. The immunoprecipitation step of the ChIP assay made use of an antibody against p65, via a protocol essentially as described elsewhere (39), but in which sodium dodecyl sulfate was replaced by deoxycholate in all buffers. After reverse cross-linking steps, the DNA was subsequently purified via a Nucleospin kit (Machery-Nagel, Bethlehem, PA). Purified DNA samples, enriched by the immunoprecipitated protein, were subjected to quantitative PCR (qPCR) analysis using primers in the NF-κB-encompassing promoter region of the IL-6 promoter. The following primers were used: AATGTGGGATTTTCCCATGA and AGCGGTTTCTGGAATTGACT. The amount of sonicated protein-DNA complexes present in the reaction before immunoprecipitation is indicated by the input controls. In the qPCR-based data, the correction for input control is implemented in the graph.
T-cell recall assay
T-cell recall responses were assessed in spleen cells isolated from mice 10 d after immunization with MOG peptide and CpdA treatment starting at d 2 after immunization. After erythrocyte lysis, cells were cultured in flat-bottom 96-well plates at a density of 7 × 105 cells per well in DMEM supplemented with 5% fetal calf serum, l-glutamine, nonessential amino acids, and antibiotics. Cultures were incubated at 37 C in the presence or absence of indicated concentrations of MOG peptide. After 48 h, culture supernatants were collected, and the concentration of IFN-γ, IL-2, and IL-17 was measured by ELISA (R&D Systems, Minneapolis, MN). For analysis of cell proliferation, cultures were pulsed with 0.5 μCi of {lsqb;3H{rsqb;thymidine per well (1 mCi/ml 3HTdR; Amersham Pharmacia Biotech, Buckinghamshire, UK) during the last 18 h. Cells were harvested onto glass fiber filter membranes by using a 96-well plate cell harvester, and thymidine incorporation was measured by scintillation counting.
Primary astrocyte and microglial cultures
Primary astrocytes were prepared from 1- to 2-d-old old mice using a modification of a previously described method (40). Briefly, the brain was dissected and meningeal tissue was stripped off. Brains were mechanically dissociated with fire-polished Pasteur pipettes, and the resulting cell suspension was passed through a sterile nylon mesh (pore size 70 μm; Falcon, BD Biosciences) in DMEM. After washing by centrifugation at 200 × g for 5 min, all cells from one brain were seeded into 25 cm2 culture flasks (Falcon) in DMEM supplemented with 20% fetal calf serum and antibiotics and grown for 10–14 d at 37 C. Under these conditions, neurons do not survive and contaminating microglial cells and oligodendrocytes were removed by intensive washing and shaking of culture flasks for 18 h (37 C, 250 rpm in orbital shaker). The resulting confluent cell monolayer consisted of more than 90% astrocytes, as confirmed by staining with glial fibrillary acidic protein (GFAP)-specific antibodies. Purified astrocytes were seeded on poly-l-lysine-treated coverslips at a density of 2 × 105 cells per six-well plate, and were pretreated with either solvent, CpdA (10−5 m), or water-soluble DEX (10−6 m) for 45 min followed by treatment with murine recombinant TNF (10 ng/ml) or IL-1β (10 ng/ml) in the cell culture medium for 30 min to 1 h. For assaying GFAP and p65 nuclear translocation, cells were analyzed by immunofluorescence using antibodies to GFAP (Chemicon, Temecula, CA) and p65 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Secondary antibodies were antimouse Alexa Fluor 594 or antirabbit Alexa Fluor 488, respectively. Cytoplasmic lysates were used for assessing phosphorylation of IκBα by Western blotting using a phospho-specific anti-IκBα antibody (Cell Signaling Technology, Danvers, MA). Microglial cells were prepared as previously described (11). Briefly, cultures were prepared in the same way as for astrocytes in DMEM supplemented with fetal calf serum, antibiotics, and L929 cell supernatant. Microglial cells were seeded at a density of 2 × 105 cells per six-well plate and were either pretreated or not with CpdA (10−5 m) or water-soluble DEX (10−6 m) for 45 min followed by treatment with murine recombinant TNF (10 ng/ml) or LPS (20 μg/ml) in the cell culture medium for 20 min, before processing for staining with CD11b and p65 antibodies.
Statistics
Results are expressed as the mean ± sem. Statistical significance between experimental groups was assessed using an unpaired two-sample Student’s t test.
Acknowledgments
We thank Carine Van Laere and Debbie Roels for animal care and the Laboratory of Clinical Biology of the University Hospital Ghent for ALT and AST measurements.
NURSA Molecule Pages:
Ligands: Dexamethasone;
Nuclear Receptors: GR.
Footnotes
G.v.L. and K.D.B. are postdoctoral researchers, and C.M. is a predoctoral researcher with the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (FWO-Vlaanderen). G.v.L. is also supported by a Marie Curie Reintegration Grant from the Sixth European Union Framework Program (FP6-ERG-2005-031063), an FWO Odysseus Grant, and grants from the Charcot Foundation. Research in the corresponding authors’ laboratories is further supported by grants from the Interuniversitaire Attractiepolen (IAP6/18), the FWO-Vlaanderen (Grant 3G010505), the Foundation against Cancer, and the Geconcerteerde Onderzoeksacties (GOA Grant 01G06B6) of the Ghent University.
Disclosure Summary: The authors have nothing to disclose.
First Published Online December 4, 2009
M.S. and N.B. contributed equally to this manuscript.
G.H., M.P., and R.B. contributed equally to this manuscript.
Abbreviations: ALT, Alanine transaminase; APP, amyloid precursor protein; AST, aspartate transaminase; ChIP, chromatin immunoprecipitation; CNS, central nervous system; CpdA, Compound A; DEX, dexamethasone; EAE, experimental autoimmune encephalomyelitis; FKBP51, FK506 binding protein 1; GC, glucocorticoid; GFAP, glial fibrillary acidic protein; GILZ, GC-induced leucine zipper; GR, glucocorticoid receptor; HPA, hypothalamic-pituitary-adrenal; IFN, interferon; LFB, luxol fast blue; LPS, lipopolysaccharide; MIP1, monocyte inflammatory protein-1; MOG, myelin oligodendrocyte glycoprotein; MKP1, MAPK phosphatase 1; MS, multiple sclerosis; NF-κB, nuclear factor-κB; qPCR, quantitative PCR.
References
- 1.Hemmer B, Archelos JJ, Hartung HP2002. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 3:291–301 [DOI] [PubMed] [Google Scholar]
- 2.McFarland HF, Martin R2007. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 8:913–919 [DOI] [PubMed] [Google Scholar]
- 3.Steinman L1996. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85:299–302 [DOI] [PubMed] [Google Scholar]
- 4.Baxter AG2007. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol 7:904–912 [DOI] [PubMed] [Google Scholar]
- 5.Farina C, Aloisi F, Meinl E2007. Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145 [DOI] [PubMed] [Google Scholar]
- 6.Mattson MP, Camandola S2001. NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107:247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mattson MP, Meffert MK2006. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ 13:852–860 [DOI] [PubMed] [Google Scholar]
- 8.Greve B, Weissert R, Hamdi N, Bettelli E, Sobel RA, Coyle A, Kuchroo VK, Rajewsky K, Schmidt-Supprian M2007. IκB kinase 2/β deficiency controls expansion of autoreactive T cells and suppresses experimental autoimmune encephalomyelitis. J Immunol 179:179–185 [DOI] [PubMed] [Google Scholar]
- 9.Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y1999. Experimental autoimmune encephalomyelitis in NF-κ B-deficient mice: roles of NF-κ B in the activation and differentiation of autoreactive T cells. J Immunol 163:2937–2943 [PubMed] [Google Scholar]
- 10.Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K2004. Antineuroinflammatory effect of NF-κB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J Immunol 173:1344–1354 [DOI] [PubMed] [Google Scholar]
- 11.van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, Lassmann H, Prinz MR, Pasparakis M2006. Inhibition of transcription factor NF-κB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol 7:954–961 [DOI] [PubMed] [Google Scholar]
- 12.Tischner D, Reichardt HM2007. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol 275:62–70 [DOI] [PubMed] [Google Scholar]
- 13.Reichardt HM, Gold R, Lühder F2006. Glucocorticoids in multiple sclerosis and experimental autoimmune encephalomyelitis. Expert Rev Neurother 6:1657–1670 [DOI] [PubMed] [Google Scholar]
- 14.De Bosscher K, Vanden Berghe W, Haegeman G2003. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24:488–522 [DOI] [PubMed] [Google Scholar]
- 15.De Bosscher K, Haegeman G2009. Minireview: latest perspectives on anti-inflammatory actions of glucocorticoids. Mol Endocrinol 23:281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G2005. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci USA 102:15827–15832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewint P, Gossye V, De Bosscher K, Vanden Berghe W, Van Beneden K, Deforce D, Van Calenbergh S, Müller-Ladner U, Vander Cruyssen B, Verbruggen G, Haegeman G, Elewaut D2008. A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol 180:2608–2615 [DOI] [PubMed] [Google Scholar]
- 18.Yemelyanov A, Czwornog J, Gera L, Joshi S, Chatterton Jr RT, Budunova I2008. Novel steroid receptor phyto-modulator compound a inhibits growth and survival of prostate cancer cells. Cancer Res 68:4763–4773 [DOI] [PubMed] [Google Scholar]
- 19.Tsutsui S, Vergote D, Shariat N, Warren K, Ferguson SS, Power C2008. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and β-arrestin-1 in monocytoid cells. FASEB J 22:786–796 [DOI] [PubMed] [Google Scholar]
- 20.Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J, Becher B, Aguzzi A2005. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11:146–152 [DOI] [PubMed] [Google Scholar]
- 21.De Keyser J, Zeinstra E, Frohman E2003. Are astrocytes central players in the pathophysiology of multiple sclerosis? Arch Neurol 60:132–136 [DOI] [PubMed] [Google Scholar]
- 22.Gimenez MA, Sim JE, Russell JH2004. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol 151:116–125 [DOI] [PubMed] [Google Scholar]
- 23.Schäcke H, Döcke WD, Asadullah K2002. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43 [DOI] [PubMed] [Google Scholar]
- 24.Newton R, Holden NS2007. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol 72:799–809 [DOI] [PubMed] [Google Scholar]
- 25.Clark AR2007. Anti-inflammatory functions of glucocorticoid- induced genes. Mol Cell Endocrinol 275:79–97 [DOI] [PubMed] [Google Scholar]
- 26.Schacke H, Zollner TM, Docke WD, Rehwinkel H, Jaroch S, Skuballa W, Neuhaus R, May E, Zugel U, Asadullah K2009. Characterization of ZK 245186, a novel, selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br J Pharmacol 158:1088–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Bosscher K, Van Craenenbroeck K, Meijer OC, Haegeman G2008. Selective transrepression versus transactivation mechanisms by glucocorticoid receptor modulators in stress and immune systems. Eur J Pharmacol 583:290–302 [DOI] [PubMed] [Google Scholar]
- 28.Gossye V, Elewaut D, Van Beneden K, Dewint P, Haegeman G, De Bosscher K 9 Feb 2009. A plant-derived glucocorticoid receptor modulator attenuated inflammation without provoking ligand-induced resistance. Ann Rheum Dis 10.1136/ard.2008.102871 [DOI] [PubMed]
- 29.Wüst S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, Lühder F, Reichardt HM2008. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol 180:8434–8443 [DOI] [PubMed] [Google Scholar]
- 30.Coghlan MJ, Jacobson PB, Lane B, Nakane M, Lin CW, Elmore SW, Kym PR, Luly JR, Carter GW, Turner R, Tyree CM, Hu J, Elgort M, Rosen J, Miner JN2003. A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol Endocrinol 17:860–869 [DOI] [PubMed] [Google Scholar]
- 31.Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K2004. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA 101:227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.López FJ, Ardecky RJ, Bebo B, Benbatoul K, De Grandpre L, Liu S, Leibowitz MD, Marschke K, Rosen J, Rungta D, Viveros HO, Yen WC, Zhi L, Negro-Vilar A, Miner JN2008. LGD-5552, an antiinflammatory glucocorticoid receptor ligand with reduced side effects, in vivo Endocrinology 149:2080–2089 [DOI] [PubMed] [Google Scholar]
- 33.Maier JV, Brema S, Tuckermann J, Herzer U, Klein M, Stassen M, Moorthy A, Cato AC2007. Dual specificity phosphatase 1 knockout mice show enhanced susceptibility to anaphylaxis but are sensitive to glucocorticoids. Mol Endocrinol 21:2663–2671 [DOI] [PubMed] [Google Scholar]
- 34.Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, Lacroix A, Batista D, Stratakis C, Hanson J, Meij B, Drouin J2006. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev 20:2871–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanisch UK2002. Microglia as a source and target of cytokines. Glia 40:140–155 [DOI] [PubMed] [Google Scholar]
- 36.De Bosscher K, Schmitz ML, Vanden Berghe W, Plaisance S, Fiers W, Haegeman G1997. Glucocorticoid-mediated repression of nuclear factor-κB-dependent transcription involves direct interference with transactivation. Proc Natl Acad Sci USA 94:13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louw A, Swart P, de Kock SS, van der Merwe KJ1997. Mechanism for the stabilization in vivo of the aziridine precursor –(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride by serum proteins. Biochem Pharmacol 53:189–197 [DOI] [PubMed] [Google Scholar]
- 38.Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, Rochford CD, Brück W, Becher B2006. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest 116:456–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clayton AL, Rose S, Barratt MJ, Mahadevan LC2000. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J 19:3714–3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarthy KD, de Vellis J1980. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902 [DOI] [PMC free article] [PubMed] [Google Scholar]